

Abstract
Multiple sclerosis (MS) is an organ-specific autoimmune disorder that is in part genetically determined. The gene encoding the α-chain of the IL-2 receptor, IL2RA, harbors alleles associated with risk to MS and other autoimmune diseases. In addition, IL2RA genetic variants correlate with the levels of a soluble form of the IL-2 receptor in subjects with type 1 diabetes and multiple sclerosis. Here, we show that the IL2RA genotypes differentially affects soluble IL-2RA (sIL-2RA) levels in MS cases vs healthy controls; the two variants associated with MS (rs12722489 and rs2104286) account for 15 and 18% of the total variance in log10-transformed sIL-2RA concentration in control subjects but less so in subjects with MS (2 and 5%), suggesting that perturbations associated with disease or treatment may influence sIL-2RA levels in subjects with MS. Whereas analyses demonstrate that sIL-2RA serum concentrations are a remarkably stable phenotype in both healthy controls and untreated MS subjects, a difference is observed between benign and malignant MS. These data indicate that, in addition to specific allelic variants at IL2RA, immunological perturbations associated with aggressive forms of the disease can influence sIL-2RA levels in serum of MS subjects. We also demonstrate, functionally, that sIL-2RA can inhibit IL-2 signaling, yet enhance T cell proliferation and expansion. In summary, we propose that before disease onset, strong genetic factors associated with disease risk dictate sIL-2RA levels that may be further modulated with onset of chronic systemic inflammation associated with MS.
Multiple sclerosis (MS)3 is an organ-specific autoimmune disorder characterized by chronic inflammation of CNS myelin where focal T cell and macrophage infiltrates result in demyelination and loss of neurological function (1). There is systemic activation of the adaptive immune system characterized by activated T cells and of the innate system with activated dendritic cells (2). Blocking T cell traffic from the circulation into the CNS with anti-VLA-4 mAbs has a strong effect on the frequency of acute neurological events (3), which occur unpredictably in a remitting-relapsing pattern. In the peripheral circulation, increased levels of cytokine receptors, including the IL-2R α-chain, have been observed (4, 5). Clinically, resolution of acute attacks or relapses is often followed by recurrent attacks and, over time, about one-half of the relapsing-remitting subjects will enter the progressive stage of the disease. This progressive phase may involve more degenerative rather than immunological changes, with a substantial degree of axonal degeneration present within the CNS (6).
Susceptibility to MS is conferred by genetic as well as largely unknown environmental factors (1, 7). The sequencing of the human genome has allowed large-scale whole genome association scans aiming at identifying risk alleles associated with risk to developing autoimmune disease. We identified single nucleotide polymorphisms (SNP) within genes encoding the IL-2Rα (IL2RA), IL-7Rα (IL7RA), CD58 (CD58) as well as other nonimmune related genes (8). Of particular interest is the overlap among autoimmune diseases at some of these loci; for example, at IL2RA, a number of SNPs also contribute to risk to type 1 diabetes (T1D) (9) and Graves’ disease (10), indicating that some pathways are shared among autoimmune disease. In the IL2RA locus, variants not only associate with disease risk but also influence the levels of sIL-2RA in the serum (9).
The IL-2/IL-2RA(CD25) pathway plays an essential role in regulating immune responses (11). IL-2 is central for both expansion and apoptosis of T cells and soluble IL-2RA (sIL-2RA) binds IL-2 at a similarly low affinity as full-length IL-2RA (12). Mitogen- and Ag-activated leukocytes release sIL-2RA into culture supernatants (13). All or part of this release of sIL-2RA from activated immune cells involves proteolytic cleavage, and thus far, an mRNA encoding this isoform has not been identified. High concentrations of soluble IL-2RA chain are found in sera from healthy subjects but are elevated in subjects with autoimmune disease, inflammation, and infection (4, 5, 14-17), which is why sIL-2RA is thus considered to be a biomarker for immune activation in the peripheral blood (12)’. sIL-2RA, like many other soluble cytokine receptors (18), can compete with cell surface IL-2RA for IL-2 binding and thus block IL-2 function, a hypothesis supported by a recent report that sIL-2RA inhibits the proliferative response of a murine CD8+ T cell line cultured in the presence of high doses of murine IL-2 (19). Although less likely, sIL-2RA might alternately complex with IL-2 and potentiate signaling, as has been demonstrated for sIL-6R-IL-6 (20) and sIL-15R-IL-15 complexes (19). The IL-2-IL-2RA system is also central for the production and function of conventional and regulatory T cells, which are critical for maintenance of immunological self tolerance and prevention of auto-reactive processes (21). Thus, there are a variety of mechanisms by which sIL-2RA may influence T cell function, but its impact on the function of human T cells is presently unclear.
Here, we have characterized the levels of sIL-2RA in the serum of healthy controls and subjects with different MS subtypes and have examined the extent to which the sIL-2RA serum levels are related to the allelic variants that have thus far been associated with MS susceptibility. In addition, we provide in vitro evidence that the levels of sIL-2RA affect human T cell function, including both effector and regulatory T cells.
Materials and Methods
Healthy controls, subject cohorts, and disease definitions
Healthy control and MS subject samples were obtained through the Partners Healthcare MS Center (Boston, MA) as part of its MS Registry project, which is approved by the Partners Healthcare institutional review board. All subjects were older than 18 years of age and met criteria of either MS per the revised McDonald diagnostic criteria (22), or clinically isolated syndrome as defined by a history of a single episode of inflammatory demyelination documented by a neurologist, lack of evidence for alternative diagnoses, and two or more periventricular or ovoid hyperintense T2 lesions of >3 mm on magnetic resonance imaging (23). Untreated subjects are defined as having no disease-modifying treatment or steroids in the preceding 4 wk (steroids), 12 wk (glatiramer acetate, IFN-β1a or -β1b, and methotrexate) or 24 wk (Cytoxan and mitoxantrone). Progressive disease is defined as progressive functional decline with deficits lasting 6 mo or more.
