

Abstract
The differentiation of CD4+ T cells into the Th2 subset is controlled by the transcription factor, GATA-3. GATA-3 is both necessary and sufficient for Th2 differentiation and works through the induction of chromatin remodeling at the Th2 effector cytokine loci. We show here that IL-4 stimulation induces GATA-3 mRNA upregulation, but the level of GATA-3 protein induced is insufficient for Th2 differentiation. The levels of GATA-3 protein and Th2 differentiation are enhanced by concomitant TCR signaling through the phosphatidylinositol 3-kinase/mammalian target of rapamycin pathway. The PI3K-mediated increase in GATA-3 protein occurs without increasing the GATA-3 mRNA level. Rather, TCR signaling through PI3K specifically enhances the translation rate of GATA-3 without affecting the protein stability. Importantly, this role of TCR signaling is independent of the effects of TCR signaling in T cell survival and expansion. Thus, TCR signaling through PI3K may play a critical role in Th2 differentiation by the specific enhancement of GATA-3 translation.
Introduction
To protect against different types of infections CD4+ T cells differentiate into functionally distinct subsets that are characterized by the cytokines they produce. The differentiation process is controlled by regulatory proteins, of which GATA-3 is the factor that is both necessary and sufficient for Th2 differentiation (1-3). GATA-3 acts as a lineage-specifying factor that is required for nervous system development, fetal liver hematopoiesis, and T cell development (4, 5). GATA-3 expression is tightly regulated during development in different tissues and small changes in the level of expression can have dramatic consequences.
The most well-recognized level of GATA-3 regulation during Th2 differentiation is transcriptional regulation. GATA-3 expression is necessary for T cell development within the thymus and GATA-3 mRNA levels are maintained at a low level in the periphery (5, 6). Activation of STAT6 by IL-4 stimulation drives transcriptional upregulation of GATA-3 mRNA (7). Additionally, the GATA-3 gene contains at least two promoters with different start sites of transcription, which allows for tissue specific expression of GATA-3 (8). Transcription of GATA-3 mRNA from the upstream exon 1A, which is primarily found in brain tissue, can be driven independently of IL-4 in T cells by Notch1 (9, 10). In addition to the regulation of expression, three proteins restrict the functional capacity of GATA-3: friend of GATA-1 (FOG), repressor of GATA (ROG), and T-bet, (11-13). An important function of GATA-3 is its ability to stabilize its own expression through transcriptional autoactivation (14). The interplay between the positive feedback loop and the negative regulators creates a threshold level of GATA-3 that must be reached for Th2 differentiation to occur. Thus, small changes in GATA-3 expression can have a dramatic impact on cell lineage commitment, as seen in humans with a genetic deficiency for GATA-3 at one allele, who suffer from hypoparathyroidism, sensorineural deafness, renal anomaly syndrome, as well as greatly reduced Th2 responses (15, 16).
In addition to the effects of IL-4R and Notch1 signaling, TCR signaling has also been implicated in GATA-3 transcriptional regulation. Changing the affinity of the TCR for pMHC complexes using altered peptide ligands or changing the peptide dose impacts the GATA-3 mRNA level (17, 18). Genetic disruption, or inhibition, of the downstream signaling molecules, NF- κB and ERK, also alters GATA-3 transcription (19, 20). However, it has been difficult to elucidate specific effects of TCR signaling on GATA-3 expression and Th2 differentiation, because TCR signaling is required for initial T cell activation, survival, and expansion.
To bypass the requirement for TCR signaling in T cell expansion we have used activated, undifferentiated T cells to determine specific contributions of IL-4R and TCR signaling towards GATA-3 expression and Th2 differentiation. We show here that expansion of activated, undifferentiated T cells is not dependant on Ag stimulation, but TCR signaling is absolutely required for Th2 differentiation. The impact of TCR signaling is not mediated through upregulation of GATA-3 transcription, as IL-4 induces GATA-3 mRNA equivalently in the presence or absence of Ag stimulation. Instead, TCR signaling is required to specifically increase the rate of GATA-3 translation, resulting in increased GATA-3 protein levels and subsequent Th2 differentiation.
Materials and Methods
T cell stimulation
CD4+ T cells were purified by negative selection from DO11.10 TCR transgenic mice. All mouse experiments were approved by the University Committee on Animal Resources at the University of Rochester. Magnetic column separation was used to enrich for >90% CD62L hi cells. T cells were stimulated with 0.2 μM OVA peptide presented by irradiated Balb/c splenocytes plus 10 U/ml hIL-2. For activated, undifferentiated T cells 10 μg/mL anti-IL-4 was added to the cultures; for Th1 cells 10 μg/mL anti-IL-4 and 10 ng/mL IL-12 was added; and for Th2 cells 20 ng/mL IL-4 was added. Activated, undifferentiated cells were restimulated on day 5 with mitomycin C- treated 6132 Pro cell transfectants expressing class II (I-Ad) and B7-1 (ProAd-B7) (21) +/- 2 μM OVA peptide and +/- 20 ng/mL IL-4. 10 U/mL hIL-2 was added to all restimulation cultures to provide for equivalent T cell expansion. Where indicated, cells were labeled with 1 μM CFSE at 37° for 5 minutes prior to restimulation. Where indicated, 10 μM LY294002 or 10 nM rapamycin was added during the restimulation. To assay for cytokine secretion, T cells were stimulated overnight with 1 μg/mL platebound anti-CD3 (2C11) and supernatants were assayed by capture ELISA for IL-2, IL-4, and IFN-γ. For intracellular cytokine staining brefeldin A was added 16-20 hours after stimulation. 4 hours later cells were collected, fixed with paraformaldehyde, and permeabilized with 0.5% saponin. Cytokine staining was performed in 0.5% saponin with PE or APC-labeled anti-IL-4 (BD/eBioscience).
Real Time PCR
Total RNA was isolated from 106 T cells using TRIzol (Invitrogen) and cDNA was made with Superscript II Reverse Transcriptase (Invitrogen). The GATA-3 real time PCR primers that spanned the exon 2-3 junction or the exon 4-5 junction and the CD3δ and lck primers were all obtained from Applied Biosystems. The custom exon 1a and exon 1b specific primers were also ordered from Applied Biosystems. The sequences for exon 1a primers are: forward-AGCTGGCCTTCAGGAGAGA and reverse-CGCTCAGAGACGGTTGCT with probe- CTTCCGATCACCAGGGCAG and for exon 1b are: forward-GCCTCCTCCTCCTCCTCTAC and reverse- CGCTCAGAGACGGTTGCT with probe-TTCCGATCACCTGAGTAGCA. Plates were run on 7900HT RT-PCR System (Applied Biosystems). GATA-3 level was normalized to CD3δ (except where indicated) relative to activated, undifferentiated T cells prior to restimulation.
Protein Analysis
Nuclear and cytosolic fractions were isolated from 3×106 T cells using NE-PER reagents (Pierce) and western blots were probed with anti-GATA-3 mAb or anti-PCNA (Santa Cruz). For all analysis a two-fold dilution curve of Th2 cell lysates was included on each blot and used to calculate fold differences between experimental samples. GATA-3 values were normalized to PCNA relative to activated, undifferentiated T cells prior to restimulation. For eIF2α experiments cytosolic lysates were isolated using an NP-40 lysis buffer (1% NP-40, 50mM Tris pH 8.0, 150mM NaCl, leupeptin, aprotinin, PMSF, and phosphatase inhibitor cocktail I (Sigma)) and probed with anti-Phospho-eIF2〈 (Ser51), anti-eIF2α (Cell Signaling Technology), and anti-lck (Santa Cruz). For biosynthetic labeling T cells were pulse labeled with 300 μCi/mL 35S trans label (Perkin Elmer) and chased for various times with pre-warmed complete media. Nuclear and cytosolic fractions were sequentially immunoprecipitated with GATA-3 and lck antibodies. The western blot and autoradiograph analysis was done on the Fluor Chem 8900 (Alpha Innotech). To determine the overall translational efficiency, the percent incorporation of 35S met/cys into newly synthesized proteins was determined following precipitation of the pulse-labeled proteins overnight at -20° with 100% ethanol.
