

Abstract
Significance: The extracellular matrix (ECM) fulfills essential functions in multicellular organisms. It provides the mechanical scaffold and environmental cues to cells. Upon cell attachment, the ECM signals into the cells. In this process, reactive oxygen species (ROS) are physiologically used as signalizing molecules. Recent Advances: ECM attachment influences the ROS-production of cells. In turn, ROS affect the production, assembly and turnover of the ECM during wound healing and matrix remodeling. Pathological changes of ROS levels lead to excess ECM production and increased tissue contraction in fibrotic disorders and desmoplastic tumors. Integrins are cell adhesion molecules which mediate cell adhesion and force transmission between cells and the ECM. They have been identified as a target of redox-regulation by ROS. Cysteine-based redox-modifications, together with structural data, highlighted particular regions within integrin heterodimers that may be subject to redox-dependent conformational changes along with an alteration of integrin binding activity. Critical Issues: In a molecular model, a long-range disulfide-bridge within the integrin β-subunit and disulfide bridges within the genu and calf-2 domains of the integrin α-subunit may control the transition between the bent/inactive and upright/active conformation of the integrin ectodomain. These thiol-based intramolecular cross-linkages occur in the stalk domain of both integrin subunits, whereas the ligand-binding integrin headpiece is apparently unaffected by redox-regulation. Future Directions: Redox-regulation of the integrin activation state may explain the effect of ROS in physiological processes. A deeper understanding of the underlying mechanism may open new prospects for the treatment of fibrotic disorders. Antioxid. Redox Signal. 20, 1977–1993.
Introduction
Cell adhesion molecules, such as integrins, mediate the interaction of cells with the extracellular matrix (ECM). Both the ECM and integrins are targets of reactive oxygen species (ROS). Hence, this review first highlights different redox-relevant aspects of ECM turnover, and then approaches the questions: (i) how integrin-mediated cell-matrix contacts regulate the redox potential of the cell and its environment, and in turn, (ii) how the redox potential by cell-produced ROS may affect integrin-based cell-matrix and intercellular contacts.
The Extracellular Matrix
Multicellular organisms are characterized by tissue diversification and the production of an ECM. The plethora of different ECM components are composed of a limited set of modular structures, likely arisen through gene duplication during evolution. For example, the von Willebrand factor-homologous A-domain (briefly A-domain) is found in various types of collagens and other matrix proteins (69). Another characteristic of ECM components is their propensity to (self)assemble into larger and insoluble supramolecular aggregates, such as fibrils and networks (Fig. 1) (13). For example, rope-like fibrils span the intercellular spaces of the connective tissue and bear tensile forces. The most abundant fibrillar protein is collagen (50, 132). Other matrix proteins, such as fibronectin, fibrillins, fibulins, and elastin, also form thread-like supramolecular aggregates (156). In the human body, 28 different types of collagens have been identified (50, 132). The common molecular feature of all collagens is their triple helical structure, which consists of three collagenous α-chains with a recurring typical Gly-X-Y tripeptide sequence. Each peptide chain folds into a left-handed polyproline II conformation (Fig. 1). Three collagenous α-chains wind around each other in a right-handed manner to form the proteolytically stable triple helix. Moreover, several collagen molecules self-assemble in a staggered manner to form supramolecular fibrils (13). Type I-collagen molecules are staggered by 67 nm against each other resulting in the electron microscopically visible, classical D-banding of collagen fibrils.
FIG. 1.

Typical components of the ECM as targets of ROS. Four typical components of ECM proteins are shown with their characteristic modules. ROS and oxygen are involved in different steps of their transcription, translation and post-translational modification, such as the hydroxylation of proline residues in collagen chains, within the cells. After secretion the ECM molecules tend to assemble into higher aggregates, which possess distinct structures or functions (as named). These aggregates can be stabilized by covalent cross-linkage via oxidative modifications, which are catalyzed by lysyl-oxidase. Fibrils withstand tensile forces, whereas the GAG-chains of proteoglycans counteract compressive forces. Together, these forces are balanced within the matrix, similar to the tensegrity of cells, and maintain the shape of tissues and organs. The GAG chains are nonenzymatically fragmented by ROS. ECM proteins are degraded by MMPs, whose activities also depend on ROS. ECM, extracellular matrix; ROS, reactive oxygen species; MMPs, matrix metalloproteinases; GAG, glycosaminoglycan. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Proteoglycans are matrix molecules bearing glycosaminoglycan (GAG) chains (1, 46, 71, 72), such as the basement membrane component perlecan, the small leucine-rich proteoglycans, decorin and biglycan, and several types of collagen (1, 71, 110). The GAG chains tend to swell as their numerous carboxy and sulfate groups are substantially hydrated. Thus, they withstand compressive forces and cause the interstitial tissue pressure, which is counteracted by collagen fibrils. Together, the tensile force-bearing collagen fibrils and the swellable proteoglycans act like a cushion, thereby sheltering organs. Furthermore, the GAG chains often tether growth factors and act as reservoir that releases/activates growth factors in a time- and space-regulated manner (1, 46). In recent years, proteoglycans, especially the small leucine-rich proteoglycans, have been pinpointed for their important roles in inflammation (71, 110).
In contrast to fibril-forming collagen types, type VI collagen assemble into a unique network of beaded microfibrils (3, 60, 163). It is ubiquitously expressed, but upregulated during inflammation and fibrosis (82), and associates with different ECM proteins, such as matrilins and fibrillins. Genetic deficiency of it leads to Bethlem myopathy and Ullrich congenital muscular dystrophy (89).
The network-forming type IV collagen (81, 86), but also type XV, and XVIII collagens, assemble into two-dimensional sheet-like structures, which constitute the structural basis of the basement membranes. A basement membrane defines the border between the connective tissue and any other tissue, and serves as both mechanical and signaling platform for epithelial and endothelial cells (86, 106, 170, 181). It also surrounds individual muscle cells and muscle fibers, as well as neurons and fat cells. To withstand the mechanical load, a basement membrane is anchored to the underlying connective tissue network via collagen VI microfibrils or via the collagen VII-comprising anchoring fibrils. Basement membranes are generally impermeable for cells, with the exception of leukocytes, which are able to penetrate the basement membrane for immunological tissue surveillance or for infiltration at sites of inflammation. Pathologically, tumor cells acquire the ability to penetrate basement membranes (135) and to survive in an unusual ECM environment, both hallmarks of cancer malignancy (57).
Basement membranes also contain laminins, nidogens/entactins, and the proteoglycan perlecan. Laminins are grouped into a family of about 16 members (56). Each laminin consists of three chains, α, β, and γ, which trimerize and form an α-helical coiled-coil motif, which can be visualized electron-microscopically as the long arm of the cruciform or Y-shaped laminin-molecule (Fig. 1). This so-called rod domain bears at its N-terminus the short arms of the individual laminin chains and at its C-terminus a globular (G) domain, which comprises the laminin α chain exclusively. Laminins associate homophilically via their N-terminal domains into a two-dimensional structure, which together with the type IV collagen-network serves an anchoring substratum for endothelial and epithelial cells. Moreover, laminins provide essential environmental signals, as, for example, mammary epithelial cells are refractory to prolactin stimulation unless they have contact to laminin (127).
Fibronectin is a dimeric molecule. Its two highly homologous chains consist of several repeats of three different modules (type I, II, and III fibronectin repeats) and are cross-linked via disulfide bridges at their C-terminal ends. Alternative splicing leads to about 20 different fibronectin variants in humans (160, 168). These splice variants differ in the presence of extra domains (EDs), EDA and EDB (called EIIIA and EIIIB in mouse) and of the type III-connecting segment (IIICS). They harbor cell-adhesive sites for immunologically relevant integrins (α9β1, α4β1, and α4β7), in addition to the binding site for the classical fibronectin receptor, α5β1 integrin, which is found in the 10th fibronectin type III repeat of all fibronectin isoforms (91). Plasma (p)-fibronectin, which is secreted by hepatocytes into the blood, contains only the IIICS cell adhesive site, whereas cellular (c)-fibronectin additionally contains both EDs and is expressed by mesenchymal cells, especially during embryogenesis and wound healing, in angiogenesis along sprouting blood vessels, and pathologically, during fibrosis and in tumor tissue (42, 160, 168). Fibronectin can also assemble into a fibrillar network, especially when it is in physical contact with cellular receptors, such as integrins and syndecan-4 (148). The ECM protein tenascin-C influences the formation of the fibronectin matrix and is expressed in granulation tissue during wound healing (160). In addition, the fibronectin matrix binds numerous growth factors. The most prominent one is transforming growth factor-β1 (TGFβ1), which forms a complex with the latency activating peptide (LAP). The latent TGFβ1-binding protein-1 (LTBP-1) links this complex to the fibronectin network (148, 168). Proteolytic cleavage of LAP or tensile forces along fibronectin fibrils release and thereby activate TGFβ1 (169).
The hypoxia-driven gene expression of several matrix proteins, the hydroxylation of collagen chains and other ECM proteins, the covalent cross-linkage of ECM proteins via oxidative modifications of amino acid side chains, and the activity regulation of matrix-degrading enzymes by ROS are redox-relevant processes, which regulate ECM turnover.
Hypoxia-inducible factor is a transcription factor that upregulates the expression of collagens, fibronectin, fibulin-5, and other matrix proteins (32, 41, 54, 61, 75, 115), as well as the expression of enzymes, which catalyze posttranslational oxidative modifications of matrix proteins, such as collagen prolyl 4-hydroxylase and lysyl oxidase (27, 39, 65, 113, 155). These two enzymes oxidize the side chains of proline and lysine residues within collagen chains and thus, stabilize the triple helix and the supramolecular assembly of collagen (48). In contrast to the collagen proline 4-hydroxylases, which stabilize the collagen triple helix, hypoxia-inducible transcription factor (HIF) proline 4-hydroxylases mark HIF-1α in a similar proline hydroxylation reaction for ubiquitinylation and proteosomal degradation. Thus, the latter serves as oxygen sensor and is less active with decreasing oxygen supply, whereas the collagen proline 4-hydroxylase is still active under lower oxygen tension because of its higher affinity for oxygen (113, 114).
The oxidative modification of ECM proteins also lead to covalent cross-linkages, which stabilize the insoluble, supramolecular scaffold structures of the ECM. Several redox-relevant cross-linkage reactions are initiated by a copper-containing enzyme lysyl-oxidase (96). It oxidizes lysine or δ-hydroxy-lysine to the respective aldehyde derivative (136). This reactive group may react with ɛ-amino-groups of other lysyl residues or the thioether group of methionine in close vicinity; thus, cross-linking collagen and other matrix molecules, for example, elastin, via an (iso)desmosine ring structure and via the more recently discovered sulfilimine bond (159). In addition, noncatalyzed oxidative cross-linkage occurs between matrix molecules, especially during aging and UV-irradiation (143).
Matrix proteins, especially the triple-helical collagen, are especially resistant to proteolysis. Therefore, matrix metalloproteinases (MMPs) are specifically employed to degrade matrix proteins during tissue remodeling, regeneration, and pathologically, in tumor invasion (47, 55, 73, 135). The matrix fragments released by MMP cleavage may influence cell behavior and constitute signaling molecules of their own (111). Some of the 24 MMP family members are able to cleave even the stable collagen triple-helix, among them MMP-1, which is redox-regulated, presumably like other MMPs (84, 120, 167, 183). The expression of some MMPs, for example, MMP-1 and MMP-2, is regulated by HIF-1α under hypoxia (61, 64, 74). However, also the endogenous tissue inhibitors of MMPs, such as TIMP-1, are expressed under the control of HIF-1α (122). Moreover, the expression of MMP-1 is highly influenced by ROS at the epigenetic and transcriptional level. H2O2 inactivates histone deacetylase-2 (HDAC-2), and it thereby keeps the MMP-1 gene promoter accessible to transcription factors, c-Fos, c-Jun, and Ets-1, which, in turn, are activated by the redox-sensitive mitogen-activated protein kinases signaling pathway (80, 121). Consequently, the increase of the basal mRNA levels of several MMPs (MMP-2, -3, -7, -10, and -12) in fibrosarcoma cells cultured in the presence of the H2O2-generating enzyme superoxide dismutase-2 (SOD-2) is accompanied by an enhanced metastatic potential (120). Restoring the redox balance by increasing the ratio of reduced to oxidized glutathione abolishes this effect in intestinal myofibroblasts (134). At the protein level, the catalytic activity of MMP-1 and other MMPs (for example, MMP-2, -7, and -9) is enhanced by oxidative activation of an internal inhibitory thiol group (53, 80, 130). Therefore, changes in the cellular redox-status may modulate both expression and enzymatic activity of MMPs. Redox-regulated matrix degradation is an important step in aging, cancer, and fibrotic diseases (26), and it is influenced by the redox-status of the cells within the connective tissue. This can partially be influenced by dietary antioxidants found in green tea leaves or the skin of red wine grapes (118, 123).
