

Abstract
There are major gaps in our understanding of the immunopathogenesis of Clostridium difficile infections (CDIs). In this study, 36 different biomarkers were examined in the stools of CDI and non-CDI patients using the Proteome Profiler human cytokine array assay and quantitative enzyme-linked immunosorbent assay. Diarrheal stools from patients with CDI (CDI-positive diarrheal stools) showed higher relative amounts of the following inflammatory markers than the diarrheal stools from CDI-negative patients (CDI-negative diarrheal stools): C5a, CD40L, granulocyte colony-stimulating factor, I-309, interleukin-13 (IL-13), IL-16, IL-27, monocyte chemoattractant protein 1, tumor necrosis factor alpha, and IL-8. IL-8 and IL-23 were present in a larger number of CDI-positive diarrheal stools than CDI-negative diarrheal stools. Th1 and Th2 cytokines were not significantly different between the CDI-positive and CDI-negative diarrheal stools. Lactoferrin and calprotectin concentrations were also higher in the CDI-positive diarrheal stools. Our results demonstrate that CDI elicits a proinflammatory host response, and we report for the first time that IL-23 is a major marker in CDI-positive diarrheal stools. IL-23 may explain the lack of a robust immunological response exhibited by a proportion of CDI patients and may relate to recurrence; the IL-23 levels induced during CDI in these patients may be inadequate to sustain the cellular immunity conferred by this cytokine in promoting the induction and proliferation of effector memory T cells.
INTRODUCTION
Clostridium difficile infection (CDI) is the leading cause of infectious antibiotic-associated diarrhea and a major problem for the elderly and immunocompromised (1). The incidence of CDI has more than doubled since 1996, making CDI the most common health care-associated bacterial infection in many hospitals in the United States (2). One of the hallmarks of CDI is inflammation of the colonic mucosa due to the actions of toxins A and B, which are released by this bacterium during infection (3). The toxins act on the actin filaments of colonic epithelial cells, in which this action disrupts tight junctions, induces apoptosis, and causes infiltration and aggregation of neutrophils (4, 5).
Recruitment and aggregation of neutrophils are early significant events in CDI, which in severe cases can lead to pseudomembranous colitis (6, 7). Only 28 to 63% of stools from CDI patients show high neutrophil counts (8), in spite of the inflammatory nature of the infection. As a result, other leukocyte-derived products excreted in feces, such as cytokines, lactoferrin, and calprotectin, have emerged as candidate biomarkers of intestinal inflammation (9, 10). Fecal lactoferrin is elevated in patients with highly inflammatory enteric diseases but not in healthy controls (6). The increase in fecal lactoferrin levels during intestinal inflammation is proportional to the level of translocation of neutrophils into the gastrointestinal tract (11). More than 60% of the total cytosolic protein content of neutrophils is calprotectin. This calcium- and zinc-binding protein plays a regulatory role in inflammation with antimicrobial and antiproliferative properties (12). Fecal calprotectin levels correlate with fecal excretion of neutrophils (13). Fecal lactoferrin and calprotectin levels help predict the increased translocation of granulocytes into the intestinal mucosa.
In this study, we sought to examine the immunopathogenesis of CDI by analyzing the major inflammatory markers present in the stools of CDI and non-CDI patients with antibiotic-associated diarrhea and hospitalized controls without diarrhea. This was done with the goal to establish the expression pattern of these biomarkers during CDI to help provide insight into the immunological pathogenesis of CDI.
MATERIALS AND METHODS
Patient population.
Stool samples were collected from patients enrolled for an ongoing study approved by the Institutional Review Board of The University of Texas Health Science Center at Houston, TX (14). All of the participating patients or their legal guardians provided written informed consent upon admission to the hospital. The patients were enrolled at a 700-bed university hospital in the Texas Medical Center in Houston, TX, whose physicians ordered a stool examination for CDI from December 2010 to June 2011.
Stool samples.
