

Summary
During assembly of the E. coli pre-replicative complex (pre-RC), initiator DnaA oligomers are nucleated from three widely separated high affinity DnaA recognition sites in oriC. Oligomer assembly is then guided by low affinity DnaA recognition sites, but is also regulated by a switch-like conformational change in oriC mediated by sequential binding of two DNA bending proteins, Fis and IHF, serving as inhibitor and activator, respectively. Although their recognition sites are separated by up to 90 bp, Fis represses IHF binding and weak DnaA interactions until accumulating DnaA displaces Fis from oriC. It remains unclear whether high affinity DnaA binding plays any role in Fis repression at a distance and it is also not known whether all high affinity DnaA recognition sites play an equivalent role in oligomer formation. To examine these issues, we developed origin-selective recombineering methods to mutate E. coli chromosomal oriC. We found that, although oligomers were assembled in the absence of any individual high affinity DnaA binding site, loss of DnaA binding at peripheral sites eliminated Fis repression, and made binding of both Fis and IHF essential. We propose a model in which interaction of DnaA molecules at high affinity sites regulates oriC DNA conformation.
Keywords: oriC, DnaA, bacterial ORC, pre-RC
Introduction
Prior to the onset of new rounds of chromosome replication, replication origins interact with initiator proteins to form at least two distinctive multi-component nucleoprotein complexes; origin recognition complexes (ORC) that serve as a scaffold to recruit additional initiator proteins, and pre-replicative complexes (pre-RC) that load DNA helicase (Mott and Berger, 2007; Katayama et al., 2010; Diffley, 2011; Leonard and Grimwade, 2011). In bacteria, a version of ORC and pre-RC are assembled on oriC from multiple copies of the highly conserved bacterial initiator protein, DnaA, reviewed in (Zakrzewska-Czerwinska et al., 2007; Leonard and Grimwade, 2011). E. coli 's ORC analog comprises DnaA bound to oriC's three high affinity DnaA recognition sites (R1, R2, and R4, Fig.1) throughout the cell cycle (Samitt et al., 1989; Nievera et al., 2006; Miller et al., 2009). The DNA bending protein Fis (Factor for inversion stimulation) also binds to oriC (Fig. 1) as a component of the ORC (Cassler et al., 1995), placing a bend in the right half of oriC (Gille et al., 1991; Filutowicz et al., 1992).
Figure 1.
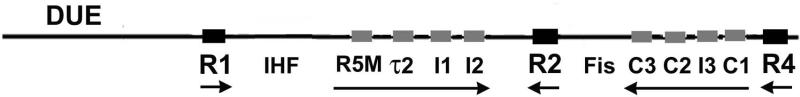
Map of E. coli oriC. The oriC region is shown, with positions of DnaA, IHF and Fis binding sites, as well as the DNA unwinding element (DUE), marked. The three high-affinity sites are designated by large black squares, and the low-affinity sites are marked by small grey rectangles. Horizontal arrows indicate orientation of sites and growth direction of DnaA oligomers. The evolution of the E. coli oriC map to this version was discussed previously (Rozgaja et al., 2011; Leonard and Grimwade, 2011).
The transition from the E coli ORC to the pre-RC depends on the ability of high affinity DnaA/oriC interactions to nucleate two DnaA oligomers, which emanate from peripherally-located DnaA at R1 and R4 towards R2, guided by precise positioning of DnaA protomers by arrays of low affinity recognition sites (Fig. 1) (Rozgaja et al., 2011). Once all low affinity sites are occupied by DnaA, the AT-rich DUE (Duplex unwinding element) in the left region of oriC unwinds, and additional DnaA-ATP binds the single stranded DNA and assists the loading of DNA helicase (Sutton et al., 1998; Mott et al., 2008; Ozaki and Katayama, 2012; Ozaki et al., 2012). DnaA bound to R1 has been proposed to play a key role in both recruitment of the additional DnaA-ATP and in helicase loading (Speck and Messer, 2001; Soultanas, 2012).
The requirement for at least some high affinity nucleation sites in pre-RC assembly is absolute, since DnaA is not capable of binding to low affinity sites in oriC without cooperative interactions (Schaper and Messer, 1995; Miller et al., 2009). It is less clear which high affinity sites are required. Although their nucleotide sequence, peripheral positions in oriC, and roles as nucleation sites suggest that R1 and R4 would play identical roles in pre-RC assembly, attempts to generate chromosomal mutants defective in DnaA binding at R1 were unsuccessful (Weigel et al., 2001), and based on this result, DnaA box R1 was proposed to be essential. In contrast, mutations knocking out binding to R4 are viable (Bates et al., 1995; Weigel et al., 2001), and even deletion of the entire right half of oriC (R2-R4) is tolerated, although these cells are no longer capable of rapid growth (Stepankiw et al., 2009). These studies suggest that the roles of R1 and R4 in pre-RC assembly might not be equivalent.
Formation of DnaA oligomers emanating from both R1 and R4 is opposed by the binding of Fis, which inhibits low affinity DnaA interactions in all of oriC (Ryan et al., 2004). Fis binding to oriC also inhibits another DNA bending protein, IHF (Integration Host Factor), from occupying its site in the left region of oriC between R1 and R5M (Ryan et al., 2004) (Fig. 1). When bound, IHF introduces a sharp bend in the left half of oriC (Polaczek, 1990; Polaczek et al., 1997), which enhances DnaA occupation at all low affinity sites (Grimwade et al., 2000). In synchronized cell cultures, as well as in vitro assays of pre-RC assembly, the switch from a Fis-bound to an IHF-bound complex is abrupt and coincident with origin unwinding (Cassler et al., 1995; Ryan et al., 2004). This conformational switch is dependent on the ability of Fis to repress both low affinity DnaA binding and IHF binding until enough DnaA-ATP has accumulated to trigger initiation (Ryan et al., 2004). The mechanism for repression is puzzling, however, since it is not obvious how Fis binding to its specific site in oriC can inhibit binding of DnaA and IHF at sites up to 90 base pairs away. Regardless, the conformational change may play an important role in precise timing of initiation, since, although both Fis and IHF are dispensable, loss of either perturbs initiation timing and synchronous initiation of replication when multiple oriC copies exist during rapid growth (Boye et al., 1993; Ryan et al., 2002).
In this study, we examine mechanisms governing the transition from ORC to pre-RC, and the interplay among DnaA, Fis and IHF during pre-RC assembly. To this end, we generated E. coli strains carrying, in the chromosomal copy of oriC, non-binding mutations at the high affinity DnaA binding sites, and at the Fis and IHF binding sites. To increase the efficiency of oriC replacement, we modified recombineering strains carrying inducible lambda recombination genes, reviewed in (Court et al., 2002), including the addition of a novel selection step that allows only cells carrying an oriC replacement to survive. We observed that loss of DnaA binding at any two high affinity sites inactivated oriC in rapidly growing cells, but loss of binding at any single high affinity site, including R1, was tolerated on the chromosome, albeit with perturbations in initiation timing. Surprisingly, when DnaA binding was eliminated at either R1 or R4, origin function became dependent on both Fis and IHF binding to oriC. We also found that low levels of DnaA bound to wild-type oriC were able to repress spontaneous unwinding in the 13-mer region, but this constraint was not observed with origins lacking a functional high affinity DnaA binding site. Loss of binding at R4 also eliminated the ability of Fis to repress DnaA and IHF binding at a distance in vitro, suggesting that high affinity sites, in addition to nucleating oligomers, may have a role in establishing a specific oriC conformation that restricts the amount of bending tolerated in oriC. Combined, the data are consistent with a model in which high affinity DnaA interactions play a key role in establishing an oriC conformation that promotes an abrupt, switch-like transition from the ORC to the pre-RC. Without these DnaA interactions, bending proteins are essential to establish a conformation that licenses oriC.
Results
DnaA binding to R1 is not essential for oriC function
Pre-RC assembly in bacteria must begin with DnaA binding to high affinity sites in oriC (Schaper and Messer, 1995; Rozgaja et al., 2011). Although E. coli oriC contains three high affinity sites (R1, R2, and R4, see Fig. 1), a previous chromosomal oriC mutagenesis study suggested an essential role for only R1 (Weigel et al., 2001). Because this determination was based largely on the inability to recover a strain carrying a non-binding mutation in R1 (Weigel et al., 2001) the possibility remained that viable R1 mutants could be made using alternative methodology. In order to test this idea, we attempted to increase the efficiency of mutating chromosomal oriC by developing methods based on the recombineering (homologous recombination-mediated genetic engineering) technique (Court et al., 2002), shown schematically in Fig. 2. In recombineering, cells expressing the lambda RED system are transformed with either a single-stranded or double-stranded DNA fragment containing the desired mutation, which is inserted into the chromosome by homologous recombination (Court et al., 2002).
Figure 2.