Results
Th2 differentiation is specifically dependent on TCR signaling
To study the specific effects of IL-4R and TCR signaling on Th2 differentiation we have generated activated, undifferentiated T helper cells. Naïve T cells from DO11.10 TCR transgenic mice are primed in the absence of polarizing cytokines and in the presence of anti-IL-4 to prevent Th2 differentiation. At day 5 these activated, undifferentiated cells make IL-2, but little to no IFN-γ and IL-4 (Fig. 1A). By comparison, cells stimulated in Th1 or Th2 conditions make high amounts of IFN-γ and IL-4, respectively (Fig. 1A). These activated, undifferentiated cells were then restimulated with IL-4, Ag, or Ag plus IL-4 for six days. The number of cells recovered at the end of the restimulation is similar in all stimulation conditions (Fig. 1B). This indicates that TCR signaling of activated, undifferentiated cells is not required for T cell expansion. The ability of T cells to expand via cytokines allows us to determine if TCR signaling is specifically required for Th2 differentiation. Restimulation with either IL-4 or Ag on their own is not sufficient to induce Th2 differentiation, as measured by IL-4 (Fig. 1C) and IL-5 (data not shown) production. Th2 differentiation only occurs after stimulation with both Ag and IL-4 (Fig. 1C). To show that the dependence on TCR signaling for Th2 differentiation is not linked to cell division, we labeled activated, undifferentiated T cells with CFSE prior to restimulation. T cells efficiently enter the cell cycle under all stimulation conditions, although restimulation with IL-4 in the absence of Ag results in slightly delayed kinetics of CFSE dilution (Fig. 2A). Over time, T cells undergo similar rounds of cell division (Fig. 2A), corresponding to the equivalent level of T cell expansion (Fig. 1B). However, little or no IL-4 producing cells are detected after stimulation with IL-4 or Ag alone (Fig. 2B). Only cells that had been stimulated with Ag plus IL-4 are capable of producing IL-4 (Fig. 2B). Thus, the defect in Th2 differentiation by cells stimulated with IL-4 or Ag is clearly not a secondary effect caused by a failure in cell division. Taken together, these data show that TCR signaling has a specific role in Th2 differentiation that is independent of the requirement for TCR signaling in T cell survival and expansion.
FIGURE 1.
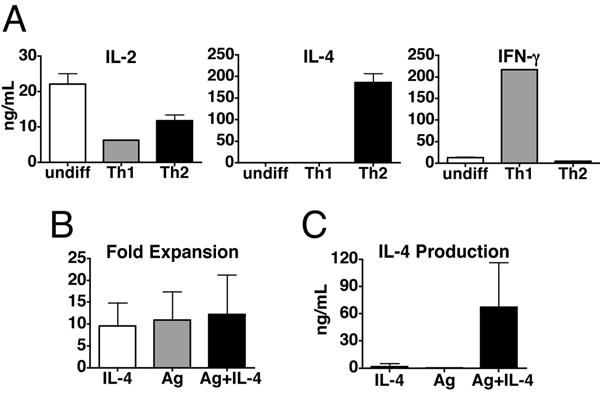
Signaling through both the TCR and IL-4R is required to drive Th2 differentiation of activated, undifferentiated T cells. A, DO11.10 CD4+ T cells were stimulated with OVA peptide presented by irradiated Balb/c splenocytes in the absence of polarizing cytokines and in the presence of anti-IL-4 to generate undifferentiated T cells (undiff), or in the presence of polarizing cytokines to drive Th1 or Th2 differentiation. On day 5 T cells were harvested and restimulated overnight on plate bound anti-CD3 and supernatants were measured for IL-2 (left), IL-4 (center), and IFN-γ (right). Data are depicted as mean + SEM. B and C, Activated, undifferentiated T cells were restimulated with IL-4, OVA peptide presented by ProAd-B7 cells (Ag), or with both Ag and IL-4 for 6 days; hIL-2 was added to all conditions. T cells were counted to determine the fold expansion during the secondary stimulation (B) and restimulated overnight on plate bound anti-CD3 to measure Th2 differentiation, defined as secretion of IL-4 (C). The data are depicted as mean + SD, n>30. In B the mean is highest in Ag+IL-4 stimulated cells but the differences are not statistically significant in either paired or unpaired t test. In C p<0.0001 for IL-4 vs Ag+IL-4 by unpaired t test.
FIGURE 2.
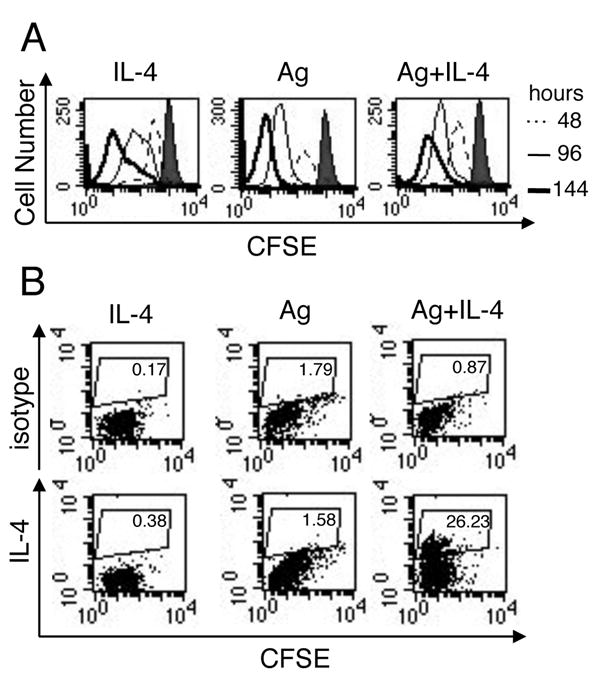
The ability of TCR signaling to promote Th2 differentiation is not mediated through enhanced cell division. A, Activated, undifferentiated T cells were CFSE labeled and then restimulated with IL-4, OVA peptide presented by ProAd-B7 cells (Ag), or with both Ag and IL-4; hIL-2 was added to all conditions. CFSE dilution was measured by flow cytometry 48 (dashed line), 96 (thin line), and 144 (thick line) hours after restimulation. Labeled T cells without any restimulation are shown (shaded) in each panel. One experiment, representative of 5, is shown. B, 6 days after restimulation T cells were stimulated overnight on plate bound anti-CD3 and stained for intracellular IL-4 (bottom); isotype control is shown for comparison (top). One experiment, representative of 6, is shown.