A total of 100 antibiotic-associated diarrheal stool samples from 100 different patients (50 CDI positive, 50 CDI negative) and 45 stool samples from hospitalized patients without diarrhea were analyzed. The stools were collected on the first day that the patients reported to the hospital and stored at −80°C until tested. Stool samples were tested by the tissue culture cytotoxicity assay and cultured on cycloserine cefoxitin fructose agar (CCFA) medium and Cdifftox agar plates (15), followed by PCR for the presence of the toxin genes (tcdA and/or tcdB). All of the stool samples classified as CDI negative were further tested and confirmed to be negative for the C. difficile toxins using the Wampole C. difficile Tox A/B II assay (Techlab, Blacksburg, VA). The CDI-positive and CDI-negative samples were matched by date of collection. The control stools were negative for C. difficile by the real-time BD GeneOhm Cdiff PCR assay for the tcdB gene and enzyme-linked immunosorbent assay (ELISA) for toxins A and B.
Cytokine assays.
A total of 50 diarrheal stool samples from patients with CDI (CDI-positive diarrheal stools) and 50 diarrheal stool samples from patients without CDI (CDI-negative diarrheal stools) were initially evaluated for the presence and relative amounts of 36 different inflammatory markers using a Proteome Profiler human cytokine array panel A kit (R&D Systems, Minneapolis, MN). The Proteome Profiler human cytokine array assay was performed according to the instructions provided by the manufacturer. Stools from the hospitalized patients without diarrhea were not evaluated by this initial assay but were included retrospectively in the quantitative ELISA (described below) for comparison. Briefly, 300 mg of each stool sample was thoroughly suspended in 1.5 ml of array buffer 5, incubated at room temperature for 15 min, and centrifuged for 5 min at 5,000 × g. The supernatant (1 ml each) was added to a cocktail of biotinylated antibodies and incubated at room temperature for 1 h. The sample-antibody mixture was subsequently incubated at 4°C for 19 h with a membrane embedded with antibodies specific to each of the 36 different inflammatory markers analyzed. Following three washes, 3 ml of a 1:1,000 dilution of streptavidin-horseradish peroxidase (HRP) was added to each membrane and the membrane was incubated at room temperature for 35 min. For detection, the membranes were probed with the Pierce ECL Western blotting substrate (Thermo Scientific, Rockford, IL) and exposed to an X-ray film. The exposed film was processed using SRX-101A medical film processor (Konica Minolta). The pixel densities of each blot (band), representing the amount of each inflammatory marker present, were determined using the ImageJ software (National Institutes of Health, Bethesda, MD).
All the images from the 100 arrays were normalized by subtracting the background and inverted to eliminate the background differences. To measure the pixel density, a fixed-size rectangular box was generated around each dot blot/band and the pixel density was measured. The same-sized rectangular box was used for all the bands in all 100 arrays performed. For analysis, the pixel density of the negative control on each array was subtracted from the pixel density obtained from each band on the array. The data were further converted and normalized into the fold change in amount by dividing the pixel density of each band by the average pixel density of the streptavidin-HRP reference spots located at three corners of each array.
Quantitative ELISAs.
The fecal concentrations of interleukin-8 (IL-8), IL-23, tumor necrosis factor alpha (TNF-α), gamma interferon (IFN-γ), and IL-13 were determined by quantitative ELISA (R&D Systems, Minneapolis, MN), using the instructions provided by the manufacturer. The relative amounts of these cytokines were determined by the initial Proteome Profiler human cytokine array assay to be significantly different between the CDI-positive and CDI-negative diarrheal stools. The concentrations of these cytokines in diarrheal stool samples from 50 CDI-positive and 50 CDI-negative patients and stools from the 45 hospitalized patients without diarrhea were determined.
Lactoferrin and calprotectin assay.
The concentrations of lactoferrin and calprotectin present in the diarrheal stool samples from 50 CDI-positive and 50 CDI-negative patients and the 45 stool samples from hospitalized patients without diarrhea were determined. Fecal lactoferrin was measured using an IBD-SCAN test (Techlab, Blacksburg, VA) with 50 mg of each stool sample. Fecal calprotectin was determined using an HK325 human calprotectin ELISA kit (Hycult Biotech, Plymouth Meeting, PA). Briefly, 100 mg of each stool sample was extracted in 5 ml extraction buffer, and the supernatant was incubated in 96-well microtiter plates coated with human calprotectin-specific antibody for 1 h at 25°C. A biotinylated tracer antibody was added to detect the calprotectin that bound to the antibodies coated on the plate surface. Then, streptavidin-peroxidase conjugate was added. Following incubation, the unbound conjugate was washed and tetramethylbenzidine was used as the detection substrate.
