

Abstract
Significance: The molecules interacting with CasL (MICAL) family members participate in a multitude of activities, including axonal growth cone repulsion, membrane trafficking, apoptosis, and bristle development in flies. An interesting feature of MICAL proteins is the presence of an N-terminal flavo-mono-oxygenase domain. This mono-oxygenase domain generates redox potential with which MICALs can either oxidize proteins or produce reactive oxygen species (ROS). Actin is one such protein that is affected by MICAL function, leading to dramatic cytoskeletal rearrangements. This review describes the MICAL-family members, and discusses their mechanisms of actin-binding and regulation of actin cytoskeleton organization. Recent Advances: Recent studies show that MICALs directly induce oxidation of actin molecules, leading to actin depolymerization. ROS production by MICALs also causes oxidation of collapsin response mediator protein-2, a microtubule assembly promoter, which subsequently undergoes phosphorylation. Critical Issues: MICAL proteins oxidize proteins through two mechanisms: either directly by oxidizing methionine residues or indirectly via the production of ROS. It remains unclear whether MICAL proteins employ both mechanisms or whether the activity of MICAL-family proteins might vary with different substrates. Future Directions: The identification of additional substrates oxidized by MICAL will shed new light on MICAL protein function. Additional directions include expanding studies toward the MICAL-like homologs that lack flavin adenine dinucleotide domains and oxidation activity. Antioxid. Redox Signal. 20, 2059–2073.
Introduction
The dynamics of actin cytoskeleton rearrangement are crucial for a range of cellular processes, including membrane trafficking, maintenance of cell shape, cell locomotion, and cytokinesis (59). Actin cytoskeleton dynamics are extremely complex and highly regulated by various actin-binding proteins that control the nucleation, elongation, capping, severing, and crosslinking of actin filaments as well as the sequestration of actin monomers (9, 75). For example, fascin and profilin family members promote actin nucleation (6), whereas actin depolymerizing factor/cofilin family members catalyze the disassembly of actin filaments (53). Actin branching and nucleation are promoted by Arp2/3 complex proteins (58), while filament severing is mediated by proteins such as cofilin (53). Filament capping is mediated by specific capping proteins such as CapZ (7). Gelsolin, on the other hand, performs both filament capping and severing (53). Several proteins catalyze the crosslinking of actin filaments, leading to gelation (filamin) and bundling (fimbrin, α-actinin) of actin filaments (44). Proteins such as profilin and thymosin bind and inhibit the spontaneous nucleation of actin monomers (77).
As expected from the intense regulation of actin cytoskeleton dynamics, the formation of highly specialized cytoskeletal networks such as filopodia, lamellipodia, stress fibers, and focal adhesions requires the coordinated action of various actin regulatory proteins (40). In addition to some of the key actin-binding proteins noted earlier, a new and unique redox potential-based protein family has recently been added. This new class of proteins, known as Molecules Interacting with CasL (MICAL) family, uses intrinsic redox potential to oxidize actin and cause disassembly of actin filaments. The actin remodeling property of the Drosophila MICAL protein (Dm MICAL) is critical for actin filament organization-related processes such as axonal growth cone repulsion and bristle development (28, 81). In this review, we will discuss the MICAL family of proteins and their complex functions and effects on the actin cytoskeleton.
Evolutionarily Conserved MICAL Proteins
MICAL family members derive their name from the first study that identified mammalian MICAL1 as a binding protein to p130 Cas family members, Cas and CasL (78). The MICAL family consists of closely related MICAL proteins as well as several partially homologous MICAL-Like (MICAL-L) proteins. MICAL-L proteins lack the N-terminal flavin mono-oxygenase domain (flavin adenine dinucleotide [FAD] domain; Fig. 1) found in MICAL proteins. All MICAL-family members have a calponin homology (CH) domain as well as a Lin11, Isl-1, and Mec-3 (LIM) domain, and most have a C-terminal coiled-coil (CC) domain (Fig. 1).
FIG. 1.

Domain architecture of MICAL family members. Domain architecture of MICAL and MICAL-L proteins found in Drosophila melanogastor (Dm), Homo sapiens (Hs), and Danio rerio (Dr). FAD, flavin adenine dinucleotide; CH domain, calponin homology domain; LIM domain, Lin11, Isl-1, Mec-3 domain; CC domain, coiled-coil domain; MICAL, Molecules Interacting with CasL; MICAL-L, MICAL-Like.
The Homo sapiens MICAL-family (Hs MICAL) consists of three MICAL proteins (MICAL1, MICAL2, and MICAL3) and two MICAL-L homologs (MICAL-L1 and MICAL-L2). Drosophila melanogaster (Dm) contains a single MICAL with at least three different splice variations (long, medium, and short; the long isoform is represented in Fig. 1) and one MICAL-L protein. In Zebrafish Danio rerio (Dr), eight MICALs have been reported, including two paralogs for MICAL2, MICAL3, and MICAL-L2 (Fig. 1) (86).
MICAL proteins are highly conserved from invertebrates to vertebrates. A phylogenetic tree constructed with known MICAL proteins (long splice variants of MICALs were used in this tree generation) is depicted in Figure 2A. In this chart, MICAL1, MICAL2, and MICAL3 are organized into different clades. For example, Dm MICAL, Dr MICAL1, and Hs MICAL1 are clustered in the same group. Phylogenetic analysis of MICAL-L proteins also showed two clades of MICAL-L1 and MICAL-L2 families (Fig. 2B). Hs MICAL-L2 is an outgroup of MICAL-L1 and Dm MICAL-L, indicating that it is more distantly related to other MICAL-family members.
FIG. 2.

Phylogenetic analysis of MICAL family members. A phylogenetic analysis of MICAL (A) and MICAL-L (B) proteins found in Drosophila melangastor (Dm), Homo sapiens (Hs), and Danio rerio (Dr) was performed using Vector NTI software and the Neighbor Joining (NJ) algorithm. In cases in which the proteins are expressed as various splice variants, the longest splice variant was used for the analysis. The NCBI protein accession number of the protein sequence used for analysis is listed.
Expression Profile
The expression profile of MICAL in invertebrate organisms has been studied primarily in Drosophila (81). Dm MICAL is expressed in the ventral neurogenic region as well as in many non-neuronal tissues during early development. During later stages of development, Dm MICAL non-neuronal expression occurs primarily in the primordial midgut and in muscles. During axonal path-finding stages, Dm MICAL is expressed in the developing brain and ventral nerve cord and is also present in neuronal cell bodies along axons and growth cones. In another Drosophila study, Dm MICAL expression was observed in a variety of tissues during embryogenesis (3). During neuromuscular development, MICAL expression was first observed in muscles before being up-regulated in the neurons of the central nervous system. However, muscle expression persisted until the end of embryogenesis. MICAL is also expressed in bristles at the bristle cell membrane and at sites of bristle branching (28).
Zebrafish have eight expressed Dr MICAL genes (86). Similar to Drosophila, Dr MICAL proteins are highly expressed in neurons and muscles. In addition, Dr MICAL2a, Dr MICAL2b, Dr MICAL3a, Dr MICAL-L2a, and Dr MICAL-L2b are expressed in heart tissue. The MICAL-L protein, Dr MICAL-L2, is expressed in blood vessels.
In chickens, three MICALs are expressed, and consistent with proposed roles in other organisms, in situ hybridization studies support the notion that MICAL3 expression in motor neurons controls the positioning of motor neuron somata (5).
Mammalian MICAL proteins have been studied in rat and human cells. In rats, MICAL proteins are expressed throughout the development of the nervous system with a highly overlapping expression profile (57, 81). While MICAL1 and MICAL3 were ubiquitously expressed in the nervous system, MICAL2 expression displayed higher selectivity, both temporally and regionally (57). For example, MICAL2 is expressed in external brain areas (i.e., cortex and hippocampus), but was absent in internal brain areas (i.e., hypothalamus and striatum). Moreover, the expression of MICAL2 was delayed in these tissues. Apart from neurons, MICAL1 and MICAL3 are also expressed in oligodendrocytes (57). Furthermore, the expression of MICALs was elevated in oligodendrocytes and meningeal fibroblasts present at the sites of spinal cord injury. Overall, the neuronal expression profile of MICAL proteins suggests a role in neural development and plasticity.
In humans, MICAL1 expression was studied from temporal lobe epilepsy patient samples (42). Temporal lobe epilepsy is a common form of epilepsy that is resistant to antiepileptic drugs. These patient samples had reduced expression of MICAL1 compared with normal/control samples. Northern blot and western blot analysis of MICAL proteins from tissue samples indicated their expression in heart, lung, kidney, liver, thymus, muscles, bone marrow, and brain, (1, 13, 78). MICAL1 is ubiquitously expressed in various cell lines, and all three MICALs are expressed in the HeLa cell line (22). In Hela, MICAL1 localizes to the cytoplasm, MICAL2 localizes to the area beneath the plasma membrane, and MICAL3 is both observed in the cytoplasm and associated with secretory vesicles (22, 23).