Scheme of the oriC-specific recombineering method. (A) A PCR fragment carrying the desired mutation in oriC (marked by white X in R1) was co-electroporated with pAL111 to transform ACL400 cells in which the RED system was induced (see Experimental Procedures for details). Recombination results in replacement of the chromosomal origin (containing the plasmid R1 origin inserted into the DUE) with the mutated oriC. If the mutated oriC is functional, loss of the plasmid R1 origin allows recombineered cells to survive in the presence of copA, and makes them sensitive to kanamycin. pAL111 confers ampicillin resistance. (B) A single-stranded DNA oligonucleotide carrying the desired mutation was electroporated into ACL401 cells in which the RED system was induced. All cells from the electroporation are plated on non-selective media and screening for the mutation is done using colony PCR. Cells are viable regardless of oriC function because this strain is deleted for dnaA and chromosome replication can only initiate from the integrated plasmid origin.
There are several obstacles facing any method for replacing oriC. One is the lack of a selectable phenotype for recombinants. Other groups have solved the selection problem by placing a drug-resistance determinant on the donor DNA (Weigel et al., 2001), but for oriC replacement this solution is not ideal, since transcription near the oriC region might affect oriC function (Baker and Kornberg, 1988; Skarstad et al., 1990). Another challenge for oriC mutagenesis is that, because oriC function is essential for cell viability, one must be able to distinguish between a failure to recover a mutation because it knocks out function, or because the mutation efficiency is too low.
To overcome these challenges, we have developed two recombineering methods to generate both functional and non-functional oriC mutations on the chromosome. For functional oriC versions, we generated a recombineering strain (ACL400) in which chromosomal oriC was inactivated by insertion of a plasmid R1 derivative that confers resistance to kanamycin (Koppes and Nordström, 1986), into the DUE (Fig. 2A). In these cells, chromosomal DNA replication is initiated by the plasmid origin. Transformation of ACL400 with a plasmid (pAL111) expressing copA, an antisense RNA that inhibits synthesis of the plasmid initiator protein RepA, prevents initiation of chromosome replication (Molin and Nordström, 1980). Thus, co-transformation with a DNA fragment carrying an oriC mutation and a plasmid expressing copA and ampicillin resistance will give ampicillin-resistant colonies only when recombination resurrects a functional oriC copy (Fig. 2A). Loss of the plasmid R1 origin also confers sensitivity to kanamycin.
Replacement of oriC in ACL400 is done in a single step, with a strong selection for recombinants. However, because lethal mutations can not be recovered by this method, we also developed an alternative recombineering strain (ACL401) in which a dnaA deletion renders the wild type copy of oriC inactive, with initiation of chromosome replication driven by a plasmid R1 origin integrated into the chromosome outside of the oriC region (Fig 2B). Any oriC mutation, regardless of activity, can be made in ACL401, but there is no simple selection for mutants. Fortunately, recombination efficiencies are high enough (1-5%) to enable identification of mutants without selection, by screening colonies by PCR, using mutation-specific (and wild-type specific) primers (for details see Experimental Procedures). After outgrowth and verification of the mutation (see below), the function of mutated origins in ACL401 can be evaluated by transforming cells with a plasmid (pAL110) expressing both dnaA (to activate oriC) and copA (to inactivate the plasmid R1 origin). Only cells harboring a functional oriC will yield colonies after this transformation.
To generate non-binding mutations in R1, R2, or R4, base pair alterations were first made on an oriC plasmid that also carries the pBR322 origin, which allows replication even when a mutation knocks out oriC function (Weigel et al., 1997). Each site was converted to 5’-GATATAGTT, the same non-binding mutation tested in previous studies by the Messer lab (Weigel et al., 1997; Langer et al., 1996; Weigel et al., 2001). The mutations on the plasmids were verified by sequence analysis.
To confirm that the mutation knocked out both high and low affinity binding, end-labeled double-stranded oligonucleotides containing the mutated site adjacent to the R4 box were incubated with DnaA, and the complexes separated by gel electrophoresis (Fig. 3A). Only one complex (formed by DnaA binding to the R4 site) was seen. In contrast, DnaA formed two complexes on probes containing the low affinity R5M site and R4 (Fig. 3B). Probes containing R5M and the mutated site did not bind any DnaA (Fig. 3C), consistent with previous studies which demonstrated that cooperative binding is required for DnaA occupation of low affinity sites (Schaper and Messer, 1995).
Figure 3.

The mutated R-box does not bind DnaA. Double-stranded DNA oligonucleotides containing R4 and the non-binding GATATAGTT site (A), R4 and the low-affinity binding site R5M (B), or R5M adjacent to the non-binding site (C) were incubated with DnaA-ATP at DnaA/DNA molar ratios of 0:1, 2:1, 10:1, and 20:1, and the resulting complexes were resolved on polyacrylamide gels. All DnaA binding sites on the probe are in the same orientation. The position of the unbound probe, and complexes resulting from 1 or 2 molecules of DnaA bound to the probe are marked.
For recombineering, PCR fragments carrying the oriC region from gidA to mioC were generated from the mutant plasmids, as well as from the wild-type oriC plasmid, and co-transformed with pAL111 into ACL400. ACL400 was used for the first attempts to generate non-binding mutations in R1 because it allows simple selection (ampicillin resistance) of recombinants. While the transformation efficiency varied somewhat among experiments, oriC mutants with defective R1, R2 or R4 replaced the integrated plasmid R1 origin with similar frequencies as wild-type oriC (Table 1). 70% to 80% of the primary colonies from all transformations were kanamycin sensitive, indicating loss of the integrated plasmid origin. While we did not examine the kanamycin-resistant cells in detail, we speculate that these colonies may have retained the integrated plasmid origin, but had insufficient copA expression to render it inactive. Typically, 50-100 of the kanamycin-sensitive colonies were tested by PCR analysis, using mutant-specific and wild-type-specific primers paired with a primer in gidA, and approximately 70% of these contained the mutated site on the correct sized fragment (712 bp for R1, 801 bp for R2, and 872 bp for R4). The remaining 30% gave no signal with the mutant-specific primer, and strong signal with the wild-type primers, suggesting that although recombineering removed the integrated plasmid R1 origin, the recombination site was located between the DUE and the position of the desired mutation. Colonies that tested positive for the mutation were passaged, and re-tested to ensure that the origin region could be amplified only with mutant-specific primers, with no fragment generated when the wild-type primer was used. Primers flanking oriC (in gidA and mioC) were also used to amplify 1432 bp encompassing the oriC region from five different isolates, and the entire region was sequenced. All five isolates carried the correct mutation. The oriC mutations were then successfully transduced into an asnAB MG1655 derivative (JEG22) to place them into cells with a clean genetic background. The presence of a single oriC copy carrying the desired mutation in the transductants was verified by: 1) PCR analysis, using mutant-specific and wild-type-specific primers paired with a primer in gidA; 2) PCR amplification of the region from gidA to mioC, followed by sequence analysis; and 3) qPCR, using primers within oriC or ter (Johnsen et al., 2011) to verify that the oriC/ter ratio was 1:1 in cells from stationary cultures. No evidence of extra copies of oriC was detected in any of the transductants, nor did we find any cells containing an oriC with both a wild-type and mutated site.
Table 1.
Analysis of clones derived from recombineering in ACL400.
| site mutateda | primary ampR colonies | percent kanS colonies | correct size PCR fragmentb | correct sequence |
|---|---|---|---|---|
| none | 230 | 82% | 100% | n.d. |
| R1 | 140 | 75% | 70% | 5/5 |
| R2 | 222 | 80% | 72% | 5/5 |
| R4 | 153 | 82% | 72% | 5/5 |
| R1/R2 | 67 | 50% | 15% | 0/5 |
| R1/R4 | 66 | 79% | 35% | 0/5 |
| R2/R4 | 120 | 79% | 30% | 0/5 |
| R1/IHF | 95 | 43% | 42% | 0/5 |
| R2/IHF | 175 | 71% | 70% | 5/5 |
| R4/IHF | 56 | 46% | 40% | 0/5 |
| R1/Fis | 75 | 35% | 31% | 0/5 |
| R2/Fis | 250 | 70% | 60% | 5/5 |
| R4/Fis | 61 | 43% | 5% | 0/1 |
R1, R2, and R4 sites were mutated to the same sequence: 5’-GATATAGTT. IHF was mutated by replacing 5’-GATCAACA with 5’-GCGATCGA and the Fis recognition site was inactivated by replacing 5’-AACTCAA with 5’-ATGTGTA (Weigel et al., 2001).
For R1, R2, and R4, PCR fragments were generated using a forward primer in gidA and mutation-specific reverse primers in R1, R2, and R4 (for details see Experimental Procedures). For double mutations, the region between gidA and mioC was amplified.
