

Abstract
Interactions between the herpesvirus entry mediator (HVEM) and the B- and T-lymphocyte attenuator (BTLA) inhibit B and T cell activation. HVEM-BTLA interactions are blocked by herpes simplex virus (HSV) glycoprotein D (gD) through binding of its N-terminal domain to the BTLA binding site of HVEM. In this study, we inserted viral antigens into the C-terminal domain of gD and expressed these antigens with plasmid or E1-deleted (replication-defective) adenovirus vectors. Viral antigens fused to gD induced T and B cell responses to the antigen that were far more potent than those elicited by the same antigen expressed without gD. The immunopotentiating effect required binding of the gD chimeric protein to HVEM. Overall, the studies demonstrate that targeting of antigen to the BTLA binding site of HVEM augments the immunogenicity of vaccines.
Interactions between HVEM, a member of the tumor necrosis factor receptor family, and LIGHT (a protein that is homologous to lymphotoxin, is inducible, competes for gD of herpesvirus, binds HVEM and is expressed on activated T lymphocytes) provide costimulatory signals and contribute to T cell induction1. HVEM also interacts with another tumor necrosis factor family member, lymphotoxin-α (ref. 2), and with BTLA (ref. 3). Interactions between HVEM and BTLA inhibit T cell activation in vitro3, defining these molecules as part of an inhibitory pathway. HSV gD, a structural component of the virus envelope that is essential for virus entry into host cells4, was the first known HVEM ligand5, and gD-HVEM (refs. 6,7) and BTLA-HVEM (refs. 3,8) contact residues have been identified. The HVEM contact residues are contained within two short segments of the hairpin loop structure in the N-terminal portion of gD (ref. 6). gD binds to HVEM at the same site as BTLA, and soluble gD can block BTLA-HVEM interactions8,9. Like BTLA, cytotoxic T-lymphocyte antigen 4 (CTLA-4) and programmed death-1 (PD-1) provide inhibitory signals for T cell activation10. The latter inhibitory receptors are not expressed on naive T cells, but they are induced after activation. In contrast, BTLA is expressed both on naive T cells and, at higher levels, on activated T cells.
Manipulation of co-inhibitory pathways can modify antigen-specific T cell responses, as has been shown by blockage of the PD-1–PD-L1 pathway10 or by treatment of tumor-bearing mice with an antibody to CTLA-411. Here we show that subunit vaccines expressing antigens as fusion proteins within HSV gD induce markedly higher antigen-specific T and B cell responses. The increase in adaptive immune responses depends on the ability of gD to bind to HVEM and can be augmented further by structural modifications of gD that increase binding to HVEM. Overall, these results show that interrupting the HVEM-BTLA inhibitory pathway during vaccination can substantially enhance immune responses and suggest a simple strategy for increasing vaccine efficacy.
RESULTS
gD chimeric proteins bind to HVEM
To evaluate whether blockade of the HVEM-BTLA pathway enhances adaptive immune responses, we inserted two distinct antigens into the C terminus of HSV-1 gD. One chimeric protein contains a truncated form of HIV-1 Gag (ref. 12) inserted after amino acid 288 of gD (Fig. 1a). The other is composed of the three oncoproteins of HPV-16, E7, E6 and E5, linked together and inserted after amino acid 244 of gD (Fig. 1b). The gD-antigen chimeric genes were inserted into DNA vaccines and E1-deleted, chimpanzee-derived adenovirus serotype 68 (AdC68) vectors13.
Figure 1.

gD chimeric proteins bind HVEM. Schematic representation of the chimeric protein gD-Gag (a), which contains the truncated form of Gag from HIV-1 clade B inserted into HSV-1 gD after amino acid 288, and the gD-E7E6E5 chimeric protein (b), which contains the HPV-16 E7, E6 and E5 oncoproteins inserted into HSV-1 gD after amino acid 244. TMR, transmembrane region. (c) ELISAs were performed with serial dilutions of protein extracts from CHO/CAR cells infected with either AdC68E7E6E5, AdC68Gag, AdC68gD, AdC68gD-E7E6E5 or AdC68gD-Gag applied on microplates coated with HVEM. Data show one representative experiment of two. The difference between gD-E7E6E5 and gD-Gag was not statistically significant (P = 0.22), but both were statistically different when compared to gD (P = 0.019, P = 0.008, respectively). (d) The effect of gD or chimeric gD on binding of LIGHT to HVEM was tested by ELISA. HVEM-coated plates were incubated with protein extracts from CHO/CAR cells infected with either AdC68E7E6E5, AdC68Gag, AdC68gD, AdC68gD-E7E6E5 or AdC68gD-Gag. Protein extract from noninfected cells was used as a negative control. Serial dilutions of human LIGHT were added and the amount of bound LIGHT was determined. Protein extracts were normalized for gD content. Differences between samples were not statistically significant (P = 0.14). (e) Confocal microscopy was carried out with B78-H1/3E5 cells, which express HVEM fused to EGFP. B78-H1/3E5 cells were infected with AdC68E7E6E5, AdC68Gag, AdC68gD, AdC68gD-E7E6E5 or AdC68gD-Gag, then stained for surface gD. AdC68gD-E7E6E– and AdC68gD-Gag–infected cells were permeabilized and then stained for gD. Bar, 7.5 μm. Green, HVEM-EGFP; red, gD; yellow, colocalization of HVEM with gD. Perm, permeabilized. (f) Comparison of gD expression on the surface of B78-H1 (HVEM−) and B78-H1/3E5 (HVEM+) cells infected with AdC68 vectors carrying gD-Gag, gD-E7E6E5, gD, Gag or E7E6E5. The dotted line shows peak fluorescence of noninfected cells.
We performed binding assays to determine whether the insertion of foreign sequences into gD affects its interactions with HVEM. Protein extracts from cells infected with AdC68 vectors encoding gD, E7E6E5, gD-E7E6E5, Gag or gD-Gag were diluted to normalize the gD content and added to plates coated with HVEM. The gD-Gag and gD-E7E6E5 chimeric proteins showed enhanced binding to HVEM compared to gD (Fig. 1c); this may reflect changes in the overall structure of gD upon modification of the C terminus. The effect of gD on LIGHT binding to HVEM was tested on HVEM-coated plates treated with saturating amounts of protein extracts containing Gag, E7E6E5, gD, gD-Gag or gD-E7E6E5 and then treated with dilutions of purified LIGHT protein. LIGHT binding was not changed by pretreatment of HVEM with any of the gD preparations (Fig. 1d). Similar results were obtained when LIGHT binding was tested on HVEM+ cells pretreated with the different protein extracts (Supplementary Fig. 1a online). In the reverse experiment, binding of human HVEM to human LIGHT was not affected by pretreatment of HVEM with gD or gD chimeric proteins (Supplementary Fig. 1b,c).
To examine how insertion of a foreign sequence within the C terminus may modify folding of gD, we modeled the structure of unligated or HVEM-bound gD-Gag (Supplementary Fig. 2a–d online). The C terminus of the unligated gD structure is anchored near the N-terminal region, masking the HVEM binding site15 (Supplementary Fig. 2b). Computational modeling of the gD-Gag structure predicted that the C terminus would be shifted away from the N-terminal portion without altering the HVEM-binding N terminus (Supplementary Fig. 2a,b). A superposition of the gD X-ray crystallographic structure over the gD-Gag model indicates that insertion of Gag into gD does not disrupt the integrity of the HVEM binding surface (Supplementary Fig. 2b,d).
