

Abstract
Enhanced permeation and retention (EPR) effect, the mechanism by which nanotherapeutics accumulate in tumors, varies in patients based on differences in the tumor and organ microenvironment. Surrogate biomarkers for the EPR effect will aid in selecting patients who will accumulate higher amounts of nanotherapeutics and show better therapeutic efficacy. Our data suggest that the differences in the vascular permeability and pegylated liposomal doxorubicin (PLD) accumulation are tumor type as well as organ-specific and significantly correlated with the relative ratio of MMP-9 to TIMP-1 in the circulation, supporting development of these molecules as biomarkers for the personalization of nanoparticle-based therapy.
Keywords: Nanotherapeutics, EPR effect, PLD, Biomarker, Transport oncophysics
1. Introduction
Rapid advances in nanotechnology have enabled the incorporation of chemotherapeutics into nanoparticles to deliver large quantities of payload at target sites [1,2]. These nanotherapeutics include liposomes, albumin-based particles, polymer-drug composites, polymeric micelles, and dendrimers. Pegylated liposomal doxorubicin (PLD) is one of the most widely used nanotherapeutics approved for the treatment of various tumor types [3]. Although nanotherapeutics can be advantageous over treatment with free/unencapsulated drug/chemotherapeutics because of the preferential accumulation of nanoparticles in tumors due to the enhanced permeation and retention (EPR) effect, the relative benefit of this effect can vary from patient to patient based differences in the tumor and organ microenvironment [4-6]. Thus, the accumulation of nanotherapeutics in the tumor can be heterogeneous, and indeed, the increase in overall survival in patients treated with PLD has been modest [7-10]. For the most part, researchers have mainly focused on the molecular mechanisms of resistance to therapy in cancer cells and have overlooked that the accumulation of therapeutics in tumor sites may also determine therapeutic efficacy [11-13].
Personalized cancer therapy based on the levels of biomarkers, such as target proteins and specific genetic mutations in tumors, have recently emerged [14-16]. The assessment of these markers facilitates the pre-selection of patients who are likely to respond to the particular therapies. We hypothesized that the therapeutic efficacy of nanotherapeutics, including PLD, may also be improved by identifying molecules that could potentially serve as surrogate biomarkers to select patients who are likely to accumulate higher amounts of PLD in their tumors and thereby achieve a better therapeutic outcome. To the best of our knowledge, promising biomarkers for the personalization of nanoparticle-based therapy have not yet been identified.
The goal of the present study was to identify candidate serum markers that can be correlated with the EPR effect for the accumulation of PLD in various tumor and organ microenvironments to personalize cancer nanotherapy. For this purpose, we used 4T1 (breast cancer), 3LL (lung cancer), and CT26 (colon cancer) mouse tumor cells growing in the mammary fat pad (mfp), under the skin, or at the tumor’s major metastatic sites, such as the brain or liver. We found that the differences in PLD accumulation in the tumors were statistically correlated with the relative ratio of Matrix metalloproteinase (MMP)-9 and its endogenous inhibitor, tissue inhibitor of metalloproteinase (TIMP)-1 in systemic circulation. To the best of our knowledge, this is the first study that describes candidate serum biomarkers to assess the vascular permeability to nanotherapeutics utilizing the EPR effect in tumors.
2. Materials and methods
2.1. Cell culture and in vitro cytotoxicity assay
4T1 murine breast cancer cells, 3LL murine lung cancer cells, and CT26 murine colon cancer cells were kindly provided by Dr. Isaiah J. Fidler (University of Texas MD Anderson Cancer Center, Houston, TX) [17,18]. The cells were maintained in complete minimal essential medium supplemented with 10% fetal bovine serum (FBS), sodium pyruvate, nonessential amino acids, L-glutamine, vitamin solution (Life Technologies, Inc., Grand Island, NY), and a penicillin–streptomycin mixture (Flow Laboratories, Rockville, MD). The cells were validated by short tandem repeat DNA fingerprinting using the AmpFℓSTR Identifier kit (Applied Biosystems, Carlsbad, CA) [19]. For cytotoxicity assay in vitro, 4T1 or 3LL cells were seeded into 96-well plates and the cultures were fed with new medium (negative control) or medium containing different concentrations of PLD. After 3 days of the incubation, the number of metabolically active cells was determined by the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide) assay.
2.2. Mice
Female BALB/C, C57/BL6 and nude/nude mice were purchased from the Animal Production Area of the National Cancer Institute, Frederick Cancer Research Facility (Frederick, MD). The animal facilities at the University of Texas MD Anderson Cancer Center were approved by the American Association for Accreditation of Laboratory Animal Care and met all current regulations and standards of the United States Department of Agriculture, United States Department of Health and Human Services, and the National Institutes of Health. The 8–12 weeks old mice were used in accordance with institutional guidelines.
2.3. Establishment of experimental brain metastases
BALB/C and C57/BL6 mice were anesthetized with an intra-peritoneal (i.p.) injection of pentobarbital sodium. 4T1, 3LL or CT26 cells were injected slowly (1 × 105 cells/50 μl) into the right common carotid artery [17].
2.4. Establishment of experimental liver metastases, subcutaneous tumors, and primary breast tumors
BALB/C and C57/BL6 mice were anesthetized by inhaling isoflurane, and a small left abdominal flank incision was made. Then a tumor cell suspension (4T1, 3LL or CT26, 1 × 105 cells/50 μl) was injected into the spleen [20]. To generate primary breast tumors, 4T1 cells (1 × 105 cells/50 μl) were injected into the mammary fat pad (mfp) of BALB/C mice. To generate subcutaneous 3LL and 4T1 tumors, 1 × 105 -cells in 50 μl were injected into the subcutaneous space of nude/nude mice.
2.5. Therapy, blood sample collection, and necropsy
Ten days after the inoculation of cells, the mice were intravenously (i.v.) injected every 7 days with phosphate buffered saline (PBS) (control) or 6 mg/kg PLD (Doxoves™-Liposomal Doxorubicin HCl, FormuMax Scientific Inc., Palo Alto, CA), a total of 3 times (n = 7, each). Batimastat (EMD Millipore, Billerica, MA), a potent broad-spectrum inhibitor of MMPs, was resuspended with PBS to a concentration of 2.5 mg/ml [21]. The mice bearing 4T1 tumors in the mfp were i.p. injected once a day with batimastat (50 mg/kg) or diluent (control) for 3 days (n = 6, each). Six hours after the final injection, these mice were i.v. injected with PLD or PBS (control) and sacrificed 24 h later. Blood samples were collected from the tail vein (100 μl) before the injection of PLD. For survival experiments, the tumor bearing mice were sacrificed when they became moribund. For therapy experiments, the mice were sacrificed 24 h, 2 or 5 days after the i.v. injection of PLD.