Statistical analysis.
The data were statistically analyzed by the Mann-Whitney two-tailed nonparametric test of significance and one-way analysis of variance using GraphPad Prism (version 5.02) software for Windows (GraphPad Software, San Diego, CA).
RESULTS
A total of 50 stool samples each from CDI-positive and CDI-negative antibiotic-associated diarrheal patients and 45 stool samples from hospitalized patients without diarrhea were collected and examined. The average ages among the study groups were 58, 56, and 63 years for CDI-positive patients, CDI-negative patients, and control patients without diarrhea, respectively. The majority of the patients enrolled were of three major ethnicities: white/Caucasians, black/African-Americans, and Hispanics/Latinos (Table 1). No other major comorbidities were noted among the study population.
TABLE 1.
Characteristics of study populationa
| Characteristic | CDI-positive patients (n = 50) | CDI-negative patients (n = 50) | Control patients with no diarrhea (n = 45) |
|---|---|---|---|
| Avg (range) age (yr) | 58 (2–91) | 56 (21–83) | 63 (22–85) |
| No. of patients by: | |||
| Ethnicity | |||
| Asian | 1 | 1 | 0 |
| B/AA | 10 | 8 | 11 |
| H/L | 4 | 6 | 9 |
| W/C | 28 | 29 | 25 |
| Other | 7 | 6 | |
| Gender | |||
| Female | 27 | 24 | 19 |
| Male | 23 | 26 | 26 |
CDI, patients with C. difficile infection; B/AA, black/African-American; H/L, Hispanic/Latino; W/C, white/Caucasian.
One of the clinical hallmarks of CDI is colonic inflammation, which is generally mediated by cytokines and other proinflammatory proteins, such as toxins A and B. To assess the essential inflammatory biomarkers that are elicited during CDI, the fecal amounts of cytokines, chemokines, and other acute-phase proteins were examined from stools collected from diarrheal and nondiarrheal patients. The CDI-positive and CDI-negative diarrheal stools were initially tested for the presence and relative amounts of key inflammatory biomarkers using the Proteome Profiler human cytokine array panel A kit (R&D Systems, Minneapolis, MN). This assay simultaneously detects 36 different inflammatory biomarkers present in a stool sample. A representative image showing the bands (dot blots) indicative of the inflammatory biomarkers detected in the samples is shown in Fig. 1. The intensities of the bands differed within and between the different stool samples evaluated, suggesting that different amounts of the biomarkers were present in the stools. Some of the 36 inflammatory markers tested were not detected, but the total number of different inflammatory biomarkers detected was also different between the groups of stools examined.
FIG 1.
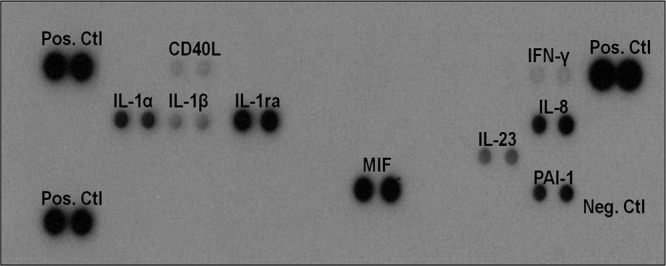
Different inflammatory biomarkers detected in the clinical stool samples examined. Stools (300 mg) from 100 antibiotic-associated diarrheal patients (50 CDI positive and 50 CDI negative) were evaluated for the presence of 36 inflammatory-associated proteins using the Proteome Profiler human cytokine array panel A kit (R&D Systems, Minneapolis, MN). Abbreviations: Pos. Ctl, positive control; Neg. Ctl, negative control; CD40L, CD40 ligand; IFN-γ, gamma interferon, IL-1α, interleukin-1α; IL-1β, interleukin-1β; IL-1ra, interleukin-1 receptor antagonist; IL-8, interleukin-8, IL-23; interleukin-23; MIF, macrophage migration inhibitory factor; PAI-1, plasminogen activator inhibitor 1.