Disease subtype definitions
Primary progressive disease is defined as disease progression from onset without a history of an initiating clinical attack. Secondary progressive disease is defined as an initially relapsing-remitting course followed by gradual progression with or without occasional relapses, minor remission, and plateaus. Relapsing-remitting disease is defined as disease characterized by clearly defined relapses with full recovery or with sequelae and residual deficit upon recovery. Periods between disease relapses are characterized by a lack of disease progression. Clinically isolated syndrome is defined as a single demyelinating event with a positive magnetic resonance imaging (two or more lesions ≥3 mm in diameter and characteristic of MS (e.g., periventricular or ovoid).
Definition of benign and malignant disease course
All subjects have a remitting-relapsing course. Benign subjects are defined as having an Expanded Disability Status Scale (EDSS) of ≤1, excluding visual function, 10–15 years after first symptom or EDSS of ≤2, excluding visual function, >15 years after first symptom. Malignant subjects are defined as having an EDSS of ≥6 within 5 years of first symptom.
sIL-2RA measurement using ELISA
ELISA measurement of sIL-2RA was performed according to the manufacturer’s recommendations (BD Biosciences). Serum samples were diluted 1/20 using PBS supplemented with 10% FBS. Microtiter plates were read using a Bio-Rad Benchmark microplate reader.
pSTAT5 phosphorylation analysis in ex vivo CD4+FoxP3+ T cells
Phosphorylation-state analysis was performed on human whole blood using BD Phosflow technology according to the manufacturer’s instructions (BD Biosciences), and as previously described (24). All human blood samples were obtained with informed consent and according to the Institutional Ethics Review Board Protocols. All blood samples were collected in sterile 10-ml lithium-heparin Monoject tubes. Four milliliters of fresh, ex vivo blood from healthy control donors were used per condition and time point. Blood samples were incubated with IL-2 (Proleukin; Chiron) or with a mixture of IL-2 and sIL-2RA (R&D Systems) in 50-ml polypropylene Falcon conical tubes for 30 min in a 37°C water bath. The neutralizing anti-IL2 mAb was purchased from R&D Systems (clone 5334). Fixation of cells and preservation of phosphorylation status were obtained by adding prewarmed BD Lyse/Fix buffer and incubation in a 37°C water bath. Permeabilization of cells was performed by incubation of cells in BD Perm Buffer III on ice for 30 min. Cells were subsequently washed twice with 2% FBS-PBS and stained using BD Staining Buffer (all reagents from BD Biosciences). Cells were stained using allophycocyanin mouse anti-human CD4 (clone RPA-T4; BD Biosciences), PE anti-human FoxP3 (clone 206D; Biolegend), and Alexa Fluor-488 mouse anti-human pSTAT5 (pY694; clone 47; BD Biosciences).
PBMCs and T cell isolation
PBMCs were isolated from heparinized venous blood by centrifugation over Ficoll-Hypaque (Amersham Pharmacia) according to standard methodologies. CD4+ T cells, isolated by negative selection using immunomagnetic beads (Miltenyi Biotec) from PBMCs, were FACS sorted on a FACS ARIA (BD Biosciences) after staining for HLA-DR (PerCP, clone L243), CD62L (allophycocyanin; clone Dreg 56), CD32 (FITC; clone 3D3), CD14 (FITC; clone M5E2), and CD116 (FITC; clone M5D12), all from BD Pharmingen; and CD25 (peripheral blood; clone BC96 from Bio-Legend) to typically >98% purity in post-sort analysis. The FITC-labeled mAbs were used as a combined mixture to ensure that no accessory cells were isolated in the target T cell population (CD4+DR−CD25−CD62Lhigh).
Cell culture
Cells were grown in serum-free X-VIVO15 medium (BioWhittaker). PBMCs, CD4+ T cells, CD8+ T cells, and monocytes were plated at 100,000 cells/well. CD4+DR−CD25−CD62Lhigh (Tresp) cells were plated at 2.5 × 103/well. T cell proliferation was assessed at 72 h by [3H]TdR incorporation (1 μCi/well) on a beta scintillation counter. Activation-induced cell death (AICD) was measured by annexin V staining using the annexin V-PE apoptosis detection kit following the manufacturer’s instructions (BD Pharmingen). Control Ig was purchased from R&D Systems.
Statistical analysis
Differences between means were tested using two-sample t tests or ANOVA, and adjustment for confound was completed using linear regression. When appropriate, a paired t test was used. sIL-2RA concentrations were log10 transformed before analysis. Values of p < 0.05 were considered significant.
Results
Serum sIL-2RA levels in healthy controls and subjects with MS
We analyzed sIL-2RA levels from 14 healthy control individuals, for which longitudinal samples taken over a period of 12 mo were available. As shown in Fig. 1A, sIL-2RA level is a remarkably stable phenotype in healthy controls. We extended this analysis to 15 untreated MS subjects with a relapsing-remitting disease course. Between 4 and 10 serum samples were available for this collection and were obtained over a maximum period of 3 years. Little fluctuation in sIL-2RA levels was observed in the longitudinal measurements of individual subjects with MS (Fig. 1B). However, some of the subjects with MS experienced marked transient elevations in sIL-2RA levels. Some of these elevations correlated with clinical or radiological evidence of disease exacerbations (data not shown). However, given the small number of such events, it is impossible to draw any conclusions as to the significance of these correlations. Furthermore, the subjects with relapsing-remitting disease course tested in this longitudinal study exhibited active disease, with multiple relapses a year.
FIGURE 1.