Binding of gD to HVEM presumably depends on secretion or cell surface expression of gD. To determine the cellular localization of the chimeric gD proteins, we infected B78-H1/3E5 cells, which express human HVEM-EGFP on their surface, with AdC68E7E6E5, AdC68Gag, AdC68gD, AdC68gD-E7E6E5 or AdC68gD-Gag, stained them with a monoclonal antibody (mAb) to gD, and analyzed the cells by confocal microscopy (Fig. 1e). HVEM colocalized with gD, gD-Gag or gD-E7E6E5 on the surface of and within infected cells. Absolute levels of gD-Gag and gD-E7E6E5 expressed on the surface of infected cells were below those of native gD (Fig. 1f). Analyses of mRNA levels showed that all vectors transcribed the chimeric genes at amounts equal to their corresponding nonchimeric versions (data not shown). Thus the lower cell surface expression of the chimeric proteins suggests their inefficient secretion, rapid re-internalization or accelerated proteolytic degradation. HVEM on the cell surface increased cell surface expression of chimeric gD (Fig. 1f), suggesting that intracellular binding of HVEM to the gD fusion proteins stabilized them or facilitated their export.
When AdC68gD- or AdC68gD-Gag–infected, HVEM-negative (HVEM−) cells were mixed with uninfected cells expressing HVEM (HVEM+), both gD and gD-Gag colocalized with HVEM on the uninfected HVEM+ cells, indicating that some of the protein was released and then bound by HVEM. This was more pronounced with gD-Gag than with gD and was not observed with gD-E7E6E5, and it may reflect increased secretion of the chimeric gD-Gag protein (Supplementary Fig. 3 online).
Expression of antigens within gD augments T cell activation
To determine whether expression of gD affects stimulation of CD8+ T cells to epitopes expressed within the gD, we performed proliferation assays. Irradiated cells from draining lymph nodes of mice intramuscularly (i.m.) immunized with AdC68 carrying gD, E7E6E5, gD-E7E6E5, Gag or gD-Gag were pulsed with low amounts of the SIINFEKL peptide and served as antigen-presenting cells carboxyfluorescein diacetate succinimidyl diester (CFSE)-labeled CD8+ T cells from OT-1 mice, which carry CD8+ T cells with a transgenic receptor specific for SIINFEKL bound to class I major histocompatibility complex (MHC) Kb. We had shown previously that i.m. application of an AdC68 vector causes an accumulation of vector-transduced mature dendritic cells within draining lymph nodes14. We therefore expected that upon application of the AdC68 vectors carrying gD with or without its fusion partners, some of the mature dendritic cells would express gD, which in turn may modulate the response of OT-1 derived CD8+ T cells to their cognate antigen bound to Kb on the same cells. OT-1 CD8+ T cells proliferated more vigorously upon co-culture with lymph node cells from mice that received any of the gD-expressing AdC68 vectors than upon co-culture with cells from uninjected mice or mice injected with a vector not expressing gD (Supplementary Fig. 4 online). The same set of lymph node lymphocytes was tested for expression of co-stimulatory molecules (CD80, CD86 and CD40) and MHC class II antigens on CD11c+ cells. Expression of these markers was similar in CD11c+ cells from mice that had received AdC68 vectors with or without gD (data not shown), indicating that gD had not affected dendritic cell maturation.
To evaluate whether blockade of the HVEM inhibitory pathway enhances adaptive immune responses in vivo, we vaccinated mice with DNA and AdC68 vectors expressing the gD chimeric proteins and then tested the T and B cell responses in comparison to those of mice injected with vectors expressing E7E6E5 or Gag without gD. Mice did not mount detectable E7-specific CD8+ T cell responses after vaccination with vaccines consisting of AdC68 encoding either E7E6E5 (Fig. 2a) or E7 alone (data not shown). In contrast, the DNA vaccine and the AdC68 vector expressing either E7 (data not shown) or the E7E6E5 fusion polypeptide within gD (Fig. 2a) induced robust E7-specific CD8+ T cell responses. Similarly, the DNA vaccine expressing the gD-Gag chimeric protein stimulated more potent Gagspecific CD8+ T cell responses than that expressing Gag only (Fig. 2a). The AdC68 vector expressing the gD-Gag chimeric protein also elicited stronger Gag-specific CD8+ (Fig. 2a) and CD4+ (Supplementary Fig. 5 online) T cell responses than that expressing Gag only, a difference especially pronounced at low vector doses (Fig. 2b). CD8+ T cell responses were maintained at stable frequencies for a year, indicating that the enhancement of the primary T cell response resulted in an increase of memory T cells (Fig. 2c).
Figure 2.

CD8+ T cell responses to vectors expressing antigens fused to gD. (a) Intracellular IFN-γ staining of E7- and Gag-specific CD8+ T cells was carried out on PBMCs from mice immunized i.m. with DNA vaccines (top graphs) or AdC68 vectors (bottom graphs) expressing either gD, E7E6E5, gD-E7E6E5, Gag or gD-Gag after stimulation. Cells were stained for surface-expressed CD8 (FITC-labeled antibody) and intracellular IFN-γ (PE-labeled antibody). PBMCs were isolated from mice 14 d after DNA vaccination or 10 d after application of AdC68 vector. The numbers in the right upper corners show frequencies of IFN-γ–producing CD8+ T cells as a percentage of all CD8+ T cells. IFN-γ+CD8+ T cell frequencies stimulated with an unrelated control peptide were below 0.2% in all groups. (b) Gag-specific CD8+ T cell frequencies were determined 10 d after immunization of mice with decreasing doses of either AdC68Gag or AdC68gD-Gag vectors. (c) The kinetics of E7-specific CD8+ T cell responses induced by the AdC68gD-E7E6E5 vector were analyzed from PBMCs of mice immunized with either AdC68E7E6E5, AdC68gD or AdC68gD-E7E6E5 vectors. (d) E7-specific IFN-γ+CD8+ responses were evaluated with splenocytes from mice immunized with DNA vaccines expressing the E7E6E5 polypeptide within either wild-type gD (pgD-E7E6E5), a mutated gD that shows loss of binding to HVEM (NBEFgD-E7E6E5) or a mutated gD that shows enhanced binding to HVEM (SgD-E7E6E5). (e) Splenocytes from mice immunized with DNA vaccines carrying E7 fused to either wild-type gD (gD-E7) or mutated gD with high affinity to HVEM (SgD-E7) were evaluated for E7-specific IFN-γ+CD8+ response.
Expression of antigens within gD increases antibody response
To analyze whether antibody responses can be enhanced by expression of the antigen within gD, we analyzed the sera of mice immunized with the AdC68 vectors expressing Gag or gD-Gag for antibodies to Gag (Table 1). The AdC68Gag vector induced only marginal levels of Gag-specific antibodies, whereas the AdC68gD-Gag vector elicited a significantly (P = 2.6 × 10−7) higher response that remained detectable for at least 11 months (Table 1). Antibody responses to gD were comparable in mice vaccinated with the AdC68gD or AdC68gD-Gag vector (data not shown).
Table 1.