ECM Regulates ROS Production
Cell anchorage to the ECM influences the cellular metabolism, including the production of ROS. Among these partially reduced oxygen species, hydrogen peroxide (H2O2) is the most persistent ROS, which is able to diffuse through lipid bilayer membranes (44, 107, 139).
Deprivation of matrix contacts triggers ROS production in many adherent cells. Two underlying pathways have been pinpointed in epithelial cells (Fig. 2) [recently reviewed by Grassian et al. (51), see also references therein]. Loss of matrix contacts decreases the uptake of nutrients (glucose, glutamine) into the cell. Additionally, the absence of ECM components and growth factors decreases the activities of phosphatidyl-inositol-3-kinase (PI3K) and protein kinase Akt (52, 138). This leads to increased expression of pyruvate dehydrogenase kinases (PDKs), all four different isoforms (PDK1–4) of which inhibit pyruvate dehydrogenase (PDH) (149). Since PDH catalyzes the gate reaction for the citric acid cycle, its inactivation turns down catabolic glucose utilization and ATP generation via this metabolic pathway. This effect is further aggravated by decreased glucose uptake by the cell (51). Moreover, a diminished activity of the pentose phosphate pathway leads to a drastic lack of reducing equivalents, especially NADPH molecules, which are usually transferred to glutathione to eventually scavenge hydrogen peroxide and other ROS. As a consequence of abolished cell-matrix contact, fatty acid oxidase (FAO) in peroxisomes metabolizes fatty acids to provide the cell with the necessary energy and therefore, is a potential source of ROS (142, 164). The peroxisomal lipid oxidation releases the by-product H2O2, which due to the lack of reducing equivalents accumulates within the cells and diffuses through the cell membrane. Thus, detached cells raise their intra- and extracellular redox potential, which underlines the fact that cell-matrix contacts are able to regulate cellular ROS production (51).
FIG. 2.
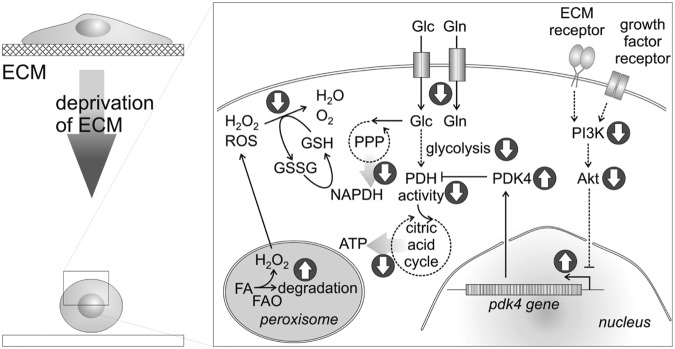
Cell contacts with the ECM affect the production of ROS. Detachment of cells from their ECM substratum and ECM deprivation, along with a lack of growth factors, decreases the activity of PI3K and consequently of protein kinase Akt. Transcription factors, repressed by Akt, are activated and upregulate the expression of PDK, especially PDK4, which phosphorylates/inactivates PDH. Together, with a diminished uptake of nutrients, glucose (Glc) and glutamine (Gln), this exacerbates fuel deprivation for the citric acid cycle and hence, a shortage of ATP. Likewise, lack of fuel for the PPP reduces the concentration of NADPH, which serves as reducing and scavenging agent for H2O2 and other ROS. To compensate the low levels of ATP and NADPH, peroxisomal FA degradation by FAO is likely turned on with a concomitant release of additional H2O2, which further increases the redox potential of ECM-deprived cells. The metabolic effect of ECM deprivation resulting in increased ROS production has recently been reviewed by Grassian et al. (51). PI3K, phosphatidyl-inositol-3-kinase; PDK, pyruvate dehydrogenase kinases; PDH, pyruvate dehydrogenase; PPP, pentose phosphate pathway; FA, fatty acid; FAO, fatty acid oxidase.
During adhesion, the generation of ROS is not homogenously distributed throughout the whole cell. By indirect measurement of ROS-driven cysteine thiol oxidation in proteins indirectly via dimedone chemistry (18, 126), we observed that high concentrations of ROS oxidize proteins in membrane protrusions at sites where a cell establishes new contacts to its surrounding matrix, presumably via integrins (29). We named these subcellular areas redox hotspots (Fig. 3). Redox hotspots are not only spatially restricted but also temporarily reversible. They return to the nonoxidized state within minutes after the initiated cell-matrix contact has been established (Fig. 3). Thus, ROS-production and concomitant protein oxidation is regulated in an adhesion-dependent manner (29). These studies were performed with vascular smooth muscle cells, which are able to secrete H2O2 in a physiological range of sub-millimolar concentrations (139). Recently, these data have been corroborated by live cell imaging of fibroblasts, in which a fluorescent redox probe was directed to focal adhesions via a paxillin tag. Thereby, it was demonstrated that a platelet-derived growth factor (PDGF)-elicited ROS signal is reduced in αVβ3 integrin-containing focal adhesions of fibroblasts attached onto fibronectin (94).
FIG. 3.

ROS production is influenced by cell adhesion in a temporally transient and spatially restricted manner. Rat aorta smooth muscle cells were seeded onto laminin-111 and the thiol-based protein oxidation was monitored by immunofluorimetric detection of dimedone adducts of cysteine sulfenic acid groups. Protein oxidation decays during the course of adhesion (A). Protein oxidation does not occur homogeneously across the cell but in spot-like areas, as seen about 10–20 min after cell-matrix contact (B). These cell areas, named redox hotspots, represent membrane protrusions, as visualized by scanning electron micrographs (C–E). The larger protrusions (filled red arrowhead in E), but not the smaller membrane wrinkles (open arrows in E) are the sites of protein oxidation. After the cell has fully spread, protein oxidation is only detected at the lamellipodia (F). The figure is adapted from the authors' recent publication (28). To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Leukocytes are also well-known for their capacity to release substantial amounts of ROS, especially in the respiratory burst, while they attack pathogens (28, 125). This ROS production in leukocytes is a regulated process. While extravasating and migrating towards the site of inflammation, leukocytes temporarily produce less ROS when in close contact with the matrix. This also indicates that cell-matrix contacts regulate ROS synthesis in a suppressive manner (185).
Tumor cells are generally characterized by increased ROS production as compared to normal tissue cells (43). Consequently, studies carried out with tumor cells, showed enhanced ROS production upon cell-matrix interaction. Using various inhibitors against different ROS-producing enzymes, Taddei et al. demonstrated an increased mitochondrial ROS-production in a fibroblastic cell line during the initial phase of cell attachment. This mitochondrial ROS production peaks at 10 min, while a second peak after 45 min is likely caused by 5-lipoxygenase (152). Recently, in invadopodia protrusions of invasive and malignant tumor cells yet another redox-relevant regulatory pathway has been identified (31). Src family members are essential signaling molecules during cell adhesion and interact with tyrosine kinase substrate (Tks) proteins-4 and -5 (Tks-4 and Tks-5). These, in turn, stimulate Nox family members to produce ROS within invadopodia (23, 30). As ROS also affect cell attachment, this may lead to a temporary stimulation of adhesion-mediated src-activation [see also the article by Giannoni and Chiarugi (47a) in this Forum].
Tissue Oxygen Tension and ROS Influence ECM Production and Deposition
Conversely, ROS and oxygen influences the production of ECM proteins at both transcriptional and (post)translational levels (115). Several genes, that code for ECM proteins, such as various collagen α-chains, fibronectin, and elastin, are transcriptionally controlled by HIFs, and hence, are preferentially expressed by cells under low oxygen tension (8, 32, 41). Among the enzymes, which post-translationally modify nascent matrix proteins, lysyl oxidase is even one of the most strongly hypoxia-regulated genes (39, 40). In addition to the hydroxylated proline and lysine residues, distinct cysteine residues within ECM-proteins are targets of ROS. Their redox modifications lead to rearrangement of inter- and intramolecular disulfide bridges and thereby to activity-relevant conformational changes (20).
While a balanced ECM production is essential for development, remodeling and regeneration, an overshooting ECM production causes excess tissue stiffness and tension. It is typical of the pathologic process of fibrosis, which can be elicited by ROS. These ROS are secreted by leukocytes after having infiltrated a site of inflammation (28, 92). Moreover, resident mesenchymal cells at the inflammation site are able to produce ROS (6).
In the classical model, developed by Gabbiani (45) and modified by Hinz (62), fibroblast differentiation into myofibroblasts is the key process of fibrosis (Fig. 4). Fibroblasts or other mesenchymal cells, for example, adventitial fibroblasts, pericytes, endothelial cells, and epithelial cells after epithelial-mesenchymal transition differentiate initially into a proto-myofibroblast with typically strong and contractile stress fibers. In a subsequent step, final differentiation into a myofibroblast goes along with abundant expression and deposition of ECM proteins (62, 63, 82). Key factors for myofibroblast differentiation are TGFβ1 and other growth factors (63), the cellular fibronectin isoform harboring both EDA and EDB domains (140, 157, 168), as well as enhanced mechanical load onto the matrix (157, 158). Markers for myofibroblasts are α-smooth muscle actin (αSMA), smooth muscle myosin heavy chain, desmin, caldesmon, and fibroblast-specific protein (FSP-1, or synonymously S100A4), although the latter is neither exclusively nor indispensably linked to the myofibroblast phenotype. According to these markers, myofibroblasts resemble smooth muscle cells.
FIG. 4.

ROS stimulates myofibroblast differentiation and fibrosis. TGFβ1, the EDA-splice variant of fibronectin and mechanical tension are well-known stimuli for myofibroblast differentiation, in which fibroblasts differentiate first into proto-myofibroblasts and further into myofibroblasts, which are characterized by increased expression of αSMA with intense formation of stress fibers, and by an excess synthesis and deposition of ECM, respectively. This fibrotic reaction is typical of inflammation elicited by leukocytes via ROS. Pathologically, the fibrotic environment of desmoplastic tumors supports tumor cell progression. Tumor cells also produce ROS, which stimulate the myofibroblast differentiation of additional fibroblasts. Thus, ROS act as an alternative stimulus for fibrosis, including the characteristic upregulation of ECM production. TGFβ1, transforming growth factor-β1; αSMA, α-smooth muscle actin; EDA, extra domain A.
Additional factors which stimulate myofibroblast differentiation are often released from immune cells infiltrating an inflamed or damaged tissue region (63). Some of these factors, such as PDGF, insulin-like growth factor, and angiotensin-II, are also known stimuli for cellular ROS production (93, 102). ROS can also elicit and influence fibrosis and ECM production at concentrations typically found at sites of inflammation (6). Hallmark matrix proteins for myofibroblast-associated fibrosis are type I and IV collagens. Type III and VI collagens, as well as tenascin-C, are also excessively deposited into the fibrotic matrix, which resembles a ontogenetically early matrix (82). ROS was reported to directly stimulate collagen synthesis in lung fibrosis (93). After ROS-stimulated expression, the alternatively spliced c-fibronectin isoform with both EDs, EDA and EDB, further stimulates myofibroblast differentiation and exacerbates fibrosis. Therefore, ROS may be considered as profibrotic agent.
Members of the nicotinamide adenine dinucleotide phosphate oxidase (NADP-oxidase, Nox) family seem to be the major source of ROS during myofibroblast differentiation. One isoform of this family, Nox4 was pinpointed as an essential player, as silencing of Nox4 expression abolishes myofibroblast differentiation and lung fibrosis (10, 17, 58). Nox4 stands apart from the other members of the Nox family [see also the article by Schröder (138a) in this Forum]. Nox proteins generally transfer single electrons from NADPH via an internal electron transfer chain to molecular oxygen, thereby converting it to the superoxide anion O2•−. In contrast, Nox4 seems to transfer two electrons onto molecular oxygen, thereby producing H2O2. The latter has a longer life-time and longer diffusion path. Because it diffuses through biological membranes, it serves as both intracellular and paracellular messenger molecule (44, 139). All Nox family members consist of several subunits, of which two subunits span the plasma membrane: the isoform-specific Nox subunit (e.g., Nox1, gp91phox=Nox2, Nox3, Nox4) and the common p22phox-subunit shared by all Nox members. Several additional, cytosolic subunits associate with the Nox complexes and regulate their enzymatic activities. However, some of these regulatory subunits are missing in the Nox4 complex. This may explain why the enzymatic activity of Nox4 is not or just poorly regulated in comparison with the other Nox members. Nevertheless, it is assumed that the constitutively active Nox4 enzyme is controlled at its expression level, whereby the translation of its mRNA into the active protein seems to be an essential step. TGFβ1, the major driver of myofibroblast differentiation, increases the expression of Nox4, and silencing Nox4 expression by siRNA technology abrogates myofibroblast differentiation (12, 58). After binding to its cognate heterodimeric receptor, TGFβ1 mainly signals via the Smad pathway (99, 100), triggering the transcription of αSMA. In addition, TGFβ1 also increases the expression of Nox4 and the p22phox subunit (12). Consequently, ROS production is enhanced and influences the Smad pathway of TGFβ1 signaling. However, the interrelation of the TGFβ1 and ROS signaling pathways has still remained elusive and may be cell-type specific (6, 12).
