

Abstract
SUMMARY
CD4+ T cells are key cells of the adaptive immune system that use T cell antigen receptors to recognize peptides that are generated in endosomes or phagosomes and displayed on the host cell surface bound to major histocompatibility complex molecules. These T cells participate in immune responses that protect hosts from microbes such as Mycobacterium tuberculosis, Cryptococcus neoformans, Leishmania major, and Salmonella enterica, which have evolved to live in the phagosomes of macrophages and dendritic cells. Here, we review studies indicating that CD4+ T cells control phagosomal infections asymptomatically in most individuals by secreting cytokines that activate the microbicidal activities of infected phagocytes but in a way that inhibits the pathogen but does not eliminate it. Indeed, we make the case that localized, controlled, persistent infection is necessary to maintain large numbers of CD4+ effector T cells in a state of activation needed to eradicate systemic and more pathogenic forms of the infection. Finally, we posit that current vaccines for phagosomal infections fail because they do not produce this “periodic reminder” form of CD4+ T cell-mediated immune control.
INTRODUCTION
The number of microbes that can cause disease in mammals is extremely large (1). Different pathogens infect and reside in different niches in the body and have various mechanisms of pathogenesis. Consequently, immune functions that are protective against one pathogen may be completely ineffective against another. To deal with this challenge, the vertebrate adaptive immune system has evolved three types of lymphocytes that survey different types of pathogen-derived antigens: B cells, which use antibodies to recognize extracellular macromolecules; CD8+ T cells, which use T cell receptors (TCRs) to recognize short peptides from cytosolic proteins; and CD4+ T cells, which use TCRs to recognize peptides from extracellular proteins that are taken up into endosomes or phagosomes. This review focuses on CD4+ T cells and their important role in protecting the host from pathogens that persistently infect the phagosomes of phagocytes.
ANTIGEN RECOGNITION BY LYMPHOCYTES
The adaptive immune system depends on lymphocytes, which exist in three main classes: B cells, CD8+ T cells, and CD4+ T cells (2). Each lymphocyte produces an antigen receptor from a composite gene assembled by somatic recombination of many possible gene segments on only one chromosome (3). This process, which occurs only in developing lymphocytes, ensures that millions of different antigen receptors can be produced but that each newly formed lymphocyte expresses many copies of a single antigen receptor. Each antigen receptor has a unique binding pocket that accommodates an antigen with a defined shape (4, 5). Since the gene segment shuffling mechanism that produces antigen receptors is relatively random (6), the antigen receptor binding pockets have random shapes. Thus, the set of lymphocytes in an individual is likely to contain only a few cells that by chance express antigen receptors with shapes that are highly complementary to the molecular shapes of antigens produced by a given microbe. The adaptive immune response to that microbe therefore depends on the expansion and differentiation of these few lymphocytes (7).
T Cell Antigen Receptor Recognition of Peptide-MHC Ligands
Immune protection from intracellular pathogens is the job of T cells (2). To understand how T cells do this job, it is important to understand the specificity of their TCRs. TCRs are composed of either alpha and beta or gamma and delta chain heterodimers that form antibody Fab-like structures on the surface of T cells (4, 8). This review focuses on alpha-beta TCRs, because the CD4+ T cells that control phagosomal infections for the most part express these receptors (9).
The ligands for alpha-beta TCRs are usually short peptides bound to host cell surface proteins called major histocompatibility complex (MHC) molecules (4). MHC molecules come in two forms: MHC class I (MHCI) molecules, which are expressed on most cells of the host and present peptides that are recognized by TCRs on CD8+ T cells, and MHCII molecules, which are constitutively expressed on B cells, dendritic cells (DCs), and macrophages and present peptides that are recognized by TCRs on CD4+ T cells (4). MHC molecules alert the host to an infection by displaying peptides from intracellular microbes on the surfaces of host cells.
In almost all cells of the body, the peptides that bind to MHCI molecules are derived exclusively from cytosolic proteins (10). Newly translated proteins are constantly being clipped by proteasomes in the cytosols of all host cells. Some of the resulting peptides are pumped by the transporter associated with antigen processing (TAP) into the endoplasmic reticulum, where 8- to 9-amino-acid peptides with certain key anchor residues bind to newly synthesized MHCI molecules (11). The resulting peptide:MHCI (p:MHCI) complexes are transported to the cell surface. Certain dendritic cells can shuttle internalized extracellular proteins from endosomes to the cytosol, where proteasomes produce peptides that enter the endoplasmic reticulum through TAP and are loaded onto MHCI molecules (12). This exceptional capacity is known as cross-presentation.
The p:MHCII ligands for TCRs on CD4+ T cells are formed from extracellular proteins that are taken up by cells that express MHCII molecules (10). Dendritic cells and macrophages take up extracellular proteins into endosomes by macropinocytosis or particles by phagocytosis. B cells are very inefficient at this process and can efficiently internalize only proteins that bind to the their surface antibody. Endosomes or phagosomes containing internalized proteins then fuse with vesicles containing proteases such as cathepsins, which cleave the internalized proteins into peptides. These vesicles also contain newly synthesized MHCII molecules that bind ∼9-amino-acid peptides that have certain key anchor residues. The p:MHCII complexes are then trafficked to the cell surface.
MHC molecules are loaded with peptides from host proteins in uninfected individuals (13). These host p:MHC complexes play a critical role in T cell development in the thymus (14). Developing T cells rearrange Tcra-V, Tcra-J, Tcrb-V, Tcrb-D, and Tcrb-J segments such that each cell expresses a unique alpha-beta TCR (15). Unlike antibodies, TCR V domains have an inherent germ line-encoded affinity for MHC molecules, which causes strong signaling and clonal deletion of many developing T cells in the thymus (16). The only cells that are not deleted are those that produce TCRs with weakened binding to self-p:MHC molecules. Weak TCR binding to self-p:MHC molecules is needed for survival and lineage commitment: thymic T cells with TCRs with low affinity for self-p:MHCI molecules become CD8+ T cells, whereas cells with TCRs with low affinity for self-p:MHCII molecules become CD4+ T cells (14). The result of this process is a diverse set of T cells, all with TCRs with weak affinity for self-p:MHC molecules, a few of which are likely to have high affinity for a host MHC molecule when complexed with a given foreign peptide.
The utility of the CD8+ T cell-MHCI system becomes apparent when considering intracellular infections such as those caused by viruses. Viral proteins are processed in the cytosol, and viral p:MHCI complexes are displayed on the cell surface of any infected cell, marking it for recognition and killing by CD8+ T cells. There is almost nowhere in the body for viruses to hide, since most cells of the body express MHCI molecules (17, 18).
Not all intracellular microbes, however, infect the cytosol; some infect the phagosomes of phagocytes, for example, Mycobacterium tuberculosis, Cryptococcus neoformans, and Salmonella species (19). These microbes are not well controlled by CD8+ T cells (9, 20, 21), probably because these microbes are not abundant in the cytosols of infected cells and therefore do not lead to efficient production of microbial p:MHCI complexes. Proteins from these microbes, however, are processed in the phagosome, loaded onto MHCII molecules, and shuttled to the cell surface, marking the infected cells for recognition by CD4+ T cells (22–26). The importance of this fundamental aspect of antigen presentation is evidenced by that fact that CD4+ T cell-deficient individuals have a preferential susceptibility to phagosomal infections (27, 28).
CD4+ T CELL RESPONSE
General Aspects of the CD4+ T Cell Response
We first review some general information about how CD4+ T cells respond to p:MHCII ligands before delving into the mechanisms used by these cells to control phagosomal infections. After leaving the thymus, a newly minted CD4+ T cell, now called a naive T cell, enters a secondary lymphoid organ (lymph nodes, spleen, and mucosal lymphoid organs) from the blood and percolates through a meshwork of MHCII-expressing dendritic cells (29). This search process optimizes the likelihood that a naive T cell will encounter the p:MHCII ligand that its TCR has a high affinity for no matter where in the body that ligand happens to be produced. The recirculation of naive T cells is facilitated by the expression of CD62L and CC chemokine receptor 7 (CCR7), which bind to ligands expressed exclusively on endothelial cells in secondary lymphoid organs (29). If a naive T cell does not encounter its high-affinity p:MHCII ligand, it leaves that secondary lymphoid organ and migrates to a different one to continue the search (30). The cell remains in the G0 phase of the cell cycle and expresses small amounts of CD44 and large amounts of CD45RA during the search process, which goes on for 2 to 3 months in mice before the cell dies (31).
The naive T cell undergoes a dramatic transformation if it encounters a dendritic cell displaying the relevant high-affinity p:MHCII ligand. This occurs during infection, as dendritic cells at the infection site take up microbial proteins and migrate to the draining lymph nodes, and free microbial proteins are carried by lymph or blood to secondary lymphoid organs for uptake by resident dendritic cells (32). In either case, dendritic cells in secondary lymphoid organs produce and display microbial p:MHCII complexes. On average, about 1 naive CD4+ T cell in a million, about 50 cells in a mouse, expresses a TCR capable of strong binding to any given microbial p:MHCII complex (33). During the relevant infection, these 50 cells interact with dendritic cells displaying the relevant microbial p:MHCII complex, receive TCR signals, produce growth factors, divide many times, and produce several hundred thousand progeny in 1 week (34, 35). These progeny are termed effector cells, which lose the naive phenotype by increasing the expression of levels CD44 and CD45RO and reducing the expression level of CD45RA (36).
Early in vitro studies showed that effector T cells differentiate into specific subsets during this period of rapid division. The best-understood effector cell subsets are known as T helper 1 (Th1), T helper 2 (Th2), T helper 17 (Th17), and T follicular helper (Tfh) cells (37). These effector cell subsets form in response to infection-specific signals from the innate immune system and perform specific functions. For example, worm infections cause epithelial cells to produce interleukin-33 (IL-33) and cause group 2 innate lymphoid cells to produce IL-4 (38). Effector T cells that are exposed to these cytokines express the GATA3 transcription factor, which enforces Th2 cell differentiation (37). When stimulated by the relevant p:MHCII ligand, Th2 cells produce IL-4, IL-5, and IL-13, which activate eosinophils and goblet cells to expel worms (39). Extracellular bacterial infections cause dendritic cells to produce IL-6 and transforming growth factor β, which cause effector cells to induce the retinoic acid-related orphan receptor γt transcription factor, which promotes Th17 cell differentiation (40). Th17 cells produce IL-17, which stimulates epithelial cells to produce chemokines that recruit neutrophils to engulf and kill bacteria. Extracellular bacterial infections also cause the production of IL-6 and IL-21, which induce the expression of the Bcl6 transcription factor, which drives Tfh differentiation in combination with inducible T cell costimulator (ICOS) signals from ICOS ligand-expressing B cells (41). Tfh cells localize to B cell-rich follicles and germinal centers and help B cells undergo antibody affinity maturation and isotype switching. Antibodies then facilitate killing of extracellular microbes via complement activation, antibody-dependent cellular cytotoxicity, or opsonization (42). Certain intracellular infections cause macrophages and dendritic cells to produce the cytokine IL-12 or type 1 interferon (IFN), which causes effector cells to express the T-bet transcription factor, which promotes Th1 cell differentiation (43). Th1 cells produce gamma interferon (IFN-γ) and tumor necrosis factor (TNF), which activate phagocytes to kill intracellular microbes (44).
CD4+ T Cell Response to Acute Infection
Generation of effector cells.
Although much of the evidence for CD4+ effector T cell differentiation comes from in vitro culture systems, recent studies of acute infections by attenuated Listeria monocytogenes bacteria or lymphocytic choriomeningitis virus (LCMV) have shed light on this process as it occurs for p:MHCII-specific T cells in vivo. Naive CD4+ T cells with TCRs specific for MHCII-bound peptides from either infection proliferate to produce effector cell populations consisting of about equal numbers of Th1 and Tfh cells in the secondary lymphoid organs by 1 week after infection (35, 45, 46). IL-12 likely drives the differentiation of Th1 cells during infection with attenuated Listeria monocytogenes bacteria (47), while type 1 IFN serves this function during LCMV infection (48). In contrast, Tfh cell formation during LCMV infection and probably Listeria monocytogenes infection depends on IL-6 and IL-21 (49).
These studies provide the important insight that two different polarized Th cell subsets, Th1 cells and Tfh cells, can differentiate simultaneously in the same infection. It is possible that individual naive T cells become activated in different niches in secondary lymphoid organs that contain either IL-12 or IL-6 and IL-21. Alternatively, some effector cells may receive ICOS signals from B cells and become Tfh cells, while others do not (45, 50). Another possibility relates to the strength of the signal that a CD4+ T cell receives through its TCR. In vitro stimulation with very low or very high doses of peptide favors the generation of IL-4-producing T cells, while moderate doses of peptide favor IFN-γ production (51, 52). In vivo studies showed that cell-mediated immunity (associated with Th1 cells) is favored at low antigen doses (53), while humoral immunity (associated with Tfh cells) is favored at high antigen doses (54). Naive T cells that experience moderate TCR stimulation during attenuated L. monocytogenes infection or after peptide injection have a tendency to become Th1 cells, while strong or prolonged TCR signaling favors Tfh cell generation (55–58). Consistent with this finding, immune control of Leishmania major infection, which depends on Th1 immunity, is induced more efficiently by immunization with a low dose of Leishmania protein than with a high dose (59). Strong TCR signaling inhibits the expression of the IL-12 receptor (60) and IL-2 receptor signaling (61), both of which are needed for maximal Th1 cell differentiation (35, 45, 62, 63). Thus, it is possible that strong TCR signaling suppresses the Th1 cell fate, thereby promoting the Tfh cell fate.