TCR signaling increases GATA-3 protein levels without affecting GATA-3 mRNA levels
Previous reports have suggested that TCR signaling components may play a role in the transcriptional regulation of GATA-3 (17-20, 22). We measured the induction of GATA-3 mRNA to determine if TCR signaling enhances GATA-3 expression during the restimulation of activated, undifferentiated T cells. Notably, we find that IL-4 stimulation drives GATA-3 mRNA induction in the presence or absence of Ag stimulation (Fig. 3A). TCR signaling is unable to drive GATA-3 mRNA on its own, or more importantly, amplify the level of GATA-3 mRNA when added to IL-4 stimulation (Fig. 3A). We believe that this induction of mRNA is reflective of transcriptional activation as there is no change in GATA-3 mRNA stability in the presence or absence of Ag stimulation (data not shown). The relative amount of GATA-3 mRNA in cells primed under Th2 conditions during initial priming is similar to the level of IL-4-induced GATA-3 mRNA, whereas Th1 cells have greatly reduced GATA-3 mRNA (Fig. 3B). Similar results were obtained for GATA-3 mRNA induction using primers that span both the exon 2/3 (Fig. 3, A and B) and the exon 4/5 junctions (data not shown). GATA-3 transcripts can be initiated by two different promoters, resulting in mRNA molecules that differ only in the exon 1 portion of their 5′ un-translated region (UTR) (8). Transcription from the downstream promoter represents the majority of the overall GATA-3 mRNA level in resting T cells (data not shown, (23)). To determine if IL-4 signaling preferentially induces one of these promoters we designed real time PCR primers that span the exon 1A and exon 1B/2 junctions. We find that IL-4 stimulation induces GATA-3 mRNA from both the upstream and downstream promoters and that Ag stimulation does not have any impact on the mRNA level from either promoter, on its own or when added to IL-4 stimulation (Fig. 3, C and D). These data indicate that IL-4 stimulation is sufficient for GATA-3 mRNA induction in the absence of concomitant TCR signaling and that the TCR-driven increase in Th2 differentiation is not mediated by an increase in GATA-3 mRNA.
FIGURE 3.
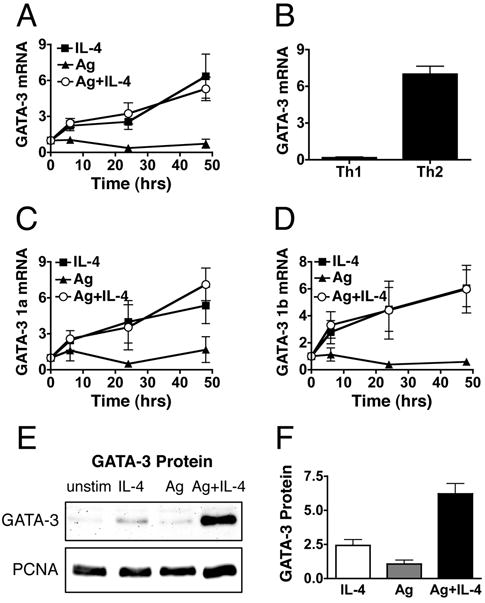
IL-4 stimulation is sufficient to drive GATA-3 mRNA, but co-engagement of TCR is required to induce maximal GATA-3 protein levels. A, Activated, undifferentiated T cells were restimulated with IL-4, OVA peptide presented by ProAd-B7 cells (Ag), or with both Ag and IL-4; hIL-2 was added to all conditions. GATA-3 mRNA was measured by real time PCR at different times following restimulation. The fold induction of GATA-3 mRNA, relative to activated, undifferentiated cells, is depicted as mean +/- SEM. B, Freshly isolated CD62L enriched DO11.10 CD4+ T cells were stimulated with OVA peptide presented by irradiated Balb/c splenocytes in the presence of Th1 or Th2 polarizing cytokines. On day 5 T cells were harvested and GATA-3 mRNA was measured by real time PCR and quantified relative to activated, undifferentiated cells. The data are depicted as mean + SEM. C and D, T cells were stimulated as in A and GATA-3 mRNA containing exon 1A (C) and exon 1B (D) was measured by real time PCR. The fold induction of GATA-3 1A and 1B mRNA, relative to activated, undifferentiated cells, is depicted as mean +/- SEM. E and F, T cells were stimulated as in A and GATA-3 protein was measured 48 hours after restimulation by western blot of nuclear lysates, which were subsequently stripped and reprobed for PCNA. GATA-3 protein level at 48 hours after restimulation, relative to unstimulated cells. The data are depicted as mean + SEM, p=0.0003 for IL-4 vs Ag+IL-4 by paired t test, n=8.
In contrast to the lack of any impact of TCR signaling on IL-4-induced GATA-3 mRNA, TCR signaling does increase GATA-3 protein levels. IL-4 stimulation induces a small increase in GATA-3 protein (Fig. 3, E and F). In contrast, the combination of Ag and IL-4 stimulation induces a greater level of GATA-3 protein than IL-4 stimulation, despite equivalent induction of GATA-3 mRNA (Fig. 3, A and F). The ability of TCR signaling to increase the level of GATA-3 protein without increasing the level of GATA-3 mRNA implicates a mechanism of TCR-dependent post-transcriptional control of GATA-3 expression.
GATA-3 translation rate is enhanced by Ag stimulation
To determine if the post-transcriptional increase is mediated by an increase in GATA-3 protein synthesis we measured the rate of GATA-3 translation by pulse labeling with 35S methionine/cysteine. A significant increase in the level of de novo synthesis of GATA-3 is detected in Ag plus IL-4 stimulated cells when compared to IL-4 stimulated cells (Fig. 4A). The increase in Ag stimulated cells is specific for GATA-3, as we detect similar levels of the control proteins lck (Fig. 4A) and MHC-I (data not shown). Furthermore, the percent incorporation of 35S met/cys into newly synthesized proteins is equivalent between IL-4 and Ag plus IL-4 stimulated cells, indicating that the overall translational efficiency is not increased by Ag stimulation (Fig. 4B). To determine the GATA-3 translation rate we measured the amount of protein produced during the pulse label (Fig. 4A) and divided by the amount of mRNA (Fig. 4C), both relative to lck. We see a consistent four-fold increase in the GATA-3 translation rate in the presence of TCR signaling (Fig. 4D). Thus, these data show that TCR signaling specifically increases the translation of GATA-3, resulting in higher GATA-3 protein levels and Th2 differentiation.
FIGURE 4.
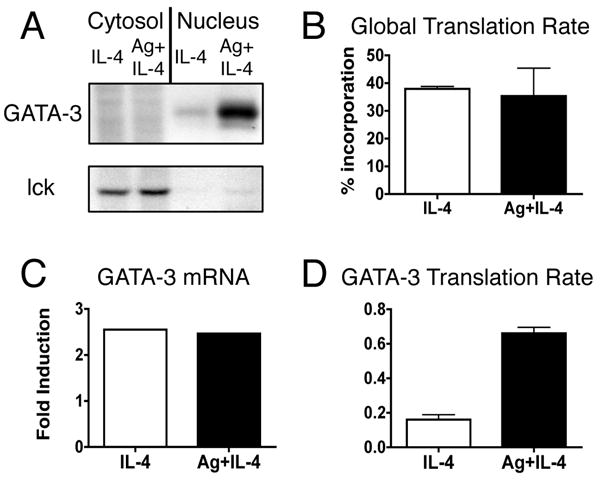
TCR signaling increases the rate of GATA-3 translation. Activated, undifferentiated T cells were restimulated with IL-4 or with both IL-4 and OVA peptide presented by ProAd-B7 cells (Ag+IL-4); hIL-2 was added to both stimulation conditions. A, 48 hours after restimulation T cells were pulsed with 35S met/cys for 10 minutes. Nuclear and cytosolic lysates were immmunoprecipitated for GATA-3 and lck and analyzed on SDS gels. One experiment, representative of 3, is shown. B, The overall translational efficiency was determined by measuring the percent incorporation of 35S met/cys into ethanol-precipitable proteins from the pulse-labeled lysates. Data are depicted as mean + SEM, n=3. C, At the time of the pulse label, mRNA was isolated from a portion of the T cells and measured by real time PCR. The fold induction of GATA-3 mRNA, relative to lck, is shown for the experiment in AD, The GATA-3 translation rate was calculated as the amount of detectable radioactivity from the pulse period divided by the amount of mRNA at the time of the pulse label, both normalized to lck. Data are depicted as mean + SD, p<0.0001 for IL-4 vs Ag+IL-4 by unpaired t test, n=3.