The intensities of the dot blots were empirically determined and expressed as pixel densities using the ImageJ software (National Institutes of Health, Bethesda, MD). Following normalization, the relative band intensities representing the relative amounts of all the inflammatory markers detected in the CDI-positive and CDI-negative diarrheal stools were determined and are shown in Fig. 2. The CDI-positive stools exhibited significantly (P < 0.05) higher relative amounts of the following biomarkers than the CDI-negative diarrheal stools: C5a, CD40L, granulocyte colony-stimulating factor (G-CSF), I-309, IL-13, IL-16, IL-27, monocyte chemoattractant protein 1 (MCP-1), TNF-α, and IL-8. On the other hand, the relative amount of IL-23 was significantly higher (P < 0.05) in the CDI-negative stools than the CDI-positive stools. Interestingly, both IL-8 and IL-23 were more frequently detected in the CDI-positive stools (88% and 90%, respectively) than the CDI-negative stools (54% and 68%, respectively) (Fig. 3).
FIG 2.

Fold change in the amount of inflammatory markers detected in CDI-positive and CDI-negative stools. Stools (300 mg) from 100 antibiotic-associated diarrheal patients (50 CDI positive and 50 CDI negative) were evaluated for the presence of 36 inflammatory biomarkers using a Proteome Profiler human cytokine array panel A kit (R&D Systems, Minneapolis, MN). Band intensities were determined and converted into pixel densities using ImageJ (National Institutes of Health, Bethesda, MD). Error bars represent the standard deviations from the mean values for each inflammatory biomarker. Significant differences between CDI-positive and CDI-negative stools are denoted by asterisks. The P values, based on a nonparametric t test, were 0.011 for C5a, 0.042 for CD40L, 0.045 for G-CSF, 0.030 for I-309, 0.001 for IL-13; 0.031 for IL-16, 0.027 for IL-27, 0.013 for MCP-1, 0.021 for TNF-α, 0.004 for IL-8, and 0.002 for IL-23. Abbreviations: GM-CSF, granulocyte colony-stimulating factor; GRO-α, growth-regulated oncogene-alpha; sICAM-1, soluble intercellular adhesion molecule 1; IP-10, interferon gamma-induced protein 10; I-TAC, interferon-inducible T cell alpha chemoattractant; MIP-1α and MIP-1β, macrophage inflammatory proteins alpha and beta, respectively; SDF-1, stromal cell-derived factor 1; sTREM-1, soluble triggering receptor expressed on myeloid cells 1; MIF, macrophage migration inhibitory factor; PAI-1, plasminogen activator inhibitor 1. The other abbreviations are defined in the text.
FIG 3.

IL-8 and IL-23 were present in the majority of CDI-positive and CDI-negative stools. (A) IL-8 and IL-23 were detected in a greater number of CDI-positive stools than CDI-negative stools. Stools (300 mg) from 100 antibiotic-associated diarrheal patients (50 CDI positive and 50 CDI negative) were evaluated for the presence of 36 inflammation-associated proteins using a Proteome Profiler human cytokine array panel A kit (R&D Systems, Minneapolis, MN). Stools from the hospitalized patients (controls) without diarrhea were not evaluated by this initial assay but were included retrospectively in the quantitative ELISA for comparison. (B) CDI-positive stools contain large amounts of IL-8 and relatively small amounts of IL-23 compared to the amounts in stools from CDI-negative diarrheal patients and hospitalized patients without diarrhea. The concentrations of IL-8 and IL-23 were determined in stools from 50 CDI-positive patients, 50 CDI-negative patients, and 45 nondiarrheal controls by quantitative ELISA (R&D Systems, Minneapolis, MN). A Mann-Whitney two-tailed test showed significant differences between CDI-positive patients and nondiarrheal controls (P = 0.0001), CDI-positive patients and CDI-negative patients with diarrhea (P = 0.002), and CDI-negative patients and nondiarrheal controls (P = 0.006) for IL-8 and CDI-positive patients and nondiarrheal controls (P = 0.001), CDI-positive patients and CDI-negative patients with diarrhea (P = 0.003), and CDI-negative patients and nondiarrheal controls (P = 0.0001) for IL-23. Horizontal bars, mean concentrations.