Fibrosis with excessive ECM production is not only triggered by ROS-secreting leukocytes. Tumor cells also secrete ROS and induce the differentiation of stromal fibroblasts into myofibroblasts (62, 124). These cancer-associated fibroblasts support the progression of such fibroblastic/desmoplastic tumors (Fig. 4).
Redox Potential and ROS Affect Cell Adhesion and Migration
In a seminal work, Go and Jones demonstrated that the extracellular redox potential in human serum varies with different (patho) physiological conditions, such as age and cigarette smoking (49). Moreover, an increased extracellular redox potential increases the expression of cell adhesion molecules, such as intercellular cell adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), as well as P- and E-selectins (49). This is of great importance for leukocytes to extravasate to an inflammation site, which is one of the initial triggers of inflammatory fibrosis (92). It comprises a cascade of subsequent steps: leukocytes first interact with endothelial cells, then adhere to the basement membrane underneath the endothelial cells and eventually penetrate it to migrate into the interstitial matrix. Cell adhesion molecules of the integrin family mediate such cell-cell and cell-matrix contacts. They consist of two subunits, α and β. Eighteen α- and eight β-subunits have been reported so far, which combine to form 24 functional receptors for ECM proteins or cell membrane-anchored adhesion molecules. During diapedesis, the production of ROS in leukocytes is suppressed in an ECM-dependent manner (185). After having reached the site of inflammation, leukocytes produce ROS in a Nox2-dependent manner, also known as the respiratory burst, which destroys pathogens (28). In addition, these ROS, including the far-reaching hydrogen peroxide, affect the infiltration of additional leukocytes, by increasing the expression of the leukocyte-adhesive molecules on endothelial cells, such as ICAM-1 and VCAM-1 (101). Moreover, the ligand-binding activity of the respective receptors, α4β1 and α4β7 integrins, is increased in a thiol-dependent manner (21). The protein disulfide isomerase inhibitor bacitracin inhibits these two integrins and thus, affects leukocyte diapedesis (112). Free cysteine thiol groups within α4β1 (90) and other integrins (104) were identified as targets for ROS on the leukocyte cell surface by protein chemical means and by proteomics.
Platelet aggregation and hemostatic thrombus formation are further examples of how adhesion is redox-regulated at the integrin level. The platelet integrin αIIbβ3 plays a crucial role in this process (9). Redox-sensitive free cysteine thiol groups within this integrin receptor and also an inhibitory effect of bacitracin on platelet aggregation support the hypothesis of a thiol-based redox-regulation of platelet activity (88, 97, 133). In fact, platelet aggregation is influenced by reducing agents, such as glutathione (4). An extensive body of literature underlines the importance of redox-regulation in integrin-mediated platelet activation, which is discussed in more detail in the article by Murphy et al. (112a), in this Forum.
Integrins as Cellular Receptors for ECM Proteins
At the molecular level, cell-matrix contacts are mediated by various cell adhesion molecules. Integrins are the most numerous and versatile family of these cell adhesion molecules [recently reviewed in Refs. (5, 68)]. Both integrin subunits, α and β, span the plasmalemma once, as type I transmembrane proteins, thereby linking various components of the ECM or cellular counter-receptors [Table 1, and reviewed in Humphries et al. (66)] with the intracellular cytoskeleton. Thus, they mediate cell-matrix and cell-cell contacts. Due to their bidirectional signaling capabilities, integrins also serve as perceptive molecules which sense their surrounding matrix and convey these environmental cues into the cell. Integrins do not signal by themselves as they lack any kinase domain. However, they recruit adaptor and signaling molecules into adhesion-dependent organelles, called adhesomes (79, 171). As key elements of adhesomes, integrins also serve as force-transducing receptors. They are docking sites for cell-spanning actin stress fibers, which together with myosins form intracellular molecular motors. Mechanical force transmission is necessary for cell anchorage to the ECM and cell migration, as well as for tissue/matrix remodeling. This is highly relevant for fibrosis as integrin-mediated forces applied to the matrix release TGFβ1, thereby accelerating the fibrotic process (169). The physiological importance of most integrins has also been shown by genetic ablation models in mice (67, 70).
Table 1.
Ligand Specificity of the Known 24 Integrins
| β1a | β2 | β3 | β4 | β5 | β6 | β7 | β8 | |
|---|---|---|---|---|---|---|---|---|
| α1b | Collagens: Col-IV, Col-I laminins (low affinity) | |||||||
| α2 | Collagens: Col-I, Col-IV, laminins (low affinity), E-Cad, C1q, CLRPs | |||||||
| α3 | Ln-332, Ln-511, Inv | |||||||
| α4 | Fn (EDA, IIICS), ICAM-1 (D1/D2), VCAM-1, MAdCAM-1, Opn, TSP | MAdCam-1, VCAM-1 (D1/D2), Fn (EDA, IIICS), Opn | ||||||
| α5 | Fn (III9+10), Inv | |||||||
| α6 | Laminins, e.g., Ln-111 (E8), Inv | Laminins, e.g., Ln-332 in hemi-desmosomes | ||||||
| α7 | Laminins, e.g., Ln-111 (E8), Inv | |||||||
| α8 | Fn, Vn, Ten-C, Opn, nephronectin | |||||||
| α9 | Fn (EDA), VCAM-1, Opn, tenascin | |||||||
| α10 | Collagens | |||||||
| α11 | Collagens | |||||||
| αE | E-Cad (D1) | |||||||
| αD | VCAM-1, ICAM-3 | |||||||
| αL | ICAM-1–5 (D1/D2) | |||||||
| αM | ICAM-1,-2,-4 Fb, iC3b, factor X | |||||||
| αX | ICAM, Fb, iC3b, collagens | |||||||
| αIIb | Fb, Fn, Vn, vWF, TSP | |||||||
| αV | Vn, Fn, Opn, LAP-TGFβ | Vn, Fn, Fb, vWF, Opn, BSP, TSP, tenascin, Fib, Fbu, gelatin, LAP-TGFβ | Vn, Opn, BSP | Fn, Opn, LAP-TGFβ | Vn, LAP-TGFβ |
Possible combinations of α- and β-subunits are indicated. Their cognate ligands are indicated in the cells of the table. This list of proposed integrin ligands is incomplete and only best-validated integrin ligands are shown. The color coding of the ligands represents the potential integrin recognition motif.
Absence of grey shading indicates integrin ligands using the peptide sequence RGD for integrin binding.
Light grey shading marks the LDV or IEV sequence within an immunoglobulin fold (D-domain), which is recognized by the immunological relevant integrins.
Dark grey shading highlights the collagen and laminin ligands, which require a characteristic three-dimensional structure (such as the collagenous triple-helix) to be recognized by their respective integrin receptor.
First row indicates the different integrin β-subunits.
First column indicates the different integrin α-subunits. Alpha-subunits that contain an A-domain are underlined.
Potential integrin ligands: BSP, bone sialoprotein; CLRP, C-type lectin-related protein (found in snake venom); Col, collagen (Roman numerals indicate the type of collagen); D, D-domain of an immunoglobulin fold (with Arabic numbers specifying its position within the molecule); E-Cad, E-cadherin; EDA, EDB, extra domains A and B, respectively, of fibronectin; Fb, fibrin(ogen); Fib, fibrillin; Fbu, fibulin; Fn, fibronectin; iC3b, immobilized complement factor 3b; ICAM, intercellular cell adhesion molecule; Inv, invasin; LAP-TGFβ, complex of latency activating peptide and TGFβ; Ln, laminin; MAdCAM, mucosal addressin cell adhesion molecule; Opn, osteopontin; Ten-C, tenascin-C; TSP, thrombospondin; VCAM-1, vascular cell adhesion molecule-1; Vn, vitronectin; vWF, von Willebrand factor; IIICS, fibronectin type III connecting sequence; RGD, arginine-glycine-aspartate; TGFβ, transforming growth factor-β.
Some of the integrin recognition sites within the ECM proteins have been elucidated for the last three decades. The best-known recognition site is the linear peptide sequence arginine-glycine-aspartate (RGD) located at the tip of a peptide loop (128, 137). The RGD-bearing peptide loop within the fibronectin type III repeat 10, together with the synergy binding site within the adjacent fibronectin type III repeat 9, is the binding site for the “classical” fibronectin receptor, integrin α5β1 (91). Originally discovered in fibronectin, this RGD peptide has been found to be the recognition site for several other integrins, now grouped into the subset of RGD-dependent integrins. The RGD-dependency of the αV-containing integrins, together with the key role of αVβ3 integrin in tumor-induced angiogenesis has spurred the development of pharmaceutical compounds mimicking the RGD motif (98). Naturally occurring hemorrhagic toxins of snake venoms also utilize RGD-containing peptide loops to block the RGD-dependent platelet integrin αIIbβ3, which usually binds to fibrin(ogen) after platelet activation and thus, forms a hemostatic thrombus. These disintegrins differ in their domain structure from a fibronectin type III repeat but share the very effective RGD-containing peptide loop (16). The pharmaceutical imitation of this structure has led to the new generation of antithrombotic drugs, which are in clinical use nowadays (24, 25).
Integrins α9β1, α4β1, and α4β7 recognize the EDA-module of c-fibronectin, and hence, are good candidate receptors to elicit the c-fibronectin-induced myofibroblast differentiation and the concomitant ROS-production (168). Interestingly, the α4-integrins, along with the β2 integrins, fulfil immunological functions. They recognize an LDV or IET-sequence within a typical immunoglobulin fold (D-domain) of their cognate ligands. Both the immunologically relevant β1 integrins, α4β1 and α4β7, and the β2-integrins (αLβ2, αMβ2, αXβ2, and αDβ2) are all expressed in leukocytes. Interestingly, the expression of β2 integrins is upregulated by ROS (117). Integrin α4β1 interacts with its ligand VCAM-1 on endothelial cells. This is an essential step for extravasation and attainment of the inflammation site (162). Due to their medical importance in inflammation, α4β1 integrin and the leukocyte integrin αLβ2 are targets for pharmaceutical integrin antagonists, which are in clinical trials to treat immunological disorders (24).
An indispensable requirement for collagens to be recognized by their cognate integrin receptors (α1β1, α2β1, α10β1, and α11β1) is their triple-helical quaternary structure, which presents the integrin binding motif as a special array of amino acid side chains to which all three collagen chains contribute (33–35, 59, 83, 184). The homotrimerically presented GFOGER-sequence (O=hydroxy-proline) was identified as a prototypic integrin recognition site in collagens (83). This RGD-independent collagen-binding is achieved by an additional A-domain of the integrin α-subunit and is accompanied by substantial conformational changes within the A-domain (38). Mediating the interaction of fibroblasts with the collagen-rich stroma, the collagen-binding integrins play an important role in fibrosis (36).
Least is known about the molecular recognition site of laminin-binding integrins. These integrins bind to the globular G-domain of laminins; however, only if the neighboring α-coiled-coil laminin rod allows a binding-competent conformation (119). The laminin-integrin interaction cannot be inhibited by RGD peptides.
Integrin Structure
Both integrin subunits comprise several modules, whereby the N-terminal domains of both subunits join to form a common head, which harbors the ECM ligand interaction site (Fig. 5). Two individual stalks (or legs) of each subunit stretch out from the head domain and connect it with a single membrane-spanning motif anchored within the plasmalemma. The common head region contains a propeller domain of the α-subunit, as well as a plexin-semaphorin-integrin (PSI) domain, a hybrid domain and a βA-domain of the β-subunit (116, 174–177). The propeller domain consists of seven blades of β-sheets and is stabilized by divalent cations complexed to the ligand-distal face of the propeller domain. Between blades 2 and 3, an additional A-domain is inserted within the α-subunits of the collagen-binding β1 integrins and the leukocyte β2 integrins. This A-domain, like the A-domain of the β-subunit, is homologous to the von Willebrand factor (vWF)-A domain and folds into a typical Rossman fold (37, 129, 131). Upon ligand binding, the A-domains undergo substantial conformational changes (38), which eventually are conveyed through the entire molecule into the stalk regions; hence, mediating signal transduction through the integrin molecule (2, 141).