2.6. Protein array analysis and measurement of MMP-9 and TIMP-1 protein levels
A Proteome Profiler™ Array Mouse Angiogenesis Array Kit (R&D Systems, Minneapolis, MN) or ELISA kits (R&D Systems) were used according to the manufacturer’s instructions to analyze the protein expression profile of the cells in vitro or measure the MMP-9 and TIMP-1 levels in the serum samples of the tumor bearing mice, respectively.
2.7. Immunohistochemical analysis to detect MMP-9, TIMP-1 and Vascular Endothelial cell Growth Factor (VEGF) in tumors and organs
Paraffin-embedded tumor sections were deparaffinized, and endogenous peroxidase was blocked with 3% hydrogen peroxide. Samples were incubated with an antibody to MMP-9 (EMD Millipore, Billerica, MA), TIMP-1 (R&D Systems) or VEGF (Santa-Cruz Biotechnology, Inc., Dallas, TX). After incubation with a peroxidase-conjugated secondary antibody (Jackson Immunoresearch, West Grove, PA), protein-antibody complexes were detected by exposure to 3,3′-Diaminobenzidine (Sigma–Aldrich Corp. St. Louis, MO).
2.8. Immunofluorescent imaging of endothelial cells (CD31), basement membrane (type IV collagen), proliferating cells (Ki67), p-glycoprotein (p-GP), macrophages (CD204), and tumor tissue perfusion
The frozen sections of the tumor tissue were immunofluorescently stained using antibodies to CD31 (BD Biosciences, San Jose, CA), type IV collagen (Abcam, Cambridge, MA), Ki67 (Abcam), p-GP (GeneTex Inc., Irvine, CA) or CD204 (AbD Serotec, Raleigh, NC). Sections were then incubated with corresponding secondary antibodies (Jackson Immunoresearch). The area of tumor tissue perfused by blood was evaluated by imaging of a lysine-fixable 70 kDa fluorescein dextran tracer (Molecular Probes, Inc. Eugene, OR) 1 min after i.v. injection. The images were captured using a laser scanning confocal microscope (Carl Zeiss MicroImaging Inc., Thornwood, NY) and analyzed using the built-in image analysis software [19]. The ratio of pixels in the whole image that has higher fluorescence intensity over the threshold (background) was shown as area fraction [22,23]. The data were shown as the average ± SD from representative sections of more than 5 images of tumors or uninvolved organs. The coverage of endothelial cells was expressed as the fractional area of endothelial cells (pseudo color in red) co-localized with basement membrane (pseudo color in green), which is indicated by the emission of yellow fluorescence, relative to the total area of endothelial cells in five randomly selected tumors.
2.9. Immunofluorescence imaging of PLD in tumors
The red fluorescence of anthracyclines enables direct visualization of doxorubicin in tissue by using confocal laser scanning microscopy. The excitation wavelength was set to 488 nm, and the doxorubicin emission was collected using a 590 nm filter [24,25].
2.10. Ex vivo whole tumor imaging
Fluorescence imaging of accumulated doxorubicin in the excised tumors was acquired and quantified using DsRed fluorescence filter in IVIS-100/Spectrum optical imaging system and the Living Image 3.1 software (Xenogen/Caliper, Mountain View, CA) [26].
2.11. Intravital microscopy (IVM) imaging of tumor vascular permeability
IVM imaging of the 4T1 tumors growing in the liver or mfp was performed while live mice were anesthetized using isoflurane. Mice received an i.v. injection of 3 kDa and 40 kDa fluorescent dextran tracers (Life Technologies, Grand Island, NY) to delineate the tumor vasculature and vascular permeability using a Nikon A1R multiphoton microscope platform (Nikon, Melville, NY) [27,28].
2.12. Statistical analysis
A Mann–Whitney U test was used to analyze the statistical differences in PLD accumulation, CD31, or Ki67 expression, MMP-9 and TIMP-1 concentrations, and the extent of endothelial cells coverage by type IV collagen in the 4T1, 3LL, and CT26 tumor models. The difference in survival was analyzed using the Kaplan–Meier method and compared using the Log-rank test. A p-value of <0.05 was considered statistically significant. A linear regression analysis was performed to evaluate the correlation between PLD accumulation and serum MMP-9/TIMP-1 ratio.
3. Results
3.1. PLD accumulation in tumors and its therapeutic efficacy
The mice bearing 4T1 or 3LL brain metastases were i.v. injected with PLD and sacrificed 24 h later to evaluate PLD accumulation in the tumors (Fig. 1A). 4T1 tumors accumulated significantly more PLD than the 3LL tumors (Fig. 1B). Both 4T1 and 3LL cells showed a similar sensitivity to PLD in vitro, as evaluated by an MTT assay (Supplementary Fig. S1A). Nevertheless, tumor-specific differences in PLD treatment efficacy were observed in vivo (Fig. 1C). The survival of the BALB/C mice bearing 4T1 brain tumors was significantly extended by treatment with PLD, whereas there was no significant improvement in the survival of C57/BL6 mice bearing 3LL brain tumors compared to the PBS control. This same trend was observed when 4T1 and 3LL tumors were grown subcutaneously in nude/nude mice (Fig. 1D). The ex vivo imaging of subcutaneous tumors with a Xenogen IVIS system confirmed a high quantity of PLD accumulation in the 4T1 tumors, but not in the 3LL tumors (Supplementary Fig. S1B). Taken together, these data suggest that the therapeutic efficacy of PLD depends on PLD accumulation.
Fig. 1.