The priming of CD4+ T cells induces components of the integrated stress response, resulting in phosphorylation of eukaryotic initiation factor (eIF) 2α at Ser 51 (24). This phosphorylation prevents exchange of GDP for GTP by eIF2B and renders initiation complexes ineffective, resulting in translational arrest of key proteins, including IL-4. Acute restimulation through the TCR relieves the stress response, resulting in dephosphorylation of eIF2α and an increase in global translationl efficiency. To determine if the stress response is contributing to the TCR-dependent increase in GATA-3 translation we measured the phosphorylation state of eIF2α at Ser 51 in activated, undifferentiated cells stimulated with IL-4, Ag, or Ag plus IL-4. At 48 hours post-stimulation, the time we detect an increase in the GATA-3 translation rate, there is no difference in the amount of eIF2α phosphorylation (Fig. 5, A and C). As a control we measured induction of eIF2α phosphorylation in fibroblast cells treated with tunicamycin, a potent inducer of the stress response, which results in a five-fold increase in phosphorylation (Fig. 5, B and D). Taken together, these data further show that the TCR-dependent translational increase is specific for GATA-3 and not due to a global enhancement of translation.
FIGURE 5.
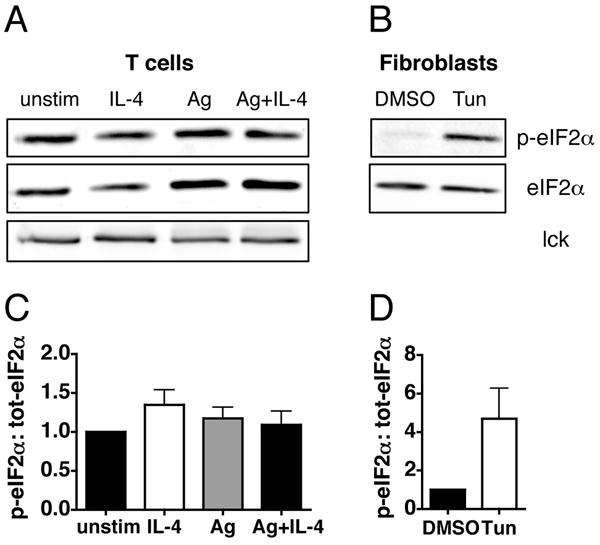
TCR signaling does not affect phosphorylation of eIF2α. A and C, Activated, undifferentiated T cells were restimulated with IL-4, OVA peptide presented by ProAd-B7 cells (Ag), or with both Ag and IL-4; hIL-2 was added to all conditions (left). B and D, Fibroblast cells were treated with 5μg/mL Tunicamycin (Tun) for 2 hours (right). Phosphorylation of eIF2α at Ser 51 was measured 48 hours after restimulation by western blot of cytosolic lysates, which were subsequently stripped and reprobed for total eIF2α and lck. The ratio of phosphorylated to total eIF2α, relative to unstimulated T cells (C) and vehicle treated fibroblast cells (D). Data are depicted as mean + SEM, n=5 for T cells, n=4 for fibroblasts.
TCR signaling does not enhance GATA-3 protein stability
It has been shown that signaling through ERK can increase GATA-3 protein stability (25). To determine if an additional TCR-mediated effect on GATA-3 protein stability also contributes to the post-transcriptional increase in GATA-3 protein we calculated the GATA-3 protein half-life with a pulse-chase assay (Fig. 6, A and B). The number of IL-4-stimulated cells was adjusted during the pulse label to compensate for the decreased GATA-3 translation in the absence of TCR signaling, so the IL-4 and Ag plus IL-4 stimulated samples have similar starting levels of GATA-3. We find that the degradation of biosynthetically labeled GATA-3 protein is not affected by Ag stimulation (Fig. 6, A and B). This result is confirmed when GATA-3 protein stability is measured after blocking new protein synthesis with cycloheximide (Fig. 6, C and D). GATA-3 has a similar rate of degradation in cells stimulated with IL-4 in the presence or absence of TCR signaling. Thus, the post-transcriptional increase in GATA-3 protein in Ag-stimulated cells is a direct effect of an increased GATA-3 translation rate and not due to an increase in GATA-3 protein stability.
FIGURE 6.
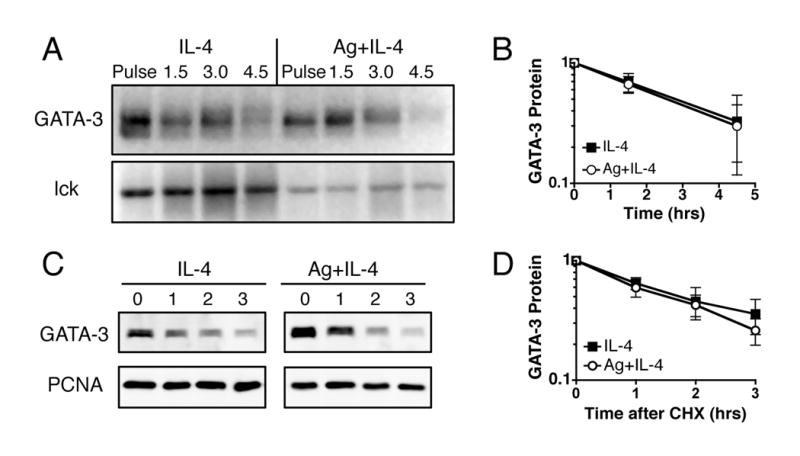
TCR signaling does not increase GATA-3 protein stability. Activated, undifferentiated T cells were restimulated with IL-4 or with both IL-4 and OVA peptide presented by ProAd-B7 cells (Ag+IL-4); hIL-2 was added to both stimulation conditions. A and B, 48 hours after restimulation T cells were pulsed with 35S met/cys for 10 minutes, then chased with unlabeled media for various times. Nuclear and cytosolic lysates were immunoprecipitated for GATA-3 and lck and analyzed on SDS gels. The cell number was adjusted so that the IL-4 and Ag plus IL-4 samples have similar starting levels of GATA-3 protein, to account for the Ag-driven increase in GATA-3 translation. A, One gel, representative of 3, is shown. B, The quantitation of GATA-3 protein level during the pulse chase experiments is depicted as mean +/- SD, n=3. For some experiments the pulse time was increased to 60 minutes. C and D, 100 μg/mL cycloheximide (CHX) was added to T cells and protein levels of GATA-3 and PCNA were measured by western blot of nuclear lysates. C, One western blot, representative of 3, is shown. D, The quantitation of GATA-3 protein level after addition of CHX is depicted as mean +/- SEM, n=3.