Functional Roles for MICAL Proteins
MICAL involvement was observed in the semaphorin/plexin signaling pathway that performs axonal growth cone repulsion (81). Various attraction and repulsion cues participate in axonal growth for proper neural development (31). Semaphorin family members cause repulsion of axons by binding to their receptors plexin and neuropilin-1 and −2 (24, 61, 79). On semaphorin binding to plexin, the cytoplasmic domain of plexin causes alterations in the growth cone cytoskeleton (51). These cytoskeletal alterations include depolymerization of actin filaments and a decreased ability of actin monomers to polymerize. While searching for the mechanism by which semaphorin signals transduce actin filament changes, MICAL was identified in a yeast-two-hybrid screen using the plexin cytoplasmic domain as bait (81). Similar to loss of function and gain of function of plexin mutants, MICAL loss-of-function mutants and gain-of-function mutants caused axonal guidance and neuromuscular defects, including increased defasciculation, projection of neurons to inappropriate regions, axon stalling, and the bypassing of target muscles (81).
Essential to the functional role of MICAL proteins in semaphorin-mediated axonal repulsion is the flavo-mono-oxygenase domain. Further confirming its role in neuronal regulation, MICAL participation was found in Sox14-regulated dendritic pruning (36). Pruning is the selective removal of neurons without causing neuronal death. Sox14 is a transcription factor and an essential participant in the dendritic pruning that is mediated by ecdysone signaling (63). Sox14 mediates dendritic pruning by promoting the expression of MICAL (36).
A large-scale mutagenesis screen in Drosophila identified a role for MICAL in the regulation of myofilament organization and synaptic structures during post-embryonic development (3). MICAL mutants had abnormally structured neuromuscular junctions and displayed penetrant defects in the patterning and arrangement of synaptic buttons. MICAL was expressed in muscle, and MICAL mutations led to disorganization of actin and myosin filaments and the failure to organize myofilaments into sarcomeres (3). SiRNA-induced loss of MICAL provided further support for the role of MICAL in organizing myofilaments in muscles. MICAL mutants also showed defects in the organization of the cytoskeleton around the synaptic buttons. These neuromuscular defects in MICAL mutants arise during post-embryonic stages, and the MICAL mutants were unable to fly (3).
In flies, loss of MICAL caused bristle development defects (28). This defect was evident by the presence of abnormal club-like or blunt tipped bristles as well as thick, bent, and twisted bristles. Moreover, over-expression of MICAL in bristles caused severe branching defects, supporting a role for MICAL in bristle morphology development. Bristle development, akin to neuronal development, is also an actin-dependent process. Correspondingly, bristles of MICAL knock-out flies had disorganized, intersecting, and larger actin bundles (28). On the contrary, over-expression of MICAL in bristles led to a complex meshwork of short actin filaments instead of bristles organized in a normal parallel pattern. Dm MICAL co-localized with actin filaments in vivo in the bristles, and its FAD and CH domains were required to induce actin and cell morphology defects. Even in vitro, Dm MICAL was capable of destabilizing actin filaments (28). This study proved that MICAL acts as a direct and specific regulator of actin dynamics that converts semaphorin induced signaling to F-actin destabilization-induced neuronal repulsion.
In chickens, MICAL3-depletion by shRNA caused motor neurons to exit from the spinal cord into the ventral root. This defect was also observed on depletion of either sema6A or plexinA2, indicating conservation of MICAL involvement in semaphorin signaling in different organisms (5).
In mammalian cell lines, both MICAL over-expression and SiRNA-mediated knock-down had a considerable impact on actin filament organization. MICAL2 over-expression caused rearrangement of actin filaments to filopodial-like protrusions, whereas MICAL1 expression did not dramatically affect the impact on the actin cytoskeleton (Fig. 3A, B) (22). Both MICAL1 and MICAL3 are auto inhibited, and either removal or mutations in the C-terminal CC domain relieves the auto inhibition (22, 23). Expression of activated (uninihibited) forms of MICAL1 and MICAL3 caused a reduction in the amount of filamentous actin.
FIG. 3.

The effect of exogenously expressed MICAL family members on the localization and organization of actin filaments. HeLa cells grown on coverslips were transiently transfected with full-length HA-MICAL1 (A), HA-MICAL2 (B), HA-MICAL-L1 (C), or GFP-MICAL-L2 (D). After 18 h, cells were fixed, permeabilized, and incubated with Rhodamine-phalloidin (D) or incubated with anti-HA antibody followed by Alexa-Fluor-568-conjugated anti-mouse secondary antibody and Alexa-Fluor-488-conjugated phalloidin (A, B) or incubated with anti-HA antibody followed by Alexa-Fluor-488-conjugated anti-rabbit secondary antibody and Rhodamine-phalloidin (C). Transfected cells are indicated by yellow stars. Colocalization between MICAL-L2-containing vesicles and actin filaments is indicated by white arrows within the inset. Scale bar: 10 μm. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
The actin remodeling property of MICAL proteins derives from their FAD domains (Fig. 4). Expression of the MICAL1 FAD and MICAL2 FAD domains in HeLa cells caused reduced levels of F-actin and rearrangement of actin to filopodial-like structures, respectively (22). Despite the high level of homology between these proteins, each of them impact F-actin in a different manner. Moreover, MICAL1 FAD domain expression also affected focal adhesion assembly and γ-tubulin organization (Fig. 5). Another study that showed MICAL1 activation induced cell contraction (67), which is possibly a result from its actin remodeling and focal adhesion loss. Depletion of MICAL proteins also impacts the actin cytoskeleton (Fig. 6). Loss of either MICAL1 or MICAL2 caused enrichment of filopodial-like actin protrusions at the cell periphery, whereas MICAL3-depletion caused an increase in the number of stress fibers and cell size (22).
FIG. 4.

Effect of the MICAL FAD domain actin filaments organization. HeLa cells grown on coverslips were transiently transfected with either truncated HA–MICAL1 (A) or HA-MICAL2 (B) containing only the FAD domain. After 18 h, cells were fixed, permeabilized, and incubated with anti-HA antibody followed by Alexa-Fluor-568-conjugated anti-mouse secondary antibody and Alexa-Fluor-488-conjugated phalloidin. Transfected cells are indicated by yellow stars. Scale bar: 10 μm. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
FIG. 5.

Effect of the MICAL FAD domain on γ-tubulin and focal adhesion plaques. HeLa cells grown on coverslips were transiently transfected with truncated HA-MICAL1 containing only the FAD domain. After 18 h, cells were fixed, permeabilized, and incubated with anti-HA antibody along with either anti-γ-tubulin or anti-phospho-paxillin followed by Alexa-Fluor-568-conjugated and Alexa-Fluor-488-conjugated secondary antibodies. Transfected cells are indicated by yellow stars. Scale bar: 10 μm. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
FIG. 6.
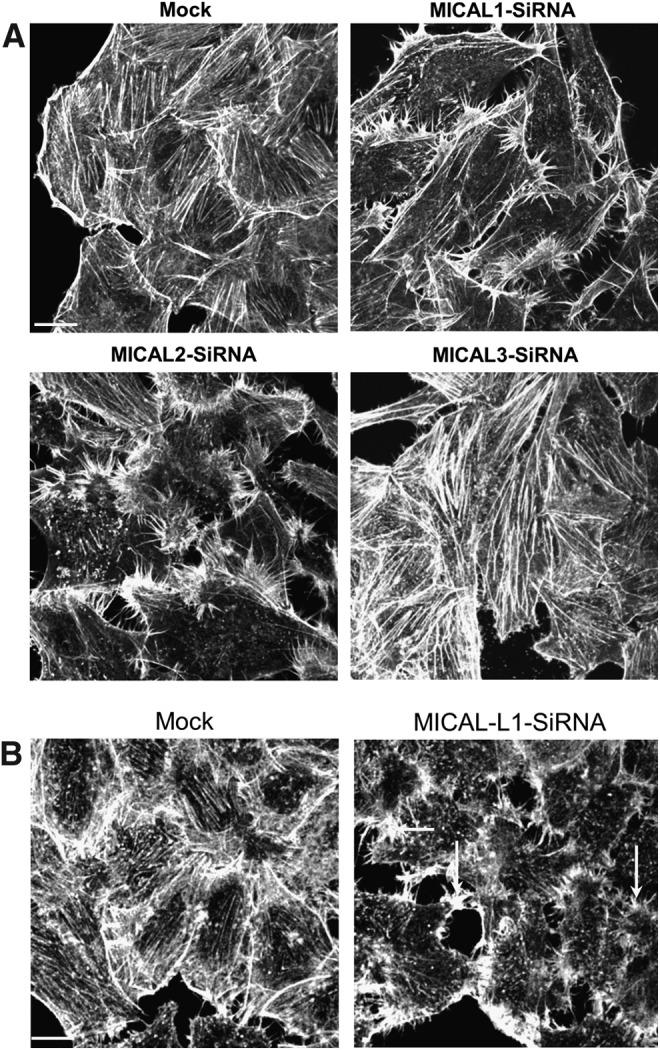
Effect MICAL-family protein depletion on actin filament localization and organization. (A) HeLa cells grown on coverslips were mock treated, treated with SiRNA against MICAL1, MICAL2, or MICAL3. After 72 h, cells were fixed, permeabilized, and incubated with Alexa-Fluor-488-conjugated phalloidin. (B) HeLa cells grown on coverslips were mock treated, treated, or with SiRNA against MICAL-L1. After 72 h, cells were fixed, permeabilized, and incubated with Alexa-Fluor-488-conjugated phalloidin. Scale bar: 10 μm.