Gag-specific antibody response after immunization with AdC68 vectors expressing gD, Gag, or gD-Gag
| Gag-specific antibody titer ± s.d.a (P value)b |
||||
|---|---|---|---|---|
| Immunization | 10 d | 7 weeks | 20 weeks | 49 weeks |
| AdC68Gag | 26 ± 8 | 35 ± 8 | 152 ± 19 | 6 ± 4 |
| AdC68gD | 3 ± 4 (0.007) |
9 ± 4 (0.0037) |
28 ± 15 (0.0004) |
8 ± 2 (0.2573) |
| AdC68gD-Gag | 287 ± 7 (2.6 × 10−7) |
740 ± 2 (6.8 × 10−9) |
1257 ± 21 (4.8 × 10−6) |
294 ± 8 (3.3 × 10−7) |
Gag-specific immunoglobulin titers were established as the reciprocal of the serum dilution that gave an absorbance twice above that of the sera from naive mice.
P values were determined with the one-tailed Student’s t-test comparing titers from mice immunized with either AdC68gD or AdC68gD-Gag with titers from mice immunized with AdC68Gag.
gD-HVEM interaction is needed to augment immune responses
To determine whether enhancement of CD8+ T cell responses by expression of an antigen within gD requires binding of gD to HVEM, we constructed DNA vaccines expressing the E7E6E5 sequence within two modified versions of gD. In the NBEFgD-E7E6E5 construct, seven amino acids at the N terminus of gD, Met11, Asn15, Leu25, Gln27, Leu28, Thr29 and Asp30, which are crucial for binding to HVEM (ref. 6), were replaced with alanine residues. In the SgD-E7E6E5 construct, the tryptophan in position 294 of gD was changed to alanine. This modification has been shown to increase binding of gD306 to HVEM (ref. 7). Mice immunized with one dose of a DNA vaccine expressing NBEFgD-E7E6E5 did not have detectable frequencies of E7-specific CD8+ T cells (Fig. 2d), whereas the frequencies of E7-specific IFN-γ+CD8+ T cells induced by pSgD-E7E6E5 were higher than those induced by pgD-E7E6E5 (Fig. 2d). To confirm this observation, we modified the pgD-E7 DNA vaccine15, which carries only E7 inserted into gD, by changing the tryptophan in position 294 to alanine. This new vector, termed pSgD-E7, stimulated significantly (P = 0.00109) higher frequencies of E7-specific CD8+ T cells compared to a plasmid vector expressing E7 within the wild-type form of gD (Fig. 2e). Taken together, these results show that binding to HVEM is essential for the immunopotentiating effect of gD, and gD mutations that increase its binding to HVEM may further augment CD8+ T cell responses.
Functionality of CD8+ T cells induced by gD chimeric proteins
We analyzed the phenotypes of vaccine-induced CD8+ T cells from mice immunized with AdC68 vectors expressing gD-Gag or Gag (Fig. 3). We measured expression of differentiation markers such as CD25, CD122, CD127, CD27, CD62L, CD69, CD103, CD43, CD44, CD54, Bcl2, BTLA, CTLA-4 and PD-1 on Gag-specific CD8+ T cells. Most of the markers tested (CD122, CD127, CD27, CD62L, CD69, CD103, CD43, CD44, CD54, Bcl2, BTLA, CTLA-4 and PD-1) were altered on antigen-specific CD8+ T cells compared to naive CD8+ T cells, and expression levels on CD8+ T cells induced by Gag or gD-Gag were identical (Fig. 3). CD27 expression was increased on a subpopulation of gD-Gag–induced Gag-specific CD8+ T cells, whereas CTLA-4 expression was marginally lower when compared to CD8+ T cells induced by Gag alone (Fig. 3). Overall, although AdC68gD-Gag elicited higher frequencies of Gag-specific CD8+ T cells than did AdC68Gag, the phenotypic profiles of the resultant effector cells were very similar.
Figure 3.
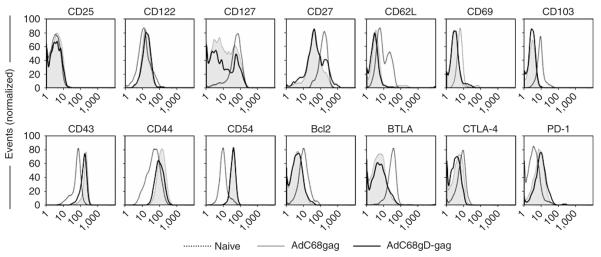
Phenotypes of Gag-specific CD8+ T cells were analyzed in PBMCs from mice immunized with either AdC68gD-Gag or AdC68Gag. PBMCs were isolated 10 d after immunization. Naive mice were used as controls. The graphs show expression levels on all CD8+ T cells from naive mice and Gag-tet+CD8+ T cells from mice immunized with either AdC68Gag or AdC68gD-Gag. Bcl-2 and CTLA-4 were stained upon permeabilizing the cell membranes. The x axis shows fluorescence intensity on a log scale.
We further assessed T cell functionality by testing whether mice vaccinated with either DNA (Fig. 4a) or AdC68 vectors (Fig. 4b) expressing E7E6E5 with or without gD were protected against challenge with TC-1 cells, which are lung epithelial cells of C57BL/6 origin transformed with v-Ha-ras and the E6 and E7 of HPV-16 (ref. 16). Mice immunized with pgD-E7E6E5 or AdC68gD-E7E6E5 were completely protected against TC-1 tumor progression, and protection was only seen upon vaccination with constructs carrying gD fusion proteins (Fig. 4a,b). Complete protection was observed at the low dose of 1 × 108 AdC68gDE7E6E5 viral particles (Fig. 4c). Mice vaccinated with DNA vaccines expressing E7 or E7E6E5 within mutant forms of gD were also tested for protection against TC-1 tumor formation. Protection correlated with CD8+ T cell responses; mice immunized with a vector expressing E7E6E5 within the gD mutant that lacks binding to HVEM were not protected, whereas mice immunized with the same antigen expressed within the gD-W294A variant were fully protected (Fig. 4d). Mice immunized once with a DNA vaccine expressing only E7 within gD developed tumors, whereas those that expressed E7 within the gD-W294A variant were completely protected (Fig. 4d).
Figure 4.
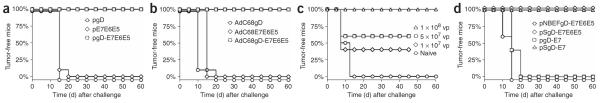
CD8+ T cells induced by gD chimeric protein are functional in vivo. Protection against TC-1 tumor cell challenge was evaluated in mice vaccinated with (a) DNA or (b) AdC68 vectors expressing either gD, E7E6E5 or gD-E7E6E5. (c) Protection from TC-1 tumor challenge in mice vaccinated with 1 × 108, 5 × 107 or 1 × 107 virus particles (vp) of AdC68gD-E7E6E5 vector. Naive mice were used as negative control. (d) Protection from TC-1 tumor challenge in mice vaccinated with DNA vaccine expressing either NBEFgD-E7E6E5, SgD-E7E6E5, gD-E7 or SgD-E7 chimeric genes. Mice were challenged 14 d and 10 d after vaccination with DNA and AdC68 vectors, respectively. Tumor development was followed for up to 60 d after challenge.