To confirm the findings made from the data obtained from the Proteome Profiler human cytokine array assay, fecal concentrations of the biomarkers whose band intensities demonstrated significant differences between the CDI-positive and CDI-negative stools were determined by quantitative ELISA (R&D Systems, Minneapolis, MN). Table 2 shows the range, mean, and median concentrations of these biomarkers (IL-8, IL-23, TNF-α, IFN-γ, and IL-13), which were measured from 50 CDI-positive and 50 CDI-negative diarrheal stool samples and 45 nondiarrheal patient stool samples. The average fecal concentration of IL-8 was significantly higher (P < 0.05) in CDI-positive patient stools (318.2 pg/ml) than in CDI-negative patient stools (84.7 pg/ml) and hospitalized patients without diarrhea (79.8 pg/ml). However, the average fecal concentration of IL-23 was significantly lower (P < 0.05) in the CDI-positive stools (722 pg/ml) than the CDI-negative stools (946.7 pg/ml) and the hospitalized patients without diarrhea (1,617 pg/ml). Interestingly, IL-23 was detected in a greater number of the CDI-positive stools than CDI-negative stools, but its concentrations were significantly lower in the CDI-positive stool samples. This is in contrast to the findings for IL-8, which was present in a larger number of the CDI-positive stools and at concentrations higher than those in the CDI-negative stools. These results suggest that IL-23 and IL-8 may be significant in the immunopathogenesis of CDI.
TABLE 2.
Fecal concentrations of IL-13, IL-8, IL-23, TNF-α, IFN-γ, lactoferrin, and calprotectin from the CDI-positive and CDI-negative diarrheal stools and nondiarrheal stools evaluateda
| Patient group | Concn (pg/ml) |
Concn (μg/ml) |
|||||
|---|---|---|---|---|---|---|---|
| IL-13 | IL-8 | IL-23 | TNF-α | IFN-γ | Lactoferrin | Calprotectin | |
| CDI positive (n = 50) | |||||||
| Mean | 323.8 | 318.2 | 722 | 59.8 | 69.7 | 43.4 | 23.8 |
| Median | 277.4 | 242 | 390.5 | 33.7 | 58 | 31.4 | 18 |
| Range | 35.7–1,150 | 13.1–1,529 | 110.0–7,069 | 2.7–163.6 | 8.7–225.7 | 3.0–155.2 | 2.8–70.2 |
| P value | 0.01 | 0.022 | 0.001 | 0.078 | 0.067 | 0.019 | 0.029 |
| CDI negative (n = 50) | |||||||
| Mean | 141.1 | 84.7 | 946.7 | 31.2 | 48.3 | 24.8 | 8.8 |
| Median | 138.8 | 83.3 | 932.6 | 25.7 | 31.5 | 6.3 | 6.5 |
| Range | 14.3–319.3 | 8.5–191.6 | 185.5–2,016 | 0.6–177.7 | 0.04–204.3 | 0.6–140.3 | 2.0–31.0 |
| P value | 0.024 | 0.034 | 0.0032 | 0.055 | 0.088 | 0.028 | 0.054 |
| Nondiarrheal control (n = 45) | |||||||
| Mean | 234.3 | 79.8 | 1,617 | 22.3 | 57.6 | 6.8 | 10.2 |
| Median | 203.5 | 52 | 768 | 18 | 50.2 | 5.6 | 8.7 |
| Range | 19.2–701.7 | 21.4–295.7 | 489.0–6,810 | 0.3–83.3 | 10.9–143.3 | 0.5–35.0 | 1.8–33.2 |
| P value | 0.06 | 0.01 | 0.041 | 0.092 | 0.002 | 0.069 | 0.073 |
Concentrations were determined by quantitative ELISA (R&D Systems, Minneapolis, MN).