The Flavo-Mono-Oxygenase Domain of MICAL Proteins
The MICAL FAD domain exhibits flavin mono-oxygenase activity. The FAD domain is present at the N-terminus of the MICAL protein and is highly conserved with high sequence homology and identity (Fig. 7A). Given that MICAL proteins vary toward the C-terminus and that the FAD is responsible for a major portion of the proteins' function, we performed a phylogenetic analysis on the FAD domains of MICAL proteins. Our analysis yielded similar results to those with the full-length MICAL proteins (compare Figs. 7B with 2A). The MICAL1 FAD and Dm MICAL FAD domain display a similar ancestral origin that correlates with a common cellular function. MICAL2 FAD and MICAL3 FAD appear to be of more recent origin. Hs MICAL2 FAD belongs to an outgroup and is more remotely related to Dm MICAL and MICAL1.
FIG. 7.

The FAD domain of MICAL proteins is highly conserved among species. Phylogenetic analysis of the FAD domain of MICAL proteins found in Drosophila melanogastor (Dm), Homo sapiens (Hs), and Danio rerio (Dr) was performed using Vector NTI software. The sequence homology and identity of various MICAL proteins against Dm MICAL and Hs MICAL1 is provided (A) along with the phylogenetic tree obtained from this analysis (B). The protein region used for analysis is indicated within brackets (accession number of the protein used is same as indicated in Fig. 2).
Flavo-mono-oxygenases are enzymes that are facilitated by a constitutively bound flavin molecule as a prosthetic group, use either NADH or NADPH as a reducing agent, and function to incorporate an oxygen atom into a substrate from molecular oxygen (56). The catalysis of insertion of a single oxygen atom into an organic substrate with high specificity is considered a complex chemical reaction. The common features found in flavoprotein mono-oxygenases are a tightly bound FAD molecule, dependence on a coenzyme such as NAD(P)H, the release of NAD(P)+ on flavin reduction, and the existence of a Rossmann Fold that facilitates the binding of the ADP moiety of the flavin molecule (82).
The crystal structure of mouse MICAL1 has been solved, and it matches all the requirements of a flavin mono-oxygenase (73). In addition to the Rossman Fold GXGXXG motif (where G is glycine and X is any amino acid), the FAD domains of MICAL proteins have other conserved motifs such as a GD (glycine-aspartic acid) motif that facilitates the binding of the ribose moiety of flavin, and a DG (aspartic acid-glycine) motif which facilitates the binding of the pyrophosphate moiety of flavin (73). Mutating the glycines in the Rossman Fold to tryptophan affects the enzymatic activity of the protein (22, 81). Similar to other mono-oxygenases, the MICAL1 FAD also lacks a binding domain for NADPH, and in order to reduce flavin, NADPH forms a transient complex (73).
Mono-oxygenases function as either a single component or two component enzymes. Two-component mono-oxygenases have a separate reductase and oxygenase component (2). In this case, the flavin molecule diffuses from the reductase component to the oxygenase component. In the course of single-component mono-oxygenase catalysis, the isoalloxazine ring of the flavin does not diffuse; instead, structural alteration of the protein facilitates the movement of flavin by several angstroms (2). Based on structural studies, MICAL is a single-component flavin mono-oxygenase (2).
By searching protein structure databases (DALI search) with the structure of the MICAL mono-oxygenase, it was revealed to have structural similarities to p-hydroxy benzoate hydroxylase (PHBH) from Pseudomonas fluorescens (48). Similar to PHBH, MICAL1 uses unbound NADPH as a cofactor (48, 73). The MICAL1 FAD domain displayed similarities to PHBH, in its ability to change conformations in the course of its catalytic cycle (48, 73). In addition, MICAL1 activity was sensitive to ionic strength and pH (90). To understand the presumed catalytic mechanism of MICAL1 mono-oxygenase function, we describe here its closest structural and functional protein, PHBH.
Catalytic Cycle of PHBH
PHBH is an extensively studied flavin mono-oxygenase that is involved in lignin catabolism (10). It is a 45-kDa protein that has a noncovalently bound flavin molecule and exists as a homo-dimer. At the expense of NADPH and O2, it catalyzes the aromatic hydroxylation of para-hydroxy benzoate (pOHB) to 3,4 dihydroxy benzoate (3,4DOHB) (Fig. 8). The catalytic cycle consists of two half reactions: a reductive half-reaction and an oxidative half-reaction (56). The part of the catalytic cycle in which the cofactor or prosthetic group of the enzyme is reduced is termed the reductive half-reaction, while the part of that reaction in which the enzyme becomes reoxidized is the oxidative half reaction. During the reductive half reaction, oxidized flavin is reduced by NADPH followed by the dissociation of NADPH (56). The oxidative half reaction involves binding of O2, reduction of reduced flavin with O2 to form a C4a-hydroperoxyflavin, oxygenation of the substrate, and regeneration of oxidized flavin (56). In this reaction, C4a-hydroperoxyflavin acts as an oxygenating reagent and performs an electrophilic aromatic substitution on the substrate, leading to oxygenation of pOHB (2, 10) (Fig. 8C, D).
FIG. 8.

Catalytic cycle of PHBH. PHBH enzyme catalyzes the conversion of pOHB to 3,4 dihydroxybenzoate (3,4DOHB) with the help of a covalently bound flavin molecule and transiently interacting NADPH cofactor. The flavin isoalloxazine (marked by a star; A) undergoes reduction by NADPH in the presence of substrate (B) and then undergoes oxidation by molecular oxygen to form a transient C4a-hydroperoxyflavin intermediate (C). The C4a-hydroperoxyflavin intermediate performs an electrophilic aromatic substitution on pOHB producing a dienone form of the product (D). The dienone form tautomerizes to 3,4DOHB and then dissociates (E). On product dissociation, new pOHB binds and the catalytic cycle continues (A). In the absence of substrate, C4a-hydroperoxyflavin undergoes rapid uncoupling producing H2O2 and regenerates oxidized flavin (C–A). PHBH, p-hydroxy benzoate hydroxylase; pOHB, para-hydroxy benzoate; 3,4DOHB, 3,4 dihydroxybenzoate.
During the reductive and oxidative cycle, the PHBH protein undergoes structural rearrangement to enable the movement of the flavin molecule (2, 10). They are termed as “out,” “in” (closed), and “open” conformations. When the flavin is in its oxidized state, pOHB (substrate) and NADPH bind to the enzyme, and the enzyme is said to be in “out” conformation. During the “out” conformation, the isoalloxazine ring (indicated by a star in Fig. 8A) of the flavin has access toward the solvent, exposing its N5 for hydride transfer (18, 68) (Fig. 8A). This conformation promotes the hydride transfer from NADPH and reduction of FAD to FADH2. On reduction of flavin, the NADP+ is released and the isoalloxazine ring moves back (46, 69). The flavin is said to be in the “in” conformation (Fig. 8B). This conformation positions the C4a of the isoalloxazine close to pOHB to facilitate the hydroxylation. The conformation rearrangement from “out” to “in” in most cases is the rate-determining step. In this conformation, the O2 binds and reacts with reduced flavin to create a transient unstable C4a-hydroperxyflavin intermediate (46, 72) (Fig. 8C). The “in” conformation isolates the flavin from the solvent and thereby prevents destabilization of the C4a-hydroperoxyflavin to H2O2 and oxidized flavin. The C4a-hydroxyflavin acts as an electrophile, and the aromatic pOHB acts as a nucleophile, leading to an electrophilic aromatic substitution (54). In this reaction, oxygen is transferred to the third position of pOHB (Fig. 8D), and the hydroxyl group in the fourth position becomes a carbonyl (54). This nonaromatic compound undergoes immediate tautomerization to the aromatic 3,4DOHB (Fig. 8E). On substrate hydroxylation, C4a-hydroxyflavin eliminates water to regenerate the oxidized flavin. At this point, the product favors the “open” conformation that enables product dissociation and permits substrate binding (85). Along with the pOHB binding, NADPH also binds, leading to transition to the “out” conformation and the cycle continues.