These results strongly suggest that no single high affinity DnaA binding site is essential for oriC function. Because this result is inconsistent with previous findings (Weigel et al., 2001), and with models in which R1 has an essential role in unwinding and in helicase loading (Speck and Messer, 2001; Soultanas, 2012), we also used short (75-85 bp) single-stranded DNA oligomers to generate the R1, R2, and R4 mutations in the ACL401 recombineering strain with the inactive oriC. In this case, the oriC-independent replication removed any selective pressure for oriC function, and the use of shorter, single-stranded oligonucleotides eliminated any chance of recombining multiple copies of the entire origin into the cells. Approximately 200 hundred colonies from recombineered cells were screened using mutant-specific and wild-type-specific primers as described above, and colonies testing positive for the presence of the mutation were detected with similar frequencies (2%-5%) for all three mutations. After passaging and rescreening the positive colonies for the presence of the mutated site and absence of the wild-type site, the origin regions (gidA-mioC) of the mutated cells were sequenced. All of the sequenced origins verified that cells carrying each of the three mutations were successfully isolated. Transforming the mutant cells with pAL110 (expressing dnaA and copA, as described above) tested function of the mutated oriC. For all three (R1, R2, and R4) mutations, colonies were obtained in this transformation, with an efficiency similar to that seen with cells containing a wild-type oriC (approximately 2.5 × 104 colonies per μg of DNA). A plasmid expressing dnaA but not copA also transformed ACL401 cells with similar efficiencies in wild-type and mutant strains, and transformation with a plasmid that expressed only copA gave no colonies in any ACL401 derivative.
Mutations disrupting the bacterial ORC perturb initiation timing
Previous studies showed that, although viable, cells carrying this non-binding mutation in R4 dramatically under-initiate chromosome replication, while the same non-binding mutation in the R2 site caused less perturbation of initiation timing (Weigel et al., 2001; Riber et al., 2009). To verify this, and to examine cells lacking a functional R1 site, we used flow cytometry to evaluate initiation timing in wild-type and mutant cells. Under rapid growth conditions, the E. coli generation time is less than the time required to complete chromosome replication (Cooper and Helmstetter, 1968), so exponentially growing cells normally contain more than one copy of oriC, which synchronously trigger chromosome replication once per cell cycle on partially duplicated chromosomes (Skarstad et al., 1986). Because all origins in the cell fire simultaneously, the origin number doubles at the time of initiation. To examine the number of replication origins, cultures are treated with rifampicin to inhibit new rounds of chromosome replication, and cephalexin, which prevents cell division (Skarstad et al., 1986). After adding the two drugs and incubating long enough to allow completion of ongoing rounds of replication, cellular DNA is stained using a fluorescent dye, and the number of chromosome equivalents in the cell, detected by flow cytometry, reflects the number of oriC copies present at the time of drug addition.
JEG22 cells (wild-type oriC), growing exponentially in minimal media supplemented with glucose and casamino acids, contain 2 (pre-initiation) or 4 (post-initiation) copies of oriC (Fig. 4A). Since there are only 2n chromosome equivalents, both copies of oriC must have fired synchronously in all cells, a key feature of normal initiation timing. In contrast, both R1 and R4 mutants, despite growing with the same doubling time as wild-type cells (τ=31 minutes), had perturbed initiation timing, including asynchronous initiations (Fig. 4B,D), with cells containing 1, 2, 3, and 4 origins. The average number of origins per cell, and origins per cell mass, were also decreased (Table 2), indicating that the mutant cells had under-initiated. The results for the R4 mutant are essentially the same as were previously reported (Weigel et al., 2001; Riber et al., 2009). Initiation timing in the R1 mutant was slightly less perturbed than in the R4 mutant. Several separate transductants of the R1 mutant, with origins generated by the two methods described above, were tested, and each showed a similar pattern in the flow cytometry experiments (not shown). The results support a model in which both R1 and R4 sites play key roles in pre-RC assembly, which is consistent with their acting as primary nucleation sites for DnaA oligomers (Rozgaja et al., 2011).
Figure 4.

DNA histograms of cells treated with cephalexin and rifampicin. JEG22 cells with wild-type oriC (A) or the indicated oriC mutations (B-D) were grown and treated as described in Experimental Procedures. The number of origins in the cells at the time of drug treatment is shown.
Table 2.
Doubling times, number of origins, and cell mass of the mutated strains
| site mutated | doubling time (min) | number of origins cell−1 | relative cell massa | origins cell mass−1 |
|---|---|---|---|---|
| none | 31 | 3.68 | 1.0 | 3.68 |
| R1 | 30 | 3.24 | 1.1 | 2.95 |
| R2 | 31 | 3.56 | 1.0 | 3.56 |
| R4 | 31 | 3.32 | 1.2 | 2.76 |
Relative cell mass was measured by scattered light of mutant strain compared to the wild type strain
Unlike R1 and R4, R2 does not seem to be a primary nucleation site, and the role for R2 in normal pre-RC assembly is less clear. Previous studies suggested that loss of DnaA binding at R2 has little effect on cellular physiology (Weigel et al., 2001). It is possible that R2 is a redundant site, playing no part in normal pre-RC assembly. If this is the case, mutants with defective R2 sites should have normal initiation timing. To re-examine this, JEG22 (R2 non-binding) cells were also analyzed by flow cytometry. The R2 mutant cells had asynchronous initiations, since some cells with three origins were detected (Fig. 4C), as has previously been reported (Weigel et al., 2001; Riber et al., 2009). The R2 mutants also had a slightly lower average number of origins compared to wild-type cells (Table 2), consistent with a slightly delayed initiation. The reason for the mild alteration of initiation timing is not yet clear. One possibility is that R2 helps stabilize the DnaA oligomers that emanate from R1 and R4, and the loss of anchoring results in less efficient initiation. Consistent with this idea, in EMSAs, when a weak DnaA recognition site is flanked on both sides by a strong site, DnaA binding to weak sites is more robust, shown by the increased proportion of fragments bound to three DnaA molecules, (Fig. 5A), relative to that observed when two weak sites are adjacent to only one strong donor site (Fig. 5B).
Figure 5.

DnaA binding to low affinity sites is improved when strong sites flank the weak site. Double-stranded DNA oligonucleotides containing R4 and R2 boxes flanking the low-affinity binding site R5M (A), or R4 adjacent to two low affinity R5M sites (B) were incubated with DnaA-ATP at DnaA/DNA molar ratios of 0:1, 0.5:1, 1:1, 2:1, 5:1, 10:1, and 20:1, and the resulting complexes were resolved on polyacrylamide gels. All DnaA binding sites on the probe are in the same orientation. The position of the unbound probe, and complexes resulting from 1, 2 or 3 molecules of DnaA bound to the probe are marked.
R2 is capable of nucleating a DnaA oligomer
Although our previous studies suggested that pre-RC assembly proceeds via growth of DnaA oligomers from R1 and R4 toward R2 (Rozgaja et al., 2011), mutants lacking the peripheral affinity DnaA binding sites are viable, demonstrating that alternative modes of pre-RC assembly must be possible in E. coli . Understanding these modes is important, since they may reveal minimum criteria for successful replication initiation. To this end, pre-RC assembly on oriCs lacking a functional R1 or R4 site was examined in vitro using high-resolution dimethysulfate (DMS) footprinting (Fig. 6). When DnaA binds to the 9mer sequence 5’-TGTGGATAA (or its variants), the binding alters the DMS modification pattern in a highly reproducible way, such that methylation of the guanine in position 2 is suppressed, while the guanine in position 4 becomes more sensitive to DMS. When supercoiled oriC plasmids carrying mutations that eliminated DnaA binding at R1 were incubated with increasing concentrations of DnaA-ATP, binding to I1 and I2 in the left region of oriC was observed (Fig. 6A, right panel), although a higher concentration of DnaA was required to fill these sites than was needed on wild-type oriC (Fig. 6A, left panel). Binding to R5M and τ2 was also detected, but, based on band intensities, it appeared that binding to R5M was more affected by the R1 mutation (manifested by reduced footprint) than was binding to I2 (Fig. 6A, graphs). This is the pattern that one would expect if a DnaA oligomer emanated from R2, and grew toward R5M, but remained unanchored. DnaA binding to low affinity sites in the right half of the R1 oriC mutant was the same when compared to wild-type oriC (data not shown). When plasmids carrying the R4 mutation were incubated with DnaA, very weak DnaA binding to the low affinity sites in the right half of oriC was detected, most apparent at the I3 site (Fig. 6B, right panel, also see graphs). DnaA binding to the left part of this mutant oriC was identical to wild-type oriC (not shown). These data are consistent with the ability of R2 to act as an alternative nucleation site for the DnaA oligomers, as was suggested by previous studies (Miller et al., 2009; Rozgaja et al., 2011). Extension from R2 was far less efficient in the right half of oriC, perhaps because there is more space between R2 and the proximal weak site C3 than between R2 and I2. The diminished right DnaA oligomer formation in the R4 mutant is also consistent with the observation that initiation timing is more perturbed in origins lacking binding to R4 than in mutants lacking binding to R1.
Figure 6.

R2 is capable of nucleating a DnaA oligomer in oriC. In vitro DMS modification patterns of wild-type (A,B left panels) and mutant oriCs (A,B right panels) are shown. Supercoiled oriC plasmids were incubated with the indicated concentrations of DnaA before treatment with DMS. Two different primers were used to analyze modifications between R1 and R2 (A) and between R2 and R4 (B). Binding site positions are marked, and the location of the G2 and G4 residues are shown, with up and down arrows indicating increased or decreased modification, respectively. Relative intensities of DMS modification at guanosine residues within the low affinity DnaA binding sites are shown in graphs below the gel scans.