FIG. 5.

Domain structure of integrins, containing (A) and lacking (B) an A-domain within their α-subunits. The propeller domain and A-domain of the α-subunit, together with the PSI, hybrid, and Aβ-domain of the β-subunit form the joint headpiece of the integrin, which binds to the extracellular ligand. Two separated stalks/legs point out of the headpiece and contain different modules lining up like pearls on strings: the thigh, genu, and the two calf domains of the α-subunit and the EGF1–4 and β-tail domain of the β-subunit. Both subunits are anchored within the cell membrane with a single-span α-helical transmembrane domain. The short cytoplasmic C-terminal domains of the two subunits interact with different cytoskeletal, adaptor, and signaling molecules, which enable the integrin to link the ECM to the cytoskeleton across the plasmalemma and to convey environmental cues by outside-in and inside-out signaling. PSI, plexin-semaphorin-integrin.
The stalk of the β-subunit is made of four EGF-domains and one small β-tail domain. They are rich in disulfide-engaged cysteine residues (76, 109, 172), whereas the stalk of the α-subunit is shaped of three β-strand-rich domains, a thigh and two calf-domains. The α-subunit leg contains only a few cysteine residues. The two calf domains share a homologous domain structure, each consisting of two β-sheets of five and four β-strands (Fig. 6). The calf-2 domains of those integrin α-subunits which lack an A-domain contain a cleavage site, the proteolysis of which results in a heavy and a light chain. They are covalently cross-linked via a disulfide-bridge between β-strands 6 and 7 (Fig. 6B). This disulfide bridge might not be necessary as the β-strands of heavy and light chains interdigitate and the numerous noncovalent interstrand bonds stabilize the two β-sheets of the calf-2 domain. The proteolytic cleavage of the integrin α-subunit is structurally interesting with respect to the newly generated, floppy N-terminus of the light chain, which allows an increased flexibility of the N-terminal β-strand 7 within the calf-2 domain (Fig. 6).
FIG. 6.

Structure of the calf-2 domain of the integrin α-subunit, either with (A) or without a proteolytic cleavage site (B). Five and four antiparallel β-strands (numbered 1, 3–10) form two β-sheets, which fold into the stable tertiary structure of the calf-2 domain. (A) In α-subunits, which contain an A-domain, the peptide chain within the calf-2 domain is not cleaved. (B) The integrin α-subunits lacking A-domains are generally cleaved in a loop between β-strands 6 and 7 of the calf-2 domain, resulting in a heavy and light chain of the α-subunit (marked in blue and green, respectively). Although there is an interchain-disulfide bridge between β-strands 6 and 7, the strong interdigitation of the β-strands is likely to hold the heavy and light chains together after the proteolytic cleavage. The proteolytic cleavage generates a new N-terminus of the light chain which allows a higher flexibility of β-strand 7. The secondary structure elements are sketched in (A, B) according to the crystal structure of the integrins, αXβ2 and αVβ3, respectively (174, 175, 176). To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
The genu region within the leg of the integrin α-subunit is of special importance for integrin activation. It acts as pivot around which the head domain and the two stalk domains can rotate relative to each other and thus, change the conformation of the integrin ectodomain between a bent and an extended/upright conformation (Fig. 7). The genu peptide of the integrin α-subunits consists of a short peptide loop, which is cross-linked by an intrachain disulfide-bridge.
FIG. 7.

The two major conformations of the integrin ectodomain: bent (A) and upright/extended (B). The genu region acts as a pivot around which the head domain and the stalks of the integrin ectodomain can hinge against each other, leading to a bent (A) and an upright (B) conformation. Low and high binding activities are ascribed to the bent and upright conformations, respectively. A disulfide bridge within the genu region of the α-subunit and the long-range disulfide bridge between the PSI and the EGF-domains of the β-subunit are indicated. Their redox-regulated cleavage is likely involved in the conformational transition of the integrin ectodomain.
As opposed to the cysteine-poor stalk domains of the α-subunit, the stalk domains of the integrin β-subunits are rich in cysteine residues. For example, there are eight cysteine residues in each EGF-domain and several ones in the β-tail domain (76, 109, 172). However, the cysteine residues within the stalk of the β integrin subunit are engaged in disulfide bridges, which stabilize the stalk domains. According to our studies, the cysteine side chains of the β1 subunit stalk are not targeted by hydrogen peroxide and hence, they cannot be redox-regulated by oxidizing agents (29).
Conformational Changes and Thiol-Based Redox-Regulation of Integrins
Both the ligand-binding head domain and the two legs play pivotal roles in integrin activation and regulation (2, 141, 145, 146, 153, 173). The head domain apparently is not susceptible to redox-regulation with the exception of one long-range disulfide bridge within the integrin β-subunit. Among the two stalks, only the leg of the integrin α-subunit, but not of the β-subunit seems to contain cysteine residues, which are sensitive to oxidizing agents. Several studies identified redox-relevant cysteine bridges via their susceptibility to reducing agents (15, 76, 77, 85). In contrast, ROS and other oxidizing agents are able to not only establish disulfide bridges, but also to prevent their formation when two vicinal disulfide bridges are concomitantly oxidized to cysteine sulfenic acid groups (29, 109). This may explain the observed different effects of ROS on thiol-based modulation and activation of integrins in comparison to studies, which employ the reductive cleavage of intramolecular disulfide bridges.
Experimental works on conformational changes within the head domain mainly highlight the A-domains within the β-subunits (βA) and the α-subunits (αA) of the collagen-binding and leukocyte integrins. While there is no evidence for activation-related conformational changes within the propeller and PSI domains of the integrin α and β-subunits, the A-domains of both integrin subunits undergo major conformational changes. The structure-function relationship of an A-domain has been unraveled by Emsley et al. (38). They demonstrated that collagen-binding to the integrin α2 A-domain causes unfolding of an αC-helix and eventually results in a translational shift of α-helices 7 and 1 against each other by almost 10 Å, a tremendous movement in molecular scale. Although A-domains from the leukocyte integrin α-subunits lack this αC-helix, they also undergo such a conformational shift of homologous α-helices (22, 147). A similar change is envisioned for the Aβ-domain of αVβ3 upon RGD-binding (177). The translational shift of the two piston-like α-helices against each other is potently amplified through a hinge between the Aβ domain and hybrid domain and leads to the transition from the bent into the upright conformation of the integrin ectodomain (2, 141, 145, 146, 153). Springer et al. (145, 146, 174, 180) proposed a mechanism of conformational signal transduction within the αA-domain-containing integrins. According to such a rope belt/pull spring mechanism, the conformational change of the αA-domain after ligand binding induces the exposure of a glutamic acid side chain at the ligand-distal face of the αA-domain, which then serves as intrinsic ligand for the Aβ-domain. Similar to ligand occupancy, this triggers the conformational change of the Aβ domain and leads to a bent-to-upright transition of the integrin receptor and to the separation of the two legs.
Despite its importance in ligand binding, there is little evidence that the head domain is redox-regulated at the level of thiol groups. The head domain, including the αA-domain, contains several cysteine residues. Yet, it does not seem to be affected by reducing agents or ROS. Moreover, mutation of cysteines within the Aβ-domain of the β3 subunit did not show any effect on the ligand-binding activity of the platelet integrin (19). An exception to this redox insensibility of the head domain is a long-range disulfide bridge between the PSI and a peptide sequence within the EGF-domains 2 and 3 of the β-subunit stalk. Its importance for activation of the platelet integrin αIIbβ3 was proven by protein-chemical, mutational and crystallographic studies (15, 144, 150, 178, 179), which demonstrated how this disulfide bridge constrains the integrin in its inactive bent conformation (Fig. 7). Such a long-range disulfide bridge of similar physiological importance also exists in the β2 subunit (182) and likely in other β integrin subunits as well (7, 144, 154). Its redox-dependent cleavage is a prerequisite for the integrin's transition into the active upright position (144, 150).
Redox-dependent conformational changes occur within the integrin legs. The high number of cysteine residues within the stalk of the β-subunit makes this region highly likely for redox-regulation (76, 109, 144, 154). However, instead of having a regulatory role, these cysteine residues are probably engaged entirely in structure-stabilizing disulfide bridges within and between the EGF-domains, although conformation-stabilizing and regulatory effects are sometimes difficult to distinguish from each other. Mutations of two individual cysteines within the EGF-domains 1 and 3 of β3 reduced the expression of αIIbβ3 integrin in Glanzmann thrombasthenia patients (105), whereas mutation of the most membrane-proximal disulfide-bridge within the β-tail-domain leads to a constitutively active platelet receptor (14). Redox-regulation by a few distinct cysteines is indicated by the work of Mor-Cohen et al. (109), as the mutation of disulfide bridged cysteine residues only affect integrin activity when performed with single cysteine residues, but not with an even number of cysteines, indicating that free thiol groups at certain positions within the leg EGF-domains may act as redox-regulated entities. However, there might be differences in the redox-regulation of EGF-domain-based cysteines between the two integrins αIIbβ3 and αVβ3, although they share the same β3 integrin subunit (108). This indicates that the partnering α-subunit also affects thiol-based redox regulation within the β integrin leg domains.
Thiol-based redox-regulation occurs within the stalk region of the integrin α-subunits. Our own work on the laminin-binding α7β1 integrin, in which we analyzed redox-modifications of this β1 integrin with hydrogen peroxide, highlighted six cysteine residues as selective targets for ROS. They are located at two sites exclusively within the integrin α7 subunit: the genu and the calf-2 domain (29). The genu region of the integrin α-subunit consists of a peptide loop, which contains a disulfide bridge. Opening this endogenous intrachain cross-link allows a higher flexibility of this pivot. This permits the integrin ectodomain to hinge from an inactive/bent to an extended/active conformation. In line with this, introducing a clamping disulfide bridge adjacent to the genu loop of the αIIb subunit yields an inactive αIIbβ3 integrin (11, 77). Although the cysteine residues within the α-subunit calf-2 domain are limited to a number of four, protein-chemical and crystallographic analysis revealed some differences in the disulfide bonding within this domain (85). These discrepancies might not only be due to methodological limitations, for example, by the fact that the integrin ectodomains are crystallized in their bent conformations (174, 176), but may reflect the disulfide-based conformational flexibility of the calf-2 domain. Therefore, we postulate the existence of three different conformations of the calf-2 domains, which differ in cross-linkage of a cysteine residue within the β-strand 6 of the heavy chain with one of the first three cysteines of the N-terminal end of the light chain (29) (Fig. 8). Newly generated by proteolytic cleavage, the N-terminus of the light chain is flexible and likely facilitates the existence and/or transitions between these conformations. As the N-terminal sequences of the α-subunit light chains flanking the three different cysteine residues mostly support β-strand folding, the conformational changes could be envisioned as sliding of the β-strands 6 and 7 against each other within the calf domain. Due to the anchoring disulfide bridge, conformational changes would require free thiol groups, which after transition can be fixed again rendering either all four cysteine residues engaged in disulfide bridges again (conformation I and III) or partially unlinked as free thiol groups (conformation II). These conformations not only vary in the length of the free N-terminal end of the light chain, but also in the size of the peptide loop between β-strands 7 and 8, which could cause far-ranging conformational transitions, including the extension of the bent to the activated integrin. Interestingly, the calf-2 domain of the integrin α-subunit forms an interface with the EGF4-β-tail domain of the β-subunit (174, 176), which is relevant for integrin activation (77, 78, 165).
FIG. 8.

Disulfide bridge-dependent conformation of the calf-2 domain. The calf-2 domain of the cleaved integrin α-subunits consists of a heavy and a light chain, marked in dark and light grey. It contains four cysteine residues. With the assumption that the cysteine residue of β-strand 6 is considered as the anchor point, it can be cross-linked to the three cysteine residues of the light chain in three different ways, resulting in conformations I–III. Interestingly, the amino acids neigboring the light chain cysteines can support β-strand folding in most α-subunits. Moreover, structural analyses of different integrin ectodomains crystallized in the bent conformation (174, 176) showed the calf-2 domain in conformation I, whereas protein chemical analyses of integrins in solution (85) indicated other disulfide patterns within the calf-2 domains, which comply with conformations II and III. This suggests that thiol-based redox-regulation may affect the conformation of the calf-2 domain, which as part of the alpha subunit leg affects the entire structure, and hence, activity of the integrin.