The rate of reduction of flavin by NADPH is increased multifold in the presence of pOHB (substrate) binding (29). In the absence of substrate, mono-oxygenases undergo a slow uncoupling reaction that eliminates H2O2 from C4a-hydroperoxyflavin without oxygenating the substrate. The C4a-hydroperoxyflavin intermediate is highly unstable and in the absence of substrate elimination of H2O2, occurs rapidly (Fig. 8C–A).
Despite the similarities between PHBH and the MICAL FAD, there are several significant differences between these two proteins. For example, the MICAL FAD region equivalent to the substrate binding cavity in PHBH is filled with tyrosine side chains (73). Moreover, MICAL does not have an “out” conformation that enables substrate access; instead, it has a channel which opens in the FAD domain that gives direct access to the active site. This channel is opposite of the putative NADPH binding site and is thought to be large enough for a substrate amino-acid side chain to enter. This structural feature of channel-running substrate access is mainly found in polyamine oxidases (4) and is not typical for mono-oxygenase-dependent hydroxylases. In addition, the catalytic activity of MICALs in the absence of substrate was several fold higher than that observed with other mono-oxygenases, including PHBH. For example, even in the absence of substrate, the mouse MICAL1 FAD domain had a high turnover of H2O2 with a Kcat of 77 s−1 and the Km of NADPH at 222 μM (48). Human MICAL1 had Kcat and Km values in the range of 3.9–4.8 s−1 and 26–41 μM, respectively (90). The large variation (greater than 10 fold) in the values derived from the two species is thought to ensue from differing experimental conditions.
Mechanism of Action
Based on the structural homology between the FAD domains and other flavoprotein mono-oxygenases, three different mechanisms were proposed to explain how MICALs might impact cytoskeletal reorganization (37, 83). The mechanisms include (i) production of reactive oxygen species (ROS), (ii) oxidation of downstream signaling molecules, or (iii) causing direct oxidation and destabilization of actin filaments. ROS are oxygen-containing reactive chemical species that include radical (superoxide, hydroxy radicals) and nonradical (hydrogen peroxide, ozone) compounds.
The notion that MICALs affect cytoskeletal reorganization by generating ROS is supported by experiments demonstrating that the mouse MICAL1-FAD domain in the presence of NADPH and molecular oxygen can produce the ROS molecule, H2O2 in vitro (48). Furthermore, H2O2 functions as a signaling molecule that modulates protein phosphorylation and also affects actin filaments directly (39, 62). Oxidation of actin can cause actin remodeling (48). In vitro, ROS molecules were able to modify cysteine, methionine, and tryptophan residues of actin, which, in turn, affect actin polymerization and also lead to actin degradation (11, 45). In support of this theory, we found that cells expressing human MICAL (as well as several MICAL truncation proteins that caused actin remodeling phenotypes), indeed, resulted in increased levels of ROS, as determined by the ROS indicator, dihydrocalcein (22). In a study by Morinaka et al., an H2O2-specific probe showed that MICALs, indeed, produce H2O2 in vivo on activation by semaphorin 3A (Sema3A), leading to oxidation of its downstream substrate protein, the collapsin response mediator protein-2 (CRMP2) (47) (Fig. 9). Oxidized CRMP2 forms a disulfide-linked homodimer that transiently complexes with thioredoxin. This complex promotes phosphorylation of CRMP2 through glycogen synthase kinase-3. CRMP2 promotes microtubule polymerization, and its phosphorylation by Sema3a, therefore, promotes microtubule disassembly that is essential for growth cone collapse (47). Furthermore, Zhou et al. expressed constitutively active MICAL1 mutants (lacking the auto-inhibitory C-terminal region) in cells and demonstrated H2O2 production in vivo (89).
FIG. 9.

Activation and function of MICAL proteins. MICAL proteins are activated either on semaphorin signaling or by the binding of proteins that relieve their auto inhibition. On activation, and in the presence of NADPH, MICAL uses its FAD domain to either oxidize proteins or produce ROS such as H2O2. MICAL proteins directly oxidize actin at methionine residues, causing depolymerization of actin filaments. Actin filaments can also be altered indirectly by the H2O2 produced by MICAL proteins. MICAL proteins regulate actin organization and on their loss, cytoskeletal organization is affected. The H2O2 produced by MICALs causes oxidation and dimerization of CRMP2, which then binds to thioredoxin (TRX) and is phosphorylated by GSK-3b. This modification of CRMP2 could promote microtubule remodeling, which along with actin remodeling promotes semaphorin-mediated growth cone collapse. CRMP2, collapsin response mediator protein-2; ROS, reactive oxygen species. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
In contrast to the ROS theory, Hung et al. showed that actin is a direct redox substrate of Dm MICAL-FAD-CH domains and that MICAL does not cause actin rearrangements through ROS production (27). MICAL uses its redox potential to oxidize methionines M44 and M47 of actin molecules to methionine sulfoxide. The oxidation of M44, present within the D loop of actin, caused severing of actin filaments. Mutations in actin M44L showed bristle morphology defects in Drosophila similar to MICAL loss of function. Supporting this study, Zucchini et al. confirmed that actin can act as a substrate for the human MICAL1-FAD domain (90). While Hung et al. showed that MICAL stably modifies actin's ability to polymerize (27), Zucchini et al. suggested that MICAL1-mediated effects on actin may be reversible (90). Based on its ability to oxidize methionine residues, MICAL proteins are also known as protein-methionine sulfoxide oxidases.
Exogenous expression of human MICAL1-FAD and human MICAL2-FAD induced different effects on actin filaments (22). The MICAL2-FAD caused a rearrangement of actin stress fibers, whereas MICAL1-FAD caused a drastic decrease in F-actin staining (Fig. 4), indicating a potential difference in the mechanism of action of the two MICALs. Since Dm MICAL requires both its FAD and CH domain to induce cell morphology changes, whereas mammalian MICALs require only their FAD domains, this further supports the notion of a difference in the mechanism of action between these MICALs (22, 28). In addition to actin, both human MICALs also regulate γ-tubulin, which is involved in microtubule nucleation in the pericentriolar region of the cell (90). CRMP2 associates with α-β-tubulin heterodimers and promotes microtubule polymerization (16). It is not clear whether the effects on γ-tubulin are caused directly by MICAL or by an indirect mechanism.
A recent proteomic screen with mouse MICAL1 identified the apoptotic proteins Nuclear-Dbf2-related kinase (NDR1 and NDR2) as binding partners (89). NDRs are serine threonine kinases that participate in apoptosis functioning downstream of mammalian Ste-20-like kinase 1 (MST1) (84). MICAL1 competes with MST1 for the C-terminal hydrophobic motif of NDRs (89). Accordingly, MICAL1 inhibits the phosphorylation of NDRs by MST1 and negatively regulates MST1-mediated apoptosis signaling. Similar to MICAL proteins, NDR kinases also participate in neurite outgrowth and cytoskeletal dynamics (25). Although MICAL's inhibitory effect on apoptosis was independent of its FAD domain, NDRs could potentially function downstream of MICAL, affecting cytoskeletal events (89). Other proteins that were identified in the proteomic screen (to identify MICAL1 binding partners) include the GTP-binding cytoskeletal protein septin-9 and the actin-binding filamin-B. Septins co-ordinate microtubule and actin dynamics and participate in Rho signaling (12, 74). Filamins are also actin-binding proteins that function as F-actin crosslinkers (49). MICAL is also a binding protein of the P130 Cas family members Cas and CasL that have been implicated in the regulation of actin dynamics (26, 33, 78). MICAL also interacts with vimentin, which can lead to cytoskeletal alterations when oxidized by H2O2 (64, 78). Although the functional significance of these proteins in MICAL-mediated cytoskeletal rearrangement is unclear, it will be interesting to see whether they are oxidized by MICAL and to determine how MICAL oxidation might alter their function.
Mono-Oxygenase Activity in Membrane Trafficking
MICAL proteins bind to a variety of different Rab proteins. A recent study showed that the mono-oxygenase activity of MICAL3 is required for vesicle docking and fusion (23). Rab6 and Rab8 cooperate to control exocytosis in the secretory pathway, with Rab6 involved in Rab8a recruitment to vesicles. Rab8, in cooperation with MICAL3, interacts with ELKS (a cortical protein that functions in docking exocytic secretory vesicles) (52), causing vesicle fusion at ELKS-positive cortical sites. MICAL3 interacts with Rab8 through its CC domain and interacts with ELKS through its C-terminal region that is distinct from its Rab8-binding region (23). The Rab6 interaction with ELKS along with the Rab8a-MICAL3-ELKS complex promotes secretory vesicle fusion.