Studies on attenuated L. monocytogenes infection also revealed that some Th1 effector cells leave secondary lymphoid organs and migrate to nonlymphoid tissues such as the liver, while Tfh cells are confined to the secondary lymphoid organs (55). Although not examined in this case, in other systems, Th1 cells express the active form of P-selectin glycoprotein 1, which binds to CD62E and CD62P, and CXC chemokine receptor 3 (CXCR3), which binds to CXC chemokine ligand 9 (CXCL9) and CXCL10 (64). CD62E CD62P, CXCL9, and CXCL10 are displayed on endothelial cells in inflamed nonlymphoid tissues, thereby facilitating the extravasation of Th1 cells (65).
Formation of memory cells.
Some of the effector cells survive acute infection to become memory cells. The number of L. monocytogenes or LCMV p:MHCII-specific effector cells in the body declines by about 90% in secondary lymphoid organs via apoptosis over a 2-week period after the peak (34, 35, 46, 66). The loss of effector cells coincides with clearance of the infection, and it was recently found that deprivation from p:MHCI molecules may cause some CD8+ T cells to die during the contraction period (67). Thus, it is possible that the 10% of effector cells that survive to become memory cells are those that happen to interact with a cell displaying the relevant p:MHC molecules during a critical period of the contraction phase. Effector cell survival into the memory phase is associated with expression of the IL-7 receptor (68) or the TNF receptor superfamily member CD27 (34), which enhances expression of prosurvival members of the Bcl-2 family.
The CD4+ memory T cells that survive the contraction period after L. monocytogenes or LCMV infection retain the CD44high phenotype of effector cells and consist of Th1 and Tfh-like subsets (35, 46, 69–71). It is likely that Th1 memory cells derive from Th1 effector cells (72) and that Tfh memory cells derive from Tfh effector cells (35, 71). Th1 memory cells lack expression of CCR7 (35) and thus resemble so-called effector memory cells (73), while Tfh-like memory cells express CCR7 (35) and resemble central memory cells (73). Most of the memory cells in either subset are not cycling (34) but are maintained for prolonged periods in the G0 or G1 phase of the cell cycle by the cytokine IL-7 (74). However, some of the memory cells in the population divide infrequently in response to the cytokine IL-15 (34, 75). In contrast, if the relevant infection occurs a second time, memory cells receive TCR signals and rapidly enter the cell cycle, with Th1 memory cells producing Th1 effector cells and Tfh memory cells producing Th1 and Tfh effector cells (35, 69, 70).
Surprisingly, the combination of IL-7-dependent survival and IL-15-dependent proliferation is not sufficient to maintain CD4+ memory T cells induced by acute infections. Levels of CD4+ memory T cells induced by attenuated L. monocytogenes or LCMV infection slowly decline, with a half-life of about 60 days in mice (34, 35, 46, 66). This decline is in contrast to the seemingly perfect numerical stability of CD8+ memory T cells and plasma B cells (76, 77). Thus, at the population level, murine CD4+ memory T cells induced by acute infections are remarkably “forgetful” compared to other memory cells. This could be due to the fact that CD4+ T cells undergo less IL-15-driven homeostatic proliferation than CD8+ T cells due to a lower expression level of the IL-15 receptor (34, 75, 78). A second, non-mutually exclusive possibility is that weak TCR signals from self-p:MHCII ligands are important for the survival of CD4+ memory T cells, and competition for these ligands reduces survival when CD4+ T cell populations specific for the same ligand are large (79, 80). In any case, murine CD4+ T cells do not conform to the textbook notion that a single transient exposure to antigen is sufficient to induce lifelong immune memory. Although human CD4+ T memory cells appear to be long-lived without the eliciting antigen (81), it is difficult to rule out the involvement of periodic TCR stimulation with environmental peptides that cross-react with the eliciting peptide (82).
CD4+ T Cell Response to Phagosomal Infection
Generation of effector cells.
Although work on attenuated L. monocytogenes and LCMV infections elucidated many interesting aspects of the in vivo CD4+ T cell response, it did not reveal much about the role that these cells play in protective immunity because CD8+ T cells are mainly responsible for protecting the host from these microbes (83, 84). The primary role of CD8+ T cells in the control of these cytosolic infections is likely related to the efficient production of microbial p:MHCI complexes by infected cells. In contrast, CD4+ T cells are probably the key controllers of primary infections by microbes such as Mycobacterium tuberculosis, Cryptococcus neoformans, Leishmania major, and Salmonella enterica (27), because these organisms replicate in the phagosomes of macrophages and dendritic cells, where p:MHCII complexes are formed (85–88). Not only are CD4+ T cells the main controllers of these infections (9, 21, 89–91), they also appear to do so with little help from other cells of the adaptive immune system. For example, despite becoming activated (92), gamma-delta T cells appear to play no positive role in controlling phagosomal infections (9, 21), and CD8+ T cells play only a small or no role (9, 20, 21, 93). Mice lacking CD1 control M. tuberculosis infection normally, demonstrating that natural killer T (NKT) cells are not required (94). In addition, a role for B cells or antibodies has not been clearly established for the control of phagosomal infections. Several groups reported that mice lacking B cells had a defect in the control of primary M. tuberculosis infection (95, 96), whereas another group reported no defect (97). Several other studies found that mice lacking B cells had mild deficits in controlling phagosomal infections, but these defects were attributed to poor priming of T cells rather than antibody production (98–100). In summary, although CD8+ T cells and B cells may play minor roles in the control of phagosomal infections, their importance in this regard pales in comparison to that of CD4+ T cells.
We now review research on the CD4+ T cell response to the phagosomal pathogens M. tuberculosis, L. major, and S. enterica. These organisms are the focus of this review because the large body of research on each one is conducive to identifying common elements of the CD4+ T cell response to phagosomal pathogens. In the case of S. enterica, special attention is given to studies of Nramp1-sufficient hosts, which generate persistent infections, unlike Nramp1-deficient hosts, which are abnormally susceptible and die very early after infection (86). Emphasis is also placed on studies in which T cells specific for MHCII-bound peptides from phagosomal pathogens were tracked, because these studies provide insight into the T cells that specifically control these infections. Because of this focus, most of the studies reviewed here were performed in mouse model systems where relevant reagents, such as p:MHCII tetramers, are available for tracking relevant CD4+ T cells (101).
All three of the prototypical phagosomal infections have similar features. In each case, the microbes enter and replicate in macrophages and dendritic cells at the initial site of infection and then spread to associated secondary lymphoid organs and eventually to blood, probably within phagocytes (9, 86, 102, 103). The number of microbes in all body sites increases steadily from a small number to a peak about 3 weeks after infection and then declines to a lower set point that is maintained for the life of the host. CD4+ T cells have no effect on the growth of the microbes for the first 2 to 3 weeks after infection but are then critical for reducing the number of microbes to the set point (9, 21, 104). Phagosomal pathogens often persist during the period of CD4+ T cell-mediated control in organoids called granulomas, which form at the initial site of infection and consist of tight clusters of phagocytes interspersed with epithelial cells and surrounded by T cells (85).
Phagosomal infections induce CD4+ T cell responses that get off to a slow start compared to the responses induced by acute bacterial and viral infections. In each case, macrophages and dendritic cells at the initial site of infection—lung for M. tuberculosis, skin for L. major, and Peyer's patches for S. enterica—take up microbes into phagosomes (103, 105–107). The infected macrophages and dendritic cells then migrate to the draining lymph nodes (105, 108–110), where activation of CD4+ T cells with specific TCRs is first detected 1 to 2 weeks after infection (111–114) (Fig. 1). The relevant p:MHCII-specific CD4+ T cells eventually produce a large number of effector cells that peak in the secondary lymphoid organs after 2 to 3 weeks, when the infection peaks. Some of the effector T cells that are generated in the secondary lymphoid organs migrate to nonlymphoid organs, including the original sites of infection (111, 114, 115) (Fig. 1). In the case of M. tuberculosis, granulomas eventually form in the lungs (116), and the T cells are contained within them (117). Granuloma-like structures also eventually form in the skin sites of L. major infection (118, 119) and the livers of S. enterica-infected mice (120).
FIG 1.
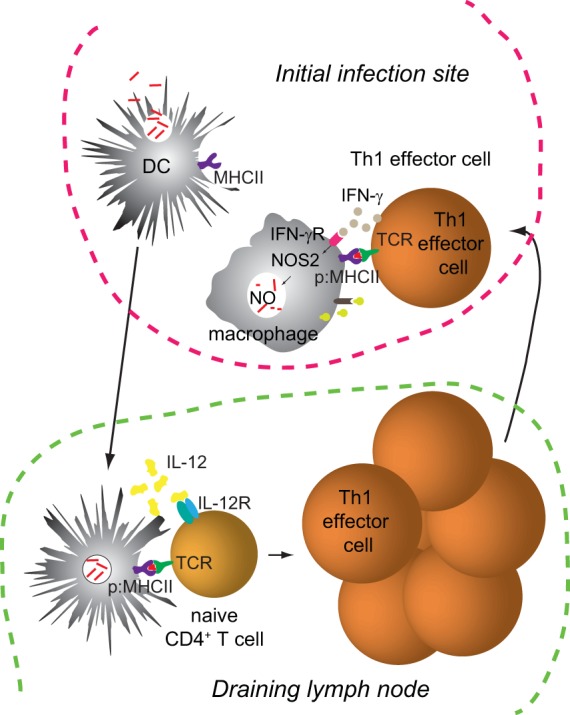
Early events after a phagosomal infection. The schematic illustrates events occurring at the initial site of infection and a draining lymph node about 1 to 2 weeks after infection.
Several factors may account for the slow tempo of the CD4+ T cell responses to phagosomal infections. First, in all three cases, it is likely that only a small number of microbes make it to the secondary lymphoid organs early after infection. The initial inoculum of M. tuberculosis or L. major organisms is low in the most common models (9, 102), and the number of S. enterica organisms that enter the mucosal lymphoid organs is also very small (121), likely because most of the bacteria die in the stomach after ingestion. In addition, it has been reported that phagosomal pathogens inhibit antigen processing and the formation of p:MHCII complexes (122–128), although this effect not been observed in all studies (129, 130). Thus, only a few of the relevant CD4+ T cells may become activated early after infection, because the number of infected phagocytes initially capable of presenting p:MHCII complexes is very small, or the infected phagocytes display very few p:MHCII complexes per cell (127). Second, M. tuberculosis p:MHCII-specific regulatory T cells become activated early after infection and slow the activation of effector T cells (114). The effector cells eventually take over, however, because the regulatory cells disappear by an IL-12-dependent mechanism (114).
Memory cell survival.
Another difference between the immune responses to persistent phagosomal infections and those to acute infections relates to the numerical stability of memory cells. The number of microbe p:MHCII-specific effector cells induced by phagosomal infections drops about 10-fold after the peak, as in the case of acute infections (111, 113, 115). Unlike the case of acute infections, however, the number of specific memory cells then remains constant in all body sites for hundreds of days (111, 113, 115). This remarkable stability is associated with low-level persistence of the phagosomal pathogen, as evidenced by the decline in the number of S. enterica p:MHCII-specific CD4+ T cells after persistent infection was cleared with antibiotics (113, 125). Thus, phagosomal pathogen p:MHCII-specific CD4+ T cell populations are maintained by a nonclassical antigen-dependent mechanism. This mechanism has also been observed for polyomavirus p:MHCII-specific CD4+ T cells in mice with persistent infection (131).
It is important to note that only a small number of cells in the specific CD4+ T cell population proliferate in response to the relevant p:MHCII ligand in secondary lymphoid organs or granulomas at any one time during the persistent phase of infection (113, 115). This phenomenon is again likely related to the fact that only a small number of infected phagocytes are present in the body during this phase. Thus, specific CD4+ memory T cells probably proliferate infrequently because they rarely encounter a phagocyte displaying the relevant p:MHCII ligand. The infrequency of this proliferation probably explains why the CD4+ T cell population is maintained at a stable level rather than increasing steadily over time (113). Indeed, p:MHCII-driven proliferation must perfectly balance the death rate to keep the population at a stable level. This model also implies that CD4+ T cells with TCRs specific for MHCII-bound peptides from phagosomal microbes spend long periods of time during persistent infection without encountering the relevant p:MHCII ligand. These long rest intervals could explain why these CD4+ T cells do not experience the activation defects exhibited by the “exhausted” T cells (113, 115) observed in individuals with systemic chronic infection (132).
Th1-mediated control of phagosomal infection.