The equilibrium between free thiol groups of cysteine residues and disulfide bridges within proteins is the basis for a reversible redox-regulation and is shifted by reducing agents, oxidants and even photolytically by UV-C light (161). Reducing agents, such as the physiological glutathionine, cleave disulfide bridges and thus, unlock cystine-stabilized tertiary structures. Oxidants, such as the membrane-permeable hydrogen peroxide and other ROS, oxidize cysteine thiolate reversibly into sulfenic acid groups or into disulfide bridges, if two cysteine residues are in close vicinity. However, concomitant oxidation of such vicinal cysteine residues into two sulfenic acid groups also prohibits disulfide cross-linkage. Despite the even number of cysteines able to pair entirely into disulfide bridges, free thiol groups have been detected in integrins under physiological conditions, indicating such a redox-relevant equilibrium between free and bridging cysteine residues (97, 104, 133). In addition to noncatalyzed redox reactions, protein disulfide isomerase and redox-relevant chaperones have been reported to associate with integrins on the cell surface, thereby regulating integrin activity (87, 151). However, the correlation between redox state and activity of integrins has not been studied conclusively yet. Moreover, other redox-related modifications of integrins, such as nitrosylation, need yet to be analyzed.
So far, the experimental data provide compelling evidence, that integrins are redox-regulated. Cysteine residues within the genu and calf-2 domain of the integrin α-subunit, as well as a long-range disulfide bridge between the head domain and the leg of the β integrin subunits have been pinpointed to be the redox-regulated sites within the integrin ectodomain. Their redox-modification influences the transition between a bent/inactive and extended/active conformation of the integrin ectodomain. Consequently, these conformational transitions within the head domain induce the physical separation of the two stalks and lead to changes within the transmembrane and cytoplasmic domains (166, 186). This restructuring then transduces a signal onto integrin-associated molecules (95, 103, 141). Thus, the redox-regulated conformational rearrangements of the integrin structure are translated into cell-matrix contact-modulating signals that eventually influence diverse physiological and pathological processes.
Concluding Remarks
In the last decade, it has been demonstrated that hydrogen peroxide and other ROS, at physiological concentrations, are crucial for cell signaling and redox-regulation, instead of just causing oxidative stress and damage. Cell-matrix interaction and ECM turnover are redox-regulated processes. While the ECM is modified by ROS, it also affects the cellular ROS production. Integrins are pivotal in cell-matrix cross-talk and are modified and thus, modulated in their activity by ROS. Conformational changes in the integrin structure suggest a mechanism of this redox-regulation of integrin activity. In future, treatment of diseases in which redox-regulated processes are involved, such as fibrosis and cancer, may benefit from understanding the redox-regulation of integrin-mediated cell-matrix interaction.
Abbreviations Used
- IIICS
fibronectin type III connecting sequence
- αSMA
α-smooth muscle actin
- A-domain
von Willebrand factor A-homologous domain
- BSP
bone sialoprotein
- CLRP
C-type lectin-related protein
- Col
collagen
- E-Cad
E-cadherin
- ECM
extracellular matrix
- ED
extra domain
- FAO
fatty acid oxidase
- Fb
fibrin(ogen)
- Fbu
fibulin
- Fib
fibrillin
- Fn
fibronectin
- FSP
fibroblast-specific protein
- GAG
glycosaminoglycan
- HDAC-2
histone deacetylase-2
- HIF
hypoxia-inducible transcription factor
- ICAM-1
intercellular cell adhesion molecule-1
- iC3b
immobilized complement factor 3b
- Inv
invasin
- LAP
latency activating peptide
- Ln
laminin
- LTBP-1
latent TGFβ1-binding protein-1
- MAdCAM
mucosal addressin cell adhesion molecule
- MMP
matrix metalloproteinase
- Nox
nicotinamide dinucleotide phosphate (NADPH)-oxidase
- Opn
osteopontin
- PDGF
platelet-derived growth factor
- PDH
pyruvate dehydrogenase
- PDK
pyruvate dehydrogenase kinase
- PI3K
phosphatidyl-inositol-3-kinase
- PPP
pentose phosphate pathway
- PSI
plexin-semaphorin-integrin
- RGD
arginine-glycine-aspartate peptide sequence (one-amino-acid-code)
- ROS
reactive oxygen species
- SOD-2
superoxide dismutase-2
- Ten-C
tenascin-C
- TGFβ1
transforming growth factor-β1
- Tks
tyrosine kinase substrate
- TSP
thrombospondin
- VCAM
vascular cell adhesion molecule
- Vn
vitronectin
- vWF
von Willebrand factor
Acknowledgments
This work was financially supported by the People Programme (Marie Curie Actions) of the European Union's Seventh Framework Programme FP7/2007–2013/under the REA grant agreement nr. 316610 (CAFFEIN) and by Deutsche Forschungsgemeinschaft (DFG, SFB815, project A6). We thank Patricia Niland, Stephan Niland, and Shaughn Daniel for proofreading the manuscript. We apologize to all experts in the field whose publications we could not cite due to the restricted number of references.
References
- 1.Afratis N, Giateli C, Nikitovic D, Tsegenidis T, Karousou E, Theocharis AD, Pavao MS, Tzanakakis GN, and Karamanos NK. Glycosaminoglycans: key players in cancer cell biology and treatment. FEBS J 279: 1177–1197, 2012 [DOI] [PubMed] [Google Scholar]
- 2.Arnaout MA, Mahalingam B, and Xiong J-P. Integrin structure, allostery, and bidirectional signaling. Annu Rev Cell Dev Biol 21: 381–410, 2005 [DOI] [PubMed] [Google Scholar]
- 3.Baldock C, Sheratt MJ, Shuttleworth CA, and Kielty CM. The supramolecular organization of collagen VI microfibrils. J Mol Biol 330: 297–307, 2003 [DOI] [PubMed] [Google Scholar]
- 4.Ball C, Vijayan KV, Nguyen T, Anthon K, Bray PF, Essex DW, and Dong JF. Glutathione regulates integrin αIIbβ3-mediated cell adhesion under flow. Thromb Haemost 100: 857–863, 2008 [PubMed] [Google Scholar]
- 5.Barczyk M, Carracedo S, and Gullberg D. Integrins. Cell Tissue Res 339: 269–280, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barnes JL. and Gorin Y. Myofibroblast differentiation during fibrosis: role of NAD(P)H oxidases. Kidney Int 79: 944–956, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beglova N, Blacklow SC, Takagi J, and Springer TA. Cysteine-rich module structure reveals a fulcrum for integrin rearrangement upon activation. Nat Struct Biol 9: 282–287, 2002 [DOI] [PubMed] [Google Scholar]
- 8.Berg JT, Breen EC, Fu Z, Mathieu-Costello O, and West JB. Alveolar hypoxia increases gene expression of extracellular matrix proteins and platelet-derived growth factor-B in lung parenchyma. Am J Respir Crit Care Med 158: 1921–1928, 1998 [DOI] [PubMed] [Google Scholar]
- 9.Bergmeier W. and Hynes RO. Extracellular matrix proteins in hemostasis and thrombosis. Cold Spring Harb Perspect Biol 4: a005132, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Block K, Eid A, Griendling KK, Lee D-Y, Wittrant Y, and Gorin Y. Nox4 NAD(P)H oxidase mediates Src-dependent tyrosine phosphorylation of PDK-1 in response to angiotensin II: role in mesangial cell hypertrophy and fibronectin expression. J Biol Chem 283: 24061–24076, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blue R, Li J, Steinberger J, Murcia M, Filizla M, and Coller BS. Effects of limiting extension at the αIIb genu on ligand binding to integrin αIIbβ3. J Biol Chem 285: 16604–17613, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bondi CD, Manickam N, Lee D-Y, Block K, Gorin Y, Abboud HE, and Barnes JL. NAD(P)H oxidase mediates TGF-β1-induced activation of kidney myofibroblasts. J Am Soc Nephrol 21: 93–102, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bruckner P. Suprastructures of extracellular matrices: paradigms of functions controlled by aggregates rather than molecules. Cell Tissue Res 339: 7–18, 2010 [DOI] [PubMed] [Google Scholar]
- 14.Butta N, Arias-Salgado EG, González-Manchón C, Ferrer M, Larrucea S, Ayuso MS, and Parrilla R. Disruptions of the β3 663–687 disulfide bridge confers constitutive activity to β3 integrins. Blood 102: 2491–2497, 2003 [DOI] [PubMed] [Google Scholar]
- 15.Calvete JJ, Henschen A, and Gonzalez-Rodriguez J. Complete localization of the intrachain disulfide bonds and the N-glycosylation points in the alpha-subunit of human platelet glycoprotein IIb. Biochem J 274: 63–71, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Calvete JJ, Marcinkiewicz C, Monleón D, Esteve V, Celda B, Juárez P, and Sanz L. Snake venom disintegrins: evolution of structure and function. Toxicon 45: 1063–1074, 2005 [DOI] [PubMed] [Google Scholar]
- 17.Carnesecchi S, Deffert C, Donati Y, Basset O, Hinz B, Preynat-Seauve O, Guichard C, Arbiser JL, Banfi B, Pache J-C, Barazzone-Argiroffo C, and Krause K-H. A key role of NOX4 in epithelial cell death during development of lung fibrosis. Antioxid Redox Signal 15: 607–619, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Charles RL, Schröder E, May G, Free P, Gaffney PRJ, Wait R, Begum S, Heads RJ, and Eaton P. Protein sulfenation as a redox sensor; proteomics studies using a novel biotinylated dimedone analogue. Mol Cell Proteomics 6: 1473–1484, 2007 [DOI] [PubMed] [Google Scholar]
- 19.Chen P, Melchior C, Brons NHC, Schlegel N, Caen J, and Kieffer N. Probing conformational changes in the I-like domain and the cysteine-rich repeat of human β3 integrins following disulfide bond disruption by cysteine mutations. J Biol Chem 276: 38628–38635, 2001 [DOI] [PubMed] [Google Scholar]
- 20.Chen VM. and Hogg PJ. Allosteric disulfide bonds in thrombosis and thrombolysis. J Thromb Haemost 4: 2533–2541, 2006 [DOI] [PubMed] [Google Scholar]
- 21.Chuang K-P, Tsai W-S, Wang Y-J, and Shieh C-C. Superoxide activates very late antigen-4 on an eosinophil cell line and increases cellular binding to vascular cell adhesion molecule-1. Eur J Immunol 33: 645–655, 2003 [DOI] [PubMed] [Google Scholar]
- 22.Corps EM, Robertson A, Dauncey MJ, and Kilshaw PJ. Role of the αI domain in ligand binding of integrin αEβ7. Eur J Immunol 33: 2599–2608, 2003 [DOI] [PubMed] [Google Scholar]
- 23.Courtneidge SA. Cell migration and invasion in human disease: the Tks adaptor proteins. Biochem Soc Trans 40: 129–132, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cox D, Brennon M, and Moran N. Integrins as therapeutic targets: lessons and opportunities. Nat Rev Drug Discov 9: 804–820, 2010 [DOI] [PubMed] [Google Scholar]
- 25.Curley GP, Blum H, and Humphries MJ. Integrin antagonists. Cell Mol Life Sci 56: 427–441, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dasgupta J, Kar S, Liu R, Joseph J, Kalyanaraman B, Remington SJ, Chen C, and Melendez JA. Reactive oxygen species control senescence-associated matrix metalloproteinase-1 through c-Jun-N-terminal kinase. J Cell Phys 225: 52–62, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Denko NC, Fontana L, A. , Hudson KM, Sutphin PD, Raychaudhuri S, Altman R, and Giaccia AJ. Investigating hypoxic tumor physiology through gene expression patterns. Oncogene 22: 5907–5914, 2003 [DOI] [PubMed] [Google Scholar]
- 28.de Oliveira-Junior EB, Bustamente J, Newburger PE, and Condino-Neto A. The human NADPH oxidase: primary and secondary defects impairing the respiratory burst function and the microbial ability of phagocytes. Scand J Immunol 73: 420–427, 2011 [DOI] [PubMed] [Google Scholar]