When semaphorin binds to plexin, it causes axonal repulsion, which not only involves cytoskeletal alterations, but also requires endocytic events causing the internalization of local membrane (43). Although MICALs are Rab-binding proteins, their role in membrane trafficking during axonal repulsion remains unexplored.
Other Domains and Motifs Found in MICAL Proteins
CH domain
CH domains are mainly found in actin-binding proteins (19, 38). In MICALs, the CH domain is adjacent to the FAD domain, whereas it is localized to the N-terminus of MICAL-L proteins. A phylogenetic analysis of the CH domain of MICAL and MICAL-L proteins indicated that the CH domain is highly conserved from Zebrafish to humans (86). CH domains are classified into three types, and MICAL and MICAL-L proteins possess a type-2 CH domain similar to that found in proteins such as smoothelin and microtubule-associated RP/EB (end binding proteins) (38). Type-2 CH domains do not possess the ability to bind directly to F-actin, but can assist type-1 CH domain-containing proteins in binding actin. The CH domains of MICAL1 and MICAL-L2 were unable to bind to F-actin (65, 76). Similar to other type-2 CH domain-containing proteins, MICALs contain a conserved phosphatidylinositol-(4,5)-bisphosphate (PIP2) binding site, and PIP2 has been implicated in F-actin regulation (15, 86). Indeed, full-length MICAL2 protein co-localizes with exogenously expressed Arf6 and PIP5K structures, suggesting that its localization to PIP2-rich membranes is likely via its CH domain (unpublished observations). Although the MICAL CH domain does not contain actin-binding ability, Dm MICAL requires the presence of the CH domain along with the FAD domain to regulate actin filaments and bristle morphology in Drosophila (28).
The solved structures of the MICAL1 and MICAL3 CH domains are highly conserved with other CH domains (30, 76). The MICAL1 CH domain consists of six alpha helices and one 310 helix (a type of helical structure with three residues per turn). The MICAL1 CH domain comprises four distinct cooperative unfolding units with a hydrophobic core. Hydrophobic interactions are predicted for the maintenance of the three-dimensional structure of the MICAL1 CH domain.
LIM domain
Another common domain shared between MICAL family members is the Lin11, Isl-1, and Mec-3 domain, termed LIM domain. LIM domains are a cytoskeletal interaction element that is capable of interacting with various proteins (88). MICAL-family members have LIM domains following the CH domain, and the distance between these two domains varies between the MICAL family members (14). MICAL2 has the largest gap between the two domains. The LIM domain of MICAL1 binds to CRMP and NDR kinase-family members (67). The CH and LIM domains of MICAL-L1 bind to CRMP2 (60). On the other hand, the LIM domain of MICAL-L2 interacts with F-actin and promotes F-actin crosslinking (60).
CC domain
Another domain found in the C-terminal region of most of the MICAL-family members is the CC domain. The CC domain is otherwise known as ezrin/radixin/moesin α-like motif. The MICAL CC was recently classified as a domain of unknown (DUF) function called the DUF3585 domain. Although CC domains are relatively common, apart from MICALs, very few proteins have defined DUF3585 domains. One DUF3585 domain-containing protein is the Eps15 homology domain binding protein 1 (EHBP1), and the EH domain-binding protein 1-like protein 1. Through their CC domains, MICALs interact with plexins, vimentin, and NDRs. MICAL and MICAL-L proteins also interact with various Rabs (Rab1, Rab8, Rab10, Rab13, Rab15, Rab35, and Rab36) through their CC domains (78) 20, 23, 60, (17, 87). Accordingly, MICAL and MICAL-L proteins are considered Rab effectors.
The MICAL-L1 CC domain associates with membranes by selectively binding to phosphatidic acid (PA) (21). An interesting MICAL CC domain function is its ability to auto inhibit various MICAL proteins. Removal of the CC domain from MICAL1 or mutations that affect the CC domain formation cause activation of the mono-oxygenase domain, leading to actin rearrangement and cell contraction (22) (67). The CC domain of MICAL1 binds to its LIM domain to mediate the auto-inhibition. CRMP binding to the MICAL1 LIM domain, therefore, relieves it from auto-inhibition (67). Similarly, MICAL-L2 is auto inhibited by the binding of its CC domain to its CH and LIM domains (66). Binding of Rab13 to MICAL-L2 relieves the auto-inhibition. In addition, on truncation of its C-terminus, MICAL3 impacted actin filament levels, further suggesting auto-inhibitive behavior (23). In this manner, despite the physical distance to the mono-oxygenase domain, the C-terminal region controls the activity of the protein. When chimeric proteins were made swapping the C-termini of MICAL1 and MICAL2, the sequence of the C-terminal region dictated its function (22). Since studies show that the MICAL2 expressed in prostate cancer cells has variations in its C-terminal region (1), the prediction is that these variant proteins might behave differently from wild-type MICAL2. For example, if these variants were to display a constitutively active MICAL2 FAD domain, this might lead to F-actin alterations, potential activation of epithelial-mesenchymal transition, and the promotion of metastasis.
Other Motifs Found in MICAL Proteins
Proline-rich domains
Proline-rich domains (PRD) are found throughout most of the MICAL-family members. MICAL1 contains PPKPP motifs before its C-terminal CC domain, and these PRD motifs interact with the Src homology 3 (SH3) domain of Cas-family members Cas and CasL (78). MICAL-L1 has a stretch of PRDs located between the LIM and CC domain. It has 14 PXXP motifs that include two Class I PXXP sites. We have demonstrated that MICAL-L1 binds to the SH3 domain of Syndapin2 via its Class I sites (21). These Class I PXXP sites are highly conserved between human and Zebrafish MICAL proteins, but are not found in Dm MICAL-L.
NPF motifs
Another motif that is conserved among the MICAL-L proteins is the asparagine-proline-phenylalanine (NPF) motif. NPF motifs are motifs that typically interact with Eps15 Homology (EH) domains (71), and NPF motifs followed by one or more acidic residues interact selectively with the C-terminal Eps15 homology domain (EHD) containing proteins (35). For example, mammalian MICAL-L1 has two NPF motifs; the first is followed by six acidic residues and it interacts with the EH domain of EHD1 (71). Dm MICAL-L has two NPF motifs, both of which are followed by acidic residues. Dr MICAL-L1a has three NPF motifs, two of which are followed by acidic residues.
MICAL-L Proteins in Actin Remodeling
MICAL-L2, due in part to its activity as a Rab13 effector, is also known as Junctional Rab13-binding (JRAB) protein (80). MICAL-L2 plays a key role in the assembly of tight and adherent junctions through regulating trafficking of occludin, claudin, and E-cadherin to the plasma membrane (80, 87). MICAL-L2 is recruited to cell-cell junctions through its binding partner and an actin binding protein, Actinin-4 (50). Apart from binding to Actinin-4, MICAL-L2 also binds to actinin-1 (65). Actinins localize to stress fibers, cellular protrusions, and cell adhesion sites, and are capable of rearranging the actin cytoskeleton through their F-actin crosslinking activity (55). Apart from indirect binding through actinins, MICAL-L2 also directly binds to F-actin, facilitating F-actin bundling and stabilizing F-actin (65). Although MICAL-L2 is endogenously expressed only in polarized cells, when exogenously expressed in HeLa cells, it is localized to vesicles aligned with actin microfilaments (Fig. 3D; insets). Furthermore, loosening of F-actin bundles was observed on MICAL-L2 loss. MICAL-L2, similar to MICAL1, is also inhibited in its activity toward actin filaments by its C-terminal CC domain (22). While this intramolecular inhibition by its CC domain inhibits its F-actin stabilization activity, its interaction with actinins suppresses F-actin crosslinking activity (65). Through its differential activity toward F-actin, it plays a key role in the establishment and maintenance of stationary cell-cell adhesions. MICAL-L2 also regulates neurite outgrowth and is involved in epithelial cell scattering (32, 66).
Although belonging to the MICAL-family, no reports have yet been made describing the involvement of MICAL-L1 in regulating actin filaments. Neither endogenous nor exogenous MICAL-L1 shows co-localization with actin filaments (Fig. 3C). However, we have observed rearrangement of actin filaments to filopodial-like structures upon MICAL-L1 depletion (Fig. 6B). The mechanism for this apparent actin rearrangement remains unclear. As noted earlier, MICAL-L1 contains a unique NPF motif that is absent in other MICALs, with which it interacts with EHD1 (35, 71). MICAL-L1 localizes to tubular recycling endosomes, and recruits both Rab8 and EHD1 to these structures (70, 71). These tubular endosomes are involved in the recycling of receptors such as integrin and transferrin back to the plasma membrane. MICAL-L1 localization to these tubular membranes depends on its CC domain and is modulated by the GTP-binding proteins, Rab35 and Arf6 (70, 71). Through its association with CRMP2, MICAL-L1 connects these tubular endosomes with the microtubule-based transporter, dynein (60).