The p:MHCII-specific CD4+ T cell populations present during the persistent phase of M. tuberculosis, S. enterica, and L. major infections have the CD44high memory cell phenotype and are dominated by Th1 cells (111, 113–115). This makes sense because all three microbes are potent inducers of IL-12 by cells of the innate immune system, and IL-12 is a major driver of Th1 cell differentiation (47, 133–135). It is less clear why Tfh cells do not form or are not maintained efficiently during these infections as in the case of acute infection with attenuated L. monocytogenes bacteria, which is also a strong IL-12 inducer (47). The fact that results for attenuated L. monocytogenes were obtained by injecting a large number of bacteria directly into the blood (35) is one possibility. This regimen provides many extracellular bacteria for uptake by antigen-specific B cells in the spleen. These B cells could present p:MHCII complexes to effector CD4+ T cells, steering them toward the Tfh fate, since antigen presentation by B cells favors Tfh cell development by providing continuously high levels of cognate p:MHCII (57). In contrast, very few free microbes likely enter the body after M. tuberculosis, S. enterica, or L. major infections, and the ones that do quickly enter a phagocyte, thereby limiting B cell access to the microbes. In this situation, very few of the effector cells would be diverted to the Tfh fate, and most would default to the Th1 fate. Poor access to antigen may also explain why B cells play little or no role in the control of phagosomal infections (97, 136–138). The dominance of Th1 cells during phagosomal infections could also be the result of the iterative stimulation of memory cells during persistent infection. Even if Tfh-like central memory cells are formed early after infection, they would be expected to yield mainly Th1 progeny after encountering the relevant p:MHCII ligand at a later time (34, 35, 69). Many rounds of this process would eventually lead to a Th1-dominated population.
Although the majority of effector cells induced by phagosomal infections are Th1 cells, some heterogeneity exists in this population. The M. tuberculosis p:MHCII-specific Th1 cell population consists of two subsets, one expressing killer cell lectin-like receptor G1 (KLRG1) and one lacking it (115). KLRG1− cells have a greater proliferative potential than KLRG1+ cells, while KLRG1+ cells are better IFN-γ and TNF producers than KLRG1− cells. KLRG1− cells may give rise to KLRG1+ cells in response to chronic TCR stimulation. The results suggest a conveyor belt model where KLRG1− Th1 cells periodically encounter their p:MHCII ligand on infected phagocytes, proliferate, and differentiate into KLRG1+ cells.
Th1 cells are important for the control of phagosomal infections because their lymphokines enhance the microbe-killing functions of phagocytes, including the production of nitric oxide (9, 139, 140). A likely scenario is that dendritic cells from the site of infection migrate to the draining lymph nodes and present pathogen p:MHCII complexes and IL-12 to naive CD4+ cells, which proliferate and become Th1 effector cells (Fig. 1). These cells then migrate to the site of infection and produce IFN-γ after encountering pathogen p:MHCII complexes on infected phagocytes. IFN-γ then binds the IFN-γ receptor on the infected phagocyte, causing the production of inducible nitric oxide synthase (NOS2) and the generation of the microbicidal compound nitric oxide (9). The finding that NOS2-deficient mice cannot control phagosomal pathogens is evidence of the importance of this pathway (9, 141–144). Synergy between IFN-γ and TNF for NOS2 induction could explain why T cells that produce both cytokines are more protective than T cells that produce IFN-γ alone (59, 145). IFN-γ and TNF also cause fusion of microbe-containing phagosomes with lysosomes, leading to microbial death due to exposure to acids, reactive oxygen species, and proteases (146, 147).
Srivastava and Ernst recently shed light on how the interaction of Th1 cells and infected phagocytes contains infection (148). They used chimeric mice that contained a mixture of MHCII-sufficient and MHCII-deficient macrophages and dendritic cells and found that the MHCII-sufficient cells controlled M. tuberculosis infection in a CD4+ T cell-dependent fashion, while the MHCII-deficient cells in the same mouse did not. The fact that the MHCII-deficient cells could not control infection indicates that the IFN-γ and TNF produced by CD4+ T cells in response to MHCII-sufficient cells could not act in a bystander fashion on nearby MHCII-deficient cells. This phenomenon could be explained by focal secretion of the cytokines into the synaptic space formed by the opposed membranes of CD4+ T cells and infected phagocytes displaying M. tuberculosis p:MHCII complexes (149).
Another way in which CD4+ T cells could contain phagosomal infections is the direct killing of infected phagocytes. Although cytotoxicity is often considered to be the job of CD8+ T cells, recent work (150) indicates that CD4+ T cells can acquire this capacity under certain conditions. Naive CD4+ T cells stimulated through the TCR, the TNF receptor family members CD134 (OX40) and CD137 (4-1BB), and the IL-2 receptor differentiate into effector cells that express the cytotoxic proteins perforin and granzyme B and kill cells displaying the relevant p:MHCII ligand (151, 152). This differentiation pathway depends on the eomesodermin transcription factor but not T-bet (151, 152). It will be of interest to determine if eomesodermin-dependent CD4+ cytotoxic T cells play a role in the control of phagosomal infections.
Some Th17 effector cells are generated during M. tuberculosis and Salmonella infections (153–155). Th17 cells might be expected to participate in the control of phagosomal infections by recruiting neutrophils. Indeed, it was reported that mice lacking the IL-17 receptor were defective in the long-term control of M. tuberculosis infection (156). On the other hand, M. tuberculosis and Salmonella infections are controlled normally in IL-23-deficient mice, in which Th17 generation does not occur (157, 158), and neutralization of IL-17 had no effect on the control of L. major infection (159). Furthermore, people with mutations that affect the expression of IL-17 or IL-17 receptor are susceptible to Candida and Staphylococcus aureus skin infections (160) but do not appear to be susceptible to the phagosomal pathogens that afflict people lacking CD4+ T cells or IL-12 receptor (27, 28, 161). Thus, the bulk of the evidence to date suggests that Th1 cells, and not Th17 cells, are critical for controlling phagosomal infections.
The curious case of herpes simplex virus.
One microbe that afflicts CD4+ T cell-deficient hosts but is not thought to persist in phagocytes is herpes simplex virus (HSV). This virus produces a lytic infection of epithelial cells in the skin and then spreads to nearby neurons, where it can persist for the life of the host (162). Local dendritic cells are also infected, at least early on (163–167). T cells are thought to play a role in keeping the infection in a latent state in neurons, although this state can sporadically break down, resulting in viral replication and a skin lesion (162). CD8+ T cells likely contribute to the control of infection by killing epithelial cells that become infected after the virus emerges from persistently infected neurons (162). The susceptibility of AIDS patients to HSV infection (27) suggests that CD4+ T cells also play a role in the control of this infection. This role may be forced by HSV shutting down the expression of MHCI molecules in dendritic cells using a protein called vhs (168, 169), thereby limiting the capacity of infected dendritic cells to stimulate CD8+ T cells. Nearby uninfected dendritic cells of the CD8α+ subtype, however, engulf dying infected cells and produce HSV peptide:MHCII complexes (163). It is therefore possible that CD4+ T cells are critical for the control of HSV infection by producing the antiviral cytokine IFN-γ in response to HSV peptide:MHCII complexes generated by CD8α+ dendritic cells that engulf dying infected cells or their debris. Thus, even though HSV is not a phagosomal infection per se, CD4+ T cells may be critical for its control because of the special role that phagosomal antigen presentation plays in this infection.
Phagosomal pathogen resistance to Th1-mediated control.
Although it is clear that CD4+ T cells control phagosomal infections, it is equally clear that infection is rarely eliminated (86, 170). Granulomas, which form in the lungs, skin, and liver in M. tuberculosis, L. major, and S. enterica infections, respectively, may be involved in this process (85, 120, 171–173). Granulomas consist of tight clusters of macrophages, some of which are infected and many of which fuse to form multinucleated giant cells (174). The macrophage clusters are surrounded by lymphocytes and interspersed with CD4+ T cells, collagen fibers, and epithelial cells. It can be argued that granulomas provide an advantage to the host by walling off the microbe in a site full of activated T cells and phagocytes (175). M. tuberculosis organisms, however, produce substances that cause host epithelial cells to secrete matrix metalloproteinase 9 (MMP9), which is critical for granuloma formation (176, 177). The fact that the pathogen appears to foster granuloma formation suggests that granulomas provide an advantage to the pathogen. Although Th1-mediated killing of infected macrophages and their microbes likely restrains the infection within granulomas, new phagocytes, which are actively recruited to granulomas, quickly engulf the dying macrophages and their microbes and become infected in the process (85, 117) (Fig. 2). Thus, granulomas are sites where M. tuberculosis organisms obtain access to a constant supply of new cells to infect. Another point in favor of the microbe is that infected phagocytes within granulomas display very small numbers of microbe p:MHCII complexes, thereby limiting IFN-γ and TNF production by Th1 cells (178, 179). It is also possible that the abnormal multinucleated giant cells found in granulomas (174) are impaired with respect to antigen processing and p:MHCII ligand production.
FIG 2.
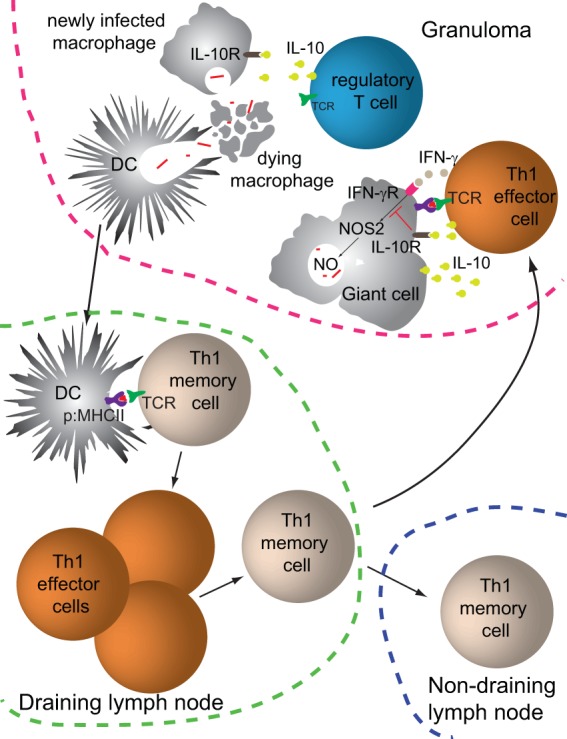
Events during the persistent phase of a phagosomal infection. The schematic illustrates events at the initial site of infection and a draining lymph node, months after the initial infection.
Another factor that limits pathogen clearance is that Th1 cells at sites of persistent phagosomal infection acquire the capacity to produce the suppressive cytokine IL-10 (111, 180) (Fig. 2). The differentiation of Th1 cells to an IL-10-producing state may depend on very high levels of IL-12 receptor signaling (181–184), which may be most likely to occur at sites of persistent infection. Importantly, IL-10 receptor signaling in macrophages inhibits MHCII expression (185, 186), thereby potentially limiting the display of microbial p:MHCII complexes by infected phagocytes in granulomas. IL-10 receptor signaling also suppresses IFN-γ and TNF production (187) and nitric oxide generation (188), which could blunt the capacity of infected macrophages to kill their microbes. This model is supported by the observation that IL-10 neutralization increases killing of phagosomal pathogens (102, 189, 190).
Regulatory T cells provide another brake on the killing of phagosomal pathogens in granulomas (Fig. 2). L. major-induced skin lesions contain abundant regulatory T cells, which are required for persistent infection (191). Some (192) but not all (111) of these regulatory T cells express TCRs that are specific for L. major p:MHCII complexes and accumulate in skin lesions through the expression of the E-cadherin binding integrin CD103 (193). These observations suggest that L. major p:MHCII-specific regulatory T cells suppress the microbe-killing functions of L. major p:MHCII-specific Th1 cells in the same lesions. M. tuberculosis-induced granulomas also contain abundant regulatory T cells (194). The TCR specificity of these cells is unclear, however, since M. tuberculosis p:MHCII-specific regulatory T cells are lost at later times after infection (114). In addition, the relevance of regulatory T cells in granulomas is uncertain, since late depletion of regulatory T cells had no effect on the number of M. tuberculosis organisms in this location (195).
Phagosomal pathogens also have intrinsic mechanisms to resist the attempts of Th1 cells to activate their phagocyte host. As mentioned above, many phagosomal microbes produce molecules that interfere with the IFN-γ-enhanced process of phagosome fusion with lysosomes (146, 171). For example, M. tuberculosis has many enzymes that detoxify microbicidal compounds, such as superoxide anion, hydrogen peroxide, and nitric oxide, that are produced in large amounts in the phagolysosomes of IFN-γ-activated phagocytes. M. tuberculosis, L. major, and S. enterica organisms also cause phagocytes to produce IL-10 (196–199) (Fig. 2), which, in addition its T cell-suppressive effects, inhibits phagosome-lysosome fusion (128, 200, 201).
Thus, phagosomal pathogens persist by resisting phagosome toxification, quickly jumping from dying phagocytes to newly recruited phagocytes, and promoting regulatory T cells and IL-10 that restrain the microbicidal functions of Th1 cells.
Concomitant immunity.
Remarkably, healthy hosts that harbor Leishmania parasites in phagocytes at a site of persistent infection are resistant to superinfection in other parts of the body (202). This situation is advantageous to the host, since unlike persistent granuloma infection, which is usually asymptomatic, systemic superinfections can be fatal. This odd state of affairs raises the question, how does the immune system eliminate infections at secondary sites but leave the initial infection intact? The answer may lie in the fact that superinfecting organisms are taken up by phagocytes that are not in the immunosuppressive environment of the granuloma and are thus susceptible to complete killing by circulating memory T cells.