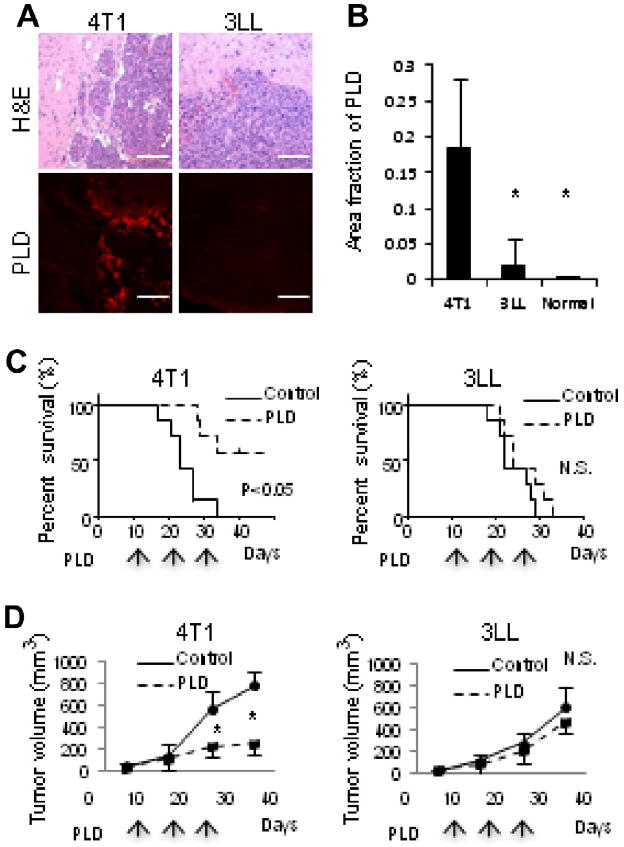
PLD accumulation and therapeutic efficacy. (A) Hematoxylin and eosin (H&E) staining and fluorescence imaging of accumulated PLD in 4T1 and 3LL tumors grown in brain. Bar indicates 100 μm. (B) Quantification of PLD accumulation in tumors. * Indicates p < 0.05 versus 4T1. The therapeutic efficacy of PLD was assessed by analyzing the survival of mice bearing tumors in the brain (C), or by measuring subcutaneous tumor growth (D). The mice were treated three times with PLD or PBS (control) at the times indicated by arrows. N.S. indicates no significant difference in the survival or tumor size. * Indicates p < 0.05 versus control.
3.2. Immunofluorescence analysis of the 4T1 and 3LL tumors
To explore the mechanisms that account for differential PLD accumulation in 4T1 and 3LL tumors, the mice were sacrificed 24 h after PLD injection and the tissue sections were stained for vascular endothelial cells using an antibody to CD31 (Fig. 2A). The amount of endothelial cells did not differ significantly between tumors (Fig. 2B). The tissue perfusion by blood was assessed by i.v. injecting fluorescein-dextran into live mice for 1 min prior to sacrifice. No significant differences in the perfusion area were observed (Supplementary Fig. S2A). Conversely, the accumulation of PLD and extravasation from vessels was significantly greater in 4T1 tumors than in 3LL tumors (Fig. 2C). The same trend was observed 2 and 5 days after the injection of PLD (Fig. 2D). The proliferation of tumor cells, evaluated by immunostaining with a Ki-67 antibody, was inhibited in areas of PLD extravasation (Fig. 2C). The quantification of Ki-67 staining revealed that cell proliferation was only significantly inhibited in the 4T1 tumors after PLD treatment (Fig. 2E). Furthermore, the expression of p-glycoprotein, which is associated with the efflux pump for various chemotherapeutics, including doxorubicin, was not significantly different between the tumors (Supplementary Fig. S2B). These data demonstrate that 4T1 and 3LL tumor-specific differences in PLD accumulation are the result of differences in the EPR effect in the tumor.
Fig. 2.
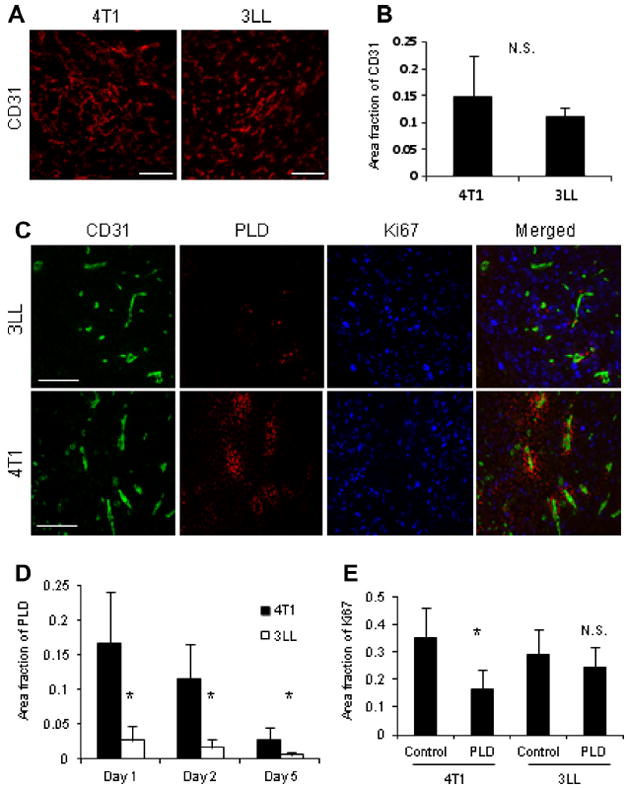
Immunofluorescence analysis of the 4T1 and 3LL tumors. (A) The amount of endothelial cells in brain tumors was evaluated by immunofluorescent analysis using a CD31 antibody. Bar indicates 100 μm. (B) Image quantification yielded no significant (N.S.) differences in the relative amount of endothelial cells. (C) A higher quantity of PLD extravasation was observed from vessels in 4T1 tumors than 3LL tumors. Bar shows 50 μm. (D) Amount of accumulated PLD to 4T1 or 3LL tumors as a function of time (* indicates p < 0.05 versus 4T1). (E) Cell proliferation, as evaluated by immunostaining with a Ki67 antibody, was significantly inhibited by PLD in 4T1 tumors (* Indicates p < 0.05 versus control), but not in 3LL tumors (N.S., no significant).
3.3. Coverage of endothelial cells by basement membrane
To identify regulators of the EPR effect, we evaluated the expression of basement membrane proteins in the tumors using an antibody to type IV collagen, a major constituent of the basement membrane (Fig. 3A). The endothelial cells were considered to be tightly covered by the basement membrane when the fluorescence of the basement membrane and endothelial cells colocalized. The percentage of tight coverage was significantly higher in 3LL tumor vessels compared to those in 4T1 tumors (Fig. 3B). Indeed, PLD extravasated only from vessels that were not tightly covered by the basement membrane (Fig. 3C).
Fig. 3.