sIL-2RA serum levels in healthy controls and MS subjects. A, Longitudinal sIL-2RA measurements in healthy control subjects. Samples were obtained over a period of 12 mo. B, Longitudinal sIL-2RA measurements in MS subjects with relapsing-remitting disease course. Samples were obtained over a maximum period of 3 years. C, sIL-2RA measurement in 68 healthy control subjects vs 284 MS subjects. Difference between means was tested using a two-sample t test with unequal variance. A summary of demographic characteristics of study participants and storage duration of samples is given in Table I. The difference remained significant (p = 0.0001) after adjusting for age, gender, and sample storage duration (data not shown).
Next, in a collection of 68 healthy controls and 264 MS subjects, we quantified serum concentrations of sIL-2RA. Consistent with previous findings (4, 5, 17), we found that sIL-2RA levels are increased in MS subjects compared with healthy controls (mean sIL-2RA concentration in healthy controls, 2.022 ng/ml and 95% confidence interval, 1.852–2.192; mean sIL-2RA concentrations in MS subjects, 2.345 ng/ml and 95% confidence interval, 2.256–2.435; p = 0.9 × 10−4; Fig. 1C). We determined the percentage of the observed variance in sIL-2RA levels that is due to the genotypes we have discovered in subjects with MS. For the two genetic variants that we have previously correlated with sIL-2RA levels, between 2 and 5% of the variance observed in the MS cases was due to IL-2RA chain genotype, whereas in the healthy control subjects, between 15 and 18% of the variance observed was due to the genotype. This led us to hypothesize that perturbations associated with disease or treatment may influence sIL-2RA levels in the MS cases because less of the variance in sIL-2RA levels could be explained by IL2RA variants in the MS subject collection than in the healthy control collection.
Severe MS disease course influences sIL-2RA levels, but treatment does not
To explore the role of clinical events in sIL-2RA expression, we studied a new set of subjects with MS; these individuals were not used in our prior genetic analysis of sIL-2RA levels. Specifically, sIL-2RA levels were quantified in 22 MS subjects with primary progressive disease, 44 MS subjects with secondary progressive disease, and 83 untreated MS subjects with a relapsing-remitting disease course along with 60 untreated subjects with clinically isolated demyelinating syndrome (subjects with a single episode of demyelination; Table I). No significant differences among the four groups were observed (p = 0.13; Fig. 2A). We then studied additional subjects with MS representing extreme forms of the disease, namely, 115 subjects with benign MS and 60 subjects with malignant MS. sIL-2RA levels were increased in subjects with the malignant form of remitting-relapsing MS (p = 0.027; Fig. 2B).
Table I.
Summary of demographic characteristics of study participants and storage duration of serum samples
| Study Population | Females (%) | Males (%) | Mean Age (yr) at Time of Sample | Mean Sample Storage Duration (yr) |
|---|---|---|---|---|
| Healthy controls | 60.3 | 39.7 | 43 (20–68)a | 2.1 (1.27–3.15) |
| MS subjects | 74.2 | 25.88 | 43 (73–18) | 2.46 (1.17–3.36) |
| MS subtypes | ||||
| CISb | 78 | 22 | 44 (20–72) | 3.06 (1.3–4.55) |
| Primary progressive | 64.3 | 35.7 | 44 (19–74) | 3.1 (1.68–4.53) |
| Secondary progressive | 97.7 | 2.3 | 43 (27–64) | 3.09 (1.3–4.5) |
| Relapsing-remitting | 83.1 | 16.9 | 43 (23–73) | 2.23 (1.32–3.82) |
| Benign MS subjects | 77 | 13 | 48 (26 –73) | 2.8 (0.36–4.54) |
| Malignant MS subjects | 50 | 50 | 44 (19–75) | 2.82 (0.39–4.48) |
Numbers in parentheses, range.
CIS, clinically isolated syndrome.
FIGURE 2.
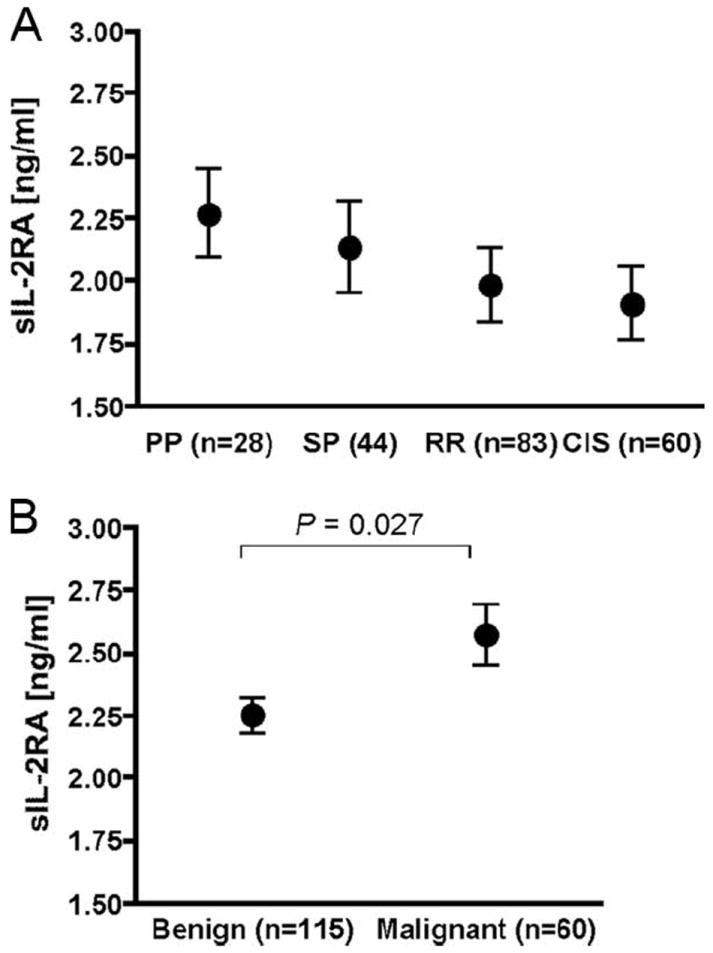
sIL-2RA serum levels in MS subjects categorized by disease subtype. A, Mean levels and 95% confidence intervals of sIL-2RA levels measured in subjects with primary progressive (PP), secondary progressive (SP), and untreated relapsing-remitting (RR) disease course as well as in untreated subjects with clinically isolated syndrome (CIS). Differences among groups were tested using an F test (p > 0.05). After adjusting for age, gender, and sample storage duration, similar results were obtained (p = 0.084). B, Mean levels and 95% confidence intervals of sIL-2RA levels measured in MS subjects with benign and malignant disease. Difference between means was tested using a two-sample t test with unequal variance. The difference remained significant (p = 0.03) after adjusting for age, gender, and sample storage duration. Disease definitions are provided in Materials and Methods. A summary of demographic characteristics of study participants and storage duration of samples is given in Table I. There is no overlap between the MS subjects tested in A and B.