Th1- and Th2-associated cytokines were examined to assess the nature of the cell-mediated response that occurs during CDI. Th1 and Th2 cytokines were detected in both CDI-positive and CDI-negative stools by the Proteome Profiler human cytokine array assay. The relative amounts of four key Th1 cytokines (IFN-γ, IL-2, IL-12, TNF-α) and five Th2 cytokines (IL-4, IL-5, IL-6, IL-10, IL-13) that were detected in both groups are shown in Fig. 4A. The CDI-positive stools generally showed larger relative amounts of both Th1 and Th2 cytokines than the CDI-negative stools. However, the differences in the relative amounts from those for the CDI-negative stools were not significant (P > 0.05), except for TNF-α and IL-13, which were present at significantly larger amounts in CDI-positive stools (P = 0.0001 for TNF-α and P = 0.016 for IL-13). These observations were confirmed when the actual concentrations of these cytokines in the stools were measured by quantitative ELISA. The average fecal concentrations of TNF-α (59.8 pg/ml), IFN-γ (69.7 pg/ml), and IL-13 (323.8 pg/ml) were higher in the CDI-positive stools than the CDI-negative stools (TNF-α, 31.2 pg/ml; IFN-γ, 48.3 pg/ml; IL-13, 141.1 pg/ml) and stools from hospitalized patients without diarrhea (TNF-α, 22.3 pg/ml; IFN-γ, 57.6 pg/ml; IL-13, 234.3 pg/ml) (Fig. 4B). These data suggest that the inflammatory response to CDI is complex and encompasses both Th1 and Th2 responses.
FIG 4.

Comparison of Th1 and Th2 cytokines in CDI-positive and CDI-negative stools. (A) Fold change in amounts of Th1 cytokines (IFN-γ, IL-2, IL-12, TNF-α) and Th2 cytokines (IL-4, IL-5, IL-6, IL-10, IL-13) obtained from the initial Proteome Profiler human cytokine array assay. Stools (300 mg) from 100 antibiotic-associated diarrheal patients (50 CDI positive and 50 CDI negative) were evaluated for the presence of 36 inflammatory proteins using a Proteome Profiler human cytokine array panel A kit (R&D Systems, Minneapolis, MN). Data are expressed as the mean of the relative band intensity of each cytokine. Stools from the hospitalized patients (controls) without diarrhea were not evaluated by this initial assay but were included retrospectively in the quantitative ELISA for comparison. Error bars represent the standard error of measurement between two replicates per sample. *, P < 0.05. (B) Concentrations of IFN-γ, TNF-α, and IL-13 in CDI-positive stools and CDI-negative stools from diarrheal patients and stools from hospitalized controls without diarrhea determined by quantitative ELISA (R&D Systems, Minneapolis, MN). The Kruskal-Wallis test showed significant differences between the means (P < 0.0001). Horizontal bars, mean concentrations.
Fecal concentrations of lactoferrin and calprotectin were determined to evaluate whether these innate inflammatory proteins play a role in CDI pathogenesis. As shown in Fig. 5, the median lactoferrin concentration in the CDI-positive stools (31.4 μg/ml) was 5-fold higher than that in the CDI-negative stools (6.3 μg/ml) and about 6-fold higher than that in the stools of hospitalized patients without diarrhea (5.6 μg/ml). The median concentration of calprotectin in the CDI-positive stools (18 μg/ml) was 3-fold higher than that in the CDI-negative stools (6.5 μg/ml) and 2-fold higher than that in the stools of hospitalized patients without diarrhea (8.7 μg/ml). The lactoferrin concentrations of 88% of the CDI-positive stools and 44% of the CDI-negative stools were higher than the average concentration in the stools of the hospitalized patients without diarrhea. Also, 80% of the CDI-positive stools and 30% of the CDI-negative stools had calprotectin concentrations higher than the average concentration in the stools of the hospitalized patients without diarrhea. As evidenced by the high levels of lactoferrin and calprotectin present in the CDI-positive stools, these data together suggest that the majority of the cases of CDI have colonic inflammation, whereas a minority of the cases of antibiotic-associated diarrhea without CDI show colonic inflammation.
FIG 5.