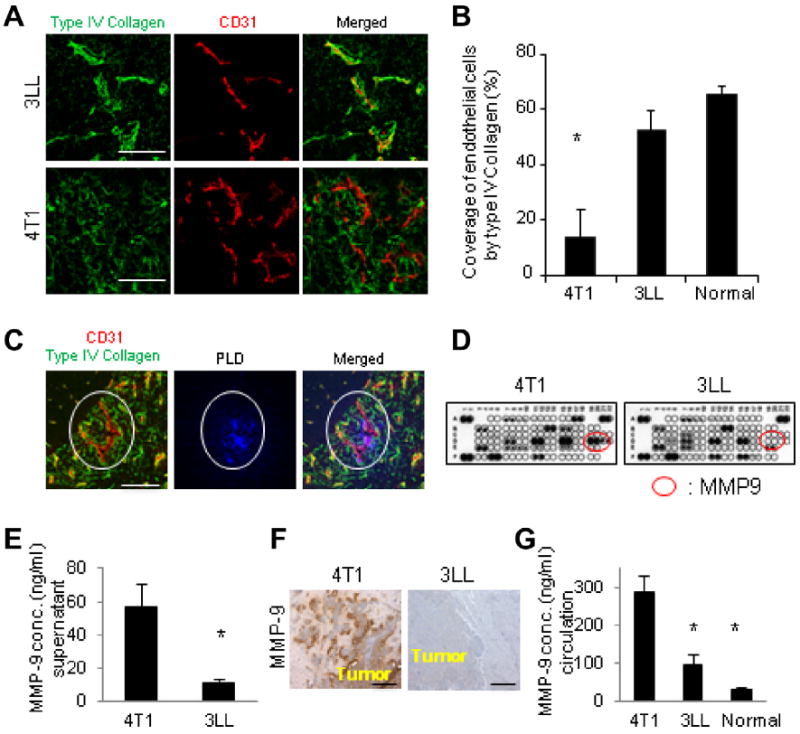
Coverage of endothelial cells by the basement membrane and differential expression of MMP-9 by 4T1 and 3LL tumors in vitro and in vivo. (A) Immunofluorescence analysis of 4T1 and 3LL tumors in brain using antibodies to type IV collagen and CD31. (B) The coverage of endothelial cells by type IV collagen was quantified. * Indicates p < 0.05 versus 3LL tumors and normal tissue. (C) PLD extravasation was observed from only vessel that was not tightly covered by basement membrane (indicated by white ovals). Bar indicates 30 μm in A and 50 μm in C. (D) Protein expression analysis of 4T1 and 3LL cells in vitro was performed using a cytokine antibody array. Secretion of MMP-9 produced by 4T1 cells was higher than that secreted by 3LL cells (indicated by red ovals). (E) MMP-9 concentrations (conc.) in culture supernatant measured by ELISA. (F) Immunohistochemical analysis of MMP-9 expression in 4T1 and 3LL brain tumors. Bar indicates 100 μm. (G) Circulating MMP-9 concentrations in the mice bearing 4T1, 3LL brain metastases, or in normal mice, measured by ELISA. * Indicates p < 0.05 versus 4T1.
3.4. Differential expression of MMP-9 in 4T1 and 3LL cells growing in vitro and in vivo
To elucidate the mechanisms that determine the coverage of endothelial cells by the basement membrane, we analyzed the proteins secreted by 4T1 and 3LL cells in vitro using a protein antibody array. Among the 53 proteins evaluated, endothelin-1, pentraxin-3, PIGF-1, CCN3 and MMP-9 were found to be produced at higher levels by 4T1 cells than 3LL cells (Fig. 3D). A growing body of evidence suggests that MMP-9, which can degrade type IV collagen, is associated with the disruption of the blood–brain barrier (BBB) following ischemic brain injury [29,30]. Thus, we proposed that the vascular permeability could be determined not only by gaps between endothelial cells but also by the amount of collagen in the basement membrane [31]. The significant difference in MMP-9 expression was independently validated by ELISA (Fig. 3E). Similarly, the MMP-9 expression was higher in 4T1 tumors than in 3LL tumors growing in the brain and the uninvolved brain in mice (Fig. 3F). Interestingly, the serum levels of MMP-9 were found to be highest in the mice bearing 4T1 brain tumors (Fig. 3G). These data suggest that the serum MMP-9 level could serve as a surrogate biomarker to indicate the vascular permeability to PLD. Conversely, the expression level of VEGF in 4T1 cells was not higher than that in 3LL cells in vitro and in vivo (Supplementary Fig. S3A and B).
3.5. MMP-9 and TIMP-1 modulate the vascular permeability to PLD in 4T1 tumors
To confirm whether MMP-9 regulates the vascular permeability to PLD, batimastat, an inhibitor of MMPs, or control PBS were injected i.p. into mice bearing 4T1 tumors in the mfp. The accumulation of PLD in 4T1 tumor was then imaged using the IVIS apparatus (Fig. 4A). The accumulation of PLD was significantly inhibited by pre-treating mice with batimastat (Fig. 4B). A confocal microscopic analysis of the tumor revealed that batimastat significantly decreased the accumulation of PLD in the tumors (Fig. 4C) by increasing the amount of type IV collagen (Fig. 4D) and the fraction of endothelial cells tightly covered by the basement membrane (Fig. 4E). Although the MMP-9 levels in circulation were not affected by batimastat (Fig. 4F), the levels of its endogenous inhibitor TIMP-1 were significantly increased (Fig. 4G). These data confirmed that MMP-9 and TIMP-1 play a pivotal role in controlling the vascular permeability to PLD.
Fig. 4.