We hypothesized that treatment of MS subjects may lower sIL-2RA levels. We thus measured sIL-2RA levels in a cohort of 15 subjects with MS for which pretreatment sera and up to 9 posttreatment serum samples obtained over a period of up to 4 years were available. No significant, prolonged effect of treatment was observed in this longitudinal study (Fig. 3 and Supplemental Fig. 1).4 Several patients experienced treatment changes over the course of the study period and/or received combination therapies. The effect of any specific treatment regimen on serum sIL-2RA levels will need to be investigated using larger sample sizes. In contrast to healthy controls or untreated MS patients, there was somewhat greater variability in sIL-2RA levels over time, which may be due to transient perturbation, disease activity, or a combination of these factors.
FIGURE 3.
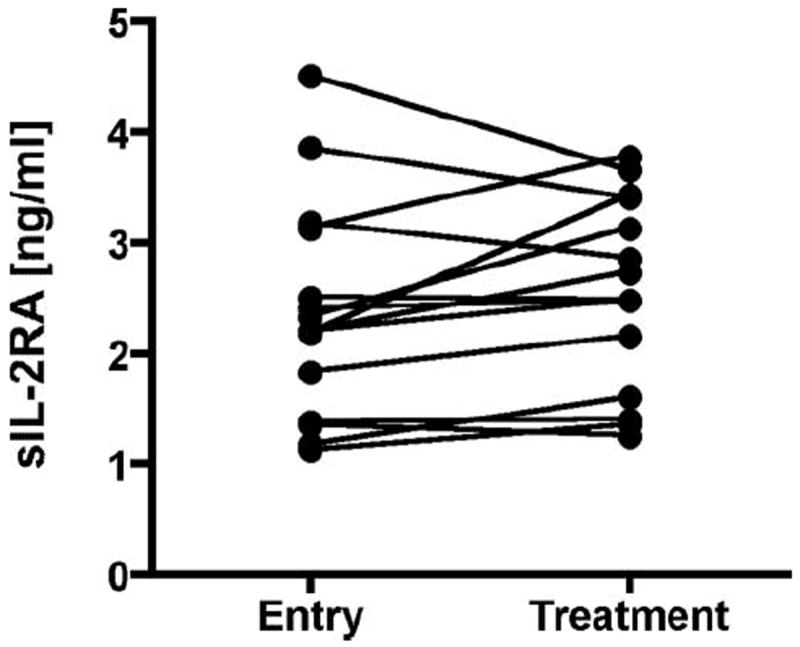
MS treatment does not affect sIL-2RA levels. sIL-2RA levels for treatment is the average of repeat measures (up to nine repeat sampling points) over a period of up to 4 years. A paired t test was used to test for a difference between the untreated value and the average treatment value. Treatment definitions are provided in Materials and Methods.
sIL-2RA inhibits IL-2-mediated signaling
We then explored a potential functional role for the sIL-2RA molecule given that little is known regarding the relevance of increased sIL-2RA levels in the pathogenesis of autoimmunity. sIL-2RA, like many other soluble cytokine receptors (18), might compete with cell surface IL-2RA for IL-2 binding and thus block IL-2 function. Alternately, sIL-2RA might complex with IL-2 and potentiate signaling, as has been demonstrated for sIL-6R-IL-6 (20) and sIL-15R-IL-15 complexes (19). However, the reported inhibition of proliferation of a murine CD8+ T cell line cultured in the presence of high doses of murine IL-2 and human sIL-2RA (19) makes this alternative hypothesis less likely.
We studied the effect of sIL-2RA on IL-2 signaling in human T cells. Because phosphorylation of signal transducer and activator of transcription 5 (STAT5) is strongly induced upon T cell activation with IL-2 (25), we measured STAT5 phosphorylation upon stimulation with IL-2 in ex vivo human CD4+ T cells. We used a flow cytometry-based phosphorylation assay (24) to detect STAT5 phosphorylation in response to IL-2 stimulation of T cells. As shown in Fig. 4A, the addition of sIL-2RA did not induce signaling in CD4+FoxP3− T responder cells or CD4+FoxP3+ regulatory T cells, which are characterized by high levels of IL-2RA cell surface expression. This indicated that sIL-2RA by itself does not exert agonistic function. In contrast, the addition of sIL-2RA strongly inhibited STAT5 phosphorylation in CD4+FoxP3− T cells and moderately in CD4+FoxP3+ T cells (Fig. 4B). This result suggests that sIL-2RA is an IL-2 antagonist.
FIGURE 4.