- 29.de Rezende FF, Martins Lima A, Niland S, Wittig I, Heide H, Schröder K, and Eble JA. Integrin α7β1 is a redox-regulated target of hydrogen peroxide in vascular smooth muscle cell adhesion Free Radic Biol Med 53: 521–531, 2012 [DOI] [PubMed] [Google Scholar]
- 30.Diaz B. and Courtneidge SA. Redox signaling at invasive microdomains in cancer cells. Free Radic Biol Med 52: 247–256, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Diaz B, Shani G, Pass I, Anderson D, Quintavalle M, and Courtneidge SA. Tks5-dependent, Nox-mediated generation of reactive oxygen species is necessary for invadopodia formation. Sci Signal 2: ra53, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Durmowicz AG, Parks WC, Hyde DM, Mecham RP, and Stenmark KR. Persistence, re-expression, and induction of pulmonary arterial fibronectin, tropoelastin, and type I procollagen mRNA expression in neonatal hypoxic pulmonary hypertension. Am J Pathol 145: 1411–1420, 1994 [PMC free article] [PubMed] [Google Scholar]
- 33.Eble JA. Collagen-binding integrins as pharmaceutical targets. Curr Pharm Des 11: 867–880, 2005 [DOI] [PubMed] [Google Scholar]
- 34.Eble JA, Golbik R, Mann K, and Kühn K. The α1β1 integrin recognition site of the basement membrane collagen molecule [α1(IV)]2α2(IV). EMBO J 12: 4795–4802, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eble JA, Kassner A, Niland S, Mörgelin M, Grifka J, and Grässel S. Collagen XVI harbors an integrin α1β1 recognition site in its C-terminal domains. J Biol Chem 281: 25745–25756, 2006 [DOI] [PubMed] [Google Scholar]
- 36.Eckes B, Zigrino P, Kessler D, Holtkötter O, Shephard P, Mauch C, and Krieg T. Fibroblast-matrix interactions in wound healing and fibrosis. Matrix Biol 19: 325–332, 2000 [DOI] [PubMed] [Google Scholar]
- 37.Emsley J, King SL, Bergelson JM, and Liddington RC. Crystal structure of the I domain from integrin α2β1. J Biol Chem 272: 28512–28517, 1997 [DOI] [PubMed] [Google Scholar]
- 38.Emsley J, Knight CG, Farndale RW, Barnes MJ, and C. LR.Structural basis of collagen recognition by integrin α2β1. Cell 100: 47–56, 2000 [DOI] [PubMed] [Google Scholar]
- 39.Erler JT, Bennewith KL, Nicolau M, Dornhöfer N, Kong C, Le Q-T, Chi J-TA, Jeffrey SS, and Giaccia AJ. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 440: 1222–1226, 2006 [DOI] [PubMed] [Google Scholar]
- 40.Fähling M, Perlewitz A, Doller A, and Thiele B-J. Regulation of collagen prolyl 4-hydroxylase and matrix metalloproteinases in fibrosarcoma cells by hypoxia. Comp Biochem Physiol C Toxicol Pharmacol 139: 119–126, 2004 [DOI] [PubMed] [Google Scholar]
- 41.Falanga V, Martin TA, Takagi H, Kirsner RS, Helfman T, Pardes J, and Ochoa MS. Low oxygen tension increases mRNA levels of α1(I) procollagen in human dermal fibroblasts J Cell Physiol 157: 408–412, 1993 [DOI] [PubMed] [Google Scholar]
- 42.Ffrench-Constant C. Alternative splicing of fibronectin—many different proteins but few different functions. Exp Cell Res 221: 261–271, 1995 [DOI] [PubMed] [Google Scholar]
- 43.Fiaschi T. and Chiarugi P. Oxidative stress, tumor microenvironment, and metabolic reprogramming: a diabolic liaison. Int J Cell Biol 2012, 762825, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Forman HJ, Maiorino M, and Ursini F. Signaling functions of reactive oxygen species. Biochemistry 49: 835–842, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gabbiani G. The myofibroblast in wound healing and fibrocontractive disease. J Pathol 200: 500–503, 2003 [DOI] [PubMed] [Google Scholar]
- 46.Gandhi NS. and Mancera RL. The structure of glycosaminoglycans and their interactions with proteins. Chem Biol Drug Des 72: 455–482, 2008 [DOI] [PubMed] [Google Scholar]
- 47.Gialeli C, Theocharis AD, and Karamanos NK. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J 278: 16–27, 2011 [DOI] [PubMed] [Google Scholar]
- 47a.Giannoni E. and Chiarugi P. Redox circuitries driving Src regulation. Antioxid Redox Signal 20: 2011–2025, 2014 [DOI] [PubMed] [Google Scholar]
- 48.Gilkes DM, Saumendra B, Chaturvedi P, Wirtz D, and Semenza GL. Hypoxia-inducible factor 1 (HIF-1) promotes extracellular matrix remodelling under hypoxic conditions by inducing P4HA1, P4HA2, and PLOD2 expression in fibroblasts. J Biol Chem 288: 10819–10829, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 49.Go Y-M. and Jones DP. Intracellular proatherogenic events and cell adhesion modulated by extracellular thiol/disulfide redox state. Circulation 111: 2973–2980, 2005 [DOI] [PubMed] [Google Scholar]
- 50.Gordon MK. and Hahn RA. Collagens. Cell Tissue Res 339: 427–457, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grassian AR, Coloff JL, and Brugge JS. Extracellular matrix regulation of metabolism and implications for tumorigenesis. Cold Spring Harb Symp Quant Biol 76: 313–324, 2011 [DOI] [PubMed] [Google Scholar]
- 52.Grassian AR, Metallo CM, Coloff JL, Stephanopoulos G, and Brugge JS. Erk regulation of pyruvate dehydrogenase flux through PDK4 modulates cell proliferation. Genes Dev 25: 1716–1733, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grote F, Flach I, Luchtefeld M, Akin E, Holland SM, Drexler H, and Schieffer B. Mechanical stretch enhances mRNA expression and proenzyme release of matrix metalloproteinase-2 (MMP-2) via NAD(P)H oxidase-related reactive oxygen species. Circ Res 92: e80–e86, 2003 [DOI] [PubMed] [Google Scholar]
- 54.Guadall A, Orriols M, Rodríguez-Calvo R, Calvayrac O, Crespo J, Aledo R, Martínez-González J, and Rodríguez C. Fibulin-5 is up-regulated by hypoxia in endothelial cells through a hypoxia-inducible factor-1 (HIF-1α)-dependent mechanism. J Biol Chem 286: 7093–7103, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hadler-Olsen E, Fadnes B, Sylte I, Uhlin-Hansen L, and Winberg J-O. Regulation of matrix metalloproteinase activity in health and disease. FEBS J 278: 28–45, 2011 [DOI] [PubMed] [Google Scholar]
- 56.Hallmann R, Horn N, Selg M, Wendler O, Pausch F, and Sorokin LM. Expression and function of laminins in the embryonic and mature vasculature. Physiol Rev 85: 979–1000, 2005 [DOI] [PubMed] [Google Scholar]
- 57.Hanahan D. and Weinberg RA. Hallmarks of cancer: the next generation. Cell 144: 646–674, 2011 [DOI] [PubMed] [Google Scholar]
- 58.Hecker L, Vittal R, Jones T, Jagirdir R, Luckhardt TR, Horowitz JC, Pennathur S, Martinez FJ, and Thannickal VJ. NADPH-oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med 15: 1077–1081, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Heino J. The collagen receptor integrins have distinct ligand recognition and signaling functions. Matrix Biol 19: 319–323, 2000 [DOI] [PubMed] [Google Scholar]
- 60.Hessle H. and Engvall E. Type VI collagen. Studies on its localization, structure, and biosynthetic form with monoclonal antibodies. J Biol Chem 259: 3955–3961, 1984 [PubMed] [Google Scholar]
- 61.Higgins DF, Kimura K, Iwano M, and Haase VH. Hypoxia-inducible factor signaling in the development of fibrosis. Cell Cycle 7: 1128–1132, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hinz B. Formation and function of the myofibroblast during tissue repair. J Invest Dermatol 127: 526–537, 2007 [DOI] [PubMed] [Google Scholar]
- 63.Hinz B, Phan SH, Thannickal VJ, Prunotto M, Desmouliére A, Varga J, De Wever O, Mareel M, and Gabbiani G. Recent developments in myofibroblast biology. Paradigms for connective tissue remodeling. Am J Pathol 180: 1340–1355, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hiraki M, Kitajima Y, Kai K, Nakamura J, Hashiguchi K, Noshiro H, and Miyazaki K. Knockdown of hypoxia-inducible factor-1α accelerates peritoneal dissemination via the upregulation of MMP-1 expression in gastric cancer cell lines. Exp Ther Med 4: 355–362, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Horino Y, Takahashi S, Miura T, and Takahashi Y. Prolonged hypoxia accelerates the posttranscriptional process of collagen synthesis in cultured fibroblasts. Life Sci 71: 3031–3045, 2002 [DOI] [PubMed] [Google Scholar]
- 66.Humphries JD, Byron A, and Humphries MJ. Integrin ligands at a glance. J Cell Sci 119: 3901–3903, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hynes RO. A reevaluation of integrins as regulators of angiogenesis. Nat Med 8: 918–921, 2002 [DOI] [PubMed] [Google Scholar]
- 68.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell 110: 673–687, 2002 [DOI] [PubMed] [Google Scholar]
- 69.Hynes RO. The extracellular matrix: not just pretty fibrils. Science 326: 1216–1219, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hynes RO, George EL, Georges EN, Guan J-L, Rayburn H, and Yang JT. Toward a genetic analysis of cell-matrix adhesion. Cold Spring Harb Symp Quant Biol 17: 249–258, 1992 [DOI] [PubMed] [Google Scholar]
- 71.Iozzo R. and Schaefer L. Proteoglycans in health and disease: novel regulatory signaling mechanisms evoked by the small leucine-rich proteoglycans. FEBS J 277: 3864–3875, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Iozzo RV. and Sanderson RD. Proteoglycans in cancer biology, tumor microenvironment and angiogenesis. J Cell Mol Med 15: 1013–1031, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Iyer PR, Patterson NL, Fields GB, and Lindsey ML. The history of matrix metalloproteinases: milestones, myths, and misperceptions. Am J Physiol Heart Circ Physiol 303: H919–H930, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jing S-W, Wang Y-D, Kuroda M, Su J-W, Sun G-G, Liu Q, Cheng Y-J, and Yang C-R. HIF-1α contributes to hypoxia-induced invasion and metastasis of esophageal carcinoma via inhibiting E-cadherin and promoting MMP-2 expression. Acta Med Okayama 65: 399–407, 2012 [DOI] [PubMed] [Google Scholar]
- 75.Kaelin WGJ. and Ratcliffe PJ. Oxygen sensing by metazoans: the central role of HIF hydroxylase pathway. Mol Cell 30: 393–402, 2008 [DOI] [PubMed] [Google Scholar]
- 76.Kamata T, Ambo H, Puzon-McLaughlin W, Tieu KK, Handa M, Ikeda Y, and Takada Y. Critical cysteine residues for regulation of integrin αIIbβ3 are clustered in the epidermal growth factor domains of the β3 subunit. Biochem J 378: 1079–1082, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kamata T, Handa M, Ito S, Sato Y, Ohtani T, Kawai Y, and Ikeda Y. Structural requirements for activation in αIIbβ3 integrin. J Biol Chem 285: 38428–38437, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kamata T, Handa M, Sato Y, Ikeda Y, and Aiso S. Membrane-proximal α/β stalk interactions differentially regulate integrin activation. J Biol Chem 280: 24775–24783, 2004 [DOI] [PubMed] [Google Scholar]
- 79.Kanchanawong P, Shtengel G, Pasapera AM, Rambko EB, Davidson MW, Hess HF, and Waterman CM. Nanoscale architecture of integrin-based cell adhesions. Nature 468: 580–584, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kar S, Subbaram S, Carrico PM, and Melendez JA. Redox-control of matrix metaloproteinase-1: a critical link between free radicals, matrix remodeling and degenerative disease. Respir Physiol Neurobiol 174: 299–306, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Khoshnoodi J, Pedchenko V, and Hudson BG. Mammalian collagen IV. Microsc Res Tech 71: 357–370, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Klingberg F, Hinz B, and White ES. The myofibroblast matrix: implications for tissue repair and fibrosis. J Pathol 229: 298–309, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Knight CG, Morton LF, Peachey AR, Tuckwell DS, Farndale RW, and Barnes MJ. The collagen-binding A-domains of integrins α1β1 and α2β1 recognize the same specific amino acid sequence, GFOGER, in native (triple-helical) collagens. J Biol Chem 275: 35–40, 2000 [DOI] [PubMed] [Google Scholar]