DISCUSSION
Antigen and secondary signals are required for activation of adaptive immune responses. Secondary signals are delivered by co-stimulatory and co-inhibitory signals, and the overall balance contributes to the quality and magnitude of the ensuing immune responses. Manipulating co-inhibitory pathways might modify immune responses and result in more efficacious vaccines. Here we tested whether expressing an antigen within gD, which binds HVEM and potentially interferes with the inhibitory BTLA pathway, can enhance adaptive immune responses.
Incorporation of the antigens into the C-terminal domain of gD markedly increased vaccine-induced T and B cell responses. This was especially pronounced for CD8+ T cell responses to E7, which expresses a low-affinity Kb-binding T cell epitope17. A single dose of vaccines expressing the E7E6E5 polypeptide failed to elicit CD8+ T cell responses or protection against challenge with an E7-expressing tumor cell line, whereas vaccines expressing the same antigen within gD induced high and sustained frequencies of E7-specific CD8+ T cells and complete protection against challenge. The HIV-1 Gag protein carries a high-affinity epitope for mice of the H-2d haplotype, and DNA vaccines or AdC68 vectors13 expressing Gag induce detectable CD8+ T cell responses. Responses to Gag are also increased upon expression of Gag within gD. The immunopotentiating effect of gD on Gag-specific T cells is more substantial when the gD-Gag chimeric protein is delivered by a DNA vaccine rather than the highly immunogenic AdC68 vector. Notwithstanding, upon dose reduction of the AdC68 vector, the results clearly show an enhancement of the CD8+ T cell frequencies. Adenoviral vectors of the common human serotype 5, tested in clinical trials, encountered dose-limiting toxicity18, and this effect is also anticipated with the chimpanzee-origin adenoviral vectors. Therefore, a further improvement of the immunogenicity of adenoviral vector vaccines that allows for a substantial dose reduction while maintaining efficacy would be valuable to lower vaccine-related side effects, reduce the cost of the vaccine and facilitate production for mass vaccination.
Insertion of antigens into gD does not affect the functionality of antigen-specific CD8+ T cells: T cells induced by the gD chimeric proteins protect against tumor cell challenge in the E7 model, are phenotypically similar to those induced in the absence of gD and differentiate efficiently into memory cells, confirming previous results with BTLA-deficient T cells19.
Antibody responses are also augmented by expressing the antigen within gD. The markedly enhanced antibody response to AdC68gD-Gag, as compared to AdC68Gag, may reflect an increased CD4+ T cell response or a direct effect of gD on B cells through inhibition of the HVEM-BTLA pathway. The latter would require secretion of the gD fusion protein or direct transduction of B cells. Direct transduction is unlikely, as B cells do not express the coxsackie adenovirus receptor (CAR) and are thus not efficiently entered by the AdC68 vector.
Binding of gD to HVEM is essential for augmentation of CD8+ T cell responses to the fusion partner, as vaccines expressing antigen within a modified gD in which the HVEM binding site had been obliterated did not induce enhanced CD8+ T cell responses. According to our molecular model, insertion of foreign sequences of certain lengths into the C terminus may effect a structural change in gD that improves its binding to HVEM, which can also be achieved through a single-amino-acid exchange in position 294 of gD (ref. 7).
Binding of gD to HVEM may not only block the BTLA pathway, but also enhance the co-stimulatory LIGHT pathway, deliver maturation signals to dendritic cells or both. Our results indicate that neither of these two mechanisms could explain the augmented T cell response; gD binding to HVEM did not affect its binding to LIGHT, and dendritic cells treated with AdC68 vectors in vivo or in vitro showed comparable maturation marker profiles regardless of gD expression by the vectors.
Manipulation or disruption of negative regulatory pathways such as the PD-1–PD-L1 pathway has been shown previously to augment T cell responses10. However, manipulation of this pathway may not readily affect primary T cell responses, as PD-1 is not expressed on naive T cells10. In contrast, low levels of BTLA are expressed on naive T cells, and these levels rapidly increase upon T cell activation and then decline20,21.
Medicinal targeting of immunoregulatory pathways can result in immunopathology. The use of a gD-antigen chimeric protein has the advantage that it does not involve systemic interruption of an inhibitory pathway but rather exerts its effects locally to the site of antigen presentation. We confirmed the spatially limited effect of gD experimentally by injecting two adenoviral vectors expressing either gD or E7E6E5 into distant anatomical sites and did not observe enhancement of antigen-specific CD8+ T cell responses (data not shown). It has been suggested previously that targeting BTLA by inhibitory antibodies or small molecules may enhance vaccine immunogenicity19. The results shown here suggest that such unconventional adjuvants may indeed be useful; however, their effect would be systemic and thus carry a higher likelihood of unwanted side effects.
In summary, the impact of diseases such as AIDS, cervical cancer, tuberculosis, malaria and chronic viral hepatitis on global human health is staggering, and vaccines against the causative infectious agents remain elusive. A major roadblock to the development of vaccines against these infectious agents is our inability to generate sufficiently strong immune responses in humans. The immunogenicity of vaccines can be enhanced through adjuvants that traditionally have targeted immunostimulatory pathways. Here we have shown that blockade of HVEM inhibitory pathway, by targeting the BTLA binding site with gD, potently enhances vaccine-induced immune responses and may thus provide a venue to improve vaccine efficacy.
METHODS
Construction of herpes simplex virus-1 gD chimeric genes
We constructed the chimeric gD-E7E6E5 construct by fusing the genes encoding HPV-16 E7, E6 and E5 to a cDNA encoding HSV-1 gD. We amplified the genes encoding E7, E6 and E5 without their respective start and stop codons by PCR using the HPV-16 genome as a template. All oligonucleotides used in this work were purchased from Integrated DNA Technologies, and their sequences are provided in Supplementary Table 1 online. We carried out separated amplification reactions with the following primer sets: E7FwApaI and E7RvNarI; E6FwNarI and E6RvNotI; and E5FwNotI and E5RvApaI. We cleaved the E7 DNA fragment with ApaI and NotI, the E6 DNA fragment with NotI and NarI, and the E5 DNA fragment with NarI and ApaI. We cloned all DNA fragments into the ApaI site of the pRE4 vector22. We confirmed the correct in-frame cloning of the E7-, E6- and E5-encoding genes by nucleotide sequencing (Wistar Sequencing Facility). We generated control vectors pE7E6E5 and AdC68E7E6E5 by PCR with pgD-E7E6E5 as template and primers E7FwHindIII and E5RvHindIII. We generated the AdC68gD control vector with pRE4 as template and primers gDFwXbaI and gDRvXbaI. To construct AdC68gD-E76E5 vector, we amplified the gD-E7E6E5 construct by PCR using the pgD-E7E6E5 vector as a template. We carried out the PCR reaction with gDFwXbaI and gDRvXbaI primers. We cleaved the gD-E7E6E5 DNA fragment with XbaI and cloned it into the XbaI site of the pShuttle vector (BD Biosciences). We confirmed the creation of the pShuttle-gD-E7E6E5 by restriction analysis and subcloned it into E1-deleted, chimpanzee-derived AdC68 using PI-SceI and I-CeuI sites as described13. We generated the gD-Gag chimeric construct by insertion of the codon-optimized truncated form of Gag from HIV-1 clade B (ref. 12) into the HSV-1 gD NarI site. We amplified the Gag gene by PCR using the pCMVGag vector as a template and primers GagFwNarI and GagRvNarI. We cleaved the DNA fragment corresponding to the Gag gene with NarI and cloned it into pShuttle-gD, then subcloned it into AdC68 vector as described above. AdC68 virus was rescued from a recombinant molecular clone on HEK293 cells as described previously13.