The concept that a localized infection is required for protection from systemic infection is called concomitant immunity and has been studied primarily with Leishmania infection. L. major parasites are deposited in the skin by a sand fly bite and are taken up by dendritic cells and macrophages, some of which migrate to the draining lymph nodes (203). The infected phagocytes process proteins from the parasite and present parasite p:MHCII ligands to naive CD4+ T cells, which expand and produce Th1 effector cells (Fig. 1). The Th1 cells migrate to the skin infection site and limit the replication of the parasites within phagocytes by stimulating nitric oxide production, as described above (Fig. 1). However, Treg cell- and IL-10-rich granuloma-like structures quickly form at this site to protect the microbes from complete elimination (102, 188, 191, 204), allowing the parasites to persist long-term (205) (Fig. 2). Th1 cells also circulate to other body sites, including nondraining lymph nodes and uninfected skin (111) (Fig. 2). Occasionally, some of the Th1 cells return to the site of infection and respond to parasite p:MHCII molecules, keeping the infection in check (Fig. 2) and maintaining the population of parasite p:MHCII-specific CD4+ T cells. In the event of a later sand fly bite, Th1 cells at the new bite site, or in the spleen if the parasites reach the blood, produce IFN-γ and TNF and kill all the parasites in these locations before new granuloma-like organoids can form to protect the parasites (204, 206) (Fig. 3). Because CD4+ memory T cells do not persist indefinitely without periodic stimulation by the relevant p:MHCII ligand (34, 35, 46, 66), the decrease in the level of cognate p:MHCII that would accompany a sterile cure of Leishmania infection would likely lead to a gradual decrease in the number of CD4+ T cells and eventual loss of immunity. Indeed, protection from superinfection is lost if the original infection is cleared (191, 207, 208).
FIG 3.

Events during superinfection of a host with a persistent phagosomal infection. The schematic illustrates events at the indicated sites of a persistently infected host, 1 week after a new infection.
While concomitant immunity is best understood in the context of Leishmania infection, it may operate for all phagosomal infections. Concomitant immunity could be responsible for containing M. tuberculosis and C. neoformans infections to lung granulomas, limiting their spread and preventing superinfection. Concomitant immunity may have evolved because it meets the needs of the microbe and the host: the microbe gets a niche to live in, and the host becomes resistant to more serious systemic forms of the infection.
Implications for vaccines.
Most vaccines consist of attenuated microbes, dead microbes, or components of microbes that are given to subjects a limited number of times. This regimen produces a situation where vaccine antigens are present in an extracellular form and are eliminated from the body shortly after administration. Transient exposure to extracellular antigen is sufficient to stimulate the production of Tfh cells, which help B cells become long-lived plasma cells that produce neutralizing antibodies for the life of the host, even after the eliciting antigens have been cleared from the body (41). Vaccines of this kind are therefore very effective for extracellular infections. Given what we now know about the immune response to phagosomal infections, it is easy see why this vaccination strategy has failed for this class of microbes. Phagosomal pathogens are not susceptible to antibodies, because they reside in phagosomes and are often transmitted to new phagocytes that engulf an infected phagocyte. In addition, transient exposure to antigen is not an effective way to induce a numerically stable population of CD4+ memory T cells over long periods of time (113). Indeed, the fact that the bacillus Calmette-Guérin (BCG) vaccine, an attenuated strain of Mycobacterium bovis, induces partial immunity to M. tuberculosis infection that fades with time (209) may be explained by the decline in the number of relevant CD4+ memory T cells as BCG antigens disappear.
The practice of “leishmanization” provides a clue as to how a vaccine for a phagosomal pathogen could work (202). People in areas where the disease is endemic have long recognized that individuals with a Leishmania-infected skin lesion rarely contract the often-fatal visceral form of the infection. This knowledge has led to leishmanization, a process whereby people inoculate themselves in an inconspicuous skin site with material from the skin lesion of an infected person. It is reasonable to suspect that this practice produces a localized persistent infection and concomitant immunity.
Although it may be effective, leishmanization is a dangerous practice in the AIDS era (210). An AIDS-related drop in the number of CD4+ T cells could lead to a loss of control of the Leishmania parasites in the skin lesion, resulting in systemic infection and death. Thus, the question becomes how to deliver antigens from phagosomal pathogens safely for a long period of time and in small amounts from a localized site, perhaps with features of a granuloma. Perhaps, it will be possible to build a better BCG vaccines, that is, more persistent vaccine strains containing suicide molecules that could be triggered in the event of AIDS. Alternatively, peptides from phagosomal pathogens could be incorporated into next-generation polymers designed to release their cargo over very long periods of time. Peptides could be incorporated to limit the formation of antibodies that would speed the clearance of the vaccine antigen. These formulations could be injected with MMP9 in an attempt to recreate a granuloma-like structure at the injection site. Success could depend on abandoning approaches aimed at elicitation of neutralizing antibodies in favor of approaches aimed at generating large, intermittently stimulated populations of Th1 cells.
Biographies

Noah J. Tubo is currently a postdoctoral fellow in Marc Jenkins' laboratory at the University of Minnesota. He received his Ph.D. in cellular and developmental biology from Harvard University in 2011, where he studied the mechanisms of lymphocyte trafficking into peripheral tissues. His current interests involve understanding the mechanisms by which naive CD4+ T cells differentiate into effector and memory cells and the implications of these processes for pathogen control and immunity.

Marc K. Jenkins received his bachelor's degree (microbiology, 1980) from the University of Minnesota and his Ph.D. (microbiology and immunology, 1985) from Northwestern University. He did postdoctoral research with Ronald Schwartz at the National Institutes of Health from 1985 to 1988. In 1988, Dr. Jenkins accepted a faculty position at the University of Minnesota in the Department of Microbiology. He is now a Distinguished McKnight Professor and the Director of the Center for Immunology. Dr. Jenkins and his colleagues investigate CD4+ T and B cell activation in vivo by directly tracking antigen-specific cells. The goal of his research is a basic understanding of lymphocyte activation that can be used to improve vaccines and prevent autoimmunity. He is the recipient of a Pew Scholar in the Biomedical Sciences Award, a Merit Award from the National Institute of Allergy and Infectious Diseases of the NIH, and the Meritorious Career Award from the American Association of Immunologists.
REFERENCES
- 1.Mascie-Taylor CG, Karim E. 2003. The burden of chronic disease. Science 302:1921–1922. 10.1126/science.1092488 [DOI] [PubMed] [Google Scholar]
- 2.Murphy KM. 2012. Janeway's immunobiology, 8th ed. Garland Science, Taylor & Francis Group, New York, NY [Google Scholar]
- 3.Eason DD, Cannon JP, Haire RN, Rast JP, Ostrov DA, Litman GW. 2004. Mechanisms of antigen receptor evolution. Semin. Immunol. 16:215–226. 10.1016/j.smim.2004.08.001 [DOI] [PubMed] [Google Scholar]
- 4.Rudolph MG, Stanfield RL, Wilson IA. 2006. How TCRs bind MHCs, peptides, and coreceptors. Annu. Rev. Immunol. 24:419–466. 10.1146/annurev.immunol.23.021704.115658 [DOI] [PubMed] [Google Scholar]
- 5.Mariuzza RA, Phillips SE, Poljak RJ. 1987. The structural basis of antigen-antibody recognition. Annu. Rev. Biophys. Biophys. Chem. 16:139–159. 10.1146/annurev.bb.16.060187.001035 [DOI] [PubMed] [Google Scholar]
- 6.Perlmutter RM, Kearney JF, Chang SP, Hood LE. 1985. Developmentally controlled expression of immunoglobulin VH genes. Science 227:1597–1601. 10.1126/science.3975629 [DOI] [PubMed] [Google Scholar]
- 7.Burnet F. 1957. A modification of Jerne's theory of antibody production using the concept of clonal selection. Aust. J. Sci. 20:67. [DOI] [PubMed] [Google Scholar]
- 8.Davis MM, Bjorkman PJ. 1988. T-cell antigen receptor genes and T-cell recognition. Nature 334:395–402. 10.1038/334395a0 [DOI] [PubMed] [Google Scholar]
- 9.Mogues T, Goodrich ME, Ryan L, LaCourse R, North RJ. 2001. The relative importance of T cell subsets in immunity and immunopathology of airborne Mycobacterium tuberculosis infection in mice. J. Exp. Med. 193:271–280. 10.1084/jem.193.3.271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Germain RN. 1994. MHC-dependent antigen processing and peptide presentation: providing ligands for T lymphocyte activation. Cell 76:287–299. 10.1016/0092-8674(94)90336-0 [DOI] [PubMed] [Google Scholar]
- 11.Suh WK, Cohen-Doyle MF, Fruh K, Wang K, Peterson PA, Williams DB. 1994. Interaction of MHC class I molecules with the transporter associated with antigen processing. Science 264:1322–1326. 10.1126/science.8191286 [DOI] [PubMed] [Google Scholar]
- 12.Joffre OP, Segura E, Savina A, Amigorena S. 2012. Cross-presentation by dendritic cells. Nat. Rev. Immunol. 12:557–569. 10.1038/nri3254 [DOI] [PubMed] [Google Scholar]
- 13.Rudensky A, Rath S, Preston-Hurlburt P, Murphy DB, Janeway CA., Jr 1991. On the complexity of self. Nature 353:660–662. 10.1038/353660a0 [DOI] [PubMed] [Google Scholar]
- 14.Starr TK, Jameson SC, Hogquist KA. 2003. Positive and negative selection of T cells. Annu. Rev. Immunol. 21:139–176. 10.1146/annurev.immunol.21.120601.141107 [DOI] [PubMed] [Google Scholar]
- 15.Davis MM. 1990. T cell receptor gene diversity and selection. Annu. Rev. Biochem. 59:475–496. 10.1146/annurev.bi.59.070190.002355 [DOI] [PubMed] [Google Scholar]
- 16.Marrack P, Scott-Browne JP, Dai S, Gapin L, Kappler JW. 2008. Evolutionarily conserved amino acids that control TCR-MHC interaction. Annu. Rev. Immunol. 26:171–203. 10.1146/annurev.immunol.26.021607.090421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Daar AS, Fuggle SV, Fabre JW, Ting A, Morris PJ. 1984. The detailed distribution of HLA-A, B, C antigens in normal human organs. Transplantation 38:287–292. 10.1097/00007890-198409000-00018 [DOI] [PubMed] [Google Scholar]
- 18.Bodmer WF. 1981. HLA structure and function: a contemporary view. Tissue Antigens 17:9–20 [DOI] [PubMed] [Google Scholar]
- 19.MacMicking JD. 2005. Immune control of phagosomal bacteria by p47 GTPases. Curr. Opin. Microbiol. 8:74–82. 10.1016/j.mib.2004.12.012 [DOI] [PubMed] [Google Scholar]
- 20.Lee SJ, Dunmire S, McSorley SJ. 2012. MHC class-I-restricted CD8 T cells play a protective role during primary Salmonella infection. Immunol. Lett. 148:138–143. 10.1016/j.imlet.2012.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hess J, Ladel C, Miko D, Kaufmann SH. 1996. Salmonella typhimurium aroA- infection in gene-targeted immunodeficient mice: major role of CD4+ TCR-alpha beta cells and IFN-gamma in bacterial clearance independent of intracellular location. J. Immunol. 156:3321–3326 [PubMed] [Google Scholar]
- 22.Harding CV, Collins DS, Slot JW, Geuze HJ, Unanue ER. 1991. Liposome-encapsulated antigens are processed in lysosomes, recycled, and presented to T cells. Cell 64:393–401. 10.1016/0092-8674(91)90647-H [DOI] [PubMed] [Google Scholar]