Functional ORCs in rapidly growing cells must contain at least two high affinity sites
Since each high affinity DnaA binding site in oriC is capable of nucleating a DnaA oligomer associated with weak arrayed recognition sites, it seemed possible that oriC might function with only one strong site, particularly if only one oligomer was required to form a functional pre-RC. However, if DnaA oligomers are unstable without a high affinity anchor site, or if their length is limited, then it is likely that two high affinity sites would be required for oriC function. To evaluate these two possible scenarios, the recombineering procedures described above were used in an attempt to introduce oriCs carrying mutations in two high affinity DnaA binding sites (R1/R2, R1/R4, and R2/R4) into the chromosome. When PCR fragments (from gidA to mioC) carrying the double mutations were co-transformed with the copA expression plasmid (pAL111) into ACL400 cells, the number of ampicillin-resistant colonies recovered was 2-3 fold lower than were obtained using the wild-type oriC fragment (Table 1). While most of these colonies were sensitive to kanamycin, screening using mutant-specific or wild-type specific primers indicated that only a portion of the PCR fragment had recombined into the chromosome, yielding cells that had a wild-type DUE region, but none, or only one, of the two mutations in oriC. Sequence analysis of the entire oriC region confirmed this result, as none of the sequenced origins contained both mutations. Based on these studies, we propose that at least two high affinity DnaA binding sites are required for oriC function under these growth conditions.
To verify this result, and to ensure that we were not missing a very rare event, the PCR fragments were used for recombineering in ACL401, which does not depend on oriC function. Using this method, cells carrying double mutations in R1/R4, R1/R2, and R2/R4 were recovered in approximately 2% of the plated cells (verified as described above). The function of each the mutated chromosomal origin was tested by transforming the cells with the dnaA/copA expression plasmid (pAL110). No colonies were obtained in this transformation (e.g. there was a transformation efficiency of <0.1% of that seen with cells containing a wild-type oriC). Taken together, these results support the requirement for at least two high affinity sites to assemble a functional pre-RC in rapidly growing cells. In slow growth conditions, the rules for DnaA oligomer formation might be different, since origins lacking the entire right half of oriC are viable, but only when they do not have to support multi-fork growth (Stepankiw et al., 2009).
High affinity sites are required to constrain oriC DNA, allowing Fis to repress DnaA binding at a distance
In vivo, the transition from the bacterial ORC to the pre-RC, is switch-like, with full assembly of the DnaA oligomers delayed until just before the time of initiation (Cassler et al., 1995; Nievera et al., 2006). The switch from ORC to pre-RC results largely from the ability of Fis to inhibit DnaA and IHF from binding to the left region of oriC (Ryan et al., 2004) until sufficient DnaA-ATP accumulates to displace Fis and relieve the repression. How Fis accomplishes this inhibition is not clear, since Fis repression must operate over a distance of up to 90 bp. One possibility is that the oriC region is contained in a constrained topological domain, so that bending in one region would make additional bends in the domain energetically unfavorable. The decreased flexibility could affect both IHF binding and nucleation of a DnaA oligomer from R1. If this scenario is true, then the question of how the domain is constrained remains. The DnaA molecules bound to the high affinity sites are logical candidates for the constraining function, since these are the only other sites observed to be occupied by proteins in the bacterial ORC (Nievera et al., 2006). To test this idea, we first looked for evidence that DnaA binding to high affinity sites could constrain oriC, by developing an assay that exploited the inherent ability of the DUE on supercoiled oriC plasmids to unwind in the absence of replication proteins (Kowalski and Eddy, 1989) under particular buffer conditions. Supercoiled oriC plasmids were cleaved with Mung Bean Nuclease (MBN), which is specific for single-stranded DNA, and primer extension revealed that the MBN cut sites were predominantly in the 13-mer region (Fig. 7), as previously reported (Kowalski and Eddy, 1989). However, when DnaA was incubated with the supercoiled plasmids prior to MBN cutting, levels of DnaA that result in occupation of high affinity sites also caused loss of the MBN sensitivity in the 13mer region (Fig. 7, first panel), indicating that DnaA repressed unwinding in the 13mer region. It is likely that the loss of unwinding was due to DnaA binding to only high affinity sites since, under the buffer conditions optimal for MBN cleavage, binding to low affinity sites is not observed. A mock incubation, in which buffer lacking DnaA was added to supercoiled plasmid, also did not change the MBN sensitivity pattern (data not shown).
Figure 7.

DnaA binding to high affinity sites constrains oriC DNA and represses spontaneous unwinding in the DUE. Supercoiled wt or mutant oriC plasmids were incubated with the indicated concentrations of DnaA, and then treated with Mung Bean Nuclease as described in Experimental Procedures. Cleavage products were revealed by primer extension with products separated on sequencing gels. The positions of right (R), middle (M) and left (L) 13-mer repeats are indicated, next to a marker lane (M) showing DMS-modified guanines.
To determine if high affinity sites acted cooperatively to constrain oriC, DnaA was incubated with plasmids lacking a functional R1, R2, or R4 site, and then treated with MBN (Fig 7, second-fourth panels). Interestingly, DnaA binding did not inhibit MBN-induced cutting in the 13mer region in any of these plasmids. These data are consistent with the idea that cooperative DnaA binding among high affinity sites in oriC topologically constrains the origin.
Based on these results, it seemed logical to test whether loss of high affinity sites also affected the ability of Fis to repress binding of DnaA at a distance. Therefore, we compared the DMS footprints of wild-type oriCs and R4-mutant oriC incubated with DnaA and Fis. (The R4 mutant was used because DnaA binding to the left half of oriC is unaffected by the mutation.) As previously reported (Ryan et al., 2004), when bound to wild-type oriC, Fis repressed DnaA binding to low affinity sites in the left region of oriC (Fig. 8A, left panel, 8B, top graph). In contrast, loss of R4 resulted in loss of this repression (Fig. 8A, right panel, 8B, bottom graph). Fis inhibition of IHF binding to the R4 mutant was also not observed, although the DMS footprint of IHF binding is subtle (SFig 1). Combined, these results are consistent with the bacterial ORC having two distinct roles in pre-RC assembly: one is to recruit additional initiator protein to nucleate oligomers in the origin, and the other is to establish a specific architecture that constrains oriC to allow Fis to repress at a distance.
Figure 8.

Fis repression of low affinity DnaA interactions at a distance requires DnaA binding to R4. Supercoiled wt (A, left panel) and mutant oriC plasmids (A, right panel) were incubated with the indicated concentrations of Fis and DnaA before treatment with DMS. A primer annealing near R4 was used to analyze modifications between R5M and R2. Low affinity binding site positions are marked. (B) Relative intensities of DMS modification at guanosine residues within DnaA binding sites are shown in graphs to the right of the gel scans.
OriCs lacking R1 or R4 require Fis and IHF for pre-RC assembly
While our results suggest that the bacterial ORC helps to establish a specific origin conformation, it remains unclear how origins lacking a high affinity site function, since presumably these mutant origins would be unable to attain the wild type architecture. One possibility is that the DNA bending proteins play an alternative role in the mutant origins, to help set up a configuration of the ORC that can progress to initiation. To test this, we generated double mutations (R1/Fis, R1/IHF, R4/Fis, R4/IHF, R2/Fis, and R2/IHF) into the recognition sites on oriC plasmids, and attempted to transfer them into chromosomal oriC (Table 1). The Fis and IHF mutations were the same as were used previously (Weigel et al., 2001). While we could recover R2/Fis and R2/IHF mutants using the one-step recombineering procedure, R1/Fis, R1/IHF, R4/Fis, and R4/IHF could be generated only in the recombineering strain (ACL401) that was not dependent on oriC. When these oriC mutant strains were transformed with the dnaA/copA expression plasmid to force replication from oriC, no colonies formed, verifying that oriCs lacking R1 or R4 require both Fis and IHF bending proteins for function.
Discussion
DnaA oligomerization along oriC DNA plays an important role in initiating new rounds of chromosomal DNA replication in bacteria (Erzberger et al., 2006; Rozgaja et al., 2011), and nucleation of these oligomers requires the high affinity interaction of DnaA with recognition sites in oriC (Miller et al., 2009). Although DnaA is highly conserved among all bacteria, the number and arrangement of oriC high affinity recognition sites for DnaA varies widely among bacterial types (Zawilak-Pawlik et al., 2005; Gao and Zhang, 2007), and it remains unclear why any particular configuration is preferred. Based on the results of our oriC-specific recombineering, we report that high affinity DnaA-oriC interactions on the E. coli chromosome are capable of at least three roles: 1) as nucleation (start) sites for DnaA oligomerization, 2) as anchor (end) sites for previously nucleated oligomers, and, unexpectedly, 3) in maintaining oriC in the proper conformation(s) for switch-like pre-RC assembly. This latter attribute adds an important new function for the bacterial ORC, and suggests that not only are there instructions for ordered DnaA oligomerization encoded into oriC DNA, but also instructions (in the form of specially-positioned recognition sites) to ensure the correct spatial orientation of oriC during pre-RC assembly.