Construction of gD mutants
We generated the SgD-E7 mutated gene construct with the QuikChange site-directed mutagenesis kit (Stratagene) as recommended by the manufacturer. Briefly, we used SgDFw and SgDRv primers designed to mutate amino acid residue 294 of gD to PCR-amplify the entire pgD-E7 vector. We then treated the reaction products with DpnI and used the resulting product to transform Escherichia coli DH5α cells. We generated the gene encoding NBEFgD-E7E6E5 by mutation of residues crucial for HVEM-gD interaction. We mutated HSV-1 gD residues 11, 15, 25, 27, 28, 29 and 30 to alanine by gene splicing by overlap extension (‘gene SOE-ing’). Briefly, we carried out two PCR reactions with the primers gDFwHindIII and NBEFgDRv or NBEFgDFw and gDRvHindIII. We used the pgD-E7E6E5 vector as a template in both PCR reactions. We used two amplified fragments as templates for a PCR reaction with gDFwHindIII and gDRvHindIII primers. We cloned the NBEFgD-E7E6E5 DNA fragment into the same pgD-E7E6E5 backbone vector. We confirmed both mutant gD sequences by sequencing the entire gene. A list of all constructs used in this study is shown in Supplementary Table 2 online.
Cloning and transient expression of human LIGHT
We amplified the LIGHT cDNA by SuperScript III OneStep RT-PCR (Invitrogen) with RNA isolated from human peripheral blood mononuclear cells (PBMCs; kindly provided by K. High and S. Murphy) and LIGHTFwBamHI and LIGHTRvEcoRI primers (Supplementary Table 1). We cleaved the LIGHT cDNA fragment with BamHI and EcoRI and cloned it into pcDNA3.1(+) (Invitrogen) that had also been digested with BamHI and EcoRI. We confirmed the LIGHT sequence by nucleotide sequencing (Wistar Sequencing Facility). For LIGHT expression, we seeded 293T cells 1 d before transfection in 75-cm2 tissue culture flasks (5 × 106 cells per flask). The next day, we transfected cells with GenePorter transfection reagent (Genlantis; 50 μl per flask) and 10 μg plasmid DNA per flask in a final volume of 5 ml. We transfected one flask with the LIGHT expression plasmid (phLIGHT) and another with the empty plasmid vector (pcDNA3.1(+)). At 5 h after transfection, we fed the cells with 5 ml DMEM and 20% FCS and incubated them for a total of 72 h.
DNA and AdC68 vector purification
We propagated DNA vaccines in E. coli DH5α cells grown in LB medium supplemented with ampicillin, and we purified the DNA with a MaxiPrep Kit (Qiagen). We determined the DNA concentrations by spectrophotometry at 260 nm (model LKB Ultrospec III, Amersham Biosciences) and confirmed them by visual inspection of ethidium bromide–stained 1% agarose gels comparing the vaccine DNAs to DNA fragments of known concentration (Invitrogen). Plasmids were kept at −20 °C until use, when the DNA concentration was adjusted to 1 μg/μl in PBS. We propagated AdC68 vectors on E1-transfected HEK293 cells and purified by CsCl gradient centrifugation as previously described13. Upon purification, the concentration of each virus vector batch was determined by measuring virus particles by spectrophotometry at 260 nm.
Cell lines
TC-1 tumor cells16 derived from lung epithelial cells of C57BL/6 mice and then transformed with v-Ha-ras and HPV-16 E6 and E7 genes were provided by T.C. Wu. We used B78-H1cells or B78-H1/3E5 mouse melanoma cells for HVEM and gD localization assays. We used 293T cells transfected with phLIGHT for HVEM binding assays. We used E1-transfected HEK293 cells to propagate AdC68 vectors. We grew all cells in DMEM supplemented with glutamine, sodium pyruvate, nonessential amino acids, HEPES buffer, antibiotics and 10% FBS (TC-1 and E1-transfected HEK293 cells) or 5% FBS (B78-H1 and B78-H1/3E5 cells; all reagents from Cellgro).
HVEM- and LIGHT-binding assays
We infected CHO/CAR cells with AdC68E7E6E5, AdC68gD, AdC68Gag, AdC68gD-E7E6E5 or AdC68gD-Gag. We used noninfected cells as control. After 72 h, we harvested the cells, suspended them in 1 ml of extraction buffer (10 mM Tris pH 8.0, 150 mM NaCl, 10 mM EDTA, 1% Nonidet P-40, 0.5% sodium deoxycholate, 1 mM phenylmethylsulfonyl fluoride) supplemented with Complete Protease Inhibitor (Roche, Mannheim), and incubated them at 4 °C for 1 h. After a spin at 12,000g for 15 min at 4 °C, protein extracts were kept at −80 °C until use. We used a capture-ELISA and western blotting to normalize the amount of gD in the extracts. ELISA plates were coated with 50 μl per well of a 10 μg/ml concentration of ID3 mAb (ref. 23) diluted in PBS. After an overnight incubation at 4 °C, we exposed the plates to blocking solution for 1 h and then to extracts diluted in blocking solution for 2 h at 25 °C. We detected captured gD by adding 50 μl of 1 μg/ml polyclonal antibody R7 per well24, followed by rabbit-specific goat antibody coupled to horseradish peroxidase (Sigma). We rinsed the plates with 20 mM citrate buffer (20 mM citric acid) pH 4.5, 2,2-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) (ABTS) peroxidase substrate (Sigma) was added, and we measured the absorbance at 405 nm with a microtiter plate reader. We normalized the amount of gD in each extract by dilution in extraction buffer. To assess receptor binding of the gD mutants, we coated ELISA plates overnight with 50 μl of human HVEM (5 μg/ml)25, exposed them to blocking solution and incubated them with normalized cell extracts diluted in blocking solution for 2 h at 25 °C. We detected bound gD as described above.
To evaluate the interference of gD chimeric proteins with HVEM-LIGHT integration, we coated plates with human HVEM and blocked as above, then added normalized protein extracts from infected cells produced as described above at saturating amounts for 1 h at 25 °C. After washing, we added 50 μl of purified human LIGHT (4 μg/ml, R&D Systems) to the wells and incubated the plates 1 h at 25 °C. We added a monoclonal human LIGHT–specific antibody (0.2 μg, R&D System) and incubated for 1 h at 25 °C , followed by a 1-h 25 °C incubation with a mouse-specific antibody coupled to alkaline phosphatase diluted 1:100 (Cappel). We incubated the plates with substrate (10 mg d-nitrophenyl phosphate disodium dissolved in 10 ml of 1 mM MgCl2, 3 mM NaN3 and 0.9 M diethanolamine, pH 9.8), and then read them in an automated ELISA reader at 405 nm (model EL311, Bio-Tek Instruments).