- 23.Harding CV, Geuze HJ. 1992. Class II MHC molecules are present in macrophage lysosomes and phagolysosomes that function in the phagocytic processing of Listeria monocytogenes for presentation to T cells. J. Cell Biol. 119:531–542. 10.1083/jcb.119.3.531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Amigorena S, Drake JR, Webster P, Mellman I. 1994. Transient accumulation of new class II MHC molecules in a novel endocytic compartment in B lymphocytes. Nature 369:113–120. 10.1038/369113a0 [DOI] [PubMed] [Google Scholar]
- 25.Tulp A, Verwoerd D, Dobberstein B, Ploegh HL, Pieters J. 1994. Isolation and characterization of the intracellular MHC class II compartment. Nature 369:120–126. 10.1038/369120a0 [DOI] [PubMed] [Google Scholar]
- 26.West MA, Lucocq JM, Watts C. 1994. Antigen processing and class II MHC peptide-loading compartments in human B-lymphoblastoid cells. Nature 369:147–151. 10.1038/369147a0 [DOI] [PubMed] [Google Scholar]
- 27.Hart CA, Beeching NJ, Duerden BI, Curry A, French N, Kariuki S, Graham SM, Gordon MA, Hoggard PG, Kewn S, Back DJ. 2000. Infections in AIDS. J. Med. Microbiol. 49:947–967 http://jmm.sgmjournals.org/content/49/11/947.long [DOI] [PubMed] [Google Scholar]
- 28.Reith W, Mach B. 2001. The bare lymphocyte syndrome and the regulation of MHC expression. Annu. Rev. Immunol. 19:331–373. 10.1146/annurev.immunol.19.1.331 [DOI] [PubMed] [Google Scholar]
- 29.von Andrian UH, Mackay CR. 2000. T-cell function and migration. N. Engl. J. Med. 343:1020–1034. 10.1056/NEJM200010053431407 [DOI] [PubMed] [Google Scholar]
- 30.Schwab SR, Cyster JG. 2007. Finding a way out: lymphocyte egress from lymphoid organs. Nat. Immunol. 8:1295–1301. 10.1038/ni1545 [DOI] [PubMed] [Google Scholar]
- 31.Sprent J, Tough DF. 1994. Lymphocyte life-span and memory. Science 265:1395–1400. 10.1126/science.8073282 [DOI] [PubMed] [Google Scholar]
- 32.Itano AA, Jenkins MK. 2003. Antigen presentation to naive CD4 T cells in the lymph node. Nat. Immunol. 4:733–739. 10.1038/ni957 [DOI] [PubMed] [Google Scholar]
- 33.Jenkins MK, Moon JJ. 2012. The role of naive T cell precursor frequency and recruitment in dictating immune response magnitude. J. Immunol. 188:4135–4140. 10.4049/jimmunol.1102661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pepper M, Linehan JL, Pagan AJ, Zell T, Dileepan T, Cleary PP, Jenkins MK. 2010. Different routes of bacterial infection induce long-lived TH1 memory cells and short-lived TH17 cells. Nat. Immunol. 11:83–89. 10.1038/ni.1826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pepper M, Pagan AJ, Igyarto BZ, Taylor JJ, Jenkins MK. 2011. Opposing signals from the bcl6 transcription factor and the interleukin-2 receptor generate T helper 1 central and effector memory cells. Immunity 35:583–595. 10.1016/j.immuni.2011.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dutton RW, Bradley LM, Swain SL. 1998. T cell memory. Annu. Rev. Immunol. 16:201–223. 10.1146/annurev.immunol.16.1.201 [DOI] [PubMed] [Google Scholar]
- 37.Zhu J, Yamane H, Paul WE. 2010. Differentiation of effector CD4 T cell populations. Annu. Rev. Immunol. 28:445–489. 10.1146/annurev-immunol-030409-101212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Licona-Limon P, Kim LK, Palm NW, Flavell RA. 2013. TH2, allergy and group 2 innate lymphoid cells. Nat. Immunol. 14:536–542. 10.1038/ni.2617 [DOI] [PubMed] [Google Scholar]
- 39.Stetson DB, Voehringer D, Grogan JL, Xu M, Reinhardt RL, Scheu S, Kelly BL, Locksley RM. 2004. Th2 cells: orchestrating barrier immunity. Adv. Immunol. 83:163–189. 10.1016/S0065-2776(04)83005-0 [DOI] [PubMed] [Google Scholar]
- 40.Basu R, Hatton RD, Weaver CT. 2013. The Th17 family: flexibility follows function. Immunol. Rev. 252:89–103. 10.1111/imr.12035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crotty S. 2011. Follicular helper CD4 T cells (TFH). Annu. Rev. Immunol. 29:621–663. 10.1146/annurev-immunol-031210-101400 [DOI] [PubMed] [Google Scholar]
- 42.Nimmerjahn F, Ravetch JV. 2007. Fc-receptors as regulators of immunity. Adv. Immunol. 96:179–204. 10.1016/S0065-2776(07)96005-8 [DOI] [PubMed] [Google Scholar]
- 43.O'Garra A, Murphy KM. 2009. From IL-10 to IL-12: how pathogens and their products stimulate APCs to induce T(H)1 development. Nat. Immunol. 10:929–932. 10.1038/ni0909-929 [DOI] [PubMed] [Google Scholar]
- 44.Martinez FO, Sica A, Mantovani A, Locati M. 2008. Macrophage activation and polarization. Front. Biosci. 13:453–461. 10.2741/2692 [DOI] [PubMed] [Google Scholar]
- 45.Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, Lao C, Crotty S. 2011. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity 34:932–946. 10.1016/j.immuni.2011.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marshall HD, Chandele A, Jung YW, Meng H, Poholek AC, Parish IA, Rutishauser R, Cui W, Kleinstein SH, Craft J, Kaech SM. 2011. Differential expression of Ly6C and T-bet distinguish effector and memory Th1 CD4(+) cell properties during viral infection. Immunity 35:633–646. 10.1016/j.immuni.2011.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hsieh CS, Heimberger AB, Gold JS, O'Garra A. 1993. Development of Th1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science 260:547–549 [DOI] [PubMed] [Google Scholar]
- 48.Dalod M, Salazar-Mather TP, Malmgaard L, Lewis C, Asselin-Paturel C, Briere F, Trinchieri G, Biron CA. 2002. Interferon alpha/beta and interleukin 12 responses to viral infections: pathways regulating dendritic cell cytokine expression in vivo. J. Exp. Med. 195:517–528. 10.1084/jem.20011672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eto D, Lao C, DiToro D, Barnett B, Escobar TC, Kageyama R, Yusuf I, Crotty S. 2011. IL-21 and IL-6 are critical for different aspects of B cell immunity and redundantly induce optimal follicular helper CD4 T cell (Tfh) differentiation. PLoS One 6:e17739. 10.1371/journal.pone.0017739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu H, Li X, Liu D, Li J, Zhang X, Chen X, Hou S, Peng L, Xu C, Liu W, Zhang L, Qi H. 2013. Follicular T-helper cell recruitment governed by bystander B cells and ICOS-driven motility. Nature 496:523–527. 10.1038/nature12058 [DOI] [PubMed] [Google Scholar]
- 51.Constant S, Pfeiffer C, Woodard A, Pasqualini T, Bottomly K. 1995. Extent of T cell receptor ligation can determine the functional differentiation of naive CD4+ T cells. J. Exp. Med. 182:1591–1596. 10.1084/jem.182.5.1591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hosken NA, Shibuya K, Heath AW, Murphy KM, O'Garra A. 1995. The effect of antigen dose on CD4+ T helper cell phenotype development in a T cell receptor-alpha beta-transgenic model. J. Exp. Med. 182:1579–1584. 10.1084/jem.182.5.1579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bretscher PA, Wei G, Menon JN, Bielefeldt-Ohmann H. 1992. Establishment of stable, cell-mediated immunity that makes “susceptible” mice resistant to Leishmania major. Science 257:539–542. 10.1126/science.1636090 [DOI] [PubMed] [Google Scholar]
- 54.Parish CR, Liew FY. 1972. Immune response to chemically modified flagellin. 3. Enhanced cell-mediated immunity during high and low zone antibody tolerance to flagellin. J. Exp. Med. 135:298–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tubo NJ, Pagan AJ, Taylor JJ, Nelson RW, Linehan JL, Ertelt JM, Huseby ES, Way SS, Jenkins MK. 2013. Single naive CD4(+) T cells from a diverse repertoire produce different effector cell types during infection. Cell 153:785–796. 10.1016/j.cell.2013.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fazilleau N, McHeyzer-Williams LJ, Rosen H, McHeyzer-Williams MG. 2009. The function of follicular helper T cells is regulated by the strength of T cell antigen receptor binding. Nat. Immunol. 10:375–384. 10.1038/ni.1704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Deenick EK, Chan A, Ma CS, Gatto D, Schwartzberg PL, Brink R, Tangye SG. 2010. Follicular helper T cell differentiation requires continuous antigen presentation that is independent of unique B cell signaling. Immunity 33:241–253. 10.1016/j.immuni.2010.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baumjohann D, Preite S, Reboldi A, Ronchi F, Ansel KM, Lanzavecchia A, Sallusto F. 2013. Persistent antigen and germinal center B cells sustain T follicular helper cell responses and phenotype. Immunity 38:596–605. 10.1016/j.immuni.2012.11.020 [DOI] [PubMed] [Google Scholar]
- 59.Darrah PA, Patel DT, De Luca PM, Lindsay RW, Davey DF, Flynn BJ, Hoff ST, Andersen P, Reed SG, Morris SL, Roederer M, Seder RA. 2007. Multifunctional TH1 cells define a correlate of vaccine-mediated protection against Leishmania major. Nat. Med. 13:843–850. 10.1038/nm1592 [DOI] [PubMed] [Google Scholar]
- 60.Schulz EG, Mariani L, Radbruch A, Hofer T. 2009. Sequential polarization and imprinting of type 1 T helper lymphocytes by interferon-gamma and interleukin-12. Immunity 30:673–683. 10.1016/j.immuni.2009.03.013 [DOI] [PubMed] [Google Scholar]
- 61.Yamane H, Zhu J, Paul WE. 2005. Independent roles for IL-2 and GATA-3 in stimulating naive CD4+ T cells to generate a Th2-inducing cytokine environment. J. Exp. Med. 202:793–804. 10.1084/jem.20051304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liao W, Lin JX, Wang L, Li P, Leonard WJ. 2011. Modulation of cytokine receptors by IL-2 broadly regulates differentiation into helper T cell lineages. Nat. Immunol. 12:551–559. 10.1038/ni.2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ballesteros-Tato A, Leon B, Graf BA, Moquin A, Adams PS, Lund FE, Randall TD. 2012. Interleukin-2 inhibits germinal center formation by limiting T follicular helper cell differentiation. Immunity 36:847–856. 10.1016/j.immuni.2012.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Groom JR, Luster AD. 2011. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol. Cell Biol. 89:207–215. 10.1038/icb.2010.158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Carlow DA, Gossens K, Naus S, Veerman KM, Seo W, Ziltener HJ. 2009. PSGL-1 function in immunity and steady state homeostasis. Immunol. Rev. 230:75–96. 10.1111/j.1600-065X.2009.00797.x [DOI] [PubMed] [Google Scholar]
- 66.Homann D, Teyton L, Oldstone MB. 2001. Differential regulation of antiviral T-cell immunity results in stable CD8+ but declining CD4+ T-cell memory. Nat. Med. 7:913–919. 10.1038/90950 [DOI] [PubMed] [Google Scholar]
- 67.Garrod KR, Moreau HD, Garcia Z, Lemaitre F, Bouvier I, Albert ML, Bousso P. 2012. Dissecting T cell contraction in vivo using a genetically encoded reporter of apoptosis. Cell Rep. 2:1438–1447. 10.1016/j.celrep.2012.10.015 [DOI] [PubMed] [Google Scholar]
- 68.Hand TW, Morre M, Kaech SM. 2007. Expression of IL-7 receptor alpha is necessary but not sufficient for the formation of memory CD8 T cells during viral infection. Proc. Natl. Acad. Sci. U. S. A. 104:11730–11735. 10.1073/pnas.0705007104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hale JS, Youngblood B, Latner DR, Mohammed AU, Ye L, Akondy RS, Wu T, Iyer SS, Ahmed R. 2013. Distinct memory CD4+ T cells with commitment to T follicular helper- and T helper 1-cell lineages are generated after acute viral infection. Immunity 38:805–817. 10.1016/j.immuni.2013.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Luthje K, Kallies A, Shimohakamada Y, Belz GT, Light A, Tarlinton DM, Nutt SL. 2012. The development and fate of follicular helper T cells defined by an IL-21 reporter mouse. Nat. Immunol. 13:491–498. 10.1038/ni.2261 [DOI] [PubMed] [Google Scholar]
- 71.Choi YS, Yang JA, Yusuf I, Johnston RJ, Greenbaum J, Peters B, Crotty S. 2013. Bcl6 expressing follicular helper CD4 T cells are fate committed early and have the capacity to form memory. J. Immunol. 190:4014–4026. 10.4049/jimmunol.1202963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Harrington LE, Janowski KM, Oliver JR, Zajac AJ, Weaver CT. 2008. Memory CD4 T cells emerge from effector T-cell progenitors. Nature 452:356–360. 10.1038/nature06672 [DOI] [PubMed] [Google Scholar]