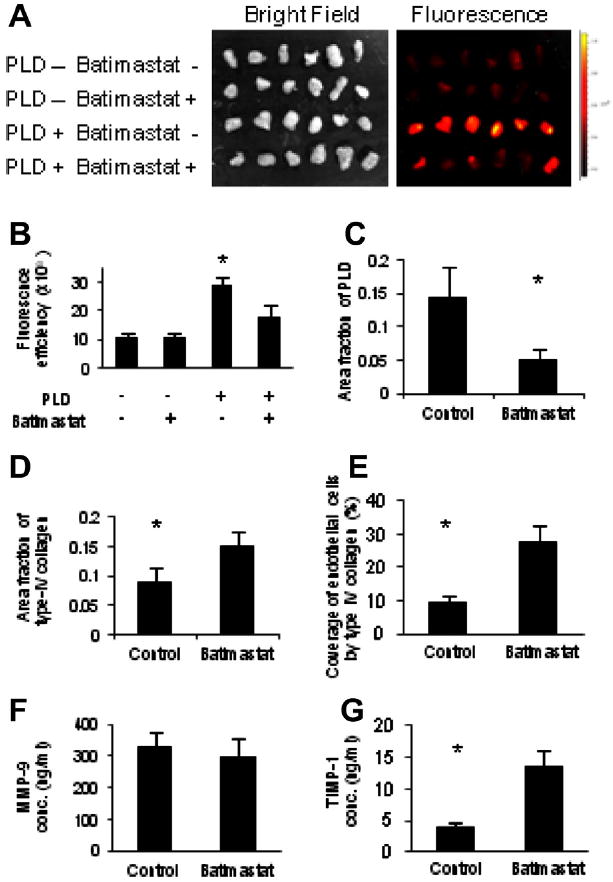
Reduction of vascular permeability to PLD by MMP inhibitor. Batimastat or PBS was injected i.p. once a day for 3 days into the mice bearing 4T1 tumor, prior to i.v. injection of PLD. (A) Accumulation of PLD to the tumors was imaged ex vivo using an IVIS apparatus. (B) Accumulation of PLD to the tumors was abrogated by pre-treatment of mice with Batimastat. * Indicates p < 0.05 versus the other treatments. Confocal microscopic imaging of the tumors and image quantification of PLD accumulation. (C) The amount of type IV collagen. (D) And the coverage of endothelial cells by basement membrane (E). (F) Measurement of serum protein levels by ELISA yielded no significant differences in MMP-9 concentration (conc.). (G) However, a significant increase in TIMP-1 concentration was observed. * Indicates p < 0.05 versus batimastat.
3.6. Organ-dependent differences in the vascular permeability to PLD
To explore the influence of the host organ microenvironment on the EPR effect in tumors, the differential PLD accumulation in 4T1 tumors growing in 3 different tumor-organ sites (brain, mfp or liver) was evaluated using confocal microscopy (Fig. 5A). As the tumor microenvironment produces an array of factors, the accumulation, differentiation and polarization of monocytes into so-called M2 macrophages inside solid tumors are frequently observed in a variety of cancers. Therefore, the infiltration of CD204+ (M2 marker) macrophages to tumor was used as a reference to identify tumors [32]. Although 4T1 tumors in the brain and mfp were accumulated with PLD, there was almost no accumulation inside the lesions of the liver metastases. Accumulation of PLD was found in numerous CD204-positive macrophages (Kupffer cells) surrounding the tumors in the liver (Fig. 5A and B). The fraction of endothelial cells tightly covered by the basement membrane was significantly higher in the 4T1 tumors growing in the liver compared to tumors in the brain or mfp (Fig. 5C, Supplementary Fig. S4). Intravital microscopy was performed to evaluate the differential transport of a fluorescence tracer injected i.v. into 4T1 tumors in the mfp and liver. Both the 3 kDa and 40 kDa dextran tracers accumulated in the mfp tumors; however, only the 3 kDa tracer accumulated in the liver tumors (Fig. 5D and E). Taken together, these data reveal that the vascular permeability in tumors is organ-specific.
Fig. 5.

Organ-specific differences in vascular permeability to PLD. The accumulation of i.v. injected PLD to the tumors was (A) imaged and (B) quantified. Bar indicates 100 μm. * Indicates p < 0.05 versus brain and mfp. 4T1 tumors growing in brain, mfp, or liver were identified by accumulation of CD204-positive tumor-associated macrophages into the tumors. Macrophages in the liver (Kupffer cells) were also positively stained with a CD204 antibody. (C) The percentage of endothelial cells tightly covered by basement membrane in the tumors growing in the different organs. * Indicates p < 0.05 versus liver. (D) Intravital microscopy of the 4T1 tumors growing in mfp or liver probed with fluorescent dextran tracers of two sizes injected i.v. (Molecular Weight (MW) = 3000 Da (red) and =40,000 Da (green)). (E) Quantification of the fluorescent intensities of the tracers indicated by the white lines in D. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.7. MMP-9 and TIMP-1 as surrogate biomarkers to indicate the vascular permeability to PLD
Because we established that high levels of serum MMP-9 were associated with increased vascular permeability, we measured the levels of circulating MMP-9 in mice bearing 4T1 tumors growing in the brain, mfp, or liver. Interestingly, the MMP-9 levels did not differ significantly among the tumor-bearing mice (Fig. 6A). An immunohistochemical analysis with a MMP-9 antibody showed that MMP-9 was strongly expressed by 4T1 cells, regardless of organ site. In contrast, the levels of TIMP-1 were highest in the mice bearing liver metastases (Fig. 6B). An immunohistochemical analysis using a TIMP-1 antibody showed TIMP-1 expression in the uninvolved tissue surrounding liver metastases. No TIMP-1 expression was detected in the normal liver (Supplementary Fig. S5A). Thus, the vascular permeability in different organ microenvironments could be determined not only by high levels of MMP-9 but also by the balance between the levels of MMP-9 and TIMP-1.
Fig. 6.