We find that no individual high affinity DnaA recognition site is essential for oriC function, and have constructed, for the first time, viable strains with a defective R1 recognition site. This result was unexpected, since R1 non-binding mutants were reported to be non-viable (Weigel et al., 2001), and deletion mutants of oriC, in which R1 is the only high affinity site retained, have unwinding and ssDNA binding activity in vitro (Ozaki and Katayama, 2012; Speck and Messer, 2001), and are viable under slow growth conditions (Stepankiw et al., 2009). Based on these reports, R1 was proposed to be essential, and required to nucleate DnaA oligomers extending into the DUE that stabilize unwound DNA and assist in the loading of DNA helicase (Hsu et al., 1994; Speck and Messer, 2001; Soultanas, 2012). However, the results from this study indicate that these activities can be accomplished using other paths. For example, the right half of oriC has been reported to play a role in helicase loading (Ozaki et al., 2012), and this role might increase in the absence of R1. Further, the left DnaA oligomer, which can be nucleated by R2, combined with the sharp DNA bend in the IHF region, is sufficient for unwinding and ssDNA binding (Ozaki and Katayama, 2012; M. Vora, manuscript in preparation). It remains unclear why strains unable to bind DnaA at R1 were not isolated in other labs, but we suggest that the increased efficiency of the origin-specific recombineering method facilitated their construction. In particular, the powerful and continuous selection (the copA expression plasmid) against chromosome replication in non-recombinant ACL 400 cells allows a significant proportion of survivors (> 50%) to carry the desired chromosomal mutation in a minimally altered strain.
In our model of pre-RC assembly, DnaA oligomers fill the gap regions between high affinity sites (Leonard and Grimwade, 2010). We found that, in origins lacking a functional R1 or R4 site, R2 acts an alternative, albeit less efficient, nucleation site for the left or right oligomer, respectively. The gap-filling model also predicts that one or both of the DnaA oligomers are normally anchored by DnaA bound to R2, and the mildly perturbed initiation timing observed in cells with a mutated R2 site is consistent with this idea. However, even though any high affinity site has the capability of nucleating a DnaA oligomer, cells containing a single high affinity site were not viable in our growth conditions, indicating that one unanchored oligomer is either not long enough or stable enough to provide origin activity. Based on this observation in E. coli, extending single DnaA oligomers over long regions of DNA may be incompatible with the function of any bacterial oriC, and large replication origins (for example, see (Zawilak-Pawlik et al., 2005) may require multiple high affinity interactions to nucleate and anchor DnaA oligomers that span the origin.
Unexpectedly, even though R2 was able to act as an alternative nucleation site, origins lacking either a functional R1 and R4 site required assistance, in the form of IHF and Fis binding, to support oriC function under rapid growth conditions. Since Fis and IHF are architectural proteins, these observations suggest that origins lacking either of the peripheral high affinity DnaA binding sites (R1 and R4) have an altered, non-functional conformation that is corrected by Fis and IHF placing bends in oriC. However, neither Fis nor IHF is normally essential, so in their absence, DnaA binding to R1 and R4 sites must be sufficient to establish a functional conformation.
The observations reported here suggest that care must be taken when evaluating the requirements for oriC function. The results from this study show that protein binding sites in oriC may have alternative functions that are revealed only when primary processes are inactivated. For example, by examining cells lacking a functional R1 or R4 site, we found alternative functions for R2 and for Fis and IHF. R2, normally an anchor site, was revealed as a secondary nucleation site, and Fis and IHF, which normally bind sequentially as part of a dynamic conformation switch, have the capability to work together to establish a functional oriC conformation. The lesson from these observations is that it may be necessary to evaluate all phenotypes produced by oriC mutagenesis to determine whether the loss of a specific oriC-protein interaction produces a local or a global effect on pre-RC assembly. Since the requirements for DnaA oligomerization may also change as a function of growth rate (Stepankiw et al., 2009), it will also be important to re-evaluate these mutants under slow growth conditions.
While the exact nature of the configuration in the bacterial ORC remains to be determined, we propose a model in which the DnaA molecules occupying high affinity sites interact to form a constrained loop/toroid (Fig. 9A). The ability of DnaA to bend DNA (Schaper and Messer, 1995) may contribute to the DnaA/DnaA interactions, by placing the DnaA molecules in closer proximity. We speculate that the interactions are via domain1, but this remains to be determined. These interactions would be a plausible target for the positive regulator of initiation DiaA, which has been shown to stabilize DnaA oligomers (Keyamura et al., 2009). This model of a loop wrapped around a core of DnaA is consistent with electron microscopy studies (Funnell et al., 1987; Crooke et al., 1993). When either R1 or R4 is missing, then the simultaneous binding of Fis and IHF help form a similar looped structure, but one with has lost constraint (Fig. 9B). If this model is correct, then the structure that is formed in the bacterial ORC has similarity to that of a proto-nucleosome. In fact, a similarly phased arrangement of nucleosome positioning proteins with looped Green monkey cell α-satellite DNA was previously reported by Strauss and Varshavsky (Strauss and Varshavsky, 1984). It appears that the earliest role of DnaA at oriC may be to generate the appropriate loop on which to assemble a nucleosome-like pre-RC (Funnell et al., 1987).
Figure 9.

Model of the E. coli ORC. (A) DnaA, shown as grey circles, binds to the marked high affinity sites on oriC DNA (black line). Bound molecules are suggested to interact via their N-terminal domains (smaller circles) to form a constrained loop. (B) When the R4 site is mutated, Fis and IHF (grey triangles) bound to their respective sites in oriC bend the DNA to form a similar loop.
An appealing feature of this model is that it provides a mechanism governing the dynamic interplay among DnaA, Fis, and IHF during pre-RC assembly (Ryan et al., 2004). Under rapid growth conditions, the constrained loop would also contain a bend induced by Fis binding. If this structure does not accommodate additional DNA bending in the region of the IHF binding site, then simultaneous interaction of Fis and IHF with oriC would be prohibited, as we have observed (Ryan et al., 2004). Also, because it is likely that bending in the IHF region is needed for R1 to nucleate the DnaA oligomer in the left half of oriC (Rozgaja et al., 2011), the Fis-induced bend would also repress growth of the DnaA oligomer in the left region of oriC (Ryan et al., 2004). Our study provides two lines of support for this model. First, we observed that the ability of Fis to repress DnaA and IHF binding at a distance was dependent on DnaA binding to R4. Second, we observed that DnaA binding to high affinity sites in wild type oriC caused a loss of supercoiled DNA-dependent unwinding in the DUE, but DnaA was not able to constrain supertwists when bound to origins lacking one of the high affinity sites.
Further studies will be necessary to assess cross-strand DnaA interactions, but a regulatory system based on dynamic DNA looping or bending (Travers, 2006) would be particularly well suited to rapidly assemble pre-RC in fast growing bacteria. By transitioning through different constrained conformational states, subassemblies of the pre-RC could permit or inhibit access of oriC binding proteins, ensuring that each conformation was a pre-requisite (and regulator) for the next assembly stage. For example, in E. coli, a loop (bound to Fis) initially made by interacting DnaA molecules at high affinity sites would be remodeled when IHF-induced bends catalyzed interactions between DnaA molecules at R1 and R5M. However, the remodeling would be possible only when sufficient DnaA was available to displace Fis and fill the rest of the available low affinity recognition sites. If regulation by DNA looping is a common attribute of bacterial replication origins, the diversity in position and number of high affinity sites among bacterial replication origins would then be determined not only by the number of anchored oligomers required to span the origin, but also by the need to produce different oriC configurations (in the form of dynamic DNA loops and bends) during pre-RC assembly that ensure correct initiation timing. Interestingly, the looped origin DNA conformation we propose for E. coli oriC may also be a common feature of eukaryotic replication origins. For example, using cryo-electron microscopy, the path of origin DNA within yeast ORC was recently shown to be a DNA loop (Sun et al., 2012) that becomes available for cross-strand interactions promoted by Cdc6.
Experimental Procedures
Bacterial strains and plasmids
Most chromosomal mutations were constructed in ACL400 (HME6, oriC::pKN1562 (clockwise), asnB::Tn10). ACL400 was made using P1 transduction (Thomason et al., 2007) to replace the wild type chromosomal oriC in HME6 (W3110 galK tyr145UAG Δ lacU169 [lambda cI857 del(cro-bioA)]) (Ellis et al., 2001) with oriC::pKN1562 (clockwise) from LK211(Koppes and Nordström, 1986). In oriC::pKN1562, the R1 plasmid origin, which drives replication and confers kanamycin resistance, was inserted into the DUE of oriC. asnB::Tn10 was moved into ACL400 from CAG18433 (E. coli Genetic Stock Center) by P1 transduction.