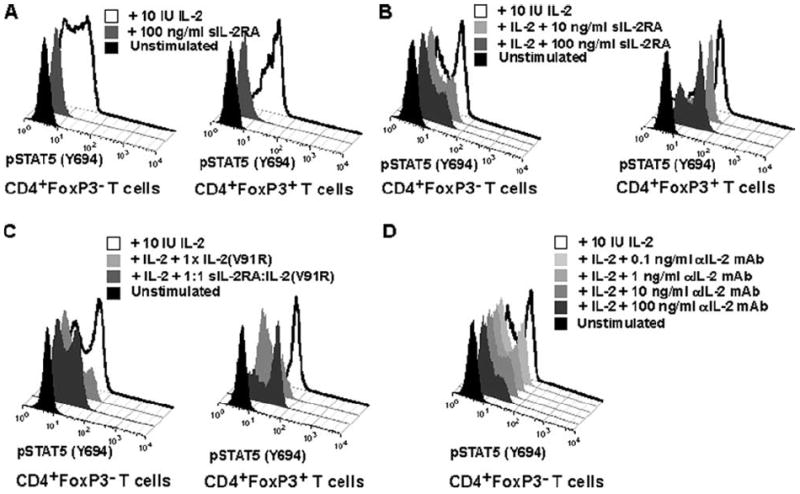
sIL-2RA inhibits IL-2-mediated signaling. A and B, FACS histograms showing pSTAT5 (Y694) phosphorylation in CD4+FoxP3− and CD4+FoxP3+ T cells obtained from fresh ex vivo blood from a healthy human subject. Background levels (black histogram), induction of phosphorylation following stimulation with 100 IU of IL-2/ml (white histogram), as well as the effect of sIL-2RA addition (gray histogram) are shown. Stimulation with IL-2 occurred for 30 min at 37°C. Results are representative of three independent experiments. C, sIL-2RA, complexed with an IL-2 antagonist, can reverse inhibition of IL-2 signaling by an IL-2 antagonist (mutated IL-2). FACS histograms showing pSTAT5 (Y694) phosphorylation in CD4+FoxP3+ in fresh ex vivo blood obtained from a healthy human subject. Background levels (black histogram) and induction of phosphorylation following stimulation with 10 IU of IL-2/ml (4 pM, open histogram) are shown as well as various experimental conditions with sIL-2RA and an IL-2 antagonist (mutated IL-2 V91R5). Mean fluorescence intensity values are: 10 IU IL-2 (39.6); IL-2 plus 1× IL-2 V91R (8.5); IL-2 plus 1:1 sIL-2RA-IL-2 V891R (30.8); unstimulated (5.9). Stimulation with IL-2 occurred for 30 min at 37°C. A 1:1 sIL-2RA:IL-2 (V91R) ratio means that sIL-2RA and IL-2 (V91R) were added in equimolar amounts (400 pM) and preincubated for 30 min at 37°C before addition to whole blood with IL-2. The addition of 1× IL-2 (V91) equals to the addition of 400 pM. Results are representative of two independent experiments. D, For comparison with the effect of sIL-2RA on pSTAT5 signaling, the inhibition of pSTAT5 signaling by a neutralizing anti (α)-IL-2 mAb is shown.
To examine whether sIL-2RA antagonism of STAT5 phosphorylation induced by IL-2 is due to the specific interaction of sIL-2RA, we used an IL-2 analog with a valine to arginine substitution at position 91. This analog has been shown to bind the IL-2 receptor without transducing signaling, effectively acting as an antagonist (34). We reasoned that by preincubating sIL-2RA with the engineered IL-2 analog at equimolar concentrations, we could selectively neutralize the IL-2 binding capacity of sIL-2RA. In Fig. 4C, we demonstrate that the ability of sIL-2RA to antagonize STAT5 phosphorylation (Fig. 4B) is lost when it is preincubated with the IL-2 analog. Furthermore, the analog by itself antagonizes STAT5 phosphorylation (Fig. 4C) Taken together, these data support the theory that sIL-2RA acts as an antagonist of IL-2 signaling through its ability to bind IL-2. A neutralizing anti-IL-2 mAb inhibited pSTAT5 signaling in a similar fashion to IL-2RA (Fig. 4D).
sIL-2RA promotes T cell activation and expansion
We have confirmed the observation by others that many cell types in the peripheral blood (T cells, monocytes, B cells) produce sIL-2RA and that the production of sIL-2RA is positively correlated with activation and proliferative responses in vitro (data not shown). To study the influence of sIL-2RA on T cell activation and expansion, sIL-2RA was added to anti-CD3-stimulated PBMCs, and T cell proliferation was measured after 72 h. We observed increases in proliferation in the presence of sIL-2RA at all doses of anti-CD3 stimulation (Fig. 5A). Analysis of eight healthy donors at an optimal amount of anti-CD3 mAb (0.5 μg/ml) demonstrated a modest yet consistent increase in proliferation (p = 0.01 using a paired, two-tailed t test). Elevated levels of IL-2 can potentiate AICD (26), and we speculated that increased sIL-2RA levels might inhibit AICD by reducing levels of IL-2. To test this hypothesis, we used FACS to obtain highly purified responder (CD4+DR−CD25−CD62Lhigh) T cells and then evaluated the influence of sIL-2RA on T cell expansion. Consistent with short-term (3 day) effects of sIL-2RA on proliferative response, we found that T cell numbers were markedly elevated at day 14 in the presence of sIL-2RA (Fig. 5, B and C), although we did not observe a significant difference in AICD as measured by annexin V staining (Fig. 5D).
FIGURE 5.

Effect of sIL-2RA on human T cell function. A, Total PBMCs were stimulated for 72 h with a range of plate-bound anti (α)-CD3 concentrations. sIL-2RA was added at a concentration of 5 ng/ml. Error bars represent SD in proliferation. Results are representative of eight independent experiments. B, FACS-sorted DR−CD25−CD62Lhigh T cells under light microscope on day 14 of culture. ×20. Cells were stimulated with anti-CD3-anti-CD28-conjugated beads and received either no treatment or 10 ng/ml sIL-2RA. C, FACS-sorted CD4+DR−CD25−CD62Lhigh T cells grown in culture for 14 days. Cells were stimulated with anti-CD3-anti-CD28-conjugated beads. Error bars represent SEM in cell number. Results are representative of three independent experiments. D, The addition of sIL-2RA does not reduce activation-induced cell death compared with addition of a control Ig (R&D Systems), as measured by the percentage of annexin V+CD4+CD25− T cells. CD4+CD25− T cells were negatively isolated using magnetic beads (Miltenyi Biotec) from PBMCs and cultured for up to 11 days. Data are representative of two independent experiments.