PI3-Kinase signaling through mTOR is required for enhanced GATA-3 translation and Th2 differentiation
The PI3K-mediated conversion of phosphatidylinositol-4,5-bisphosphate (PI(4,5)P2) to PI(3,4,5)P3 serves as a membrane-recruitment site for PH-domain containing proteins including Akt, PDK1, Itk, and Vav (26). Signaling through Akt leads to activation of the mTOR kinase, a protein central to cell growth and the regulation of both gene specific and global translation. To determine if the PI3K/mTOR pathway is involved in GATA-3 translation we measured GATA-3 mRNA and protein levels in the absence of PI3K signaling. Inhibition of PI3K with the inhibitor LY294002 has no effect on GATA-3 mRNA levels, but specifically decreases the level of GATA-3 protein (Fig. 7, A-C). These data suggest that PI3K signaling is required for the TCR-dependent increase in GATA-3 translation. To determine whether the decrease in GATA-3 protein is due to a specific translational defect or reflective of a global effect of PI3K we measured de novo protein synthesis by 35S met/cys pulse labeling. Consistent with the role of PI3K signaling in global translational efficiency there is a slight decrease in the overall translation rate in the presence of the PI3K inhibitor (Fig. 7, D). However, in addition to of this global effect there is a specific decrease in GATA-3 translation (Fig. 7, E and F). The reduction in GATA-3 translation is greater than the reduction in global translation and when GATA-3 is normalized, for both mRNA and protein, to lck, we detect a specific decrease in the GATA-3 translation rate (Fig. 7, E and F). Thus, the addition of LY294002 specifically decreases the GATA-3 translation rate, indicating that the TCR-dependent specific increase in GATA-3 translation is mediated by signaling through PI3K. To determine if this pathway is dependent on mTOR T cells were treated with rapamycin. Addition of rapamycin to Ag plus IL-4 stimulated cells does not effect the level of GATA-3 mRNA (Fig.7 G). Rather, the level of GATA-3 protein is decreased in the absence of mTOR signaling (Fig. 7 H and I), indicating that mTOR signaling is required for the TCR-mediated translational upregulation of GATA-3. Taken together, these data indicate that the TCR-mediated induction of GATA-3 translation is dependent on PI3K signaling through mTOR.
FIGURE 7.
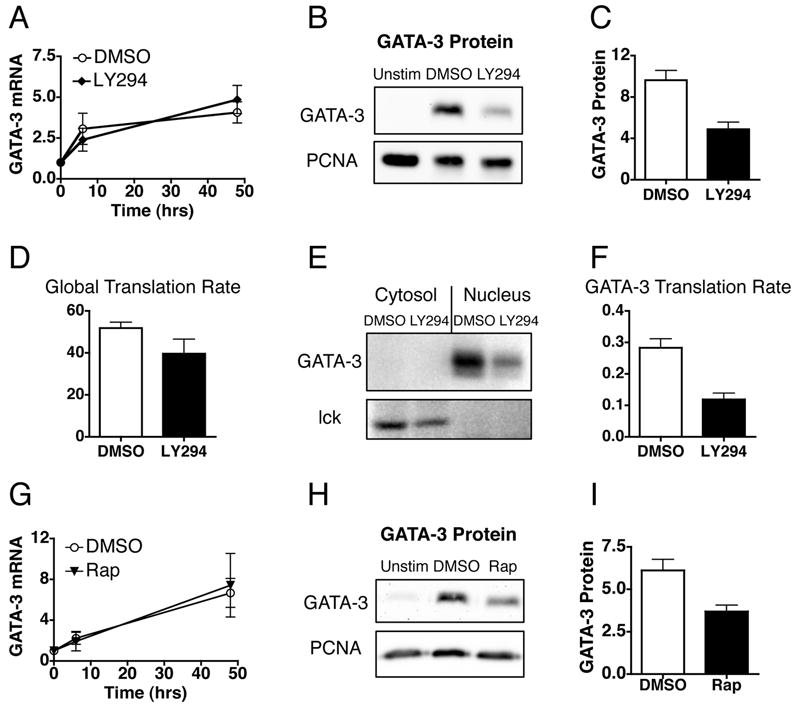
PI3-Kinase signaling through mTOR is required for enhanced GATA-3 translation. Activated, undifferentiated T cells were restimulated with both IL-4 and OVA peptide presented by ProAd-B7 cells in the presence of 10μM LY294002 (LY294), 10nM rapamycin (Rap), or vehicle control DMSO; hIL-2 was added to all stimulation conditions. A, GATA-3 mRNA was measured by real time PCR at different times following restimulation. The fold induction of GATA-3 mRNA, relative to activated, undifferentiated cells, is depicted as mean +/- SEM, n≥5 for each time point. B, GATA-3 protein was measured 48 hours after restimulation by western blot of nuclear lysates. C, The amount of GATA-3 protein relative to unstimulated cells is depicted as mean + SEM, p=0.0018 for DMSO vs LY294 treated samples by paired t test, n=4. D and E, 48 hours after restimulation T cells were pulsed with 35S met/cys for 10 minutes. D, The overall translational efficiency was determined by measuring the percent incorporation of 35S met/cys into ethanol-precipitable proteins from the pulse-labeled lysates. The data are depicted as mean + SEM, n=2. E, Nuclear and cytosolic lysates were immmunoprecipitated for GATA-3 and lck and analyzed on SDS gels. F, The GATA-3 translation rate was calculated as the amount of detectable radioactivity from the pulse period divided by the amount of mRNA at the time of the pulse label, both normalized to lck. The data are depicted as mean + SEM, n=2. G, GATA-3 mRNA was measured as in A and is depicted as mean +/- SEM, n=3. H and I, GATA-3 protein was measured as in B and is depicted as mean + SEM, p=.0041 for DMSO vs Rap treated samples by paired t test, n=5.
The induction of Th2 responses in vivo has been shown to be dependent on PI3K signaling. However, these effects may be T-cell independent, as alteration of PI3K signaling has effects on DC IL-12 production, mast cell and eosinophil recruitment, and airway hyperreactivity in asthma models (27-30). In vitro the effect of PI3K inhibition on Th2 differentiation is concomitant with a decrease in cell division, making it unclear if the effect is specific for the Th2 program or a secondary consequence of the block in T cell expansion (31). Consistent with this, we detect major decreases in cell expansion after stimulation of naïve T cells in the presence of LY294002 or rapamycin (data not shown). However, using activated, undifferentiated T cells allows us to measure Th2 differentiation while bypassing this effect of PI3K inhibition on cell expansion. Although there is a slight decrease in cell division, addition of LY294002 or rapamycin has no effect on T cell expansion of previously activated cells (Fig. 8, A and C). In contrast to the minimal effects on cell expansion, the amount of Th2 differentiation is greatly reduced with LY294002 or rapamycin treatment (Fig. 8, B and C). These data indicate that there is a specific defect in Th2 differentiation in the absence of PI3K/mTOR signaling, and that this defect correlates with a failure to upregulate GATA-3 translation.
FIGURE 8.
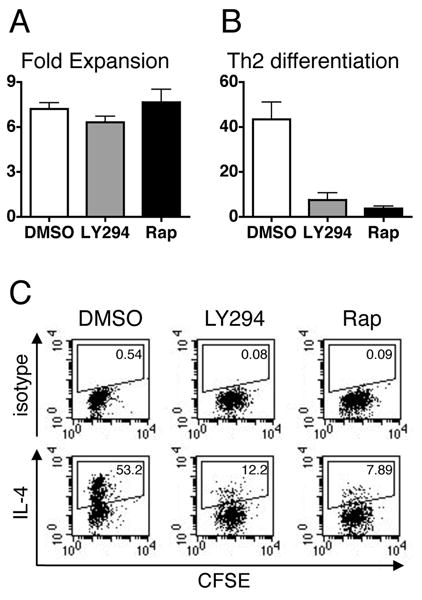
Inhibition of PI3-Kinase or mTOR signaling inhibits Th2 differentiation. Activated, undifferentiated T cells were restimulated for 6 days with both IL-4 and OVA peptide presented by ProAd-B7 cells in the presence of 10μM LY294002 (LY294), 10nM rapamycin (Rap), or vehicle control DMSO; hIL-2 was added to all stimulation conditions. T cells were counted to determine the fold expansion during the secondary stimulation (A) and restimulated overnight on plate bound anti-CD3 to measure Th2 differentiation, defined as secretion of IL-4 (B). The data are depicted as mean + SEM, n≥6. In A there is no statistical difference between any of the samples. In B p=.0015 for DMSO vs. LY294 and p=.0051 for DMSO vs Rap by paired t test. C, Activated, undifferentiated cells were stained with CFSE and stimulated as in A and B. 6 days after restimulation T cells were stimulated overnight on plate bound anti-CD3 and stained for intracellular IL-4 (bottom); isotype control is shown for comparison (top). One experiment, representative of 4, is shown. The mean fluorescence intensity for IL-4 positive cells is 266.40 for DMSO treatment, 173.86 for LY294, and 130.19 for Rap.