A proteomic screen to identify proteins that bind to specific SH3 domains showed that MICAL-L1 can interact with Syndapin family members (41). We have demonstrated that Syndapin2, MICAL-L1, and EHD1 form a tripartite complex, and that MICAL-L1 and Syndapin2 cooperate in the generation of these tubular recycling endosomes (21). Both MICAL-L1 and Syndapin2 are PA binding proteins. PA recruits the two proteins that initiate tubular membrane generation and the trafficking of receptors through the recycling endosomes. Syndapin2 associates directly with the actin binding proteins Wiskott–Aldrich syndrome protein (WASP) and Rac1 (8, 34). Syndapin2 controls Rac1 signaling and Rac1 controls Sydapin2, localizing to tubular membranes. Accordingly, the effect of MICAL-L1-depletion on actin filaments could be an indirect one, mediated by the Syndapin2 binding proteins, Rac and/or WASP.
Conclusion
Although MICAL-family proteins were identified just a decade ago, evidence suggests that they play important regulatory functions in actin cytoskeleton organization, neuronal growth, neuromuscular development, apoptosis, bristle development (in flies), and membrane trafficking. Key to MICAL protein function is the FAD domain, whose role in generating ROS may be central to its actin regulatory activity. In addition, the presence of various protein-protein interaction motifs and domains in MICALs, they often serve as scaffolding proteins. For example, MICAL-L1 acts as a scaffolding protein at recycling endosomes, connecting key regulatory proteins to endosomes (20). The degree to which other MICAL proteins are involved in scaffolding activity remains unclear, but with the identification of a growing number of MICAL interaction partners, it is anticipated that the diverse functions of MICAL-family proteins will be further clarified.
Abbreviations Used
- 3,4 DOHB
3,4 dihydroxy benzoate
- CC
coiled-coil
- CH
calponin homology
- CRMP2
collapsin response mediator protein-2
- Dm
Drosophila melanogaster
- Dr
Danio rerio
- DUF
domain of unknown function
- EHBP1
Eps15 homology domain binding protein 1
- EHD
Eps15 homology domain
- ELKS
E-twenty six (ETS) domain-containing protein
- FAD
flavin adenine dinucleotide
- Hs
homo sapiens
- JRAB
junctional Rab13-binding
- LIM
Lin11, Isl-1 and Mec-3
- MICAL
molecules interacting with CasL
- MICAL-L
MICAL-like
- MST1
mammalian Ste-20-like kinase
- NADP+
nicotinamide adenine dinucleotide phosphate
- NADPH
reduced NADP+
- NDR
nuclear-Dbf2-related kinase
- NPF
asparagine-proline-phenylalanine
- PA
phosphatidic acid
- PHBH
p-hydroxy benzoate hydroxylase
- PIP2
phosphatidylinositol-(4,5)-bisphosphate
- pOHB
para-hydroxy benzoate
- PRD
proline rich domains
- ROS
reactive oxygen species
- Sema3A
semaphorin 3A
- SH3
Src homology 3
- WASP
Wiskott–Aldrich syndrome protein
Acknowledgments
The authors gratefully acknowledge funding from the following sources: National Institutes of Health (NIGMS) Grant R01GM087455, The Nebraska Center for Cellular Signaling Grant 5P20GM103489-10, and The Nebraska Department of Health.
References
- 1.Ashida S, Furihata M, Katagiri T, Tamura K, Anazawa Y, Yoshioka H, Miki T, Fujioka T, Shuin T, Nakamura Y, and Nakagawa H. Expression of novel molecules, MICAL2-PV (MICAL2 prostate cancer variants), increases with high Gleason score and prostate cancer progression. Clin Cancer Res 12: 2767–2773, 2006 [DOI] [PubMed] [Google Scholar]
- 2.Ballou DP, Entsch B, and Cole LJ. Dynamics involved in catalysis by single-component and two-component flavin-dependent aromatic hydroxylases. Biochem Biophys Res Commun 338: 590–598, 2005 [DOI] [PubMed] [Google Scholar]
- 3.Beuchle D, Schwarz H, Langegger M, Koch I, and Aberle H. Drosophila MICAL regulates myofilament organization and synaptic structure. Mech Dev 124: 390–406, 2007 [DOI] [PubMed] [Google Scholar]
- 4.Binda C, Coda A, Angelini R, Federico R, Ascenzi P, and Mattevi A. A 30-angstrom-long U-shaped catalytic tunnel in the crystal structure of polyamine oxidase. Structure 7: 265–276, 1999 [DOI] [PubMed] [Google Scholar]
- 5.Bron R, Vermeren M, Kokot N, Andrews W, Little GE, Mitchell KJ, and Cohen J. Boundary cap cells constrain spinal motor neuron somal migration at motor exit points by a semaphorin-plexin mechanism. Neural Dev 2: 21, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chesarone MA. and Goode BL. Actin nucleation and elongation factors: mechanisms and interplay. Curr Opin Cell Biol 21: 28–37, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cooper JA. and Sept D. New insights into mechanism and regulation of actin capping protein. Int Rev Cell Mol Biol 267: 183–206, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Kreuk BJ, Nethe M, Fernandez-Borja M, Anthony EC, Hensbergen PJ, Deelder AM, Plomann M, and Hordijk PL. The F-BAR domain protein PACSIN2 associates with Rac1 and regulates cell spreading and migration. J Cell Sci 124: 2375–2388, 2011 [DOI] [PubMed] [Google Scholar]
- 9.dos Remedios CG, Chhabra D, Kekic M, Dedova IV, Tsubakihara M, Berry DA, and Nosworthy NJ. Actin binding proteins: regulation of cytoskeletal microfilaments. Physiol Rev 83: 433–473, 2003 [DOI] [PubMed] [Google Scholar]
- 10.Entsch B, Cole LJ, and Ballou DP. Protein dynamics and electrostatics in the function of p-hydroxybenzoate hydroxylase. Arch Biochem Biophys 433: 297–311, 2005 [DOI] [PubMed] [Google Scholar]
- 11.Fedorova M, Kuleva N, and Hoffmann R. Identification of cysteine, methionine and tryptophan residues of actin oxidized in vivo during oxidative stress. J Proteome Res 9: 1598–1609, 2010 [DOI] [PubMed] [Google Scholar]
- 12.Field CM. and Kellogg D. Septins: cytoskeletal polymers or signalling GTPases? Trends Cell Biol 9: 387–394, 1999 [DOI] [PubMed] [Google Scholar]
- 13.Fischer J, Weide T, and Barnekow A. The MICAL proteins and rab1: a possible link to the cytoskeleton? Biochem Biophys Res Commun 328: 415–423, 2005 [DOI] [PubMed] [Google Scholar]
- 14.Friedberg F. Alternative splicing for members of human mosaic domain superfamilies. I. The CH and LIM domains containing group of proteins. Mol Biol Rep 36: 1059–1081, 2009 [DOI] [PubMed] [Google Scholar]
- 15.Fukami K, Furuhashi K, Inagaki M, Endo T, Hatano S, and Takenawa T. Requirement of phosphatidylinositol 4,5-bisphosphate for alpha-actinin function. Nature 359: 150–152, 1992 [DOI] [PubMed] [Google Scholar]
- 16.Fukata Y, Itoh TJ, Kimura T, Menager C, Nishimura T, Shiromizu T, Watanabe H, Inagaki N, Iwamatsu A, Hotani H, and Kaibuchi K. CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat Cell Biol 4: 583–591, 2002 [DOI] [PubMed] [Google Scholar]
- 17.Fukuda M, Kanno E, Ishibashi K, and Itoh T. Large scale screening for novel rab effectors reveals unexpected broad Rab binding specificity. Mol Cell Proteomics 7: 1031–1042, 2008 [DOI] [PubMed] [Google Scholar]
- 18.Gatti DL, Palfey BA, Lah MS, Entsch B, Massey V, Ballou DP, and Ludwig ML. The mobile flavin of 4-OH benzoate hydroxylase. Science 266: 110–114, 1994 [DOI] [PubMed] [Google Scholar]
- 19.Gimona M, Djinovic-Carugo K, Kranewitter WJ, and Winder SJ. Functional plasticity of CH domains. FEBS Lett 513: 98–106, 2002 [DOI] [PubMed] [Google Scholar]