To assess the effect of gD binding to HVEM on the binding of HVEM to cell-bound LIGHT, we transfected 293T cells with a plasmid vector expressing human LIGHT or an empty pcDNA3.1(+) vector. 72 h after transfection, we stained aliquots of the cells for 30 min on ice with 0.2 μg of a human LIGHT–specific mAb (R&D Systems) and then with a phycoerythrin (PE)-labeled mouse-specific IgG (Sigma) for 30 min on ice to assess expression of LIGHT. We treated the remaining cells (2 × 105 cells per sample) for 30 min on ice with a mixture of 5 μg/ml of human HVEM and normalized extracts containing gDE7E6E5, gDGag, gD, E7E6E5 or Gag (at a higher dilution from that in Figure 1c), which had been preincubated for 30 min on ice. We then washed the cells with PBS, treated them with 0.6 μg/ml rabbit human HVEM–specific antibody for 30 min on ice and then with a PE-labeled antibody to rabbit IgG (Sigma) for an additional 30 min on ice. We recovered the cells from then plates by treatment with a 1:5,000 solution of Versene (Invitrogen) and then analyzed them by flow cytometry.
To determine the effect of gD on LIGHT binding to HVEM, we treated HVEM+ B78-H1/3E5 cells with normalized protein extracts from infected cells produced as described above for 1 h at 25 °C. We washed the cells and then treated them with 50 μl of purified human LIGHT (4 μg/ml, R&D Systems) for 1 h at 25 °C. We added a monoclonal human LIGHT–specific antibody (0.2 μg, R&D Systems) at a 1:100 dilution and incubated for 1 h at 25 °C, followed by a 1-h 25 °C incubation with an mouse-specific antibody coupled to PE (Sigma). We analyzed cell suspensions by flow cytometry to determine presence of LIGHT.
Flow cytometry analysis
We performed flow cytometry analyses with either an EPICS XL (Beckman-Coulter) or a FACSCalibur (BD Biosciences). We analyzed data with FlowJo software, version 7.1.2 (Tree Star).
Confocal microscopy
We infected B78-H1/3E5 cells, which express HVEM fused to enhanced GFP (HVEM-EGFP)26, or B78-H1 cells with either AdC68E7E6E5, AdC68gD, AdC68Gag, AdC68gD-E7E6E5 or AdC68gD-Gag. After 48 h, we stained the cells directly or permeabilized them with Cytofix/Cytoperm (BD Biosciences) and then stained them with gD-specific mAbs (ID3, ref. 23, and DL-6, ref. 24; both 0.1 μg) followed by antibody to mouse IgG conjugated to Texas Red (microscopy) or PE (FACS; both Sigma). To assess binding of gD chimeric protein to HVEM in trans, we infected CHO/CAR cells with AdC68 vectors for 5 h and then extensively washed them (10 times) with PBS. We used noninfected cells as a control. We cultured CHO/CAR cells with B78H1/3E5 cells for 48 h at a 4:1 ratio and then stained them with the gD monoclonal antibodies as described above. We performed confocal microscopy with a Leica TCS SP2 confocal microscope at 400× magnification and Leica Confocal software version 2.61. We analyzed cell suspensions by flow cytometry to determine the presence of gD.
Molecular modeling of gD-Gag
We constructed the three-dimensional models of gD-Gag with the MODELLER package (Accelrys) by combining the structures of individual protein domains as determined by X-ray crystallography. The receptor-bound form of the gD-Gag model was based upon the HSV-1 gD HVEM complex (1JMA)27, chain A, residues 1–259; SIV Gag (1ECW)28, residues 1–119; and HIV-1 Gag (1E6J)29, chain P, residues 11–220. The gD-Gag unligated form was based upon the cyclophilin A–HIV-1 chimera complex (1M9D)30, chain A, residues 1–15; HSV-1 gD (2C36)7, chain A, residues 23–256; SIV Gag (1ECW), residues 1–119; and HIV-1 Gag (1E6J), chain P, residues 11–220. We prepared ribbon representations within the Swiss-PdbViewer program and rendered them with the Persistence of Vision Ray Tracer program (POV-Ray 2004, version 3.6).
In vitro T cell proliferation assay
We harvested cells from draining popliteal lymph nodes of naive and mice i.m. immunized 48 h earlier with 1 × 1011 virus particles of either AdC68E7E6E5, AdC68Gag, AdC68gD, AdC68gD-E7E6E5 or AdC68gD-Gag, then irradiated them with 2,000 rad. We isolated CD8+ cells from the spleens of OT-1 mice by negative selection with magnetic beads (Miltenyi Biotec) and labeled them with 2 μM CFSE (Peprotech). We cultured a total of 1 × 106 irradiated lymph node cells with 1 × 105 CD8+ CFSE-labeled OT-1 cells in the presence of either SIINFEKL peptide (5 μg/ml, Alpha Diagnostic International) or control peptide AMQMLKETI (10 μg/ml, AnaSpec) in 96-well plates for 72 h. We stained the cells with antibody to CD8 conjugated to peridinin–chlorophyll protein complex (PerCP) and antibody to T cell receptor Vα2 conjugated to PE (both BD Biosciences) for 30 min on ice. We examined the cells by flow cytometry Additionally, we stained cells harvested from the draining lymph node with CD11c-allophycocyanin in combination with CD40-PE, CD80-PE, CD86-PE and I-Ab-PE (All BD Biosciences) for 30 min on ice and then examined them by flow cytometry.
Mice and immunization
We purchased female BALB/c and C57BL/6 mice at 6–8 weeks of age from Charles River Laboratories and housed them at the Animal Facility of the Wistar Institute. We performed all procedures involving handling and killing of mice using approved protocols in accordance with recommendations for the proper use and care of laboratory animals at the Institutional Animal Care and Use Committee of the Wistar Institute. We vaccinated groups of three to ten BALB/c or C57BL/6 mice i.m. with the DNA vaccines or E1-deleted AdC68 vectors into the tibialis anterior muscle of each hind limb. We gave the DNA vaccine at 100 μg divided into two 50-μl aliquots. We inoculated AdC68 vectors at 1 × 1010 virus particles per mouse unless stated otherwise.
Intracellular cytokine staining
We performed intracellular IFN-γ staining with PBMCs and cells from spleens 2 weeks after DNA vaccination or 10 d after application of the AdC68 vector unless stated otherwise. We washed the cells twice with L-15 medium (Cellgro), and treated them for 5 min on ice with ACK lysing buffer (Invitrogen) to rupture red blood cells, washed them again, and suspended them in DMEM (Cellgro) supplemented with 2% FBS. Samples were cultured at 1 × 106 cells per well for 5 h at 37 °C in 96-well round-bottom microtiter plates (Costar) in 200 μl of DMEM supplemented with 2% FBS and 1 μM 2-mercaptoethanol. We added brefeldin A (GolgiPlug; BD Biosciences) at a concentration of 1 μl/ml. We used the E7-specific RAHYNIVTF peptide, which carries the immunodominant epitope of E7 for mice of the H-2 Kb haplotype, or the AMQMLKETI peptide (both AnaSpec), which carries the immunodominant MHC class I epitope of Gag for mice of the H-2d haplotype, for peptide stimulation at a concentration of 3 μg/ml. We used the V3 peptide delineated from the sequence of the envelope protein of HIV-1 clade B (VVEDEGCTNLSGF) and the SIINFEKL peptide (both Alpha Diagnostic International) as control peptides. After washing, we incubated the cells for 30 min at 4 °C with 100 μl of a 1:100 dilution of FITC-conjugated mAb to mouse CD8a (BD Biosciences). We washed the cells once with PBS and then permeabilized them with Cytofix/Cytoperm (BD Biosciences) for 20 min at 4 °C, washed them twice with Perm/Wash buffer (BD Biosciences) and incubated them in the same buffer for 30 min at 4 °C with 50 μl of a 1:100 dilution of a PE-labeled monoclonal antibody to mouse IFN-γ (BD Biosciences). After washing, we suspended the cells in PBS and examined them by flow cytometry. Then we determined the percentages of antigen-specific CD8+ T cells that stained positive for IFN-γ over all CD8+ T cells.