- 73.Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. 1999. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401:708–712. 10.1038/44385 [DOI] [PubMed] [Google Scholar]
- 74.Schluns KS, Kieper WC, Jameson SC, Lefrancois L. 2000. Interleukin-7 mediates the homeostasis of naive and memory CD8 T cells in vivo. Nat. Immunol. 1:426–432. 10.1038/80868 [DOI] [PubMed] [Google Scholar]
- 75.Purton JF, Tan JT, Rubinstein MP, Kim DM, Sprent J, Surh CD. 2007. Antiviral CD4+ memory T cells are IL-15 dependent. J. Exp. Med. 204:951–961. 10.1084/jem.20061805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Slifka MK, Antia R, Whitmire JK, Ahmed R. 1998. Humoral immunity due to long-lived plasma cells. Immunity 8:363–372. 10.1016/S1074-7613(00)80541-5 [DOI] [PubMed] [Google Scholar]
- 77.Murali-Krishna K, Altman JD, Suresh M, Sourdive DJ, Zajac AJ, Miller JD, Slansky J, Ahmed R. 1998. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity 8:177–187. 10.1016/S1074-7613(00)80470-7 [DOI] [PubMed] [Google Scholar]
- 78.van Leeuwen EM, Sprent J, Surh CD. 2009. Generation and maintenance of memory CD4(+) T cells. Curr. Opin. Immunol. 21:167–172. 10.1016/j.coi.2009.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hataye J, Moon JJ, Khoruts A, Reilly C, Jenkins MK. 2006. Naive and memory CD4+ T cell survival controlled by clonal abundance. Science 312:114–116. 10.1126/science.1124228 [DOI] [PubMed] [Google Scholar]
- 80.Singh NJ, Bando JK, Schwartz RH. 2012. Subsets of nonclonal neighboring CD4+ T cells specifically regulate the frequency of individual antigen-reactive T cells. Immunity 37:735–746. 10.1016/j.immuni.2012.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hammarlund E, Lewis MW, Hansen SG, Strelow LI, Nelson JA, Sexton GJ, Hanifin JM, Slifka MK. 2003. Duration of antiviral immunity after smallpox vaccination. Nat. Med. 9:1131–1137. 10.1038/nm917 [DOI] [PubMed] [Google Scholar]
- 82.Su LF, Kidd BA, Han A, Kotzin JJ, Davis MM. 2013. Virus-specific CD4(+) memory-phenotype T cells are abundant in unexposed adults. Immunity 38:373–383. 10.1016/j.immuni.2012.10.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rahemtulla A, Fung-Leung WP, Schilham MW, Kundig TM, Sambhara SR, Narendran A, Arabian A, Wakeham A, Paige CJ, Zinkernagel RM, Miller RG, Mak TW. 1991. Normal development and function of CD8+ cells but markedly decreased helper cell activity in mice lacking CD4. Nature 353:180–184. 10.1038/353180a0 [DOI] [PubMed] [Google Scholar]
- 84.Kaufmann SH, Ladel CH. 1994. Role of T cell subsets in immunity against intracellular bacteria: experimental infections of knock-out mice with Listeria monocytogenes and Mycobacterium bovis BCG. Immunobiology 191:509–519. 10.1016/S0171-2985(11)80457-2 [DOI] [PubMed] [Google Scholar]
- 85.Ramakrishnan L. 2012. Revisiting the role of the granuloma in tuberculosis. Nat. Rev. Immunol. 12:352–366. 10.1038/nri3211 [DOI] [PubMed] [Google Scholar]
- 86.Monack DM, Mueller A, Falkow S. 2004. Persistent bacterial infections: the interface of the pathogen and the host immune system. Nat. Rev. Microbiol. 2:747–765. 10.1038/nrmicro955 [DOI] [PubMed] [Google Scholar]
- 87.Alexander J, Satoskar AR, Russell DG. 1999. Leishmania species: models of intracellular parasitism. J. Cell Sci. 112:2993–3002 [DOI] [PubMed] [Google Scholar]
- 88.Johnston SA, May RC. 2013. Cryptococcus interactions with macrophages: evasion and manipulation of the phagosome by a fungal pathogen. Cell. Microbiol. 15:403–411. 10.1111/cmi.12067 [DOI] [PubMed] [Google Scholar]
- 89.Scanga CA, Mohan VP, Yu K, Joseph H, Tanaka K, Chan J, Flynn JL. 2000. Depletion of CD4(+) T cells causes reactivation of murine persistent tuberculosis despite continued expression of interferon gamma and nitric oxide synthase 2. J. Exp. Med. 192:347–358. 10.1084/jem.192.3.347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Johanns TM, Ertelt JM, Rowe JH, Way SS. 2010. Regulatory T cell suppressive potency dictates the balance between bacterial proliferation and clearance during persistent Salmonella infection. PLoS Pathog. 6:e1001043. 10.1371/journal.ppat.1001043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Erb K, Blank C, Ritter U, Bluethmann H, Moll H. 1996. Leishmania major infection in major histocompatibility complex class II-deficient mice: CD8+ T cells do not mediate a protective immune response. Immunobiology 195:243–260. 10.1016/S0171-2985(96)80043-X [DOI] [PubMed] [Google Scholar]
- 92.Lockhart E, Green AM, Flynn JL. 2006. IL-17 production is dominated by gammadelta T cells rather than CD4 T cells during Mycobacterium tuberculosis infection. J. Immunol. 177:4662–4669 http://www.jimmunol.org/content/177/7/4662.long [DOI] [PubMed] [Google Scholar]
- 93.Overath P, Harbecke D. 1993. Course of Leishmania infection in beta 2-microglobulin-deficient mice. Immunol. Lett. 37:13–17. 10.1016/0165-2478(93)90126-M [DOI] [PubMed] [Google Scholar]
- 94.Behar SM, Dascher CC, Grusby MJ, Wang CR, Brenner MB. 1999. Susceptibility of mice deficient in CD1D or TAP1 to infection with Mycobacterium tuberculosis. J. Exp. Med. 189:1973–1980. 10.1084/jem.189.12.1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Maglione PJ, Xu J, Chan J. 2007. B cells moderate inflammatory progression and enhance bacterial containment upon pulmonary challenge with Mycobacterium tuberculosis. J. Immunol. 178:7222–7234 http://www.jimmunol.org/content/178/11/7222.long [DOI] [PubMed] [Google Scholar]
- 96.Vordermeier HM, Venkataprasad N, Harris DP, Ivanyi J. 1996. Increase of tuberculous infection in the organs of B cell-deficient mice. Clin. Exp. Immunol. 106:312–316. 10.1046/j.1365-2249.1996.d01-845.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Johnson CM, Cooper AM, Frank AA, Bonorino CB, Wysoki LJ, Orme IM. 1997. Mycobacterium tuberculosis aerogenic rechallenge infections in B cell-deficient mice. Tuber. Lung Dis. 78:257–261. 10.1016/S0962-8479(97)90006-X [DOI] [PubMed] [Google Scholar]
- 98.Bosio CM, Gardner D, Elkins KL. 2000. Infection of B cell-deficient mice with CDC 1551, a clinical isolate of Mycobacterium tuberculosis: delay in dissemination and development of lung pathology. J. Immunol. 164:6417–6425 http://www.jimmunol.org/content/164/12/6417.long [DOI] [PubMed] [Google Scholar]
- 99.Nanton MR, Way SS, Shlomchik MJ, McSorley SJ. 2012. B cells are essential for protective immunity against Salmonella independent of antibody secretion. J. Immunol. 189:5503–5507. 10.4049/jimmunol.1201413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ronet C, Voigt H, Himmelrich H, Doucey MA, Hauyon-La Torre Y, Revaz-Breton M, Tacchini-Cottier F, Bron C, Louis J, Launois P. 2008. Leishmania major-specific B cells are necessary for Th2 cell development and susceptibility to L. major LV39 in BALB/c mice. J. Immunol. 180:4825–4835 http://www.jimmunol.org/content/180/7/4825.long [DOI] [PubMed] [Google Scholar]
- 101.Moon JJ, Chu HH, Hataye J, Pagan AJ, Pepper M, McLachlan JB, Zell T, Jenkins MK. 2009. Tracking epitope-specific T cells. Nat. Protoc. 4:565–581. 10.1038/nprot.2009.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Belkaid Y, Hoffmann KF, Mendez S, Kamhawi S, Udey MC, Wynn TA, Sacks DL. 2001. The role of interleukin (IL)-10 in the persistence of Leishmania major in the skin after healing and the therapeutic potential of anti-IL-10 receptor antibody for sterile cure. J. Exp. Med. 194:1497–1506. 10.1084/jem.194.10.1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mastroeni P, Grant AJ. 2011. Spread of Salmonella enterica in the body during systemic infection: unravelling host and pathogen determinants. Expert Rev. Mol. Med. 13:e12. 10.1017/S1462399411001840 [DOI] [PubMed] [Google Scholar]
- 104.Locksley RM, Wakil AE, Corry DB, Pingel S, Bix M, Fowell DJ. 1995. The development of effector T cell subsets in murine Leishmania major infection. Ciba Found. Symp. 195:110–117 [DOI] [PubMed] [Google Scholar]
- 105.Wolf AJ, Linas B, Trevejo-Nunez GJ, Kincaid E, Tamura T, Takatsu K, Ernst JD. 2007. Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo. J. Immunol. 179:2509–2519 http://www.jimmunol.org/content/179/4/2509.long [DOI] [PubMed] [Google Scholar]
- 106.Peters NC, Egen JG, Secundino N, Debrabant A, Kimblin N, Kamhawi S, Lawyer P, Fay MP, Germain RN, Sacks D. 2008. In vivo imaging reveals an essential role for neutrophils in leishmaniasis transmitted by sand flies. Science 321:970–974. 10.1126/science.1159194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tam MA, Rydstrom A, Sundquist M, Wick MJ. 2008. Early cellular responses to Salmonella infection: dendritic cells, monocytes, and more. Immunol. Rev. 225:140–162. 10.1111/j.1600-065X.2008.00679.x [DOI] [PubMed] [Google Scholar]
- 108.Chackerian AA, Alt JM, Perera TV, Dascher CC, Behar SM. 2002. Dissemination of Mycobacterium tuberculosis is influenced by host factors and precedes the initiation of T-cell immunity. Infect. Immun. 70:4501–4509. 10.1128/IAI.70.8.4501-4509.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Moll H, Fuchs H, Blank C, Rollinghoff M. 1993. Langerhans cells transport Leishmania major from the infected skin to the draining lymph node for presentation to antigen-specific T cells. Eur. J. Immunol. 23:1595–1601. 10.1002/eji.1830230730 [DOI] [PubMed] [Google Scholar]
- 110.Voedisch S, Koenecke C, David S, Herbrand H, Forster R, Rhen M, Pabst O. 2009. Mesenteric lymph nodes confine dendritic cell-mediated dissemination of Salmonella enterica serovar Typhimurium and limit systemic disease in mice. Infect. Immun. 77:3170–3180. 10.1128/IAI.00272-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pagan AJ, Peters NC, Debrabant A, Ribeiro-Gomes F, Pepper M, Karp CL, Jenkins MK, Sacks DL. 2013. Tracking antigen-specific CD4+ T cells throughout the course of chronic Leishmania major infection in resistant mice. Eur. J. Immunol. 43:427–438. 10.1002/eji.201242715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wolf AJ, Desvignes L, Linas B, Banaiee N, Tamura T, Takatsu K, Ernst JD. 2008. Initiation of the adaptive immune response to Mycobacterium tuberculosis depends on antigen production in the local lymph node, not the lungs. J. Exp. Med. 205:105–115. 10.1084/jem.20071367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nelson RW, McLachlan JB, Kurtz JR, Jenkins MK. 2013. CD4+ T cell persistence and function after infection are maintained by low-level peptide:MHC class II presentation. J. Immunol. 190:2828–2834. 10.4049/jimmunol.1202183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shafiani S, Dinh C, Ertelt JM, Moguche AO, Siddiqui I, Smigiel KS, Sharma P, Campbell DJ, Way SS, Urdahl KB. 2013. Pathogen-specific Treg cells expand early during Mycobacterium tuberculosis infection but are later eliminated in response to interleukin-12. Immunity 38:1261–1270. 10.1016/j.immuni.2013.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Reiley WW, Shafiani S, Wittmer ST, Tucker-Heard G, Moon JJ, Jenkins MK, Urdahl KB, Winslow GM, Woodland DL. 2010. Distinct functions of antigen-specific CD4 T cells during murine Mycobacterium tuberculosis infection. Proc. Natl. Acad. Sci. U. S. A. 107:19408–19413. 10.1073/pnas.1006298107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ernst JD. 2012. The immunological life cycle of tuberculosis. Nat. Rev. Immunol. 12:581–591. 10.1038/nri3259 [DOI] [PubMed] [Google Scholar]
- 117.Egen JG, Rothfuchs AG, Feng CG, Winter N, Sher A, Germain RN. 2008. Macrophage and T cell dynamics during the development and disintegration of mycobacterial granulomas. Immunity 28:271–284. 10.1016/j.immuni.2007.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ridley DS, Ridley MJ. 1983. The evolution of the lesion in cutaneous leishmaniasis. J. Pathol. 141:83–96. 10.1002/path.1711410109 [DOI] [PubMed] [Google Scholar]
- 119.Belkaid Y, Mendez S, Lira R, Kadambi N, Milon G, Sacks D. 2000. A natural model of Leishmania major infection reveals a prolonged “silent” phase of parasite amplification in the skin before the onset of lesion formation and immunity. J. Immunol. 165:969–977 http://www.jimmunol.org/content/165/2/969.long [DOI] [PubMed] [Google Scholar]