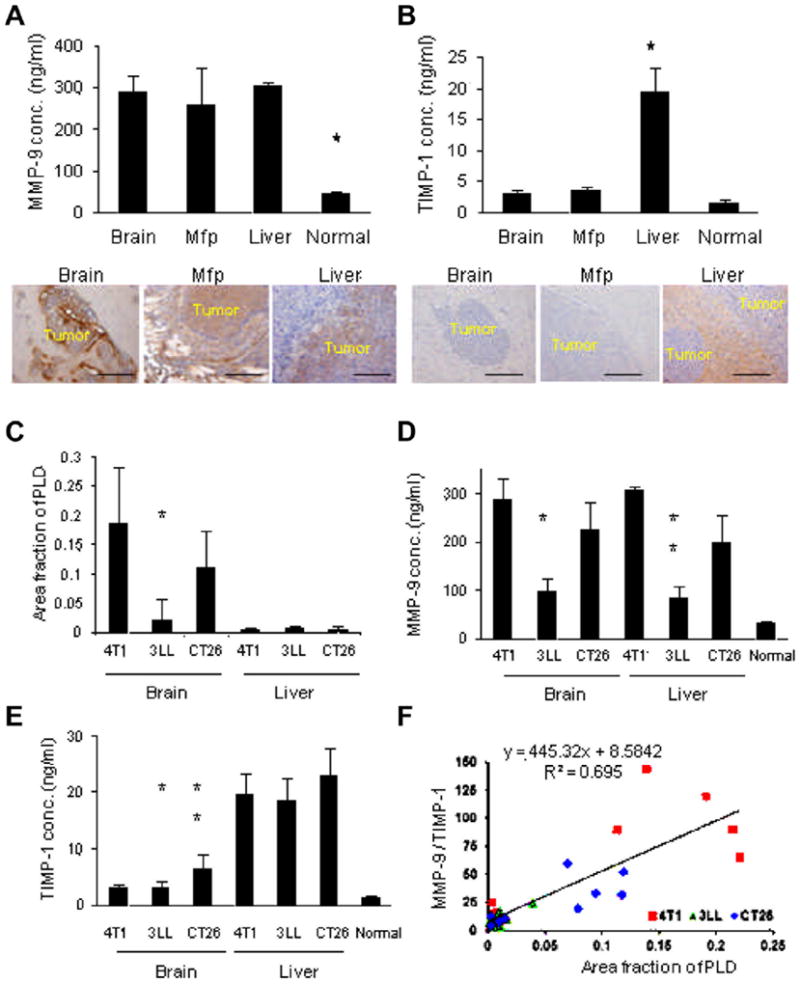
Organ specific differences in MMP-9 and TIMP-1concentration. (A) Serum concentration (conc.) of MMP-9 in the mice bearing 4T1 tumors in brain, mfp, or liver was measured by ELISA (graph). * Indicates p < 0.05 versus the others. MMP-9 expression in the 4T1 tumors growing in these organs was examined by immunohistochemical analysis using antibody to MMP-9. (B) Serum concentration (conc.) of TIMP-1 in these mice was measured using ELISA (graph). * Indicates p < 0.05 versus the others. TIMP-1 expression in these 4T1 tumors was examined by immunohistochemical analysis using antibody to TIMP-1. Bar indicates 100 μm. (C) PLD accumulation in the tumors is shown. * p < 0.05, 3LL brain tumors versus CT26 brain tumors. There was no significant difference in PLD accumulation to liver, regardless of the tumor type. (D) The serum concentration (conc.) of MMP-9 in the tumor bearing mice. * p < 0.05, 3LL brain tumors versus CT26 brain tumors; ** p < 0.05, 3LL liver tumors versus CT26 liver tumors. (E) The serum concentration (conc.) of TIMP-1. * p < 0.05, 3LL brain tumors versus 3LL liver tumors; ** p < 0.05, CT26 brain tumors versus CT26 liver tumors. (F) There was a significant correlation between PLD accumulation to the tumors with circulating MMP-9/TIMP-1 ratio evaluated by the linear regression analysis (R2 = 0.695).
Additionally, PLD accumulation in tumors and the relative ratio of serum MMP-9 to TIMP-1 were independently studied in mice bearing 3LL tumors in the liver and CT26 tumors in the brain or liver to confirm that these findings are not limited to 4T1 cells (Fig. 6C–E). The levels of PLD accumulation showed a statistically significant correlation with serum MMP-9/TIMP-1 ratio as evaluated by a linear regression analysis (R2 = 0.695) (Fig. 6F). This significant correlation was confirmed in all tumor types studied (Supplementary Fig. S5B–D).
4. Discussion
Clinicians have long recognized intra- and inter-patient differences in the responses of tumors treated with the same therapeutics. Recent advances in biology and genetics have enabled the design of personalized cancer therapy. As an example, the addition of trastuzumab to standard chemotherapy has been shown to improve the median survival of metastatic breast cancer patients who have epidermal growth factor receptor type-2 (HER-2) over-expressing tumors [14]. Additionally, the KRAS mutation status is a strong candidate marker for predicting the survival and response of patients with metastatic colorectal cancer who have been treated with cetuximab (epidermal growth factor receptor antibody) [15]. To apply these ‘‘personalized therapies’’ to more tumor types, efforts are focusing on the development of surrogate markers that can identify patients likely to respond to specific therapies. In this study we investigate for the first time that biomarkers of vascular permeability can serve as predictors of therapeutic response to nano-drugs. To the best of our knowledge, biomarkers to assist in the prediction of the therapeutic efficacy of nanotherapeutics that utilize the EPR effect have not been reported. The therapeutic efficacy of nanotherapeutics depends not only on the resistance mechanisms to therapeutics in tumor cells but also on the amount of nanotherapeutics that are transported to the tumor from systemic circulation. Thus, understanding the transport differentials in cancer, termed ‘‘transport oncophysics’’, can provide a novel framework to improve cancer therapy [33,34]. In the current study, we found that the EPR effect in tumor was dependent not only on the tumor type, but also on the organ site where the tumor is located. These data indicate that the tumor microenvironment plays a major role in ‘‘transport oncophysics’’ and relevant (orthotopic) animal models of cancer should be utilized to evaluate therapeutic efficacy of nanotherapeutics.
MMPs play a fundamental role in extracellular matrix (ECM) remodeling and degradation of proteins [35]. Among the protein components of the ECM, type IV collagen, a substrate to MMP-9, constitutes approximately 50% of all basement membrane proteins [36]. The activities of existing MMPs are modulated by both the activation of pro-enzymes and the inhibition of active enzymes by endogenous inhibitors such as TIMPs [37]. Therefore, the balance between the levels of active MMP-9 and TIMP-1 is critical in controlling proteolysis in the ECM.
We identified increased MMP-9 levels produced by 4T1 cells as compared to 3LL cells in vitro in cell culture supernatant, as well as in vivo in the tumor tissue and the circulation of tumor-bearing mice. These data suggested that the MMP-9 levels in blood can be used as a surrogate biomarker for the EPR effect. The pivotal role of MMP-9 in modulating the EPR effect is supported by the fact that pre-treatment of tumor-bearing mice with an MMP inhibitor, batimastat, increased the amount of type IV collagen, the coverage of endothelial cells by basement membrane, and reduced the PLD accumulation in 4T1 tumors. Nevertheless, elevated MMP-9 levels alone do not appear sufficient to induce PLD accumulation in the tumor, as demonstrated by 4T1 cells grown in the liver. Although the amount of PLD accumulated in tumors was significantly lower when they were growing in the liver, the levels of serum MMP-9 did not differ significantly among mice with tumors growing in different sites. Interestingly, the level of serum TIMP-1 was significantly higher in the mice bearing tumors grown in liver compared to other sites. Thus, high levels of MMP-9 in conjunction with high TIMP-1 levels may fail to sufficiently increase vascular permeability. Although the concentration of TIMP-1 in circulation was significantly lower compared to that of MMP-9, we speculate that the local concentrations and activities of these molecules near the vessel walls may be different. Surprisingly, the levels of VEGF expression were not correlated with PLD accumulation in our study. This data indicate that a single serum protein cannot predict the EPR effect in tumor, due to the heterogeneous nature of tumor microenvironment. Therefore, a combination of candidate proteins in the circulation will be necessary to estimate the EPR effect with sufficient statistical power.