ACL401 was used to construct oriC mutations that could not be made in ACL400. To make ACL401, P1 transduction was used to move the λ RED system from DY329 (Yu et al., 2000), which carries a tetracycline resistance marker in nadA near the RED genes, into EH3827 (ΔdnaA, asnB, asnA, ilv, kan, zia::pKN500) (Hansen and Yarmolinsky, 1986). EH3827 lacks DnaA, and replicates from a chromosomal copy of the R1 plasmid origin, which permits EH3827 to carry inactive versions of oriC.
JEG22 was made by transducing asnA::kan from JW3722-1 (E. coli Genetic Stock Center), and asnB::Tn10 from CAG18433 (E. coli Genetic Stock Center) into MG1655. For flow cytometry, oriC mutations were moved into JEG22 from ACL400 or ACL401 by P1 transduction and selecting for the ability to grow in the absence of asparagine.
pOC170 (3852 bp) (Langer et al., 1996) carries both the pBR322 replication origin as well as oriC and the bla gene to confer ampicillin resistance. pMW110 (7762 bp) carries the E. coli dnaA gene expressed from its own promoter (Pierucci et al., 1987), as well as the bla gene. pAL110 (8016 bp) is derived from pMW110, and carries both the dnaA gene and the copA gene. To make pAL110, a 254 bp fragment containing copA was amplified from LK211 (Koppes and Nordström, 1986). Primers are shown in Table S1. The fragment was inserted into the unique SnaB restriction site of pMW110 using blunt-end ligation. pAL111 is derived from pAL110, but carries an inactivated dnaA gene, made by EcoR1 digestion and religation to remove the dnaA promoter region.
Chemicals, oligonucleotides, proteins, and enzymes
Reagent grade chemicals were purchased from IBI, Fisher Scientific, or Sigma. Media components were from Difco. Oligonucleotides were purchased from Invitrogen or Integrated DNA Technologies. All enzymes were from New England Biolabs or Bioline. Sequencing gel reagents were purchased from National Diagnostics. Amino-terminal His10-tagged DnaA was isolated as described by Li and Crooke (Li and Crooke, 1999). Li and Crooke (1999) tested the functionality of His10-DnaA and found it equivalent to wild-type DnaA in oriC binding, adenine nucleotide binding, interaction with acidic phospholipids, initiating DNA replication in vitro, and suppression of a dnaA(ts) mutant at non-permissive temperature. We previously found that His10-DnaA has the same unwinding activity as wild type DnaA (Ryan et al., 2002), and gives identical DMS footprints to those seen with wild-type DnaA (compare the footprints in this study with those published in Ryan et al., 2002). Purified Fis was provided by Reid Johnson (UCLA). Purified IHF was provided by Steve Goodman (Ohio State).
Mutagenesis of oriC
pOC170 was used as the starting material for all plasmid mutant oriC constructions. Supercoiled plasmid was isolated using the QIAPrep Spin plasmid preparation kit (Qiagen). Site-directed mutagenesis was performed as described (McGarry et al., 2004), with all mutations being verified by sequence analysis (done by University of Florida, ICBR). High affinity DnaA recognition sites were inactivated by changing 5’-TTATCCACA (R1 and R4) or 5’-TTATACACA (R2) to 5’-GATATAGTT (Weigel et al., 2001). The IHF recognition site was inactivated by replacing 5’-GATCAACA with 5’-GCGATCGA and the Fis recognition site was inactivated by replacing 5’-AACTCAA with 5’-ATGTGTA (Weigel et al., 2001). Previous studies demonstrated that these mutations eliminated binding (Roth et al., 1994; Grimwade et al., 2000; Ryan et al., 2004). PCR fragments (1432 bp) containing verified mutant origins were amplified using primers (Table S1) extending from gidA to mioC and used for recombineering (see below).
To create chromosomal oriC mutations, ACL400 was grown at 32°C in Luria-Bertani broth until the culture reached a density (OD600) of 0.2. Induction of λ RED and recombineering was performed as previously described (Sharan et al., 2009) except PCR fragments carrying the mutant oriC were co-electroporated with pAL111. After 1 hour outgrowth in non-selective media, cells were plated on LB agar containing 50 μg ml−1 ampicillin and incubated at 32°C. All colonies were then checked for kanamycin sensitivity, since replacement of the inserted R1 plasmid origin with the PCR fragment containing oriC removes the kanamycin resistance gene. OriC replacement was verified by PCR, using a primer that selectively annealed to the mutated or wild-type oriC region and OR9 (see supplemental information for primer sequences). The OR9 (gidA)-OR10 (mioC) PCR fragment was amplified from colonies testing positive for the mutated site and negative for the wild-type site, and sequenced.
Mutations in chromosomal oriC that eliminated function, as well as the R1, R2, and R4 non-binding mutations, were generated in ACL401 by recombineering with PCR fragments, or, in the case of single site mutations, with short single-stranded oligonucleotides as described above, except that pAL111 was not used, and colonies were plated onto LB agar plates. After overnight growth at 32°C, 200 colonies were randomly picked and analyzed by colony PCR, using mutant-specific or wild-type-specific primers. Colonies giving an amplification product using the mutant primer were then grown in culture to segregate chromosomes, re-plated onto LB agar, and rechecked by PCR. OriC replacement was verified by sequencing the oriC region, as described above. To determine if the mutated origin was functional, cells were transformed with pAL110, which expresses copA to inactivate the integrated R1 origin, and dnaA to activate oriC. Cells containing plasmid and a viable oriC were selected for growth on LB agar containing 50 μg ml−1 ampicillin.
After sequence verification, all functional oriC mutations were moved into JEG22 using P1 transduction, selecting for growth on minimal agar plates supplemented with glucose (0.2%) and tetracycline (12 μg ml−1). Colony PCR, using wild-type or mutation-specific primers, were used to verify transfer of the mutation, and the presence of the mutated origin was verified by sequencing the oriC region, as describe above. OriC/ter ratios were determined by qPCR as described in (Johnsen et al., 2011). Briefly, genomic DNA was isolated from overnight cultures of stationary phase cells using the BioRad AquaPure Genomic DNA Isolation kit. After digestion with EcoR1, 10 ng of DNA was used as a template in 25 μl reactions with 2.5 μl of each primer (forward, 9 μM; reverse, 9 μM; and probe, 2.5 μM; see Table S1 for sequence) and 12.5 μl of 2X Taq Mix (New England Biolobs). Reactions to amplify oriC and ter were done separately. Amplification was done using the BioRad MyIQ ICycler, which provided the fluorescence threshold values, which were then used to calculate the relative values of template. Reactions were performed in triplicate. The fluorescent threshold values were averaged and the average used to calculate the oriC/ter ratio. Standard deviations were under 5%.
Dimethyl Sulfate Footprinting
DMS modification of DNA (0.75 μg) in vitro was performed as described (Ryan et al., 2004). DnaA was preincubated in reaction buffer with 5 mM ATP for 5 min before addition to reactions at the concentrations indicated in the figures. In some reaction plasmids were first incubated with Fis, followed by DnaA. DMS-treated samples were extended with radiolabelled primer as described (Ryan et al., 2004). Two primers were used, a left primer hybridizing at bases 272–290 to analyze top strand modifications of plasmid template, and a right primer hybridizing at bases 124–142 to analyze bottom-strand modifications. Extension products were resolved on 6% polyacrylamide sequencing gels, and dried gels were scanned on a Bio-Rad Molecular Imager FX PhosphorImager. Representative scans are shown in the figures. Images were analyzed by using Bio-Rad QUANTITY ONE software. Intensities of bands in binding sites (relative to the total intensity of all selected bands) were calculated. Deviations in band intensities among experiments were < 10%.
EMSA
Complementary single stranded oligonucleotides (sequences shown in Table S1) were annealed, and doubled stranded oligonucleotides were isolated and labeled as described (Rozgaja et al., 2011). Binding reactions were set up in a mixture containing 20 mM HEPES•KOH pH 8, 2.5 mM magnesium acetate, 1 mM EDTA, 4 mM DTT, 100 mM ATP, 5 mg ml-1 BSA, 0.2% Triton X-100, 5% glycerol, 100 ng poly(dI-dC), 4 nM of end-labeled fragment, and 2, 4, 8, 20, 40, 80 or 200 nM of DnaA-ATP. Reactions were incubated at 37°C for 8 min, and then loaded onto a 7% polyacrylamide gel. Complexes were separated by electrophoresis, using 45 mM Tris-Borate, 1 mM EDTA as the running buffer. Dried gels were scanned on a Bio-Rad Molecular Image FX PhosphorImager.
Mung Bean Nuclease Digestion
Supercoiled plasmids were incubated in 20 μl reactions with DnaA-ATP in 10 mM Tris, 1 mM EDTA, pH 7.0, at the concentrations indicated in the Figures. Mung Bean Nuclease (0.1 unit) was added, and the samples were incubated at 38°C for 15 minutes. The reactions were stopped using 100 μl of 3M ammonium acetate, 250 μg ml−1 tRNA. Samples were precipitated, and extended as described above for DMS-treated samples.