- 20.Giridharan SS, Cai B, Naslavsky N, and Caplan S. Trafficking cascades mediated by Rab35 and its membrane hub effector, MICAL-L1. Commun Integr Biol 5: 384–387, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giridharan SS, Cai B, Vitale N, Naslavsky N, and Caplan S. Cooperation of MICAL-L1, syndapin2 and phosphatidic acid in tubular recycling endosome biogenesis. Mol Biol Cell 24: 1776–1790, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giridharan SS, Rohn JL, Naslavsky N, and Caplan S. Differential regulation of actin microfilaments by human MICAL proteins. J Cell Sci 125: 614–624, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grigoriev I, Yu KL, Martinez-Sanchez E, Serra-Marques A, Smal I, Meijering E, Demmers J, Peranen J, Pasterkamp RJ, van der Sluijs P, Hoogenraad CC, and Akhmanova A. Rab6, Rab8, and MICAL3 cooperate in controlling docking and fusion of exocytotic carriers. Curr Biol 21: 967–974, 2011 [DOI] [PubMed] [Google Scholar]
- 24.He Z, Wang KC, Koprivica V, Ming G, and Song HJ. Knowing how to navigate: mechanisms of semaphorin signaling in the nervous system. Sci STKE 2002: re1, 2002 [DOI] [PubMed] [Google Scholar]
- 25.Hergovich A, Stegert MR, Schmitz D, and Hemmings BA. NDR kinases regulate essential cell processes from yeast to humans. Nat Rev Mol Cell Biol 7: 253–264, 2006 [DOI] [PubMed] [Google Scholar]
- 26.Honda H, Nakamoto T, Sakai R, and Hirai H. p130(Cas), an assembling molecule of actin filaments, promotes cell movement, cell migration, and cell spreading in fibroblasts. Biochem Biophys Res Commun 262: 25–30, 1999 [DOI] [PubMed] [Google Scholar]
- 27.Hung RJ, Pak CW, and Terman JR. Direct redox regulation of F-actin assembly and disassembly by Mical. Science 334: 1710–1713, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hung RJ, Yazdani U, Yoon J, Wu H, Yang T, Gupta N, Huang Z, van Berkel WJ, and Terman JR. Mical links semaphorins to F-actin disassembly. Nature 463: 823–827, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Husain M. and Massey V. Kinetic studies on the reaction of p-hydroxybenzoate hydroxylase. Agreement of steady state and rapid reaction data. J Biol Chem 254: 6657–6666, 1979 [PubMed] [Google Scholar]
- 30.Jin X, Zhang J, Dai H, Sun H, Wang D, Wu J, and Shi Y. Investigation of the four cooperative unfolding units existing in the MICAL-1 CH domain. Biophys Chem 129: 269–278, 2007 [DOI] [PubMed] [Google Scholar]
- 31.Kalil K. and Dent EW. Touch and go: guidance cues signal to the growth cone cytoskeleton. Curr Opin Neurobiol 15: 521–526, 2005 [DOI] [PubMed] [Google Scholar]
- 32.Kanda I, Nishimura N, Nakatsuji H, Yamamura R, Nakanishi H, and Sasaki T. Involvement of Rab13 and JRAB/MICAL-L2 in epithelial cell scattering. Oncogene 27: 1687–1695, 2008 [DOI] [PubMed] [Google Scholar]
- 33.Kawauchi K, Tan WW, Araki K, Abu Bakar FB, Kim M, Fujita H, Hirata H, and Sawada Y. p130Cas-dependent actin remodelling regulates myogenic differentiation. Biochem J 445: 323–332, 2012 [DOI] [PubMed] [Google Scholar]
- 34.Kessels MM. and Qualmann B. Syndapins integrate N-WASP in receptor-mediated endocytosis. EMBO J 21: 6083–6094, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kieken F, Sharma M, Jovic M, Giridharan SS, Naslavsky N, Caplan S, and Sorgen PL. Mechanism for the selective interaction of C-terminal Eps15 homology domain proteins with specific Asn-Pro-Phe-containing partners. J Biol Chem 285: 8687–8694, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kirilly D, Gu Y, Huang Y, Wu Z, Bashirullah A, Low BC, Kolodkin AL, Wang H, and Yu F. A genetic pathway composed of Sox14 and Mical governs severing of dendrites during pruning. Nat Neurosci 12: 1497–1505, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kolk SM. and Pasterkamp RJ. MICAL flavoprotein monooxygenases: structure, function and role in semaphorin signaling. Adv Exp Med Biol 600: 38–51, 2007 [DOI] [PubMed] [Google Scholar]
- 38.Korenbaum E. and Rivero F. Calponin homology domains at a glance. J Cell Sci 115: 3543–3545, 2002 [DOI] [PubMed] [Google Scholar]
- 39.Lee K. and Esselman WJ. Inhibition of PTPs by H(2)O(2) regulates the activation of distinct MAPK pathways. Free Radic Biol Med 33: 1121–1132, 2002 [DOI] [PubMed] [Google Scholar]
- 40.Lee SH. and Dominguez R. Regulation of actin cytoskeleton dynamics in cells. Mol Cells 29: 311–325, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Linkermann A, Gelhaus C, Lettau M, Qian J, Kabelitz D, and Janssen O. Identification of interaction partners for individual SH3 domains of Fas ligand associated members of the PCH protein family in T lymphocytes. Biochim Biophys Acta 1794: 168–176, 2009 [DOI] [PubMed] [Google Scholar]
- 42.Luo J, Xu Y, Zhu Q, Zhao F, Zhang Y, Peng X, Wang W, and Wang X. Expression pattern of Mical-1 in the temporal neocortex of patients with intractable temporal epilepsy and pilocarpine-induced rat model. Synapse 65: 1213–1221, 2011 [DOI] [PubMed] [Google Scholar]
- 43.Mann F. and Rougon G. Mechanisms of axon guidance: membrane dynamics and axonal transport in semaphorin signalling. J Neurochem 102: 316–323, 2007 [DOI] [PubMed] [Google Scholar]
- 44.Matsudaira P. Actin crosslinking proteins at the leading edge. Semin Cell Biol 5: 165–174, 1994 [DOI] [PubMed] [Google Scholar]
- 45.Milzani A, Rossi R, Di Simplicio P, Giustarini D, Colombo R, and DalleDonne I. The oxidation produced by hydrogen peroxide on Ca-ATP-G-actin. Protein Sci 9: 1774–1782, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moran GR, Entsch B, Palfey BA, and Ballou DP. Electrostatic effects on substrate activation in para-hydroxybenzoate hydroxylase: studies of the mutant lysine 297 methionine. Biochemistry 36: 7548–7556, 1997 [DOI] [PubMed] [Google Scholar]
- 47.Morinaka A, Yamada M, Itofusa R, Funato Y, Yoshimura Y, Nakamura F, Yoshimura T, Kaibuchi K, Goshima Y, Hoshino M, Kamiguchi H, and Miki H. Thioredoxin mediates oxidation-dependent phosphorylation of CRMP2 and growth cone collapse. Sci Signal 4: ra26, 2011 [DOI] [PubMed] [Google Scholar]
- 48.Nadella M, Bianchet MA, Gabelli SB, Barrila J, and Amzel LM. Structure and activity of the axon guidance protein MICAL. Proc Natl Acad Sci USA 102: 16830–16835, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakamura F, Stossel TP, and Hartwig JH. The filamins: organizers of cell structure and function. Cell Adh Migr 5: 160–169, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nakatsuji H, Nishimura N, Yamamura R, Kanayama HO, and Sasaki T. Involvement of actinin-4 in the recruitment of JRAB/MICAL-L2 to cell-cell junctions and the formation of functional tight junctions. Mol Cell Biol 28: 3324–3335, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Negishi M, Oinuma I, and Katoh H. Plexins: axon guidance and signal transduction. Cell Mol Life Sci 62: 1363–1371, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nomura H, Ohtsuka T, Tadokoro S, Tanaka M, and Hirashima N. Involvement of ELKS, an active zone protein, in exocytotic release from RBL-2H3 cells. Cell Immunol 258: 204–211, 2009 [DOI] [PubMed] [Google Scholar]
- 53.Ono S. Mechanism of depolymerization and severing of actin filaments and its significance in cytoskeletal dynamics. Int Rev Cytol 258: 1–82, 2007 [DOI] [PubMed] [Google Scholar]
- 54.Ortiz-Maldonado M, Ballou DP, and Massey V. Use of free energy relationships to probe the individual steps of hydroxylation of p-hydroxybenzoate hydroxylase: studies with a series of 8-substituted flavins. Biochemistry 38: 8124–8137, 1999 [DOI] [PubMed] [Google Scholar]
- 55.Otey CA. and Carpen O. Alpha-actinin revisited: a fresh look at an old player. Cell Motil Cytoskeleton 58: 104–111, 2004 [DOI] [PubMed] [Google Scholar]
- 56.Palfey BA. and McDonald CA. Control of catalysis in flavin-dependent monooxygenases. Arch Biochem Biophys 493: 26–36, 2010 [DOI] [PubMed] [Google Scholar]
- 57.Pasterkamp RJ, Dai HN, Terman JR, Wahlin KJ, Kim B, Bregman BS, Popovich PG, and Kolodkin AL. MICAL flavoprotein monooxygenases: expression during neural development and following spinal cord injuries in the rat. Mol Cell Neurosci 31: 52–69, 2006 [DOI] [PubMed] [Google Scholar]