Discussion
Here, we characterized the levels of a biomarker of peripheral inflammation, sIL-2RA, in healthy controls and subjects with MS. It has been previously shown that genetic variants in the IL2RA locus correlate with the levels of sIL-2RA (9). Here we show that the IL2RA genotype more strongly affects sIL-2RA levels in healthy control subjects than in subjects with MS. Although longitudinal analyses demonstrated that sIL-2RA serum concentrations were a remarkably stable phenotype in both healthy controls and untreated MS subjects, a difference was observed between benign and malignant MS, indicating that in addition to specific allelic variants at IL2RA, immunological perturbations associated with extreme disease course can influence sIL-2RA levels. Whereas we observed that sIL-2RA at concentrations observed in sera can enhance the expansion and proliferation of CD4+ T cells cultured ex vivo, sIL-2RA inhibited IL-2 signaling in CD4+FoxP3+ T cells as measured by STAT5 phosphorylation. These data indicate that before disease onset, strong genetic factors associated with disease risk dictate sIL-2RA levels that are likely to modulate disease onset, potentially affecting the function of multiple lymphocyte subsets.
Our data agree with previous reports of increased levels of sIL-2RA in MS subjects compared with controls (4, 5, 17). In our longitudinal analyses, sIL-2RA serum concentrations were found to be a remarkably stable phenotype in both healthy controls and untreated MS subjects with relapsing-remitting disease course. These data are contrary to what was previously observed in a small study of 60 MS subjects with relapsing-remitting MS and 33 healthy controls, which showed fluctuations over time (27). Although no differences were observed in our collection of serum samples from subjects classified into the more commonly used categories of primary progressive, secondary progressive, and relapsing-remitting MS and clinically isolated syndrome, future studies with larger sample sizes might show subtle differences. We extended our study to subjects with different forms of MS and show a difference between the two extreme forms of MS, benign and malignant. These data indicate that in addition to specific allelic variants at IL2RA, extreme disease course may influence sIL-2RA levels in the serum of MS subjects.
A significant fraction of the variance in sIL-2RA is explained by the IL2RA variants tested thus far in healthy controls: the two variants previously associated with MS (rs12722489 and rs2104286) account for 15 and 18% of the total variance in log10-transformed sIL-2RA concentration in our healthy control collection, whereas only 2 and 5% of the variance are explained in subjects with MS. This is comparable with findings in a sample of 1351 T1D case plasma samples, where the 2 IL2RA variants (rs41295061 and rs11594656) accounted for 2.3 and 6.6%, respectively (9). In this large sample of T1D case samples, the effect of various covariates (including subject age, disease duration, collection month of sample) on sIL-2RA plasma concentration was also assessed. The studied covariates were found to account for 11.3% of the total variance in log10 sIL-2RA concentrations (9). Thus, the contribution of other factors, including additional genetic variants in immune response genes, will need to be determined.
Upon activation and entry into cell cycle the IL-2RA molecule is proteolytically cleaved off the surface of many cell types, including T cells, B cells, and monocytes (13, 28-30). However, it is as yet unclear how different cell types respond to human sIL-2RA and what its potential immunomodulatory role may be. Our group first demonstrated that although the frequency of CD4+CD25high regulatory T cells is normal, the cells are dysfunctional in subjects with MS (31). Given the importance of IL-2 to regulatory T cell function and the potential for sIL-2RA to neutralize available IL-2, we hypothesized that genetic variants at IL2RA that correlate with sIL-2RA may act by modulating levels of IL-2 and thereby affect suppressor cell function and thereby inflammatory disease. We have demonstrated that sIL-2RA can enhance the proliferation/expansion of responder CD4+ T cells (Fig. 5, A–C). Recent findings suggest an autoregulatory feedback loop for IL-2, in which IL-2 inhibits its own production (32, 33). Thus, the level of sIL-2RA may affect this autoinhibitory loop and hence IL-2 production. Moreover, it should be recognized that alterations in concentration or kinetics of production of sIL-2RA might affect both responder and regulatory T cells in the context of an autoimmune disease. In this regard, we observed an inhibitory effect of sIL-2RA on IL-2 signaling by CD4+FoxP3+ T cells as measured by STAT5 phosphorylation.
As yet, three associations for T1D susceptibility that are independent of each other and two independent associations for MS susceptibility have been discovered. Despite clear associations and correlations between genetic variants in IL2RA and both autoimmune disease susceptibility (T1D, MS and Graves’ disease; Refs. 8-10) and sIL-2RA levels (9), correlating disease susceptibility and sIL-2RA levels is not trivial. In the original report of correlations between sIL-2RA levels and T1D risk alleles (9), the SNP with the strongest association to T1D risk was not the SNP with the strongest correlation to sIL-2RA. Moreover, it was suggested that T1D risk alleles correlate with reduced sIL-2RA levels (9). In contrast, we have shown that the MS risk alleles correlate with increased levels.6 Indeed, a combined genetic and phenotypic analysis of T1D and MS alleles indicates that a novel IL2RA association with T1D harbors a risk allele that correlates with increased rather than decreased sIL-2RA levels).6 These data also emphasize the value of comparative analysis of susceptibility loci in multiple autoimmune diseases. Thus, although many autoimmune diseases may share common susceptibility loci, it is apparent now that there may be both allelic and phenotypic heterogeneity within these loci among these diseases, including IL2RA (35). These data indicate that the net level of sIL-2RA could be determined by several variants at IL2RA and that susceptibility to autoimmunity at the IL2RA locus will be defined by a complex interplay of many variants. Significantly, although IL2RA genotype is associated with several autoimmune diseases, the extent to which sIL-2RA influences, and is influenced by, these disease processes may differ.