- 120.Guilloteau L, Buzoni-Gatel D, Blaise F, Bernard F, Pepin M. 1991. Phenotypic analysis of splenic lymphocytes and immunohistochemical study of hepatic granulomas after a murine infection with Salmonella abortusovis. Immunology 74:630–637 [PMC free article] [PubMed] [Google Scholar]
- 121.Vazquez-Torres A, Jones-Carson J, Baumler AJ, Falkow S, Valdivia R, Brown W, Le M, Berggren R, Parks WT, Fang FC. 1999. Extraintestinal dissemination of Salmonella by CD18-expressing phagocytes. Nature 401:804–808. 10.1038/44593 [DOI] [PubMed] [Google Scholar]
- 122.Noss EH, Harding CV, Boom WH. 2000. Mycobacterium tuberculosis inhibits MHC class II antigen processing in murine bone marrow macrophages. Cell. Immunol. 201:63–74. 10.1006/cimm.2000.1633 [DOI] [PubMed] [Google Scholar]
- 123.Pancholi P, Mirza A, Bhardwaj N, Steinman RM. 1993. Sequestration from immune CD4+ T cells of mycobacteria growing in human macrophages. Science 260:984–986. 10.1126/science.8098550 [DOI] [PubMed] [Google Scholar]
- 124.Wick MJ, Harding CV, Twesten NJ, Normark SJ, Pfeifer JD. 1995. The phoP locus influences processing and presentation of Salmonella typhimurium antigens by activated macrophages. Mol. Microbiol. 16:465–476. 10.1111/j.1365-2958.1995.tb02411.x [DOI] [PubMed] [Google Scholar]
- 125.Halici S, Zenk SF, Jantsch J, Hensel M. 2008. Functional analysis of the Salmonella pathogenicity island 2-mediated inhibition of antigen presentation in dendritic cells. Infect. Immun. 76:4924–4933. 10.1128/IAI.00531-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mitchell EK, Mastroeni P, Kelly AP, Trowsdale J. 2004. Inhibition of cell surface MHC class II expression by Salmonella. Eur. J. Immunol. 34:2559–2567. 10.1002/eji.200425314 [DOI] [PubMed] [Google Scholar]
- 127.Fruth U, Solioz N, Louis JA. 1993. Leishmania major interferes with antigen presentation by infected macrophages. J. Immunol. 150:1857–1864 [PubMed] [Google Scholar]
- 128.Bobadilla K, Sada E, Jaime ME, Gonzalez Y, Ramachandra L, Rojas RE, Pedraza-Sanchez S, Michalak C, Gonzalez-Noriega A, Torres M. 2013. Human phagosome processing of Mycobacterium tuberculosis antigens is modulated by interferon-gamma and interleukin-10. Immunology 138:34–46. 10.1111/imm.12010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ramachandra L, Smialek JL, Shank SS, Convery M, Boom WH, Harding CV. 2005. Phagosomal processing of Mycobacterium tuberculosis antigen 85B is modulated independently of mycobacterial viability and phagosome maturation. Infect. Immun. 73:1097–1105. 10.1128/IAI.73.2.1097-1105.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Majlessi L, Combaluzier B, Albrecht I, Garcia JE, Nouze C, Pieters J, Leclerc C. 2007. Inhibition of phagosome maturation by mycobacteria does not interfere with presentation of mycobacterial antigens by MHC molecules. J. Immunol. 179:1825–1833 http://www.jimmunol.org/content/179/3/1825.long [DOI] [PubMed] [Google Scholar]
- 131.Lin E, Kemball CC, Hadley A, Wilson JJ, Hofstetter AR, Pack CD, Lukacher AE. 2010. Heterogeneity among viral antigen-specific CD4+ T cells and their de novo recruitment during persistent polyomavirus infection. J. Immunol. 185:1692–1700. 10.4049/jimmunol.0904210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wherry EJ. 2011. T cell exhaustion. Nat. Immunol. 12:492–499. 10.1038/ni.2035 [DOI] [PubMed] [Google Scholar]
- 133.Cooper AM, Magram J, Ferrante J, Orme IM. 1997. Interleukin 12 (IL-12) is crucial to the development of protective immunity in mice intravenously infected with Mycobacterium tuberculosis. J. Exp. Med. 186:39–45. 10.1084/jem.186.1.39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jouanguy E, Doffinger R, Dupuis S, Pallier A, Altare F, Casanova JL. 1999. IL-12 and IFN-gamma in host defense against mycobacteria and Salmonella in mice and men. Curr. Opin. Immunol. 11:346–351. 10.1016/S0952-7915(99)80055-7 [DOI] [PubMed] [Google Scholar]
- 135.von Stebut E, Belkaid Y, Jakob T, Sacks DL, Udey MC. 1998. Uptake of Leishmania major amastigotes results in activation and interleukin 12 release from murine skin-derived dendritic cells: implications for the initiation of anti-Leishmania immunity. J. Exp. Med. 188:1547–1552. 10.1084/jem.188.8.1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.DeKrey GK, Jones JJ, Mbow ML, Brodskyn CI, Titus RG. 2003. Requirement of B cells for delayed type hypersensitivity-like pathology after secondary infection with Leishmania major in resistant C57BL/6 mice. Am. J. Trop. Med. Hyg. 69:481–483 http://www.ajtmh.org/content/69/5/481.long [PubMed] [Google Scholar]
- 137.Mastroeni P, Simmons C, Fowler R, Hormaeche CE, Dougan G. 2000. Igh-6(-/-) (B-cell-deficient) mice fail to mount solid acquired resistance to oral challenge with virulent Salmonella enterica serovar Typhimurium and show impaired Th1 T-cell responses to Salmonella antigens. Infect. Immun. 68:46–53. 10.1128/IAI.68.1.46-53.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.McSorley SJ, Jenkins MK. 2000. Antibody is required for protection against virulent but not attenuated Salmonella enterica serovar Typhimurium. Infect. Immun. 68:3344–3348. 10.1128/IAI.68.6.3344-3348.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hernandez-Pando R, Orozco H, Aguilar D. 2009. Factors that deregulate the protective immune response in tuberculosis. Arch. Immunol. Ther. Exp. (Warsz.) 57:355–367. 10.1007/s00005-009-0042-9 [DOI] [PubMed] [Google Scholar]
- 140.Flynn JL, Scanga CA, Tanaka KE, Chan J. 1998. Effects of aminoguanidine on latent murine tuberculosis. J. Immunol. 160:1796–1803 [PubMed] [Google Scholar]
- 141.MacMicking JD, North RJ, LaCourse R, Mudgett JS, Shah SK, Nathan CF. 1997. Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 94:5243–5248. 10.1073/pnas.94.10.5243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Alam MS, Akaike T, Okamoto S, Kubota T, Yoshitake J, Sawa T, Miyamoto Y, Tamura F, Maeda H. 2002. Role of nitric oxide in host defense in murine salmonellosis as a function of its antibacterial and antiapoptotic activities. Infect. Immun. 70:3130–3142. 10.1128/IAI.70.6.3130-3142.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Stenger S, Donhauser N, Thuring H, Rollinghoff M, Bogdan C. 1996. Reactivation of latent leishmaniasis by inhibition of inducible nitric oxide synthase. J. Exp. Med. 183:1501–1514. 10.1084/jem.183.4.1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wei XQ, Charles IG, Smith A, Ure J, Feng GJ, Huang FP, Xu D, Muller W, Moncada S, Liew FY. 1995. Altered immune responses in mice lacking inducible nitric oxide synthase. Nature 375:408–411. 10.1038/375408a0 [DOI] [PubMed] [Google Scholar]
- 145.Lindenstrom T, Agger EM, Korsholm KS, Darrah PA, Aagaard C, Seder RA, Rosenkrands I, Andersen P. 2009. Tuberculosis subunit vaccination provides long-term protective immunity characterized by multifunctional CD4 memory T cells. J. Immunol. 182:8047–8055. 10.4049/jimmunol.0801592 [DOI] [PubMed] [Google Scholar]
- 146.Shaler CR, Horvath C, Lai R, Xing Z. 2012. Understanding delayed T-cell priming, lung recruitment, and airway luminal T-cell responses in host defense against pulmonary tuberculosis. Clin. Dev. Immunol. 2012:628293. 10.1155/2012/628293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Schaible UE, Sturgill-Koszycki S, Schlesinger PH, Russell DG. 1998. Cytokine activation leads to acidification and increases maturation of Mycobacterium avium-containing phagosomes in murine macrophages. J. Immunol. 160:1290–1296 [PubMed] [Google Scholar]
- 148.Srivastava S, Ernst JD. 2013. Direct recognition of infected cells by CD4 T cells is required for control of intracellular Mycobacterium tuberculosis in vivo. J. Immunol. 191:1016–1020. 10.4049/jimmunol.1301236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Huse M, Lillemeier BF, Kuhns MS, Chen DS, Davis MM. 2006. T cells use two directionally distinct pathways for cytokine secretion. Nat. Immunol. 7:247–255. 10.1038/ni1304 [DOI] [PubMed] [Google Scholar]
- 150.Marshall NB, Swain SL. 2011. Cytotoxic CD4 T cells in antiviral immunity. J. Biomed. Biotechnol. 2011:954602. 10.1155/2011/954602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Qui HZ, Hagymasi AT, Bandyopadhyay S, St Rose MC, Ramanarasimhaiah R, Menoret A, Mittler RS, Gordon SM, Reiner SL, Vella AT, Adler AJ. 2011. CD134 plus CD137 dual costimulation induces eomesodermin in CD4 T cells to program cytotoxic Th1 differentiation. J. Immunol. 187:3555–3564. 10.4049/jimmunol.1101244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Curran MA, Geiger TL, Montalvo W, Kim M, Reiner SL, Al-Shamkhani A, Sun JC, Allison JP. 2013. Systemic 4-1BB activation induces a novel T cell phenotype driven by high expression of eomesodermin. J. Exp. Med. 210:743–755. 10.1084/jem.20121190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Khader SA, Cooper AM. 2008. IL-23 and IL-17 in tuberculosis. Cytokine 41:79–83. 10.1016/j.cyto.2007.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lee SJ, McLachlan JB, Kurtz JR, Fan D, Winter SE, Baumler AJ, Jenkins MK, McSorley SJ. 2012. Temporal expression of bacterial proteins instructs host CD4 T cell expansion and Th17 development. PLoS Pathog. 8:e1002499. 10.1371/journal.ppat.1002499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Khader SA, Bell GK, Pearl JE, Fountain JJ, Rangel-Moreno J, Cilley GE, Shen F, Eaton SM, Gaffen SL, Swain SL, Locksley RM, Haynes L, Randall TD, Cooper AM. 2007. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat. Immunol. 8:369–377. 10.1038/ni1449 [DOI] [PubMed] [Google Scholar]
- 156.Freches D, Korf H, Denis O, Havaux X, Huygen K, Romano M. 2013. Mice genetically inactivated in interleukin-17A receptor are defective in long-term control of Mycobacterium tuberculosis infection. Immunology 140:220–231. 10.1111/imm.12130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Khader SA, Pearl JE, Sakamoto K, Gilmartin L, Bell GK, Jelley-Gibbs DM, Ghilardi N, deSauvage F, Cooper AM. 2005. IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen-specific IFN-gamma responses if IL-12p70 is available. J. Immunol. 175:788–795 http://www.jimmunol.org/content/175/2/788.long [DOI] [PubMed] [Google Scholar]
- 158.Mayuzumi H, Inagaki-Ohara K, Uyttenhove C, Okamoto Y, Matsuzaki G. 2010. Interleukin-17A is required to suppress invasion of Salmonella enterica serovar Typhimurium to enteric mucosa. Immunology 131:377–385. 10.1111/j.1365-2567.2010.03310.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Gonzalez-Lombana C, Gimblet C, Bacellar O, Oliveira WW, Passos S, Carvalho LP, Goldschmidt M, Carvalho EM, Scott P. 2013. IL-17 mediates immunopathology in the absence of IL-10 following Leishmania major infection. PLoS Pathog. 9:e1003243. 10.1371/journal.ppat.1003243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.McDonald DR. 2012. TH17 deficiency in human disease. J. Allergy Clin. Immunol. 129:1429–1435. 10.1016/j.jaci.2012.03.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.de Jong R, Altare F, Haagen IA, Elferink DG, Boer T, van Breda Vriesman PJ, Kabel PJ, Draaisma JM, van Dissel JT, Kroon FP, Casanova JL, Ottenhoff TH. 1998. Severe mycobacterial and Salmonella infections in interleukin-12 receptor-deficient patients. Science 280:1435–1438. 10.1126/science.280.5368.1435 [DOI] [PubMed] [Google Scholar]
- 162.Iwasaki A. 2010. Antiviral immune responses in the genital tract: clues for vaccines. Nat. Rev. Immunol. 10:699–711. 10.1038/nri2836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Nopora K, Bernhard CA, Ried C, Castello AA, Murphy KM, Marconi P, Koszinowski U, Brocker T. 2012. MHC class I cross-presentation by dendritic cells counteracts viral immune evasion. Front. Immunol. 3:348. 10.3389/fimmu.2012.00348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Salio M, Cella M, Suter M, Lanzavecchia A. 1999. Inhibition of dendritic cell maturation by herpes simplex virus. Eur. J. Immunol. 29:3245–3253 [DOI] [PubMed] [Google Scholar]