- 58.Pollard TD. Regulation of actin filament assembly by Arp2/3 complex and formins. Annu Rev Biophys Biomol Struct 36: 451–477, 2007 [DOI] [PubMed] [Google Scholar]
- 59.Pollard TD. and Cooper JA. Actin, a central player in cell shape and movement. Science 326: 1208–1212, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rahajeng J, Giridharan SS, Naslavsky N, and Caplan S. Collapsin response mediator protein-2 (Crmp2) regulates trafficking by linking endocytic regulatory proteins to dynein motors. J Biol Chem 285: 31918–31922, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Raper JA. Semaphorins and their receptors in vertebrates and invertebrates. Curr Opin Neurobiol 10: 88–94, 2000 [DOI] [PubMed] [Google Scholar]
- 62.Rhee SG, Bae YS, Lee SR, and Kwon J. Hydrogen peroxide: a key messenger that modulates protein phosphorylation through cysteine oxidation. Sci STKE 2000: pe1, 2000 [DOI] [PubMed] [Google Scholar]
- 63.Ritter AR. and Beckstead RB. Sox14 is required for transcriptional and developmental responses to 20-hydroxyecdysone at the onset of Drosophila metamorphosis. Dev Dyn 239: 2685–2694, 2010 [DOI] [PubMed] [Google Scholar]
- 64.Rogers KR, Morris CJ, and Blake DR. Oxidation of thiol in the vimentin cytoskeleton. Biochem J 275 (Pt 3): 789–791, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sakane A, Abdallah AA, Nakano K, Honda K, Ikeda W, Nishikawa Y, Matsumoto M, Matsushita N, Kitamura T, and Sasaki T. Rab13 small G protein and junctional Rab13-binding protein (JRAB) orchestrate actin cytoskeletal organization during epithelial junctional development. J Biol Chem 287: 42455–42468, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sakane A, Honda K, and Sasaki T. Rab13 regulates neurite outgrowth in PC12 cells through its effector protein, JRAB/MICAL-L2. Mol Cell Biol 30: 1077–1087, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schmidt EF, Shim SO, and Strittmatter SM. Release of MICAL autoinhibition by semaphorin-plexin signaling promotes interaction with collapsin response mediator protein. J Neurosci 28: 2287–2297, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schreuder HA, Mattevi A, Obmolova G, Kalk KH, Hol WG, van der Bolt FJ, and van Berkel WJ. Crystal structures of wild-type p-hydroxybenzoate hydroxylase complexed with 4-aminobenzoate,2,4-dihydroxybenzoate, and 2-hydroxy-4-aminobenzoate and of the Tyr222Ala mutant complexed with 2-hydroxy-4-aminobenzoate. Evidence for a proton channel and a new binding mode of the flavin ring. Biochemistry 33: 10161–10170, 1994 [DOI] [PubMed] [Google Scholar]
- 69.Schreuder HA, Prick PA, Wierenga RK, Vriend G, Wilson KS, Hol WG, and Drenth J. Crystal structure of the p-hydroxybenzoate hydroxylase-substrate complex refined at 1.9 A resolution. Analysis of the enzyme-substrate and enzyme-product complexes. J Mol Biol 208: 679–696, 1989 [DOI] [PubMed] [Google Scholar]
- 70.Sharma M, Giridharan SS, Rahajeng J, Caplan S, and Naslavsky N. MICAL-L1: An unusual Rab effector that links EHD1 to tubular recycling endosomes. Commun Integr Biol 3: 181–183, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sharma M, Giridharan SS, Rahajeng J, Naslavsky N, and Caplan S. MICAL-L1 links EHD1 to tubular recycling endosomes and regulates receptor recycling. Mol Biol Cell 20: 5181–5194, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shoun H, Beppu T, and Arima K. On the stable enzyme-substrate complex of p-hydroxybenzoate hydroxylase. Evidences for the proton uptake from the substrate. J Biol Chem 254: 899–904, 1979 [PubMed] [Google Scholar]
- 73.Siebold C, Berrow N, Walter TS, Harlos K, Owens RJ, Stuart DI, Terman JR, Kolodkin AL, Pasterkamp RJ, and Jones EY. High-resolution structure of the catalytic region of MICAL (molecule interacting with CasL), a multidomain flavoenzyme-signaling molecule. Proc Natl Acad Sci USA 102: 16836–16841, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Spiliotis ET. Regulation of microtubule organization and functions by septin GTPases. Cytoskeleton (Hoboken) 67: 339–345, 2010 [DOI] [PubMed] [Google Scholar]
- 75.Stossel TP, Fenteany G, and Hartwig JH. Cell surface actin remodeling. J Cell Sci 119: 3261–3264, 2006 [DOI] [PubMed] [Google Scholar]
- 76.Sun H, Dai H, Zhang J, Jin X, Xiong S, Xu J, Wu J, and Shi Y. Solution structure of calponin homology domain of Human MICAL-1. J Biomol NMR 36: 295–300, 2006 [DOI] [PubMed] [Google Scholar]
- 77.Sun HQ, Kwiatkowska K, and Yin HL. Actin monomer binding proteins. Curr Opin Cell Biol 7: 102–110, 1995 [DOI] [PubMed] [Google Scholar]
- 78.Suzuki T, Nakamoto T, Ogawa S, Seo S, Matsumura T, Tachibana K, Morimoto C, and Hirai H. MICAL, a novel CasL interacting molecule, associates with vimentin. J Biol Chem 277: 14933–14941, 2002 [DOI] [PubMed] [Google Scholar]
- 79.Tamagnone L, Artigiani S, Chen H, He Z, Ming GI, Song H, Chedotal A, Winberg ML, Goodman CS, Poo M, Tessier-Lavigne M, and Comoglio PM. Plexins are a large family of receptors for transmembrane, secreted, and GPI-anchored semaphorins in vertebrates. Cell 99: 71–80, 1999 [DOI] [PubMed] [Google Scholar]
- 80.Terai T, Nishimura N, Kanda I, Yasui N, and Sasaki T. JRAB/MICAL-L2 is a junctional Rab13-binding protein mediating the endocytic recycling of occludin. Mol Biol Cell 17: 2465–2475, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Terman JR, Mao T, Pasterkamp RJ, Yu HH, and Kolodkin AL. MICALs, a family of conserved flavoprotein oxidoreductases, function in plexin-mediated axonal repulsion. Cell 109: 887–900, 2002 [DOI] [PubMed] [Google Scholar]
- 82.van Berkel WJ, Kamerbeek NM, and Fraaije MW. Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts. J Biotechnol 124: 670–689, 2006 [DOI] [PubMed] [Google Scholar]
- 83.Ventura A. and Pelicci PG. Semaphorins: green light for redox signaling? Sci STKE 2002: pe44, 2002 [DOI] [PubMed] [Google Scholar]
- 84.Vichalkovski A, Gresko E, Cornils H, Hergovich A, Schmitz D, and Hemmings BA. NDR kinase is activated by RASSF1A/MST1 in response to Fas receptor stimulation and promotes apoptosis. Curr Biol 18: 1889–1895, 2008 [DOI] [PubMed] [Google Scholar]
- 85.Wang J, Ortiz-Maldonado M, Entsch B, Massey V, Ballou D, and Gatti DL. Protein and ligand dynamics in 4-hydroxybenzoate hydroxylase. Proc Natl Acad Sci USA 99: 608–613, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xue Y, Kuok C, Xiao A, Zhu Z, Lin S, and Zhang B. Identification and expression analysis of mical family genes in zebrafish. J Genet Genomics 37: 685–693, 2010 [DOI] [PubMed] [Google Scholar]
- 87.Yamamura R, Nishimura N, Nakatsuji H, Arase S, and Sasaki T. The interaction of JRAB/MICAL-L2 with Rab8 and Rab13 coordinates the assembly of tight junctions and adherens junctions. Mol Biol Cell 19: 971–983, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zheng Q. and Zhao Y. The diverse biofunctions of LIM domain proteins: determined by subcellular localization and protein-protein interaction. Biol Cell 99: 489–502, 2007 [DOI] [PubMed] [Google Scholar]
- 89.Zhou Y, Adolfs Y, Pijnappel WW, Fuller SJ, Van der Schors RC, Li KW, Sugden PH, Smit AB, Hergovich A, and Pasterkamp RJ. MICAL-1 is a negative regulator of MST-NDR kinase signaling and apoptosis. Mol Cell Biol 31: 3603–3615, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zucchini D, Caprini G, Pasterkamp RJ, Tedeschi G, and Vanoni MA. Kinetic and spectroscopic characterization of the putative monooxygenase domain of human MICAL-1. Arch Biochem Biophys 515: 1–13, 2011 [DOI] [PubMed] [Google Scholar]