- 84.Koch S, Volkmar CM, Kolb-Bachofen V, Korth HG, Kirsch M, Horn AH, Sticht H, Pallua N, and Suschek CV. A new redox-dependent mechanism of MMP-1 activity control comprising reduced low-molecular-weight thiols and oxidizing radicals. J Mol Med 87: 261–272, 2009 [DOI] [PubMed] [Google Scholar]
- 85.Krokhin OV, Cheng K, Sousa SL, Ens W, Standing KG, and Wilkins JA. Mass spectrometric based mapping of disulfide bonding patterns of integrin α chains. Biochemistry 42: 12950–12959, 2003 [DOI] [PubMed] [Google Scholar]
- 86.Kühn K. Basement membrane (type IV) collagen. Matrix Biol 14: 439–445, 1995 [DOI] [PubMed] [Google Scholar]
- 87.Lahav J, Gofer-Dadosh N, Luboshitz J, Hess O, and Shaklai M. Protein disulfide isomerase mediates integrin-dependent adhesion. FEBS Lett 475: 89–92, 2000 [DOI] [PubMed] [Google Scholar]
- 88.Lahav J, Jurk K, Hess O, Barnes MJ, Farndale RW, Luboshitz J, and Kehrel BE. Sustained integrin ligation involves extracellular free sulfhydryls and enzymatically catalyzed disulfide exchange. Blood 100: 2472–2478, 2002 [DOI] [PubMed] [Google Scholar]
- 89.Lampe AK. and Bushby KMD. Collagen VI related muscle disorders. J Med Genet 42: 673–685, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Laragione T, Bonetto V, Casoni F, Massignan T, Bianchi G, Gianazza E, and Ghezzi P. Redox regulation of surface protein thiols: identification of integrin α-4 as a molecular target by using redox proteomics. Proc Natl Acad Sci USA 100: 14737–14741, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Leahy DJ, Aukhil I, and Erickson HP. 2.0 A crystal structure of a four-domain segment of human fibronectin encompassing the RGD loop and synergy region. Cell 84: 155–164, 1996 [DOI] [PubMed] [Google Scholar]
- 92.Ley K, Laudanna C, Cybulsky MI, and Noushargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol 7: 678–689, 2007 [DOI] [PubMed] [Google Scholar]
- 93.Lijnen PJ, van Pelt JF, and Fagard RH. Stimulation of reactive oxygen species and collagen synthesis by angiotensin II in cardiac fibroblasts. Cardiovasc Ther 30: e1–e8, 2010 [DOI] [PubMed] [Google Scholar]
- 94.Lin L-J, Grimme JM, Sun J, Lu S, Gai L, Cropek DM, and Wang Y. The antagonistic roles of PDGF and integrin αvβ3 in regulating ROS production at focal adhesions. Biomaterials 34: 3807–3815, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Luo B-H, Springer TA, and Takagi J. A specific interface between integrin transmembrane helices and affinity for ligand. PLoS Biol 2: 0776–0786, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mäki JM. Lysyl oxidase: an oxidative enzyme and effector of cell function. Cell Mol Life Sci 63: 2304–2316, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Manickam N, Ahmad SS, and Essex DW. Vicinal thiols are required for activation of the αIIbβ3 platelet integrin. J Thromb Haemost 9: 1207–1215, 2011 [DOI] [PubMed] [Google Scholar]
- 98.Mas-Moruno C, Rechenmacher F, and Kessler H. Cilengitide: the first anti-angiogenic small molecule drug candidate, design, synthesis and clinical evaluation. Anticancer Agents Med Chem 10: 753–768, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Massagué J. TGFβ signalling in context. Nat Rev Mol Cell Biol 13: 616–630, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Massagué J, Seoane J, and Wotton D. Smad transcription factors. Genes Dev 19: 2783–2810, 2005 [DOI] [PubMed] [Google Scholar]
- 101.Matheny HE, Deem TL, and Cook-Mills JM. Lymphocyte migration through monolayers of endothelial cell lines involves VCAM-1 signaling via endothelial cell NADPH oxidase. J Immunol 164: 6550–6559, 2000 [DOI] [PubMed] [Google Scholar]
- 102.Meng D, Dan-Dan L, and Fang J. Insulin-like growth factor-1 induces reactive oxygen species production and cell migration through Nox4 and Rac1 in vascular smooth muscle cells. Cardiovasc Res 80: 299–308, 2008 [DOI] [PubMed] [Google Scholar]
- 103.Metcalf DG, Moore DT, Wu Y, Kielec JM, Molnar K, Valentine KG, Wand AJ, Bennett JS, and DeGrado WF. NMR analysis of the αIIbβ3 cytoplasmic interaction suggests a mechanism for integrin activation. Proc Natl Acad Sci USA 107: 22481–22486, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Metcalfe C, Cresswell P, Ciaccia L, Thomas B, and Barclay AN. Labile disulfide bonds are common at the leukocyte cell surface. Open Biol 1: 110010, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Milet-Marsal S, Breillat C, Peyruchaud O, Nurden P, Combrié R, Nurden A, and Bourre F. Two different β3 cysteine substitutions alter αIIbβ3 maturation and result in Glanzmann thrombasthenia. Thromb Haemost 88: 104–110, 2002 [PubMed] [Google Scholar]
- 106.Miner JH. The glomerular basement membrane. Exp Cell Res 318: 973–978, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mishina NM, Tyurin-Kuzmin PA, Markvicheva KN, Vorotnikov AV, Tkachuk V, Laketa V, Schultz C, Lukyanov S, and Belousov VV. Does cellular hydrogen peroxide diffuse or act locally? Antioxid Redox Signal 14: 1–7, 2011 [DOI] [PubMed] [Google Scholar]
- 108.Mor-Cohen R, Rosenberg N, Einav Y, Zelzion E, Landau M, Averbukh Y, and Seligsohn U. Unique disulfide bonds in epidermal growth factor (EGF) domains of β3 affect structure and function of αIIbβ3 and αvβ3 integrins in different manner. J Biol Chem 287: 8879–8891, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mor-Cohen R, Rosenberg N, Landau M, Lahav J, and Seligsohn U. Specific cysteines in β3 are involved in disulfide bond exchange-dependent and—independent activation of αIIbβ3. J Biol Chem 283: 19235–19244, 2008 [DOI] [PubMed] [Google Scholar]
- 110.Moreth K, Iozzo RV, and Schaefer L. Small leucine-rich proteoglycans orchestrate crosstalk during inflammation. Cell Cycle 11: 2084–2091, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mott J. and Werb Z. Regulation of matrix biology by matrix metalloproteinases. Curr Opin Cell Biol 16: 558–564, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mou Y, Ni H, and Wilkins JA. The selective inhibition of β1 and β7 integrin-mediated lymphocyte adhesion by bacitracin. J Immunol 161: 6323–6329, 1998 [PubMed] [Google Scholar]
- 112a.Murphy DD, Reddy EC, Moran N and O'Neill S. Regulation of platelet activity in a changing redox environment. Antioxid Redox Signal 20: 2074–2089, 2014 [DOI] [PubMed] [Google Scholar]
- 113.Myllyharju J. Prolyl 4-hydroxylases, key enzymes in the synthesis of collagens and regulation of the response to hypoxia, and their role as treatment targets. Ann Med 40: 402–417, 2008 [DOI] [PubMed] [Google Scholar]
- 114.Myllyharju J. and Koivunen P. Hypoxia-inducible factor prolyl 4-hydroxylases: common and specific roles. Biol Chem 394: 435–448, 2013 [DOI] [PubMed] [Google Scholar]
- 115.Myllyharju J. and Schipani E. Extracellular matrix genes as hypoxia-inducible targets. Cell Tissue Res 339: 19–29, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nagae M, Re S, Mihara E, Nogi T, Sugita Y, and Takagi J. Crystal structure of α5β1 integrin ectodomain: atomic details of the fibronectin receptor. J Cell Biol 197: 131–140, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nagata M. Inflammatory cells and oxygen radicals. Curr Drug Targets Inflamm Allergy 4: 503–504, 2005 [DOI] [PubMed] [Google Scholar]
- 118.Nakamuta M, Higashi N, Kohijama M, Fukushima M, Ohta S, Kotoh K, Kobayashi N, and Enjoji M. Epigallocatechin-3-gallate, a polyphenol component of green tea, suppresses both collagen production and collagenase activity in hepatic stellate cells. Int J Mol Med 16: 677–681, 2005 [PubMed] [Google Scholar]
- 119.Navdaev A, Heitmann V, deSantana Evangelista K, Mörgelin M, Wegener J, and Eble JA. The C-terminus of the γ2 chain but not of the β3 chain of laminin-332 is indirectly but indispensably necessary for integrin-mediated cell reactions. Exp Cell Res 314: 489–497, 2008 [DOI] [PubMed] [Google Scholar]
- 120.Nelson KK, Ranganathan AC, Mansouri J, Rodriguez AM, Providence KM, Rutter JL, Pumiglia K, Bennett JA, and Melendez JA. Elevated SOD2 activity augments matrix metalloproteinase expression: evidence for the involvement of endogenous hydrogen peroxide in regulating metastasis. Clin Cancer Res 9: 424–432, 2003 [PubMed] [Google Scholar]
- 121.Nelson KK, Subbaram S, Connor KM, Dasgupta J, Ha XF, Meng TC, Tonks NK, and Melendez JA. Redox-dependent matrix-metalloproteinase-1 expression is regulated by JNK through Ets and AP-1 promoter motifs. J Biol Chem 281: 14100–14110, 2006 [DOI] [PubMed] [Google Scholar]
- 122.Norman JT, Clark IM, and Garcia PL. Regulation of TIMP-1 expression by hypoxia in kidney fibroblasts. Ann N Y Acad Sci 878: 503–505, 1999 [DOI] [PubMed] [Google Scholar]
- 123.Orbe J, Rodriguez JA, Arias R, Belzunce M, Nespereira B, Perez-Ilzarbe M, Roncal C, and Paramo JA. Antioxidant vitamins increase the collagen content and reduce MMP-1 in a porcine model of atherosclerosis: implication for plaque stabilization. Atherosclerosis 167: 45–53, 2003 [DOI] [PubMed] [Google Scholar]
- 124.Otranto M, Sarrazy V, Bonté F, Hinz B, Gabbiani G, and Desmoulière A. The role of the myofibroblast in tumor stroma remodelling. Cell Adh Migr 6: 203–2012, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Petry A, Weitnauer M, and Görlach A. Receptor activation of NADPH oxidases. Antioxid Redox Signal 13: 467–487, 2010 [DOI] [PubMed] [Google Scholar]
- 126.Poole LB. and Nelson KJ. Discovering mechanisms of signaling-mediated cysteine oxidation. Curr Opin Chem Biol 12: 18–24, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Prince JM, Klinowska TCM, Marshman E, Lowe ET, Mayer U, Miner J, Aberdam D, Vestweber D, Gusterson B, and Streuli CH. Cell-matrix interactions during development and apoptosis of the mouse mammary gland in vivo. Dev Dyn 223: 497–516, 2002 [DOI] [PubMed] [Google Scholar]
- 128.Pytela R, Pierschbacher MD, Argraves S, Suzuki S, and Ruoslahti E. Arginine-glycine-aspartic acid adhesion receptors. Methods Enzymol 144: 475–489, 1987 [DOI] [PubMed] [Google Scholar]
- 129.Qu A. and Leahy DJ. Crystal structure of the I-domain from the CD11a/CD18 (LFA-1, αLβ2) integrin. Proc Natl Acad Sci USA 92: 10277–10281, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Rajagopalan S, Meng P, Ramasamy S, Harrison DG, and Galis ZS. Reactive oxygen species produced by macrophage-derived foam cells regulate the activity of vascular matrix metalloproteinases in vitro. Implications for atherosclerotic plaque stability. J Clin Invest 98: 2572–2579, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Randi AM. and Hogg N. I domain of β2 integrin lymphocyte function-associated antigen-1 contains a binding site for ligand intercellular adhesion molecule-1. J Biol Chem 269: 12395–12398, 1994 [PubMed] [Google Scholar]
- 132.Ricard-Blum S. The collagen family. Cold Spring Harb Perspect Biol 3: 1–19, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Robinson A, O'Neill S, Kiernan A, O'Donoghue N, and Moran N. Bacitracin reveals a role for multiple thiol isomerases in platelet function. Br J Haematol 132: 339–349, 2005 [DOI] [PubMed] [Google Scholar]