Thus, the EPR effect on the accumulation of PLD in tumors may be determined not only by the concentration of MMP-9 but also by the balance between MMP-9 and TIMP-1. These factors could be used as surrogate biomarkers for selecting patients who are likely to accumulate higher amounts of PLD in their tumors and, thus, respond better to PLD therapy. Indeed, we found that the accumulation of PLD in tumors significantly correlated with the ratio of serum MMP-9 to TIMP-1 levels. The application of these biomarkers could be expanded to other types of nanotherapeutics (e.g., albumin based particles, polymeric micelles, dendrimers and others) that rely on the EPR effect. However, predicting the EPR effect by only measuring the levels of these enzymes in circulation would still be difficult, especially in patients with multiple tumors in multiple organs. In parallel with measuring serum MMP-9/TIMP-1 levels, it may be necessary to develop novel imaging modalities to evaluate local levels of MMP-9 and TIMP-1 or the vascular permeability of each tumor.
In conclusion, our data confirm that the EPR effect depends both on the tumor type and organ site. This effect is controlled by the extent to which endothelial cells are covered by the basement membrane, which in turn is influenced by the levels of MMP-9 and TIMP-1. These findings can potentially be translated to the clinical setting, and the differences in the EPR effect of tumors should be considered in planning the therapeutic strategy for each patient who is a candidate for nanotherapy. Further studies focused on evaluating these potential biomarkers will help to personalize nanotherapeutic cancer therapy.
Supplementary Material
Acknowledgments
This study was supported by the National Cancer Institute (U54CA143837). The authors thank Dr. Isaiah J. Fidler (MD Anderson Cancer Center, Houston, TX) for scientific review of the manuscript.
Appendix A. Supplementary material
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.canlet.2013.11.015.
Footnotes
Conflict of Interest
The authors have no conflict of interest in this study to disclose.
References
- 1.Ferrari M. Nanogeometry: beyond drug delivery. Nat Nanotechnol. 2008;3:131–132. doi: 10.1038/nnano.2008.46. [DOI] [PubMed] [Google Scholar]
- 2.Wang AZ, Langer R, Farokhzad OC. Nanoparticle delivery of cancer drugs. Annu Rev Med. 2012;63:185–198. doi: 10.1146/annurev-med-040210-162544. [DOI] [PubMed] [Google Scholar]
- 3.Duggan ST, Keating GM. Pegylated liposomal doxorubicin: a review of its use in metastatic breast cancer, ovarian cancer, multiple myeloma and AIDS-related Kaposi’s sarcoma. Drugs. 2011;71:2531–2558. doi: 10.2165/11207510-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 4.Chauhan VP, Stylianopoulos T, Boucher Y, Jain RK. Delivery of molecular and nanoscale medicine to tumors: transport barriers and strategies. Annu Rev Chem Biomol Eng. 2012;2:281–298. doi: 10.1146/annurev-chembioeng-061010-114300. [DOI] [PubMed] [Google Scholar]
- 5.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65:271–284. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 6.Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7:771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 7.Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7:653–664. doi: 10.1038/nrclinonc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gordon AN, Fleagle JT, Guthrie D, Parkin DE, Gore ME, Lacave AJ. Recurrent epithelial ovarian carcinoma: a randomized phase III study of pegylated liposomal doxorubicin versus topotecan. J Clin Oncol. 2001;19:3312–3322. doi: 10.1200/JCO.2001.19.14.3312. [DOI] [PubMed] [Google Scholar]
- 9.O’Brien ME, Wigler N, Inbar M, Rosso R, Grischke E, Santoro A, Catane R, Kieback DG, Tomczak P, Ackland SP, Orlandi F, Mellars L, Alland L, Tendler C. CAELYX breast cancer study group, reduced cardiotoxicity and comparable efficacy in a phase III trial of pegylated liposomal doxorubicin HCl (CAELYX/Doxil) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Ann Oncol. 2004;15:440–449. doi: 10.1093/annonc/mdh097. [DOI] [PubMed] [Google Scholar]
- 10.Northfelt DW, Dezube BJ, Thommes JA, Miller BJ, Fischl MA, Friedman-Kien A, Kaplan LD, Du Mond C, Mamelok RD, Henry DH. Pegylated-liposomal doxorubicin versus doxorubicin, bleomycin, and vincristine in the treatment of AIDS-related Kaposi’s sarcoma: results of a randomized phase III clinical trial. J Clin Oncol. 1998;16:2445–2451. doi: 10.1200/JCO.1998.16.7.2445. [DOI] [PubMed] [Google Scholar]
- 11.Sanhai WR, Sakamoto JH, Canady R, Ferrari M. Seven challenges for nanomedicine. Nat Nanotechnol. 2008;3:242–244. doi: 10.1038/nnano.2008.114. [DOI] [PubMed] [Google Scholar]
- 12.Baluk P, Morikawa S, Haskell A, Mancuso M, McDonald DM. Abnormalities of basement membrane on blood vessels and endothelial sprouts in tumors. Am J Pathol. 2003;163:1801–1815. doi: 10.1016/S0002-9440(10)63540-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maeda H. Macromolecular therapeutics in cancer treatment: the EPR effect and beyond. J Control Release. 2009;164:138–144. doi: 10.1016/j.jconrel.2012.04.038. [DOI] [PubMed] [Google Scholar]
- 14.Amar S, Roy V, Perez EA. Treatment of metastatic breast cancer: looking towards the future. Breast Cancer Res Treat. 2009;114:413–422. doi: 10.1007/s10549-008-0032-3. [DOI] [PubMed] [Google Scholar]
- 15.Lièvre A, Bachet JB, Boige V, Cayre A, Le Corre D, Buc E, Ychou M, Bouché O, Landi B, Louvet C, André T, Bibeau F, Diebold MD, Rougier P, Ducreux M, Tomasic G, Emile JF, Penault-Llorca F, Laurent-Puig P. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol. 2008;26:374–379. doi: 10.1200/JCO.2007.12.5906. [DOI] [PubMed] [Google Scholar]
- 16.Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11:865–878. doi: 10.1038/nrc3143. [DOI] [PubMed] [Google Scholar]