Discussion
The transcription factor GATA-3 is a tightly regulated protein that is essential for Th2 responses (1). In order to understand the signaling pathways that lead to GATA-3 upregulation, we have developed stimulation conditions that allow us to separately study the contributions of IL-4R and TCR signaling. We have identified a role of TCR signaling in GATA-3 translation that is independent of the role of TCR signaling in initial T cell activation, survival, and expansion. This translational increase is specific for GATA-3 and responsible for the TCR-dependent increase in GATA-3 protein that is associated with increased Th2 differentiation.
The majority of data investigating GATA-3 expression has focused on transcriptional upregulation. It was initially shown that addition of IL-4 to activated T cells promotes Th2 differentiation and GATA-3 transcription through the activation of the transcription factor, STAT6 (7, 32, 33). While the role of STAT6 in promoting GATA-3 transcription is well established, the previous experiments have been done in the presence of concomitant TCR signaling, making it difficult to determine if STAT6 is working independently or in concert with molecules downstream of the TCR. A role for TCR signaling in regulating GATA-3 induction and subsequent Th2 differentiation was first implicated by altering TCR signaling through the use of altered peptide ligands or changing the Ag dose (17, 18). Additionally, T cells from p50 or Bcl-3 deficient mice have a defect in GATA-3 induction and Th2 differentiation in the presence of exogenous IL-4, suggesting a role for NF- κB signaling in conjunction with STAT6 for GATA-3 upregulation (19, 34). However, the NF- κB pathway is activated downstream of many different receptors and genetic disruption of this pathway results in developmental defects both within and outside the T cell compartment. Furthermore, inhibition of TCR signaling using knockout animals or by altering the peptide dose or affinity may simply reduce the early TCR-dependent production of IL-4, rather than directly affect GATA-3 induction. It was recently shown that TCR activation can induce GATA-3 mRNA in the absence of IL-4 through the transcription factor TCF-1 (22). Though our data in both naïve (data not shown) and previously activated cells, as well as previous reports, have shown a dependence on IL-4 and STAT6, this recent finding suggests that under some conditions signaling through the TCR may be sufficient to induce GATA-3 mRNA upregulation. Thus, TCR signaling can impact GATA-3 mRNA upregulation in multiple ways: direct induction independent of STAT6, in conjunction with IL-4R signaling, or indirectly through induction of IL-4. We have designed our experiments to restrict the effects of TCR signaling to modulation of Th2 differentiation, and under these conditions we find that TCR signaling does not have any impact on GATA-3 mRNA upregulation. Despite the many ways in which TCR signaling can potentially impact GATA-3 mRNA, these results highlight that none of these pathways are required as IL-4 is capable of driving GATA-3 mRNA independently of Ag stimulation. While we cannot rule out any long-lasting effect of the initial stimulation, we show that in the absence of acute TCR stimulation, IL-4 stimulation is sufficient to drive GATA-3 mRNA induction and that addition of TCR signaling does not increase the level of IL-4-driven GATA-3 mRNA.
Priming of naïve CD4+ T cells both in vivo and in vitro can lead to proliferating cells that produce IL-2, but neither IL-4 nor IFN-γ (35, 36). These cells maintain a flexible differentiation state and can be pushed into different subsets depending on the nature of the secondary challenge. This flexibility could be compromised if exposure to polarizing cytokines in the absence of cognate antigen resulted in GATA-3 protein upregulation. Our data establish a clear requirement for concomitant TCR and IL-4R signaling to direct Th2 responses for primed, undifferentiated T cells with IL-4R signaling important for upregulation of GATA-3 mRNA and TCR signaling for enhancement of GATA-3 translation. This establishes the translational control of GATA-3 as a key point of signal integration associated with Th2 differentiation, providing a layer of protection against aberrant GATA-3 production, which could lead to Th2-biased immune responses and atopic diseases such as asthma.
In addition to the signaling pathways that modulate transcriptional activation of GATA-3, it has been reported that ERK signaling can modulate GATA-3 protein stability (25). Targeted deletion of growth factor independent-1 (Gfi-1), whose induction is reduced by inhibition of ERK, results in decreased stability of GATA-3 protein and subsequent Th2 differentiation (37). However, we find that Ag stimulation has no effect on GATA-3 protein stability. Furthermore, inhibition of ERK activation did not alter the level of TCR-induced GATA-3 protein (data not shown). Although it remains possible that under some stimulation conditions TCR signaling may contribute towards GATA-3 stability through ERK activation, this mechanism is not required for TCR signaling to enhance GATA-3 protein levels. The more significant contribution of TCR signaling towards GATA-3 protein levels is through the enhancement of the GATA-3 translation rate.
Translational control of gene expression can be mediated through effects on the global translational machinery or through gene specific effects. TCR signaling has been shown to impact global translational efficiency following initial T cell activation through effects on the expression level and functional activity of translation initiation factors (24, 38, 39). Specifically, the dephosphorylation of eIF2α following acute TCR stimulation has been shown to enhance translational activity (24). The ability of TCR signaling to promote dephosphorylation of eIF2α is likely a time-restricted event that is dependent on the strength of the signaling. Accordingly, we find that there is no change in the phosphorylation state of eIF2α in the presence or absence of Ag stimulation at the time we detect an increase in the GATA-3 translation rate. Rather, we find that Ag stimulation specifically induces translation of GATA-3, without affecting phosphorylation of eIF2α, global translational efficiency, or the translation of control proteins.
In addition to the effects on eIF2α, TCR signaling can also impact both global and gene-specific translation through PI3K and mTOR. The best-characterized mechanism of mTOR-dependent control of translation is through phosphorylation of eIF4E-BP, an inhibitor of ribosome recruitment to the 5′ end of an mRNA molecule (40). This phosphorylation decreases affinity of eIF4E-BP for eIF4E, allowing eIF4G to bind eIF4E and recruit the ribosome to initiate translation. This process appears to be more complex than initially suggested however, as overexpression of eIF4E selectively enhances translation of mRNA molecules that contain secondary structures within their 5′UTR (41). GATA-3 transcripts contain unusually long 5′UTRs which contain many potential structural elements. The PI3K pathway effect on eIF4E-BP phosphorylation may have a larger effect on these structured 5′UTR containing mRNA molecules, including GATA-3. An additional step necessary only for translation of mRNA with structured 5′UTR is the eIF4A helicase-dependent unwinding of mRNA (42). The activity of eIF4A can be regulated by the PI3K/mTOR pathway through the phosphorylation-dependent binding of eIF4B to eIF4A (43). Reduction of this phosphorylation through PI3K inhibition could lead to decreased translation of GATA-3 and other mRNA with structured 5′UTR. Global translation is also enhanced by PI3K/mTOR signaling through enhanced generation of ribosome components. This enhanced translation is dependent on mTOR and the presence of a 5′ terminal oligopyrimidine (TOP) tract, a polypyrimidine stretch adjacent to the 5′ end of mRNA molecule (44). Interestingly, GATA-3 mRNA molecules contains multiple polypyrimidine stretches within its 5′UTR. Though these elements are not in the correct position to be classified as a TOP, these regions could confer mTOR-dependent translational control.