Flow Cytometry
JEG22 strains, carrying wild-type or mutant oriCs were grown in minimal salts media (Helmstetter and Krajewski, 1982) supplemented with 0.2% glucose, 0.2% uracil, and 0.2% casamino acids to an OD450 of 0.2. Cells were then treated for 4 hours with 300 μg/ml rifampicin to stop new initiation events, and 15 μg ml−1 cephalexin, to stop cell division (Skarstad et al., 1986). All ongoing rounds of DNA replication are completed in the 4 hour run-out period, yielding cells with fully replicated chromosomes. The number of chromosomes is equivalent to the number of oriC copies in the cell at the time of addition. After drug treatment, 1 ml of cells were mixed with 9 ml of 70% ethanol and stored at 4°C. Prior to flow cytometric analysis, 1 ml of cells was pelleted, washed with 50 mM Tris-Cl, pH 7.5. 150 mM NaCl (TBS), and resuspended in 1 ml of TBS containing 0.5 μl Vybrant DyeCycle Green (Invitrogen/Molecular Probes) (final concentration of dye is 2.5 μM). Stained cells (3000 cells ml−1) were analyzed using an Accuri C6 personal flow cytometer, and data from 10,000 cells were collected. Forward scatter was used for cell mass measurement. CFlow software was used to calculate the percentage of cells in each chromosomal DNA peak.
Supplementary Material
Acknowledgements
We are extremely grateful to Donald Court (NCI), Nadim Majdalani (NCI) and Dhruba Chattaraj (NIH), for providing bacterial strains and assistance with the recombineering procedures. We also thank Reid Johnson (UCLA) for providing us with purified Fis protein, Steve Goodman (Ohio State University) for providing us with purified IHF protein, and Elliott Crooke (Georgetown University) for providing the His10-DnaA overproduction plasmid. This work was supported by NIH-GM54042.
References
- Baker TA, Kornberg A. Transcriptional activation of initiation of replication from the E. coli chromosomal origin: an RNA-DNA hybrid near oriC. Cell. 1988;55:113–123. doi: 10.1016/0092-8674(88)90014-1. [DOI] [PubMed] [Google Scholar]
- Bates DB, Asai T, Cao Y, Chambers MW, Cadwell GW, Boye E, Kogoma T. The DnaA box R4 in the minimal oriC is dispensable for initiation of Escherichia coli chromosome replication. Nucleic acids research. 1995;23:3119–3125. doi: 10.1093/nar/23.16.3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boye E, Lyngstadaas A, Lobner-Olesen A, Skarstad K, Wold S. Regulation of DNA replication in Escherichia coli. In: Fanning E, Knippers R, Winnacker E-L, editors. Regulation of DNA replication in Escherichia coli. Springer; Berlin: 1993. pp. 15–26. [Google Scholar]
- Cassler MR, Grimwade JE, Leonard AC. Cell cycle-specific changes in nucleoprotein complexes at a chromosomal replication origin. Embo J. 1995;14:5833–5841. doi: 10.1002/j.1460-2075.1995.tb00271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper S, Helmstetter CE. Chromosome replication and the division cycle of Escherichia coli B/r. J Mol Biol. 1968;31:519–540. doi: 10.1016/0022-2836(68)90425-7. [DOI] [PubMed] [Google Scholar]
- Court DL, Sawitzke JA, Thomason LC. Genetic engineering using homologous recombination. Annu Rev Genet. 2002;36:361–388. doi: 10.1146/annurev.genet.36.061102.093104. [DOI] [PubMed] [Google Scholar]
- Crooke E, Thresher R, Hwang DS, Griffith J, Kornberg A. Replicatively active complexes of DnaA protein and the Escherichia coli chromosomal origin observed in the electron microscope. J Mol Biol. 1993;233:16–24. doi: 10.1006/jmbi.1993.1481. [DOI] [PubMed] [Google Scholar]
- Diffley JF. Quality control in the initiation of eukaryotic DNA replication. Philos Trans R Soc Lond B Biol Sci. 2011;366:3545–3553. doi: 10.1098/rstb.2011.0073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis HM, Yu D, DiTizio T, Court DL. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc Natl Acad Sci U S A. 2001;98:6742–6746. doi: 10.1073/pnas.121164898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erzberger JP, Mott ML, Berger JM. Structural basis for ATP-dependent DnaA assembly and replication-origin remodeling. Nat Struct Mol Biol. 2006;13:676–683. doi: 10.1038/nsmb1115. [DOI] [PubMed] [Google Scholar]
- Filutowicz M, Ross W, Wild J, Gourse RL. Involvement of Fis protein in replication of the Escherichia coli chromosome. J Bacteriol. 1992;174:398–407. doi: 10.1128/jb.174.2.398-407.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funnell BE, Baker TA, Kornberg A. In vitro assembly of a prepriming complex at the origin of the Escherichia coli chromosome. J Biol Chem. 1987;262:10327–10334. [PubMed] [Google Scholar]
- Gao F, Zhang CT. DoriC: a database of oriC regions in bacterial genomes. Bioinformatics. 2007;23:1866–1867. doi: 10.1093/bioinformatics/btm255. [DOI] [PubMed] [Google Scholar]
- Gille H, Egan JB, Roth A, Messer W. The FIS protein binds and bends the origin of chromosomal DNA replication, oriC, of Escherichia coli. Nucleic acids research. 1991;19:4167–4172. doi: 10.1093/nar/19.15.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimwade JE, Ryan VT, Leonard AC. IHF redistributes bound initiator protein, DnaA, on supercoiled oriC of Escherichia coli. Mol Microbiol. 2000;35:835–844. doi: 10.1046/j.1365-2958.2000.01755.x. [DOI] [PubMed] [Google Scholar]
- Hansen EB, Yarmolinsky MB. Host participation in plasmid maintenance: dependence upon dnaA of replicons derived from P1 and F. Proc Natl Acad Sci USA. 1986;83:4423–4427. doi: 10.1073/pnas.83.12.4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmstetter CE, Krajewski CA. Initiation of chromosome replication in dnaA and dnaC mutants of Escherichia coli B/r F. J Bacteriol. 1982;149:685–693. doi: 10.1128/jb.149.2.685-693.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu J, Bramhill D, Thompson CM. Open complex formation by DnaA initiation protein at the Escherichia coli chromosomal origin requires the 13-mers precisely spaced relative to the 9-mers. Mol Microbiol. 1994;11:903–911. doi: 10.1111/j.1365-2958.1994.tb00369.x. [DOI] [PubMed] [Google Scholar]
- Johnsen L, Flatten I, Morigen, Dalhus B, Bjoras M, Waldminghaus T, Skarstad K. The G157C mutation in the Escherichia coli sliding clamp specifically affects initiation of replication. Mol Microbiol. 2011;79:433–446. doi: 10.1111/j.1365-2958.2010.07453.x. [DOI] [PubMed] [Google Scholar]
- Katayama T, Ozaki S, Keyamura K, Fujimitsu K. Regulation of the replication cycle: conserved and diverse regulatory systems for DnaA and oriC. Nat Rev Microbiol. 2010;8:163–170. doi: 10.1038/nrmicro2314. [DOI] [PubMed] [Google Scholar]
- Keyamura K, Abe Y, Higashi M, Ueda T, Katayama T. DiaA dynamics are coupled with changes in initial origin complexes leading to helicase loading. J. Biol. Chem. 2009;284:25038–25050. doi: 10.1074/jbc.M109.002717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppes L, Nordström K. Insertion of an R1 plasmid into the origin of replication of the E. coli chromosome: random timing of replication of the hybrid chromosome. Cell. 1986;44:117–124. doi: 10.1016/0092-8674(86)90490-3. [DOI] [PubMed] [Google Scholar]
- Kowalski D, Eddy MJ. The DNA unwinding element: a novel, cis-acting component that facilitates opening of the Escherichia coli replication origin. EMBO J. 1989;8:4335–4344. doi: 10.1002/j.1460-2075.1989.tb08620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer U, Richter S, Roth A, Weigel C, Messer W. A comprehensive set of DnaA-box mutations in the replication origin, oriC, of Escherichia coli. Mol Microbiol. 1996;21:301–311. doi: 10.1046/j.1365-2958.1996.6481362.x. [DOI] [PubMed] [Google Scholar]
- Leonard AC, Grimwade JE. Regulating DnaA complex assembly: it is time to fill the gaps. Curr Opin Microbiol. 2010;13:766–772. doi: 10.1016/j.mib.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard AC, Grimwade JE. Regulation of DnaA assembly and activity: taking directions from the genome. Annu Rev Microbiol. 2011;65:19–35. doi: 10.1146/annurev-micro-090110-102934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Crooke E. Functional analysis of affinity-purified polyhistidine-tagged DnaA protein. Protein Expr Purif. 1999;17:41–48. doi: 10.1006/prep.1999.1094. [DOI] [PubMed] [Google Scholar]