- 165.Kruse M, Rosorius O, Kratzer F, Stelz G, Kuhnt C, Schuler G, Hauber J, Steinkasserer A. 2000. Mature dendritic cells infected with herpes simplex virus type 1 exhibit inhibited T-cell stimulatory capacity. J. Virol. 74:7127–7136. 10.1128/JVI.74.15.7127-7136.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Coffin RS, Thomas SK, Thomas NS, Lilley CE, Pizzey AR, Griffiths CH, Gibb BJ, Wagstaff MJ, Inges SJ, Binks MH, Chain BM, Thrasher AJ, Rutault K, Latchman DS. 1998. Pure populations of transduced primary human cells can be produced using GFP expressing herpes virus vectors and flow cytometry. Gene Ther. 5:718–722. 10.1038/sj.gt.3300634 [DOI] [PubMed] [Google Scholar]
- 167.Mikloska Z, Bosnjak L, Cunningham AL. 2001. Immature monocyte-derived dendritic cells are productively infected with herpes simplex virus type 1. J. Virol. 75:5958–5964. 10.1128/JVI.75.13.5958-5964.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Samady L, Costigliola E, MacCormac L, McGrath Y, Cleverley S, Lilley CE, Smith J, Latchman DS, Chain B, Coffin RS. 2003. Deletion of the virion host shutoff protein (vhs) from herpes simplex virus (HSV) relieves the viral block to dendritic cell activation: potential of vhs- HSV vectors for dendritic cell-mediated immunotherapy. J. Virol. 77:3768–3776. 10.1128/JVI.77.6.3768-3776.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Smiley JR. 2004. Herpes simplex virus virion host shutoff protein: immune evasion mediated by a viral RNase? J. Virol. 78:1063–1068. 10.1128/JVI.78.3.1063-1068.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Ruby T, McLaughlin L, Gopinath S, Monack D. 2012. Salmonella's long-term relationship with its host. FEMS Microbiol. Rev. 36:600–615. 10.1111/j.1574-6976.2012.00332.x [DOI] [PubMed] [Google Scholar]
- 171.Ehlers S, Schaible UE. 2012. The granuloma in tuberculosis: dynamics of a host-pathogen collusion. Front. Immunol. 3:411. 10.3389/fimmu.2012.00411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.McElrath MJ, Murray HW, Cohn ZA. 1988. The dynamics of granuloma formation in experimental visceral leishmaniasis. J. Exp. Med. 167:1927–1937. 10.1084/jem.167.6.1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Tuon FF, Fernandes ER, Pagliari C, Duarte MI, Amato VS. 2010. The expression of TLR9 in human cutaneous leishmaniasis is associated with granuloma. Parasite Immunol. 32:769–772. 10.1111/j.1365-3024.2010.01243.x [DOI] [PubMed] [Google Scholar]
- 174.Russell DG. 2007. Who puts the tubercle in tuberculosis? Nat. Rev. Microbiol. 5:39–47. 10.1038/nrmicro1538 [DOI] [PubMed] [Google Scholar]
- 175.Saunders BM, Cooper AM. 2000. Restraining mycobacteria: role of granulomas in mycobacterial infections. Immunol. Cell Biol. 78:334–341. 10.1046/j.1440-1711.2000.00933.x [DOI] [PubMed] [Google Scholar]
- 176.Taylor JL, Hattle JM, Dreitz SA, Troudt JM, Izzo LS, Basaraba RJ, Orme IM, Matrisian LM, Izzo AA. 2006. Role for matrix metalloproteinase 9 in granuloma formation during pulmonary Mycobacterium tuberculosis infection. Infect. Immun. 74:6135–6144. 10.1128/IAI.02048-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Volkman HE, Pozos TC, Zheng J, Davis JM, Rawls JF, Ramakrishnan L. 2010. Tuberculous granuloma induction via interaction of a bacterial secreted protein with host epithelium. Science 327:466–469. 10.1126/science.1179663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Bold TD, Banaei N, Wolf AJ, Ernst JD. 2011. Suboptimal activation of antigen-specific CD4+ effector cells enables persistence of M. tuberculosis in vivo. PLoS Pathog. 7:e1002063. 10.1371/journal.ppat.1002063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Egen JG, Rothfuchs AG, Feng CG, Horwitz MA, Sher A, Germain RN. 2011. Intravital imaging reveals limited antigen presentation and T cell effector function in mycobacterial granulomas. Immunity 34:807–819. 10.1016/j.immuni.2011.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.O'Garra A, Vieira P. 2007. T(H)1 cells control themselves by producing interleukin-10. Nat. Rev. Immunol. 7:425–428. 10.1038/nri2097 [DOI] [PubMed] [Google Scholar]
- 181.Gerosa F, Paganin C, Peritt D, Paiola F, Scupoli MT, Aste-Amezaga M, Frank I, Trinchieri G. 1996. Interleukin-12 primes human CD4 and CD8 T cell clones for high production of both interferon-gamma and interleukin-10. J. Exp. Med. 183:2559–2569. 10.1084/jem.183.6.2559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Windhagen A, Anderson DE, Carrizosa A, Williams RE, Hafler DA. 1996. IL-12 induces human T cells secreting IL-10 with IFN-gamma. J. Immunol. 157:1127–1131 [PubMed] [Google Scholar]
- 183.Meyaard L, Hovenkamp E, Otto SA, Miedema F. 1996. IL-12-induced IL-10 production by human T cells as a negative feedback for IL-12-induced immune responses. J. Immunol. 156:2776–2782 [PubMed] [Google Scholar]
- 184.Shaw MH, Freeman GJ, Scott MF, Fox BA, Bzik DJ, Belkaid Y, Yap GS. 2006. Tyk2 negatively regulates adaptive Th1 immunity by mediating IL-10 signaling and promoting IFN-gamma-dependent IL-10 reactivation. J. Immunol. 176:7263–7271 http://www.jimmunol.org/content/176/12/7263.long [DOI] [PubMed] [Google Scholar]
- 185.Ding L, Linsley PS, Huang LY, Germain RN, Shevach EM. 1993. IL-10 inhibits macrophage costimulatory activity by selectively inhibiting the up-regulation of B7 expression. J. Immunol. 151:1224–1234 [PubMed] [Google Scholar]
- 186.de Waal Malefyt R, Abrams J, Bennett B, Figdor CG, de Vries JE. 1991. Interleukin 10 (IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J. Exp. Med. 174:1209–1220. 10.1084/jem.174.5.1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Gazzinelli RT, Wysocka M, Hieny S, Scharton-Kersten T, Cheever A, Kuhn R, Muller W, Trinchieri G, Sher A. 1996. In the absence of endogenous IL-10, mice acutely infected with Toxoplasma gondii succumb to a lethal immune response dependent on CD4+ T cells and accompanied by overproduction of IL-12, IFN-gamma and TNF-alpha. J. Immunol. 157:798–805 [PubMed] [Google Scholar]
- 188.Vouldoukis I, Becherel PA, Riveros-Moreno V, Arock M, da Silva O, Debre P, Mazier D, Mossalayi MD. 1997. Interleukin-10 and interleukin-4 inhibit intracellular killing of Leishmania infantum and Leishmania major by human macrophages by decreasing nitric oxide generation. Eur. J. Immunol. 27:860–865. 10.1002/eji.1830270409 [DOI] [PubMed] [Google Scholar]
- 189.Redford PS, Boonstra A, Read S, Pitt J, Graham C, Stavropoulos E, Bancroft GJ, O'Garra A. 2010. Enhanced protection to Mycobacterium tuberculosis infection in IL-10-deficient mice is accompanied by early and enhanced Th1 responses in the lung. Eur. J. Immunol. 40:2200–2210. 10.1002/eji.201040433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Arai T, Hiromatsu K, Nishimura H, Kimura Y, Kobayashi N, Ishida H, Nimura Y, Yoshikai Y. 1995. Effects of in vivo administration of anti-IL-10 monoclonal antibody on the host defence mechanism against murine Salmonella infection. Immunology 85:381–388 [PMC free article] [PubMed] [Google Scholar]
- 191.Belkaid Y, Piccirillo CA, Mendez S, Shevach EM, Sacks DL. 2002. CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature 420:502–507. 10.1038/nature01152 [DOI] [PubMed] [Google Scholar]
- 192.Suffia IJ, Reckling SK, Piccirillo CA, Goldszmid RS, Belkaid Y. 2006. Infected site-restricted Foxp3+ natural regulatory T cells are specific for microbial antigens. J. Exp. Med. 203:777–788. 10.1084/jem.20052056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Suffia I, Reckling SK, Salay G, Belkaid Y. 2005. A role for CD103 in the retention of CD4+CD25+ Treg and control of Leishmania major infection. J. Immunol. 174:5444–5455 http://www.jimmunol.org/content/174/9/5444.long [DOI] [PubMed] [Google Scholar]
- 194.Scott-Browne JP, Shafiani S, Tucker-Heard G, Ishida-Tsubota K, Fontenot JD, Rudensky AY, Bevan MJ, Urdahl KB. 2007. Expansion and function of Foxp3-expressing T regulatory cells during tuberculosis. J. Exp. Med. 204:2159–2169. 10.1084/jem.20062105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Ozeki Y, Sugawara I, Udagawa T, Aoki T, Osada-Oka M, Tateishi Y, Hisaeda H, Nishiuchi Y, Harada N, Kobayashi K, Matsumoto S. 2010. Transient role of CD4+CD25+ regulatory T cells in mycobacterial infection in mice. Int. Immunol. 22:179–189. 10.1093/intimm/dxp126 [DOI] [PubMed] [Google Scholar]
- 196.Barnes PF, Chatterjee D, Abrams JS, Lu S, Wang E, Yamamura M, Brennan PJ, Modlin RL. 1992. Cytokine production induced by Mycobacterium tuberculosis lipoarabinomannan. Relationship to chemical structure. J. Immunol. 149:541–547 [PubMed] [Google Scholar]
- 197.Barnes PF, Lu S, Abrams JS, Wang E, Yamamura M, Modlin RL. 1993. Cytokine production at the site of disease in human tuberculosis. Infect. Immun. 61:3482–3489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Uchiya K, Groisman EA, Nikai T. 2004. Involvement of Salmonella pathogenicity island 2 in the up-regulation of interleukin-10 expression in macrophages: role of protein kinase A signal pathway. Infect. Immun. 72:1964–1973. 10.1128/IAI.72.4.1964-1973.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Kane MM, Mosser DM. 2001. The role of IL-10 in promoting disease progression in leishmaniasis. J. Immunol. 166:1141–1147 http://www.jimmunol.org/content/166/2/1141.long [DOI] [PubMed] [Google Scholar]
- 200.O'Leary S, O'Sullivan MP, Keane J. 2011. IL-10 blocks phagosome maturation in Mycobacterium tuberculosis-infected human macrophages. Am. J. Respir. Cell Mol. Biol. 45:172–180. 10.1165/rcmb.2010-0319OC [DOI] [PubMed] [Google Scholar]
- 201.Via LE, Fratti RA, McFalone M, Pagan-Ramos E, Deretic D, Deretic V. 1998. Effects of cytokines on mycobacterial phagosome maturation. J. Cell Sci. 111:897–905 [DOI] [PubMed] [Google Scholar]
- 202.Greenblatt CL. 1988. Cutaneous leishmaniasis: the prospects for a killed vaccine. Parasitol. Today 4:53–54. 10.1016/0169-4758(88)90067-1 [DOI] [PubMed] [Google Scholar]
- 203.Kaye P, Scott P. 2011. Leishmaniasis: complexity at the host-pathogen interface. Nat. Rev. Microbiol. 9:604–615. 10.1038/nrmicro2608 [DOI] [PubMed] [Google Scholar]
- 204.Mendez S, Reckling SK, Piccirillo CA, Sacks D, Belkaid Y. 2004. Role for CD4(+) CD25(+) regulatory T cells in reactivation of persistent leishmaniasis and control of concomitant immunity. J. Exp. Med. 200:201–210. 10.1084/jem.20040298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Aebischer T, Moody SF, Handman E. 1993. Persistence of virulent Leishmania major in murine cutaneous leishmaniasis: a possible hazard for the host. Infect. Immun. 61:220–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Sacks D, Anderson C. 2004. Re-examination of the immunosuppressive mechanisms mediating non-cure of Leishmania infection in mice. Immunol. Rev. 201:225–238. 10.1111/j.0105-2896.2004.00185.x [DOI] [PubMed] [Google Scholar]
- 207.Uzonna JE, Wei G, Yurkowski D, Bretscher P. 2001. Immune elimination of Leishmania major in mice: implications for immune memory, vaccination, and reactivation disease. J. Immunol. 167:6967–6974 http://www.jimmunol.org/content/167/12/6967.long [DOI] [PubMed] [Google Scholar]
- 208.Zaph C, Uzonna J, Beverley SM, Scott P. 2004. Central memory T cells mediate long-term immunity to Leishmania major in the absence of persistent parasites. Nat. Med. 10:1104–1110. 10.1038/nm1108 [DOI] [PubMed] [Google Scholar]
- 209.Ottenhoff TH, Kaufmann SH. 2012. Vaccines against tuberculosis: where are we and where do we need to go? PLoS Pathog. 8:e1002607. 10.1371/journal.ppat.1002607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Alvar J, Aparicio P, Aseffa A, Den Boer M, Canavate C, Dedet JP, Gradoni L, Ter Horst R, Lopez-Velez R, Moreno J. 2008. The relationship between leishmaniasis and AIDS: the second 10 years. Clin. Microbiol. Rev. 21:334–359. 10.1128/CMR.00061-07 [DOI] [PMC free article] [PubMed] [Google Scholar]