- 17.Brantley EC, Guo L, Zhang C, Lin Q, Yokoi K, Langley RR, Kruzel E, Maya M, Kim SW, Kim SJ, Fan D, Fidler IJ. Nitric oxide-mediated tumoricidal activity of murine microglial cells. Transl Oncol. 2010;3:380–388. doi: 10.1593/tlo.10208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shinohara H, Killion JJ, Bucana CD, Yano S, Fidler IJ. Oral administration of the immunomodulator JBT-3002 induces endogenous interleukin 15 in intestinal macrophages for protection against irinotecan-mediated destruction of intestinal epithelium. Clin Cancer Res. 1999;5:2148–2156. [PubMed] [Google Scholar]
- 19.Yokoi K, Godin B, Oborn CJ, Alexander JF, Liu X, Fidler IJ, Ferrari M. Porous silicon nanocarriers for dual targeting tumor associated endothelial cells and macrophages in stroma of orthotopic human pancreatic cancers. Cancer Lett. 2013;334:319–327. doi: 10.1016/j.canlet.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ozawa S, Shinohara H, Kanayama HO, Bruns CJ, Bucana CD, Ellis LM, Davis DW, Fidler IJ. Suppression of angiogenesis and therapy of human colon cancer liver metastasis by systemic administration of interferon-alpha. Neoplasia. 2001;3:154–164. doi: 10.1038/sj.neo.7900128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Low JA, Johnson MD, Bone EA, Dickson RB. The matrix metalloproteinase inhibitor batimastat (BB-94) retards human breast cancer solid tumor growth but not ascites formation in nude mice. Clin Cancer Res. 1996;2:1207–1214. [PubMed] [Google Scholar]
- 22.Ren Y, Cheung HW, von Maltzhan G, Agrawal A, Cowley GS, Weir BA, Boehm JS, Tamayo P, Karst AM, Liu JF, Hirsch MS, Mesirov JP, Drapkin R, Root DE, Lo J, Fogal V, Ruoslahti E, Hahn WC, Bhatia SN. Targeted tumor-penetrating siRNA nanocomplexes for credentialing the ovarian cancer oncogene ID4. Sci Transl Med. 2012;4:47–112. doi: 10.1126/scitranslmed.3003778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Diop-Frimpong B, Chauhan VP, Krane S, Boucher Y, Jain RK. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc Natl Acad Sci USA. 2011;108:2909–2914. doi: 10.1073/pnas.1018892108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vaage J, Donovan D, Uster P, Working P. Tumour uptake of doxorubicin in polyethylene glycol-coated liposomes and therapeutic effect against a xenografted human pancreatic carcinoma. Br J Cancer. 1997;75:482–486. doi: 10.1038/bjc.1997.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Erikson A, Tufto I, Bjønnum AB, Bruland ØS, Davies CdeL. The impact of enzymatic degradation on the uptake of differently sized therapeutic molecules. Anticancer Res. 2008;28:3557–3566. [PubMed] [Google Scholar]
- 26.Cruz-Monserrate Z, Abd-Elgaliel WR, Grote T, Deng D, Ji B, Arumugam T, Wang H, Tung CH, Logsdon CD. Detection of pancreatic cancer tumours and precursor lesions by cathepsin E activity in mouse models. Gut. 2012;61:1315–1322. doi: 10.1136/gutjnl-2011-300544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van de Ven AL, Wu M, Lowengrub J, McDougall SR, Chaplain MA, Cristini V, Ferrari M, Frieboes HB. Integrated intravital microscopy and mathematical modeling to optimize nanotherapeutics delivery to tumors. AIP Adv. 2012;2:11208. doi: 10.1063/1.3699060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van de Ven AL, Kim P, Haley O, Fakhoury JR, Adriani G, Schmulen J, Moloney P, Hussain F, Ferrari M, Liu X, Yun SH, Decuzzi P. Rapid tumoritropic accumulation of systemically injected plateloid particles and their biodistribution. J Control Release. 2012;158:148–155. doi: 10.1016/j.jconrel.2011.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bonoiu A, Mahajan SD, Ye L, Kumar R, Ding H, Yong KT, Roy I, Aalinkeel R, Nair B, Reynolds JL, Sykes DE, Imperiale MA, Bergey EJ, Schwartz SA, Prasad PN. MMP-9 gene silencing by a quantum dot-siRNA nanoplex delivery to maintain the integrity of the blood brain barrier. Brain Res. 2009;1282:142–155. doi: 10.1016/j.brainres.2009.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fujimura M, Gasche Y, Morita-Fujimura Y, Massengale J, Kawase M, Chan PH. Early appearance of activated matrix metalloproteinase-9 and blood-brain barrier disruption in mice after focal cerebral ischemia and reperfusion. Brain Res. 1999;842:92–100. doi: 10.1016/s0006-8993(99)01843-0. [DOI] [PubMed] [Google Scholar]
- 31.Moulton KS, Olsen BR, Sonn S, Fukai N, Zurakowski D, Zeng X. Loss of collagen XVIII enhances neovascularization and vascular permeability in atherosclerosis. Circulation. 2004;110:1330–1336. doi: 10.1161/01.CIR.0000140720.79015.3C. [DOI] [PubMed] [Google Scholar]
- 32.Tang X. Tumor-associated macrophages as potential diagnostic and prognostic biomarkers in breast cancer. Cancer Lett. 2013;332:3–10. doi: 10.1016/j.canlet.2013.01.024. [DOI] [PubMed] [Google Scholar]
- 33.Ferrari M. Frontiers in cancer nanomedicine: directing mass transport through biological barriers. Trends Biotechnol. 2010;28:181–188. doi: 10.1016/j.tibtech.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Michor F, Liphardt J, Ferrari M, Widom J. What does physics have to do with cancer? Nat Rev Cancer. 2011;11:657–670. doi: 10.1038/nrc3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Malemud CJ. Matrix metalloproteinases (MMPs) in health and disease: an overview. Front Biosci. 2006;11:1696–1701. doi: 10.2741/1915. [DOI] [PubMed] [Google Scholar]
- 36.Reichel CA, Rehberg M, Bihari P, Moser CM, Linder S, Khandoga A, Krombach F. Gelatinases mediate neutrophil recruitment in vivo: evidence for stimulus specificity and a critical role in collagen IV remodeling. J Leukoc Biol. 2008;83:864–874. doi: 10.1189/jlb.1007666. [DOI] [PubMed] [Google Scholar]
- 37.Hornebeck W, Lambert E, Petitfrère E, Bernard P. Beneficial and detrimental influences of tissue inhibitor of metalloproteinase-1 (TIMP-1) in tumor progression. Biochimie. 2005;87:377–383. doi: 10.1016/j.biochi.2004.09.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.