- McGarry KC, Ryan VT, Grimwade JE, Leonard AC. Two discriminatory binding sites in the Escherichia coli replication origin are required for DNA strand opening by initiator DnaA-ATP. Proc Natl Acad Sci USA. 2004;101:2811–2816. doi: 10.1073/pnas.0400340101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DT, Grimwade JE, Betteridge T, Rozgaja T, Torgue JJ, Leonard AC. Bacterial origin recognition complexes direct assembly of higher-order DnaA oligomeric structures. Proc Natl Acad Sci USA. 2009;106:18479–18484. doi: 10.1073/pnas.0909472106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molin S, Nordström K. Control of plasmid R1 replication: functions involved in replication, copy number control, incompatibility, and switch-off of replication. J Bacteriol. 1980;141:111–120. doi: 10.1128/jb.141.1.111-120.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mott ML, Berger JM. DNA replication initiation: mechanisms and regulation in bacteria. Nat Rev Microbiol. 2007;5:343–354. doi: 10.1038/nrmicro1640. [DOI] [PubMed] [Google Scholar]
- Mott ML, Erzberger JP, Coons MM, Berger JM. Structural synergy and molecular crosstalk between bacterial helicase loaders and replication initiators. Cell. 2008;135:623–634. doi: 10.1016/j.cell.2008.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nievera C, Torgue JJ, Grimwade JE, Leonard AC. SeqA blocking of DnaA-oriC interactions ensures staged assembly of the E. coli pre-RC. Mol Cell. 2006;24:581–592. doi: 10.1016/j.molcel.2006.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki S, Katayama T. Highly organized DnaA-oriC complexes recruit the single-stranded DNA for replication initiation. Nucleic Acids Res. 2012;40:1648–1665. doi: 10.1093/nar/gkr832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki. S, Noguchi Y, Hayashi Y, Miyazaki E, Katayama T. Differentiation of the DnaA-oriC-subcomplex for DNA unwinding in a replication initiation complex. J Biol Chem. 2012;287:37458–37471. doi: 10.1074/jbc.M112.372052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierucci O, Helmstetter CE, Rickert M, Weinberger M, Leonard AC. Overexpression of the dnaA gene in Escherichia coli B/r: chromosome and minichromosome replication in the presence of rifampin. J Bacteriol. 1987;169:1871–1877. doi: 10.1128/jb.169.5.1871-1877.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polaczek P. Bending of the origin of replication of E. coli by binding of IHF at a specific site. New Biol. 1990;2:265–271. [PubMed] [Google Scholar]
- Polaczek P, Kwan K, Liberies DA, Campbell JL. Role of architectural elements in combinatorial regulation of initiation of DNA replication in Escherichia coli. Mol Microbiol. 1997;26:261–275. doi: 10.1046/j.1365-2958.1997.5701931.x. [DOI] [PubMed] [Google Scholar]
- Riber L, Fujimitsu K, Katayama T, Løbner-Olesen A. Loss of Hda activity stimulates replication initiation from I-box, but not R4 mutant origins in Escherichia coli. Mol Microbiol. 2009;71:107–122. doi: 10.1111/j.1365-2958.2008.06516.x. [DOI] [PubMed] [Google Scholar]
- Roth A, Urmoneit B, Messer W. Functions of histone-like proteins in the initiation of DNA replication at oriC of Escherichia coli. Biochimie. 1994;76:917–923. doi: 10.1016/0300-9084(94)90016-7. [DOI] [PubMed] [Google Scholar]
- Rozgaja TA, Grimwade JE, Iqbal M, Czerwonka C, Vora M, Leonard AC. Two oppositely oriented arrays of low-affinity recognition sites in oriC guide progressive binding of DnaA during Escherichia coli pre-RC assembly. Mol Microbiol. 2011;82:475–488. doi: 10.1111/j.1365-2958.2011.07827.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan VT, Grimwade JE, Camara JE, Crooke E, Leonard AC. Escherichia coli prereplication complex assembly is regulated by dynamic interplay among Fis, IHF and DnaA. Mol Microbiol. 2004;51:1347–1359. doi: 10.1046/j.1365-2958.2003.03906.x. [DOI] [PubMed] [Google Scholar]
- Ryan VT, Grimwade JE, Nievera CJ, Leonard AC. IHF and HU stimulate assembly of pre-replication complexes at Escherichia coli oriC by two different mechanisms. Mol Microbiol. 2002;46:113–124. doi: 10.1046/j.1365-2958.2002.03129.x. [DOI] [PubMed] [Google Scholar]
- Samitt CE, Hansen FG, Miller JF, Schaechter M. In vivo studies of DnaA binding to the origin of replication of Escherichia coli. EMBO J. 1989;8:989–993. doi: 10.1002/j.1460-2075.1989.tb03462.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaper S, Messer W. Interaction of the initiator protein DnaA of Eschericha coli with its DNA target. J Biol Chem. 1995;270:17622–17626. doi: 10.1074/jbc.270.29.17622. [DOI] [PubMed] [Google Scholar]
- Sharan SK, Thomason LC, Kuznetsov SG, Court DL. Recombineering: a homologous recombination-based method of genetic engineering. Nat Protoc. 2009;4:206–223. doi: 10.1038/nprot.2008.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarstad K, Baker TA, Kornberg A. Strand separation required for initiation of replication at the chromosomal origin of E.coli is facilitated by a distant RNA--DNA hybrid. EMBO J. 1990;9:2341–2348. doi: 10.1002/j.1460-2075.1990.tb07406.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarstad K, Boye E, Steen HB. Timing of initiation of chromosome replication in individual Escherichia coli cells. EMBO J. 1986;5:1711–1717. doi: 10.1002/j.1460-2075.1986.tb04415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soultanas P. Loading mechanisms of ring helicases at replication origins. Mol Microbiol. 2012;84:6–16. doi: 10.1111/j.1365-2958.2012.08012.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speck C, Messer W. Mechanism of origin unwinding: sequential binding of DnaA to double- and single-stranded DNA. EMBO J. 2001;20:1469–1476. doi: 10.1093/emboj/20.6.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepankiw N, Kaidow A, Boye E, Bates D. The right half of the Escherichia coli replication origin is not essential for viability, but facilitates multi-forked replication. Mol Microbiol. 2009;74:467–479. doi: 10.1111/j.1365-2958.2009.06877.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss F, Varshavsky A. A protein binds to a satellite DNA repeat at three specific sites that would be brought into mutual proximity by DNA folding in the nucleosome. Cell. 1984;37:889–901. doi: 10.1016/0092-8674(84)90424-0. [DOI] [PubMed] [Google Scholar]
- Sun J, Kawakami H, Zech J, Speck C, Stillman B, Li H. Cdc6-induced conformational changes in ORC bound to origin DNA revealed by cryo-electron microscopy. Structure. 2012;20:534–544. doi: 10.1016/j.str.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MD, Carr KM, Vicente M, Kaguni JM. Escherichia coli DnaA protein. The N-terminal domain and loading of DnaB helicase at the E. coli chromosomal origin. J Biol Chem. 1998;273:34255–34262. doi: 10.1074/jbc.273.51.34255. [DOI] [PubMed] [Google Scholar]
- Thomason LC, Costantino N, Court DL. E. coli genome manipulation by P1 transduction. Curr Protoc Mol Biol. 2007 doi: 10.1002/0471142727.mb0117s79. Chapter 1: Unit 1.17. [DOI] [PubMed] [Google Scholar]
- Travers A. DNA topology: dynamic DNA looping. Curr Biol. 2006;16:R838–R840. doi: 10.1016/j.cub.2006.08.070. [DOI] [PubMed] [Google Scholar]
- Weigel C, Messer W, Preiss S, Welzeck M, Morigen, Boye E. The sequence requirements for a functional Escherichia coli replication origin are different for the chromosome and a minichromosome. Mol Microbiol. 2001;40:498–507. doi: 10.1046/j.1365-2958.2001.02409.x. [DOI] [PubMed] [Google Scholar]
- Weigel C, Schmidt A, Rückert B, Lurz R, Messer W. DnaA protein binding to individual DnaA boxes in the Escherichia coli replication origin, oriC. EMBO J. 1997;16:6574–6583. doi: 10.1093/emboj/16.21.6574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Ellis HM, Lee EC, Jenkins NA, Copeland NG, Court DL. An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci U S A. 2000;97:5978–5983. doi: 10.1073/pnas.100127597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakrzewska-Czerwinska J, Jakimowicz D, Zawilak-Pawlik A, Messer W. Regulation of the initiation of chromosomal replication in bacteria. FEMS Microbiol Rev. 2007;31:378–387. doi: 10.1111/j.1574-6976.2007.00070.x. [DOI] [PubMed] [Google Scholar]
- Zawilak-Pawlik A, Kois A, Majka J, Jakimowicz D, Smulczyk-Krawczyszyn A, Messer W, Zakrzewska-Czerwińska J. Architecture of bacterial replication initiation complexes: orisomes from four unrelated bacteria. Biochem J. 2005;389:471–481. doi: 10.1042/BJ20050143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.