- 134.Romagnoli C, Marcucci T, Picariello L, Tonelli F, Vincenzini MT, and Iantomasi T. Role of N-acetylcysteine and GSH redox system on total and active MMP-2 in intestinal myofibroblasts of Crohn's disease patients. Int J Colorectal Dis 28, 915–924, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Rowe RG. and Weiss SJ. Breaching the basement membrane: who, when and how? Trends Cell Biol 18: 560–574, 2008 [DOI] [PubMed] [Google Scholar]
- 136.Rucker RB. and Murray J. Cross-linking amino acids in collagen and elastin. Am J Clin Nutr 31: 1221–1236, 1978 [DOI] [PubMed] [Google Scholar]
- 137.Ruoslahti E. RGD and other recognition sequences for integrins. Annu Rev Cell Dev Biol 12: 697–715, 1996 [DOI] [PubMed] [Google Scholar]
- 138.Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY, Puigserver P, and Brugge JS. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 461: 109–113, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138a.Schröder K. NADPH oxidases in redox regulation of cell adhesion and migration. Antioxid Redox Signal 20: 2043–2058, 2014 [DOI] [PubMed] [Google Scholar]
- 139.Schröder E. and Eaton P. Hydrogen peroxide as an endogenous mediator and exogenous tool in cardiovascular research: issues and considerations. Curr Opin Pharmacol 8: 153–159, 2008 [DOI] [PubMed] [Google Scholar]
- 140.Serini G, Bochaton-Piallat , Ropraz P, Geinoz A, Borsi L, Zardi L, and Gabbiani G. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-β1. J Cell Biol 142: 873–881, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Shattil SJ, Kim C, and Ginsberg MH. The final steps in integrin activation: the end game. Nat Rev Mol Cell Biol 11: 288–300, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sheik FG, Pahan K, Khan M, Barbosa E, and Singh I. Abnormality in catalse import into peroxisomes leads to severe neurological disorder. Proc Natl Acad Sci USA 95: 2961–2966, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sherratt MJ. Tissue elasticity and the ageing elastic fibre. Age 31: 305–325, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Shi M-J, Sundramurthy K, Liu B, Tan S-M, Law SKA, and Lescar J. The crystal structure of the plexin-semaphorin-integrin domain/hybrid domain/I-EGF1 segment from the human integrin β2 subunit at 1.8-A resolution. J Biol Chem 280: 30586–30593, 2005 [DOI] [PubMed] [Google Scholar]
- 145.Shimaoka M. and Springer TA. Therapeutic antagonists and conformational regulation of integrin function. Nat Rev Drug Discov 2: 703–717, 2003 [DOI] [PubMed] [Google Scholar]
- 146.Shimaoka M, Takagi J, and Springer TA. Conformational regulation of integrin structure and function. Annu Rev Biophys Biol Struct 31: 485–516, 2002 [DOI] [PubMed] [Google Scholar]
- 147.Shimaoka M, Xiao T, Liu J-H, Yang Y, Dong Y, Jun C-D, McCormack A, Zhang R, Joachimiak A, and Takagi J. Structures of the αL I domain and its complex with ICAM-1 reveal a shape-shifting pathway for integrin regulation. Cell 112: 99–111, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Singh P, Carraher C, and Schwarzbauer JE. Assembly of fibronectin extracellular matrix. Annu Rev Cell Dev Biol 26: 397–419, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sudgen MC. and Holness MJ. Mechanisms underlying regulation of the expression and activities of the mammalian pyruvate dehydrogenase kinases. Arch Physiol Biochem 112: 139–149, 2006 [DOI] [PubMed] [Google Scholar]
- 150.Sun Q-H, Liu C-Y, Wang R, Paddock C, and Newman PJ. Disruption of the long-range GPIIIa Cys5-Cys435 disulfide bond results in the production of constitutively active GPIIb-GPIIIa (αIIbβ3) integrin complexes. Blood 100: 2094–2101, 2002 [DOI] [PubMed] [Google Scholar]
- 151.Swiatkowska M, Szymanski J, Padula G, and Cierniewski C. Interaction and functional association of protein disulfide isomerase with αvβ3 integrin on endothelial cells. FEBS J 275: 1813–1823, 2008 [DOI] [PubMed] [Google Scholar]
- 152.Taddei ML, Parri M, Mello T, Catalano A, Levine AD, Raugei G, Ramponi G, and Chiarugi P. Integrin-mediated cell adhesion and spreading engage different sources of reactive oxygen species. Antioxid Redox Signal 9: 469–481, 2007 [DOI] [PubMed] [Google Scholar]
- 153.Takagi J. and Springer TA. Integrin activation and structural rearrangement. Immunol Rev 186: 141–163, 2002 [DOI] [PubMed] [Google Scholar]
- 154.Takagi J, Beglova N, Yalamanchili P, Blacklow SC, and Springer TA. Definition of the EGF-like, closely interacting modules that bear activation epitopes in integrin β subunits. Proc Natl Acad Sci USA 98: 11175–11180, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Takahashi Y, Takahashi S, Shiga Y, Yoshimi T, and Miura T. Hypoxic induction of prolyl 4-hydroxylase α(I) in cultured cells. J Biol Chem 275: 14139–14146, 2000 [DOI] [PubMed] [Google Scholar]
- 156.Timpl R, Sasaki T, Kostka G, and Chu M-L. Fibulins: a versatile family of extracellular matrix proteins. Nat Rev Mol Cell Biol 4: 479–489, 2003 [DOI] [PubMed] [Google Scholar]
- 157.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, and Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol 3: 349–363, 2002 [DOI] [PubMed] [Google Scholar]
- 158.Tomasek JJ, Vaughan MB, Kropp BP, Gabbiani G, Martin MD, Haaksma CJ, and Hinz B. Contraction of myofibroblasts in granulation tissue is dependent on Rho/Rho kinase/myosin light chain phosphatase activity. Wound Repair Regen 14: 313–320, 2006 [DOI] [PubMed] [Google Scholar]
- 159.Vanacore R, Ham A-JL, Voehler M, Sanders CR, Conrads TP, Veenstra TD, Sharpless KB, Dawson PE, and Hudson BG. A sulfilimine bond identified in collagen IV. Science 325: 1230–1234, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Van Obberghen-Schilling E, Tucker RT, Saupe F, Gasser I, Cseh B, and Orend G. Fibronectin and tenascin-C: accomplices in vascular morphogenesis during development and tumor growth. Int J Dev Biol 55: 511–525, 2011 [DOI] [PubMed] [Google Scholar]
- 161.Verhaar R, Dekkers DWC, DeCuyper IM, Ginsberg MH, de Korte D, and Verhoeven AJ. UV-C irradiation disrupts platelet surface disulfide bonds and activates the platelet integrin αIIbβ3. Blood 112: 4935–4939, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Vestweber D. Novel insights into leukocyte extravasation. Curr Opin Hematol 19: 212–217, 2012 [DOI] [PubMed] [Google Scholar]
- 163.von der Mark H, Aumailley M, Wick G, Fleischmajer R, and Timpl R. Immunochemistry, genuine size and long-spacing fibrils: relation to type VI collagen. Eur J Biochem 142: 493–502, 1984 [DOI] [PubMed] [Google Scholar]
- 164.Wanders RJ. Peroxisomes, lipid metabolism, and peroxisomal disorders. Mol Genet Metab 83: 16–27, 2004 [DOI] [PubMed] [Google Scholar]
- 165.Wang JH, Fu G, and Luo BH. Dissociation of the α-subunit calf-2 domain and the β-subunit I-EGF4 domain in integrin activation and signaling. Biochem 49: 10158–10165, 2010 [DOI] [PubMed] [Google Scholar]
- 166.Wang W. and Luo B-H. Structural basis of integrin transmembrane activation. J Cell Biochem 109: 447–452, 2010 [DOI] [PubMed] [Google Scholar]
- 167.Wenk J, Brenneisen P, Wlaschek M, Poswig A, Briviba K, Oberley TD, and Scharffetter-Kochanek K. Stable overexpression of manganese superoxide dismutase in mitochondria identifies hydrogen peroxide as a major oxidant in the AP-1-mediated induction of matrix-degrading metalloprotease-1. J Biol Chem 274: 25869–25876, 1999 [DOI] [PubMed] [Google Scholar]
- 168.White ES. and Muro AF. Fibronectin splice variants: understanding their multiple roles in health and disease using engineered mouse models. IUBMB Life 63: 538–546, 2011 [DOI] [PubMed] [Google Scholar]
- 169.Wipff P-J, Rifkin DB, Meister J-J, and Hinz B. Myofibroblast contraction activates latent TGF-β1 from the extracellular matrix. J Cell Biol 179: 1311–1323, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Wiradjaja F, DiTommaso T, and Smyth I. Basement membranes in development and disease. Birth Defects Res C Embryo Today 90: 8–31, 2010 [DOI] [PubMed] [Google Scholar]
- 171.Worth DC. and Parsons M. Advances in imaging cell-matrix adhesions. J Cell Sci 123: 3629–3638, 2010 [DOI] [PubMed] [Google Scholar]
- 172.Wouters MA, Rigoutsos I, Chu CK, Feng LL, Sparrow DB, and Dunwoodie SL. Evolution of distinct EGF domains with specific functions. Protein Sci 14: 1091–1103, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Xiao T, Takagi J, Coller BS, Wang JH, and Springer TA. Structural basis for allostery in integrins and binding to fibrinogen-mimetic therapeutics. Nature 432: 59–67, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Xie C, Zhu J, Chen X, Mi L, Nishida N, and Springer TA. Structure of an integrin with an αI domain, complement receptor type 4. EMBO J 29: 666–679, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Xiong J-P, Mahalingam B, Alonso JL, Borelli LA, Rui X, Anand S, Hyman BT, Rysiok T, Müller-Pompalla D, Goodman SL, and Arnaout MA. Crystal structure of the complete integrin αVβ3 ectodomain plus an α/β transmembrane fragment. J Cell Biol 186: 589–600, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Xiong J-P, Stehle T, Diefenbach B, Zhang R, Dunker R, Scott DL, Joachimiak A, Goodman SL, and Arnaout MA. Crystal structure of the extracellular segment of integrin αVβ3. Science 294: 339–345, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Xiong J-P, Stehle T, Zhang R, Joachimiak A, Frech M, Goodman S, and Arnaout MA. Crystal structure of the extracellular segment of integrin αVβ3 in complex with Arg-Gly-Asp ligand. Science 296: 151–155, 2002 [DOI] [PubMed] [Google Scholar]
- 178.Yan B. and Smith JW. Mechanism of integrin activation by disulfide bond reduction. Biochem 40: 8861–8867, 2001 [DOI] [PubMed] [Google Scholar]
- 179.Yan B, Hu DD, Knowles SK, and Smith JW. Probing chemical and conformational differences in the resting and active conformers of platelet αIIbβ3. J Biol Chem 275: 7249–7290, 1999 [DOI] [PubMed] [Google Scholar]
- 180.Yang W, Shimaoka M, Salas A, Takagi J, and Springer TA. Intersubunit signal transmission in integrins by a receptor-like interaction with a pull spring. Proc Natl Acad Sci USA 101: 2906–2911, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Yurchenco P. and Patton BL. Developmental and pathogenic mechanisms of basement membrane assembly. Curr Pharm Des 15: 1277–1294, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Zang Q. and Springer TA. Amino acid residues in the PSI domain and cysteine-rich repeats of the integrin β2 subunit that restrain activation of the integrin axβ2. J Biol Chem 276: 6922–6929, 2001 [DOI] [PubMed] [Google Scholar]
- 183.Zaw KK, Yokoyama Y, Abe M, and Ishikawa O. Catalase restores the altered mRNA expression of collagen and matrix metalloproteinases by dermal fibroblasts exposed to reactive oxygen species. Eur J Dermatol 16: 375–379, 2006 [PubMed] [Google Scholar]
- 184.Zhang W-M, Käpylä J, Puranen JS, Knight CG, Tiger C-F, Pentikäinen OT, Johnson MS, Farndale RW, Heino J, and Gullberg D. α11β1 integrin recognizes the GFOGER sequence in interstitial collagens. J Biol Chem 279: 7270–7277, 2003 [DOI] [PubMed] [Google Scholar]
- 185.Zhao T, Benard V, Bohl BP, and Bokoch GM. The molecular basis for adhesion-mediated suppression of reactive oxygen species generation by human neutrophils. J Clin Invest 112: 1732–1740, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Zhu J, Carman CV, Kim M, Shimaoka M, Springer TA, and Luo B-H. Requirement of α and β subunit transmembrane helix separation for integrin outside-in signaling Blood 110: 2475–2483, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]