Further experiments are needed to determine if these potential mechanisms are responsible for the TCR-mediated enhancement of GATA-3 translation. Understanding how the different transcriptional and post-transcriptional regulatory elements cooperate to mediate the fine control of GATA-3 expression will be key to understanding how this pathway may be misregulated in atopic diseases or can be targeted for immune manipulation.
Acknowledgments
The authors thank Deborah Fowell for critical reading of the manuscript, Bart Eisfelder for initiating experiments to assess the functional outcome of TCR signaling during Th2 differentiation, the URMC Functional Genomics Center for running all real time PCR plates, and the NCI Clinical Repository for providing human interleukin 2 and antibodies to murine IL-4.
Abbreviations
- mTOR
mammalian target of rapamycin
- UTR
un-translated region
- eIF
eukaryotic initiation factor
Footnotes
This work was supported by NIH grant R01AI48237 to J. Miller.
K. Cook was supported by NIH training grant T32HL66988.
The authors have no conflicting financial interests.
References
- 1.Zhu J, Min B, Hu-Li J, Watson CJ, Grinberg A, Wang Q, Killeen N, Urban JF, Jr, Guo L, Paul WE. Conditional deletion of Gata3 shows its essential function in T(H)1-T(H)2 responses. Nat Immunol. 2004;5:1157–1165. doi: 10.1038/ni1128. [DOI] [PubMed] [Google Scholar]
- 2.Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 1997;89:587–596. doi: 10.1016/s0092-8674(00)80240-8. [DOI] [PubMed] [Google Scholar]
- 3.Zhang DH, Cohn L, Ray P, Bottomly K, Ray A. Transcription factor GATA-3 is differentially expressed in murine Th1 and Th2 cells and controls Th2-specific expression of the interleukin-5 gene. J Biol Chem. 1997;272:21597–21603. doi: 10.1074/jbc.272.34.21597. [DOI] [PubMed] [Google Scholar]
- 4.Pandolfi PP, Roth ME, Karis A, Leonard MW, Dzierzak E, Grosveld FG, Engel JD, Lindenbaum MH. Targeted disruption of the GATA3 gene causes severe abnormalities in the nervous system and in fetal liver haematopoiesis. Nat Genet. 1995;11:40–44. doi: 10.1038/ng0995-40. [DOI] [PubMed] [Google Scholar]
- 5.Ting CN, Olson MC, Barton KP, Leiden JM. Transcription factor GATA-3 is required for development of the T-cell lineage. Nature. 1996;384:474–478. doi: 10.1038/384474a0. [DOI] [PubMed] [Google Scholar]
- 6.George KM, Leonard MW, Roth ME, Lieuw KH, Kioussis D, Grosveld F, Engel JD. Embryonic expression and cloning of the murine GATA-3 gene. Development. 1994;120:2673–2686. doi: 10.1242/dev.120.9.2673. [DOI] [PubMed] [Google Scholar]
- 7.Kurata H, Lee HJ, O'Garra A, Arai N. Ectopic expression of activated Stat6 induces the expression of Th2-specific cytokines and transcription factors in developing Th1 cells. Immunity. 1999;11:677–688. doi: 10.1016/s1074-7613(00)80142-9. [DOI] [PubMed] [Google Scholar]
- 8.Asnagli H, Afkarian M, Murphy KM. Cutting edge: Identification of an alternative GATA-3 promoter directing tissue-specific gene expression in mouse and human. J Immunol. 2002;168:4268–4271. doi: 10.4049/jimmunol.168.9.4268. [DOI] [PubMed] [Google Scholar]
- 9.Amsen D, Antov A, Jankovic D, Sher A, Radtke F, Souabni A, Busslinger M, McCright B, Gridley T, Flavell RA. Direct regulation of Gata3 expression determines the T helper differentiation potential of Notch. Immunity. 2007;27:89–99. doi: 10.1016/j.immuni.2007.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fang TC, Yashiro-Ohtani Y, Del Bianco C, Knoblock DM, Blacklow SC, Pear WS. Notch directly regulates Gata3 expression during T helper 2 cell differentiation. Immunity. 2007;27:100–110. doi: 10.1016/j.immuni.2007.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou M, Ouyang W, Gong Q, Katz SG, White JM, Orkin SH, Murphy KM. Friend of GATA-1 represses GATA-3-dependent activity in CD4+ T cells. J Exp Med. 2001;194:1461–1471. doi: 10.1084/jem.194.10.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miaw SC, Choi A, Yu E, Kishikawa H, Ho IC. ROG, repressor of GATA, regulates the expression of cytokine genes. Immunity. 2000;12:323–333. doi: 10.1016/s1074-7613(00)80185-5. [DOI] [PubMed] [Google Scholar]
- 13.Hwang ES, Szabo SJ, Schwartzberg PL, Glimcher LH. T helper cell fate specified by kinase-mediated interaction of T-bet with GATA-3. Science. 2005;307:430–433. doi: 10.1126/science.1103336. [DOI] [PubMed] [Google Scholar]
- 14.Ouyang W, Lohning M, Gao Z, Assenmacher M, Ranganath S, Radbruch A, Murphy KM. Stat6-independent GATA-3 autoactivation directs IL-4-independent Th2 development and commitment. Immunity. 2000;12:27–37. doi: 10.1016/s1074-7613(00)80156-9. [DOI] [PubMed] [Google Scholar]
- 15.Skapenko A, Leipe J, Niesner U, Devriendt K, Beetz R, Radbruch A, Kalden JR, Lipsky PE, Schulze-Koops H. GATA-3 in human T cell helper type 2 development. J Exp Med. 2004;199:423–428. doi: 10.1084/jem.20031323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van Esch H, Groenen P, Nesbit MA, Schuffenhauer S, Lichtner P, Vanderlinden G, Harding B, Beetz R, Bilous RW, Holdaway I, Shaw NJ, Fryns JP, Van de Ven W, Thakker RV, Devriendt K. GATA3 haplo-insufficiency causes human HDR syndrome. Nature. 2000;406:419–422. doi: 10.1038/35019088. [DOI] [PubMed] [Google Scholar]
- 17.Brogdon JL, Leitenberg D, Bottomly K. The potency of TCR signaling differentially regulates NFATc/p activity and early IL-4 transcription in naive CD4+ T cells. J Immunol. 2002;168:3825–3832. doi: 10.4049/jimmunol.168.8.3825. [DOI] [PubMed] [Google Scholar]
- 18.Ise W, Totsuka M, Sogawa Y, Ametani A, Hachimura S, Sato T, Kumagai Y, Habu S, Kaminogawa S. Naive CD4+ T cells exhibit distinct expression patterns of cytokines and cell surface molecules on their primary responses to varying doses of antigen. J Immunol. 2002;168:3242–3250. doi: 10.4049/jimmunol.168.7.3242. [DOI] [PubMed] [Google Scholar]
- 19.Das J, Chen CH, Yang L, Cohn L, Ray P, Ray A. A critical role for NF-kappa B in GATA3 expression and TH2 differentiation in allergic airway inflammation. Nat Immunol. 2001;2:45–50. doi: 10.1038/83158. [DOI] [PubMed] [Google Scholar]