Enzyme-linked immunosorbent assay for antibodies to Gag
We tested the sera from vaccinated or naive mice on plates coated with purified Gag protein. Briefly, we coated 96-well round-bottom Maxisorb (Nunc) plates with 0.2 μg of Gag p24 HIV-1 (Immuno Diagnostics) diluted in 100 μl of coating buffer (15 mM Na2CO3, 35 mM NaHCO3 and 3 mM NaN3, pH 9.6) overnight at 4 °C. The next day, we blocked the plates with 200 μl of PBS containing 3% BSA for 2 h at 25 °C. We serially diluted serum samples in PBS supplemented with 3% BSA in triplicate at 100 μl per well on the Gag-coated plates and incubated for 1 h at 25 °C. We washed the plates and added a 1:200 dilution of alkaline phosphatase–conjugated goat antibody specific for mouse immunoglobulins (Cappel) to each well, and then we incubated the plates for 1 h at 25 °C. After washing, we incubated the plates with substrate (10 mg d-nitrophenyl phosphate disodium dissolved in 10 ml of a 1 mM MgCl2, 3 mM NaN3, and 0.9 M diethanolamine, pH 9.8 solution) and then read the plates in an automated ELISA reader at 405 nm (model EL311, Bio-Tek Instruments).
T cell enzyme-linked immunospot assay
We performed an IFN-γ–capture enzyme-linked immunospot assay (ELISPOT) with 96-well Millipore polyvinylidene difluoride plates coated with mouse IFN-γ–capture antibody (BD Biosciences) diluted in PBS and incubated overnight at 4 °C. After washing with PBS, we blocked the plates with complete RPMI-1640 (Mediatech) medium supplemented with 10% FBS for 2 h at 37 °C. We added lymphocytes in triplicate and stimulated them with the 11-mer peptide NPPIPVGELIY, which carries the immunodominant CD4 epitope of Gag for mice of the H-2d haplotype, and with mouse antibodies to the co-stimulatory molecules CD28 and CD49d (both BD Biosciences) for 18–20 h at 37 °C with 5% CO2. We removed the cells and washed the plates with 0.01% Tween (Sigma) in PBS and then incubated them with biotin-labeled secondary antibody (BD Biosciences) in 5% FBS in 0.01% Tween/PBS for 2 h at 25 °C. We washed the plates, added streptavidin–alkaline phosphatase (MAbtech AB) and incubated at 25 °C for 1 h. We developed the spots by adding 5-bromo-4-chloro-3-indolyl phosphate–nitro blue tetrazolium developer (Pierce) to each well and incubating for 5 min at 25 °C. We washed the plates in water and dried them before counting the spots with the C.T.L. Series 3A Analyzer and ImmunoSpot 3.2 (Cellular Technology). We subtracted data from unstimulated cells and used this as background control, and we subtracted these values from sample values before plotting.
Tetramer and T cell phenotyping
We detected antigen-specific CD8+ T cells with allophycocyanin-labeled MHC class I tetramers carrying the AMQMLKETI peptide of Gag (MHC Tetramer Core Facility). We treated PBMCs and splenocytes isolated 10 d after immunization with AdC68 vectors as described for intracellular cytokine staining. We stained the samples for 30 min at 25 °C with Gag-tet and antibody to CD8a-PerCP in combination with the following antibodies: CD25-PE, CD122-PE, CD127-PE, CD27-PE, BTLA-PE (eBioscience), PD1-PE, CD62L-FITC, CD69-FITC, CD103-FITC, CD43-FITC, CD44-FITC and CD54-FITC (all from BD Biosciences, unless indicated otherwise). For Bcl2 and CTLA-4 staining, we washed the cells, permeabilized them for 30 min at 4 °C with Cytofix/Cytoperm (BD Biosciences) and then stained them with Bcl2-PE or CTLA-FITC (BD Biosciences) antibodies. We performed flow cytometric analyses with at least 100,000 viable cells live-gated.
TC-1 cell challenge
We challenged groups of ten C57BL/6 mice subcutaneously with 1 × 105 TC-1 cells suspended in 100 μl serum-free medium injected under the skin of one rear flank. We challenged the mice 14 and 10 d after vaccination with DNA vaccine and AdC68 vector, respectively. We monitored tumor growth by visual inspection and palpation three times a week. We scored mice were as tumor-bearing when tumors attained sizes of approximately 1–2 mm in diameter. We killed the mice once the tumors exceeded a diameter of 1 cm. We followed tumor growth for a period of 60 d after challenge.
Statistical analysis
We conducted experiments using three to ten mice per group. We assayed samples tested by ELISA in triplicate. Results show the means ± s.d. We conducted intracellular cytokine staining with PBMCs from individual mice, whereas we performed tetramer and marker staining with pooled samples. We looked for significant differences between groups with the one-tailed Student’s t-test or ANOVA.
Supplementary Material
ACKNOWLEDGMENTS
This work was sponsored by a US National Institutes of Health grant (AI-052271) to H.C.E.; a US National Institute of Allergy and Infectious Diseases grant (AI-18289) to J.C.W., G.H.C. and R.J.E.; and institutional grants to the Wistar Institute including a US National Cancer Institute Cancer Core Grant (CA10815) and the Commonwealth Universal Research Enhancement Program from the Pennsylvania Department of Health. M.O.L. was supported with a fellowship from the Cancer Research and Prevention Foundation. We thank the MHC Tetramer Core Facility (Emory University Vaccine Center, Atlanta, Georgia) for providing the Gag-tetramer, T.C. Wu (Johns Hopkins University, Baltimore, Maryland) for providing TC-1 cells, K. High and S. Murphy (University of Pennsylvania, Philadelphia, Pennsylvania) for human PBMC RNA samples, W. Giles-Davis for excellent technical assistance, J. Faust and M. Farabaugh for assistance with flow cytometry, J. Hayden and F. Keeney for assistance with confocal microscopy, and C. Cole and C. Barth for preparation of the manuscript.
Footnotes
Note: Supplementary information is available on the Nature Medicine website.
AUTHOR CONTRIBUTIONS M.O.L. planned and performed most of the experiments and wrote the manuscript; N.T. and S.E.H. planned some experiments and contributed to writing of the manuscript; J.C.W. and S.-W.L. planned and performed some of the experiments; J.J.R. performed the molecular modeling; E.J.W. provided technical advice; G.H.C. and R.J.E. provided advice; and H.C.E. supervised all experimental procedures and helped to write the manuscript.