Future large-scale studies will need to address the overlap and relationship between variants associated with disease risk and correlated with sIL-2RA levels. Even though we demonstrate a functional role of sIL-2RA on immune responses, sIL-2RA may not be the primary causal factor regulated by IL2RA genetic variants: given that sIL-2RA is a cleavage product of surface IL-2RA, an individual’s sIL-2RA level may simply reflect of the individual’s surface expression of IL-2RA or ability to up-regulate IL-2RA expression.
Supplementary Material
Acknowledgments
Sample collection in the MS Registry project was supported by a collaboration with Millenium Pharmaceuticals. We thank Ms. Mira Weiner for her work in subject recruitment and sample collection.
Footnotes
This work was funded by the National Multiple Sclerosis Society, the National Institutes of Health-National Institute of Allergy and Infectious Diseases (P01 AI39671) and the National Institutes of Health (R01 NS049477). Further support was provided by a JDRF Postdoctoral Fellowship (to L.M.M.), a grant by the American Cancer Society (to D.E.A.), a Harry Weaver Neuroscience Scholar award by the National Multiple Sclerosis Society (to P.L.D.), and Jacob Javits Merit Award NS2427 (to D.A.H.) from the National Institute of Neurological Disorders and Stroke.
Abbreviations used in this paper: MS, multiple sclerosis; SNP, single nucleotide polymorphism; T1D, type 1 diabetes; sIL-2RA, soluble IL-2RA; EDSS, Expanded Disability Status Scale; AICD, activation-induced cell death.
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Hafler DA, Slavik JM, Anderson DE, O’Connor KC, De Jager P, Baecher-Allan C. Multiple sclerosis. Immunol Rev. 2005;204:208–231. doi: 10.1111/j.0105-2896.2005.00240.x. [DOI] [PubMed] [Google Scholar]
- 2.Hafler DA, Fox DA, Manning ME, Schlossmann SF, Reinherz EL, Weiner HL. In vivo activated T lymphocytes in the peripheral blood and cerebrospinal fluid of patients with multiple sclerosis. N Engl J Med. 1985;312:1405–1411. doi: 10.1056/NEJM198505303122201. [DOI] [PubMed] [Google Scholar]
- 3.Yednock TA, Cannon C, Fritz LC, Sanchez-Madrid F, Steinman L, Karin N. Prevention of experimental autoimmune encephalomyelitis by antibodies against α4β1 integrin. Nature. 1992;356:63–66. doi: 10.1038/356063a0. [DOI] [PubMed] [Google Scholar]
- 4.Greenberg SJ, Marcon L, Hurwitz BJ, Waldmann TA, Nelson DL. Elevated levels of soluble interleukin-2 receptors in multiple sclerosis. N Engl J Med. 1988;319:1019–1020. doi: 10.1056/NEJM198810133191517. [DOI] [PubMed] [Google Scholar]
- 5.Gallo P, Piccinno MG, Pagni S, Argentiero V, Giometto B, Bozza F, Tavolato B. Immune activation in multiple sclerosis: study of IL-2, sIL-2R, and γ-IFN levels in serum and cerebrospinal fluid. J Neurol Sci. 1989;92:9–15. doi: 10.1016/0022-510x(89)90171-8. [DOI] [PubMed] [Google Scholar]
- 6.Trapp BD, Ransohoff R, Rudick R. Axonal pathology in multiple sclerosis: relationship to neurologic disability. Curr Opin Neurol. 1999;12:295–302. doi: 10.1097/00019052-199906000-00008. [DOI] [PubMed] [Google Scholar]
- 7.Hauser SL, Oksenberg JR. The neurobiology of multiple sclerosis: genes, inflammation, and neurodegeneration. Neuron. 2006;52:61–76. doi: 10.1016/j.neuron.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 8.The International Multiple Sclerosis Genetics Consortium. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med. 2007;357:851–862. doi: 10.1056/NEJMoa073493. [DOI] [PubMed] [Google Scholar]
- 9.Lowe CE, Cooper JD, Brusko T, Walker NM, Smyth DJ, Bailey R, Bourget K, Plagnol V, Field S, Atkinson M, et al. Large-scale genetic fine mapping and genotype-phenotype associations implicate polymorphism in the IL2RA region in type 1 diabetes. Nat Genet. 2007;39:1074–1082. doi: 10.1038/ng2102. [DOI] [PubMed] [Google Scholar]
- 10.Brand OJ, Lowe CE, Heward JM, Franklyn JA, Cooper JD, Todd JA, Gough SC. Association of the interleukin-2 receptor α (IL-2Rα)/CD25 gene region with Graves’ disease using a multilocus test and tag SNPs. Clin Endocrinol. 2007;66:508–512. doi: 10.1111/j.1365-2265.2007.02762.x. [DOI] [PubMed] [Google Scholar]
- 11.Malek TR, Bayer AL. Tolerance, not immunity, crucially depends on IL-2. Nat Rev Immunol. 2004;4:665–674. doi: 10.1038/nri1435. [DOI] [PubMed] [Google Scholar]
- 12.Robb RJ, Kutny RM. Structure-function relationships for the IL 2-receptor system, IV: analysis of the sequence and ligand-binding properties of soluble Tac protein. J Immunol. 1987;139:855–862. [PubMed] [Google Scholar]
- 13.Rubin LA, Galli F, Greene WC, Nelson DL, Jay G. The molecular basis for the generation of the human soluble interleukin 2 receptor. Cytokine. 1990;2:330–336. doi: 10.1016/1043-4666(90)90062-x. [DOI] [PubMed] [Google Scholar]