- 20.Yamane H, Zhu J, Paul WE. Independent roles for IL-2 and GATA-3 in stimulating naive CD4+ T cells to generate a Th2-inducing cytokine environment. J Exp Med. 2005;202:793–804. doi: 10.1084/jem.20051304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zuckerman LA, Pullen L, Miller J. Functional consequences of costimulation by ICAM-1 on IL-2 gene expression and T cell activation. J Immunol. 1998;160:3259–3268. [PubMed] [Google Scholar]
- 22.Yu Q, Sharma A, Oh SY, Moon HG, Hossain MZ, Salay TM, Leeds KE, Du H, Wu B, Waterman ML, Zhu Z, Sen JM. T cell factor 1 initiates the T helper type 2 fate by inducing the transcription factor GATA-3 and repressing interferon-gamma. Nat Immunol. 2009;10:992–999. doi: 10.1038/ni.1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scheinman EJ, Avni O. Transcriptional regulation of GATA3 in T helper cells by the integrated activities of transcription factors downstream of the interleukin-4 receptor and T cell receptor. J Biol Chem. 2009;284:3037–3048. doi: 10.1074/jbc.M807302200. [DOI] [PubMed] [Google Scholar]
- 24.Scheu S, Stetson DB, Reinhardt RL, Leber JH, Mohrs M, Locksley RM. Activation of the integrated stress response during T helper cell differentiation. Nat Immunol. 2006;7:644–651. doi: 10.1038/ni1338. [DOI] [PubMed] [Google Scholar]
- 25.Yamashita M, Shinnakasu R, Asou H, Kimura M, Hasegawa A, Hashimoto K, Hatano N, Ogata M, Nakayama T. Ras-ERK MAPK cascade regulates GATA3 stability and Th2 differentiation through ubiquitin-proteasome pathway. J Biol Chem. 2005;280:29409–29419. doi: 10.1074/jbc.M502333200. [DOI] [PubMed] [Google Scholar]
- 26.Koyasu S. The role of PI3K in immune cells. Nat Immunol. 2003;4:313–319. doi: 10.1038/ni0403-313. [DOI] [PubMed] [Google Scholar]
- 27.Duan W, Aguinaldo Datiles AM, Leung BP, Vlahos CJ, Wong WS. An anti-inflammatory role for a phosphoinositide 3-kinase inhibitor LY294002 in a mouse asthma model. Int Immunopharmacol. 2005;5:495–502. doi: 10.1016/j.intimp.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 28.Fukao T, Tanabe M, Terauchi Y, Ota T, Matsuda S, Asano T, Kadowaki T, Takeuchi T, Koyasu S. PI3K-mediated negative feedback regulation of IL-12 production in DCs. Nat Immunol. 2002;3:875–881. doi: 10.1038/ni825. [DOI] [PubMed] [Google Scholar]
- 29.Fukao T, Yamada T, Tanabe M, Terauchi Y, Ota T, Takayama T, Asano T, Takeuchi T, Kadowaki T, Hata Ji J, Koyasu S. Selective loss of gastrointestinal mast cells and impaired immunity in PI3K-deficient mice. Nat Immunol. 2002;3:295–304. doi: 10.1038/ni768. [DOI] [PubMed] [Google Scholar]
- 30.Myou S, Leff AR, Myo S, Boetticher E, Tong J, Meliton AY, Liu J, Munoz NM, Zhu X. Blockade of inflammation and airway hyperresponsiveness in immune-sensitized mice by dominant-negative phosphoinositide 3-kinase-TAT. J Exp Med. 2003;198:1573–1582. doi: 10.1084/jem.20030298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okkenhaug K, Patton DT, Bilancio A, Garcon F, Rowan WC, Vanhaesebroeck B. The p110delta isoform of phosphoinositide 3-kinase controls clonal expansion and differentiation of Th cells. J Immunol. 2006;177:5122–5128. doi: 10.4049/jimmunol.177.8.5122. [DOI] [PubMed] [Google Scholar]
- 32.Le Gros G, Ben-Sasson SZ, Seder R, Finkelman FD, Paul WE. Generation of interleukin 4 (IL-4)-producing cells in vivo and in vitro: IL-2 and IL-4 are required for in vitro generation of IL-4-producing cells. J Exp Med. 1990;172:921–929. doi: 10.1084/jem.172.3.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaplan MH, Schindler U, Smiley ST, Grusby MJ. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity. 1996;4:313–319. doi: 10.1016/s1074-7613(00)80439-2. [DOI] [PubMed] [Google Scholar]
- 34.Corn RA, Hunter C, Liou HC, Siebenlist U, Boothby MR. Opposing roles for RelB and Bcl-3 in regulation of T-box expressed in T cells, GATA-3, and Th effector differentiation. J Immunol. 2005;175:2102–2110. doi: 10.4049/jimmunol.175.4.2102. [DOI] [PubMed] [Google Scholar]
- 35.Sad S, Mosmann TR. Single IL-2-secreting precursor CD4 T cell can develop into either Th1 or Th2 cytokine secretion phenotype. J Immunol. 1994;153:3514–3522. [PubMed] [Google Scholar]
- 36.Wang X, Mosmann T. In vivo priming of CD4 T cells that produce interleukin (IL)-2 but not IL-4 or interferon (IFN)-gamma, and can subsequently differentiate into IL-4- or IFN-gamma-secreting cells. J Exp Med. 2001;194:1069–1080. doi: 10.1084/jem.194.8.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shinnakasu R, Yamashita M, Kuwahara M, Hosokawa H, Hasegawa A, Motohashi S, Nakayama T. Gfi1-mediated stabilization of GATA3 protein is required for Th2 cell differentiation. J Biol Chem. 2008;283:28216–28225. doi: 10.1074/jbc.M804174200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mao X, Green JM, Safer B, Lindsten T, Frederickson RM, Miyamoto S, Sonenberg N, Thompson CB. Regulation of translation initiation factor gene expression during human T cell activation. J Biol Chem. 1992;267:20444–20450. [PubMed] [Google Scholar]
- 39.Morley SJ, Rau M, Kay JE, Pain VM. Increased phosphorylation of eukaryotic initiation factor 4 alpha during early activation of T lymphocytes correlates with increased initiation factor 4F complex formation. Eur J Biochem. 1993;218:39–48. doi: 10.1111/j.1432-1033.1993.tb18349.x. [DOI] [PubMed] [Google Scholar]
- 40.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 41.Koromilas AE, Lazaris-Karatzas A, Sonenberg N. mRNAs containing extensive secondary structure in their 5′ non-coding region translate efficiently in cells overexpressing initiation factor eIF-4E. EMBO J. 1992;11:4153–4158. doi: 10.1002/j.1460-2075.1992.tb05508.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rogers GW, Jr, Komar AA, Merrick WC. eIF4A: the godfather of the DEAD box helicases. Prog Nucleic Acid Res Mol Biol. 2002;72:307–331. doi: 10.1016/s0079-6603(02)72073-4. [DOI] [PubMed] [Google Scholar]
- 43.Shahbazian D, Roux PP, Mieulet V, Cohen MS, Raught B, Taunton J, Hershey JW, Blenis J, Pende M, Sonenberg N. The mTOR/PI3K and MAPK pathways converge on eIF4B to control its phosphorylation and activity. EMBO J. 2006;25:2781–2791. doi: 10.1038/sj.emboj.7601166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jefferies HB, Fumagalli S, Dennis PB, Reinhard C, Pearson RB, Thomas G. Rapamycin suppresses 5′TOP mRNA translation through inhibition of p70s6k. EMBO J. 1997;16:3693–3704. doi: 10.1093/emboj/16.12.3693. [DOI] [PMC free article] [PubMed] [Google Scholar]