COMPETING INTERESTS STATEMENT The authors declare competing financial interests: details accompany the full-text HTML version of the paper at http://www.nature.com/naturemedicine/.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions
References
- 1.Granger SW, Rickert S. LIGHT-HVEM signaling and the regulation of T cell–mediated immunity. Cytokine Growth Factor Rev. 2003;14:289–296. doi: 10.1016/s1359-6101(03)00031-5. [DOI] [PubMed] [Google Scholar]
- 2.Sarrias MR, et al. The three HveA receptor ligands, gD, LT-α and LIGHT bind to distinct sites on HveA. Mol. Immunol. 2000;37:665–673. doi: 10.1016/s0161-5890(00)00089-4. [DOI] [PubMed] [Google Scholar]
- 3.Sedy JR, et al. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat. Immunol. 2005;6:90–98. doi: 10.1038/ni1144. [DOI] [PubMed] [Google Scholar]
- 4.Highlander SL, et al. Neutralizing monoclonal antibodies specific for herpes simplex virus glycoprotein D inhibit virus penetration. J. Virol. 1987;61:3356–3364. doi: 10.1128/jvi.61.11.3356-3364.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Montgomery RI, Warner MS, Lum BJ, Spear PG. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell. 1996;87:427–436. doi: 10.1016/s0092-8674(00)81363-x. [DOI] [PubMed] [Google Scholar]
- 6.Connolly SA, et al. Structure-based mutagenesis of herpes simplex virus glycoprotein D defines three critical regions at the gD-HveA/HVEM binding interface. J. Virol. 2003;77:8127–8140. doi: 10.1128/JVI.77.14.8127-8140.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krummenacher C, et al. Structure of unliganded HSV gD reveals a mechanism for receptor-mediated activation of virus entry. EMBO J. 2005;24:4144–4153. doi: 10.1038/sj.emboj.7600875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Compaan DM, et al. Attenuating lymphocyte activity: the crystal structure of the BTLA-HVEM complex. J. Biol. Chem. 2005;280:39553–39561. doi: 10.1074/jbc.M507629200. [DOI] [PubMed] [Google Scholar]
- 9.Cheung TC, et al. Evolutionarily divergent herpesviruses modulate T cell activation by targeting the herpesvirus entry mediator cosignaling pathway. Proc. Natl. Acad. Sci. USA. 2005;102:13218–13223. doi: 10.1073/pnas.0506172102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barber DL, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 11.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 12.Schneider R, Campbell M, Nasioulas G, Felber BK, Pavlakis GN. Inactivation of the human immunodeficiency virus type 1 inhibitory elements allows Rev-independent expression of Gag and Gag/protease and particle formation. J. Virol. 1997;71:4892–4903. doi: 10.1128/jvi.71.7.4892-4903.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fitzgerald JC, et al. A simian replication-defective adenoviral recombinant vaccine to HIV-1 Gag. J. Immunol. 2003;170:1416–1422. doi: 10.4049/jimmunol.170.3.1416. [DOI] [PubMed] [Google Scholar]
- 14.Hensley SE, Giles-Davis W, McCoy KC, Weninger W, Ertl HC. Dendritic cell maturation, but not CD8+ T cell induction, is dependent on type I IFN signaling during vaccination with adenovirus vectors. J. Immunol. 2005;175:6032–6041. doi: 10.4049/jimmunol.175.9.6032. [DOI] [PubMed] [Google Scholar]
- 15.Lasaro MO, Diniz MO, Reyes-Sandoval A, Ertl HC, Ferreira LC. Anti-tumor DNA vaccines based on the expression of human papillomavirus-16 E6/E7 oncoproteins genetically fused with the glycoprotein D from herpes simplex virus-1. Microbes Infect. 2005;7:1541–1550. doi: 10.1016/j.micinf.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 16.Lin KY, et al. Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Res. 1996;56:21–26. [PubMed] [Google Scholar]
- 17.He Z, et al. Viral recombinant vaccines to the E6 and E7 antigens of HPV-16. Virology. 2000;270:146–161. doi: 10.1006/viro.2000.0271. [DOI] [PubMed] [Google Scholar]
- 18.Kresge KJ. Renewed promise. Annual AIDS vaccine meeting highlights recent data from clinical trials and lessons on recruitment and retention of volunteers. IAVI Rep. 2005;9:18–20. [PubMed] [Google Scholar]
- 19.Krieg C, Boyman O, Fu YX, Kaye J. B and T lymphocyte attenuator regulates CD8+ T cell-intrinsic homeostasis and memory cell generation. Nat. Immunol. 2007;8:162–171. doi: 10.1038/ni1418. [DOI] [PubMed] [Google Scholar]
- 20.Hurchla MA, et al. B and T lymphocyte attenuator exhibits structural and expression polymorphisms and is highly induced in anergic CD4+ T cells. J. Immunol. 2005;174:3377–3385. doi: 10.4049/jimmunol.174.6.3377. [DOI] [PubMed] [Google Scholar]
- 21.Han P, Goularte OD, Rufner K, Wilkinson B, Kaye J. An inhibitory Ig super-family protein expressed by lymphocytes and APCs is also an early marker of thymocyte positive selection. J. Immunol. 2004;172:5931–5939. doi: 10.4049/jimmunol.172.10.5931. [DOI] [PubMed] [Google Scholar]
- 22.Cohen GH, et al. Expression of herpes simplex virus type 1 glycoprotein D deletion mutants in mammalian cells. J. Virol. 1988;62:1932–1940. doi: 10.1128/jvi.62.6.1932-1940.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Friedman HM, Cohen GH, Eisenberg RJ, Seidel CA, Cines DB. Glycoprotein C of herpes simplex virus 1 acts as a receptor for the C3b complement component on infected cells. Nature. 1984;309:633–635. doi: 10.1038/309633a0. [DOI] [PubMed] [Google Scholar]
- 24.Isola VJ, et al. Fine mapping of antigenic site II of herpes simplex virus glycoprotein D. J. Virol. 1989;63:2325–2334. doi: 10.1128/jvi.63.5.2325-2334.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whitbeck JC, et al. Glycoprotein D of herpes simplex virus (HSV) binds directly to HVEM, a member of the tumor necrosis factor receptor superfamily and a mediator of HSV entry. J. Virol. 1997;71:6083–6093. doi: 10.1128/jvi.71.8.6083-6093.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bender FC, et al. Specific association of glycoprotein B with lipid rafts during herpes simplex virus entry. J. Virol. 2003;77:9542–9552. doi: 10.1128/JVI.77.17.9542-9552.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carfi A, et al. Herpes simplex virus glycoprotein D bound to the human receptor HveA. Mol. Cell. 2001;8:169–179. doi: 10.1016/s1097-2765(01)00298-2. [DOI] [PubMed] [Google Scholar]
- 28.Rao Z, et al. Crystal structure of SIV matrix antigen and implications for virus assembly. Nature. 1995;378:743–747. doi: 10.1038/378743a0. [DOI] [PubMed] [Google Scholar]
- 29.Monaco-Malbet S, et al. Mutual conformational adaptations in antigen and antibody upon complex formation between an Fab and HIV-1 capsid protein p24. Structure. 2000;8:1069–1077. doi: 10.1016/s0969-2126(00)00507-4. [DOI] [PubMed] [Google Scholar]
- 30.Howard BR, Vajdos FF, Li S, Sundquist WI, Hill CP. Structural insights into the catalytic mechanism of cyclophilin A. Nat. Struct. Biol. 2003;10:475–481. doi: 10.1038/nsb927. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.