- 14.Kim HP, Imbert J, Leonard WJ. Both integrated and differential regulation of components of the IL-2/IL-2 receptor system. Cytokine Growth Factor Rev. 2006;17:349–366. doi: 10.1016/j.cytogfr.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 15.Makis AC, Galanakis E, Hatzimichael EC, Papadopoulou ZL, Siamopoulou A, Bourantas KL. Serum levels of soluble interleukin-2 receptor α (sIL-2Rα) as a predictor of outcome in brucellosis. J Infect. 2005;51:206–210. doi: 10.1016/j.jinf.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 16.Giordano C, Galluzzo A, Marco A, Panto F, Amato MP, Caruso C, Bompiani GD. Increased soluble interleukin-2 receptor levels in the sera of type 1 diabetic patients. Diabetes Res. 1988;8:135–138. [PubMed] [Google Scholar]
- 17.Adachi K, Kumamoto T, Araki S. Interleukin-2 receptor levels indicating relapse in multiple sclerosis. Lancet. 1989;1:559–560. doi: 10.1016/s0140-6736(89)90103-7. [DOI] [PubMed] [Google Scholar]
- 18.Rose-John S, Heinrich PC. Soluble receptors for cytokines and growth factors: generation and biological function. Biochem J. 1994;300(Pt. 2):281–290. doi: 10.1042/bj3000281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rubinstein MP, Kovar M, Purton JF, Cho JH, Boyman O, Surh CD, Sprent J. Converting IL-15 to a superagonist by binding to soluble IL-15Rα. Proc Natl Acad Sci USA. 2006;103:9166–9171. doi: 10.1073/pnas.0600240103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones SA, Horiuchi S, Topley N, Yamamoto N, Fuller GM. The soluble interleukin 6 receptor: mechanisms of production and implications in disease. FASEB J. 2001;15:43–58. doi: 10.1096/fj.99-1003rev. [DOI] [PubMed] [Google Scholar]
- 21.Sakaguchi S, Ono M, Setoguchi R, Yagi H, Hori S, Fehervari Z, Shimizu J, Takahashi T, Nomura T. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev. 2006;212:8–27. doi: 10.1111/j.0105-2896.2006.00427.x. [DOI] [PubMed] [Google Scholar]
- 22.McDonald WI, Compston A, Edan G, Goodkin D, Hartung HP, Lublin FD, McFarland HF, Paty DW, Polman CH, Reingold SC, et al. Recommended diagnostic criteria for multiple sclerosis: guidelines from the international panel on the diagnosis of multiple sclerosis. Ann Neurol. 2001;50:121–127. doi: 10.1002/ana.1032. [DOI] [PubMed] [Google Scholar]
- 23.Miller D, Barkhof F, Montalban X, Thompson A, Filippi M. Clinically isolated syndromes suggestive of multiple sclerosis: II. Non-conventional MRI, recovery processes, and management. Lancet Neurol. 2005;4:341–348. doi: 10.1016/S1474-4422(05)70095-8. [DOI] [PubMed] [Google Scholar]
- 24.Maier LM, Anderson DE, De Jager PL, Wicker LS, Hafler DA. Allelic variant in CTLA4 alters T cell phosphorylation patterns. Proc Natl Acad Sci USA. 2007;104:18607–18612. doi: 10.1073/pnas.0706409104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leonard WJ, O’Shea JJ. Jaks and STATs: biological implications. Annu Rev Immunol. 1998;16:293–322. doi: 10.1146/annurev.immunol.16.1.293. [DOI] [PubMed] [Google Scholar]
- 26.Waldmann TA. The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nat Rev Immunol. 2006;6:595–601. doi: 10.1038/nri1901. [DOI] [PubMed] [Google Scholar]
- 27.Freedman MS, Muth KL, Trotter JL, Yoshizawa CN, Antel JP. Prospective serial analysis of interleukin-2 and soluble interleukin-2 receptor in relapsing-remitting multiple sclerosis. Neurology. 1992;42:1596–1601. doi: 10.1212/wnl.42.8.1596. [DOI] [PubMed] [Google Scholar]
- 28.Rubin LA, Kurman CC, Fritz ME, Biddison WE, Boutin B, Yarchoan R, Nelson DL. Soluble interleukin 2 receptors are released from activated human lymphoid cells in vitro. J Immunol. 1985;135:3172–3177. [PubMed] [Google Scholar]
- 29.Kniep EM, Strelow I, Lohmann-Matthes ML. The monocyte interleukin-2 receptor light chain: production of cell-associated and soluble interleukin-2 receptor by monocytes. Immunology. 1992;75:299–304. [PMC free article] [PubMed] [Google Scholar]
- 30.Holter W, Goldman CK, Casabo L, Nelson DL, Greene WC, Waldmann TA. Expression of functional IL 2 receptors by lipopolysaccharide and interferon-γ stimulated human monocytes. J Immunol. 1987;138:2917–2922. [PubMed] [Google Scholar]
- 31.Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med. 2004;199:971–979. doi: 10.1084/jem.20031579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gong D, Malek TR. Cytokine-dependent Blimp-1 expression in activated T cells inhibits IL-2 production. J Immunol. 2007;178:242–252. doi: 10.4049/jimmunol.178.1.242. [DOI] [PubMed] [Google Scholar]
- 33.Villarino AV, Tato CM, Stumhofer JS, Yao Z, Cui YK, Hennighausen L, O’Shea JJ, Hunter CA. Helper T cell IL-2 production is limited by negative feedback and STAT-dependent cytokine signals. J Exp Med. 2007;204:65–71. doi: 10.1084/jem.20061198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu DV, Maier LM, Hafler DA, Wittrup KD. Engineered interleukin-2 antagonists for inhibition of regulatory T cells. doi: 10.1097/CJI.0b013e3181b528da. Submitted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maier LM, Lowe CE, Cooper J, Downes K, Anderson DE, Severson C, Clark PM, Healy B, Walker N, Aubin C, et al. IL2RA genetic heterogeneity in multiple sclerosis and type 1 diabetes susceptibility and soluble interleukin-2 receptor production. PLoS Genetics. doi: 10.1371/journal.pgen.1000322. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.