CDI-positive stools contain larger amounts of lactoferrin and calprotectin than CDI-negative diarrheal stools and stools from hospitalized patients (controls) without diarrhea. The concentrations of lactoferrin and calprotectin were measured in stools from 50 CDI-positive and CDI-negative patients and hospitalized patients without diarrhea. Fecal lactoferrin and calprotectin concentrations were determined using an IBD-SCAN test (Techlab, Blacksburg, VA) and an HK325 human calprotectin ELISA kit (Hycult Biotech, Plymouth Meeting, PA), respectively. Median lactoferrin concentrations were 31.4 μg/ml (CDI-positive patient stools), 6.3 μg/ml (CDI-negative patient stools), and 5.6 μg/ml (stools from hospitalized patients without diarrhea). Median calprotectin concentrations were 18.0 μg/ml (CDI-positive patient stools), 6.5 μg/ml (CDI-negative patient stools), and 8.7 μg/ml (stools from hospitalized patients without diarrhea). One-way analysis of variance showed significant differences between the means of the CDI-positive patients, CDI-negative patients, and nondiarrheal controls for both lactoferrin and calprotectin (P < 0.0001).
DISCUSSION
Major gaps still remain in our understanding of the immunopathogenesis of CDI, despite its increasing prevalence rates and poor patient outcomes. The relative amounts of 36 major biomarkers in stools collected from CDI and non-CDI patients were evaluated. Initially, 50 CDI-positive and 50 CDI-negative stool samples obtained from hospitalized patients with antibiotic-associated diarrhea were assessed using the Proteome Profiler human cytokine array assay. The fecal concentrations of the biomarkers that were found by the initial Proteome Profiler human cytokine array assay to be significantly different between the CDI-positive and CDI-negative stools were measured by quantitative ELISA for comparison with those in hospitalized patients without diarrhea. We have provided data demonstrating that the immunopathogenesis of CDI is complex and that CDI elicits both Th1 and Th2 responses, with an increased expression of proinflammatory proteins.
IL-8 and IL-23 appeared to be important in the immunopathogenesis of CDI. These two cytokines were detected in more CDI-positive stools than CDI-negative stools. The average concentration of IL-8 in the CDI-positive stools was significantly (P < 0.05) higher than that in the stools from CDI-negative patients and hospitalized patients without diarrhea. IL-8 is a chemoattractant involved in the recruitment of neutrophils to sites of infection and has been implicated to play a key role in the pathogenesis of CDI (16). Increased levels of IL-8 are associated with more severe forms of CDI (6). Moreover, a single nucleotide polymorphism (SNP) in the promoter region of the IL-8 gene that increases its expression is associated with susceptibility to CDI (17). These reports are consistent with our data showing high levels of IL-8 in the majority of CDI-positive stools compared to levels in stools from the CDI-negative patients and hospitalized patients without diarrhea.
Remarkably, the average concentration of IL-23 in the CDI-positive stools was lower than that in the CDI-negative stools and stools from hospitalized patients without diarrhea, even though IL-23 was present in a higher number of the CDI-positive stools. IL-23 is produced by activated macrophages and dendritic cells and plays an important role in host defense against bacterial infections and the development of chronic inflammation (18). During bacterial infection, antigen-stimulated dendritic cells and macrophages produce IL-23, which promotes the development of Th17 cells, leading to enhanced priming of memory T cells (18, 19). This results in the induction and production of a variety of inflammatory mediators that trigger potent inflammatory responses. IL-23 also stimulates the generation of proinflammatory cytokines, such as IL-1, IL-6, IFN-γ, and TNF-α, through its effects on dendritic cells and macrophages (20–22). Our data demonstrate for the first time that the average fecal concentration of IL-23 in the stools of CDI-positive patients was significantly (P < 0.05) lower than that in the stools of hospitalized patients without diarrhea and CDI-negative patients. This suggests that the amount of IL-23 produced during CDI may be inadequate to sustain the cellular immunity conferred by this cytokine in promoting the induction and proliferation of effector memory T cells. Thus, decreased production of IL-23 may explain the lack of a robust immunological response exhibited by a proportion of CDI patients and may also relate to recurrence. Perhaps boosting the level of IL-23 may help activate the cellular immune response required for a robust response to CDI. Our data appear to contrast to the findings of Buonomo et al., who recently reported increased IL-23 levels in a small number of colon biopsy specimens from CDI patients and two murine CDI models (23). However, both studies identified IL-23 to be important in CDI. The present study involved a larger number of patients with CDI of variable severity than the number in the smaller human and animal studies reported by Buonomo et al. (23). The patient populations between the two studies as well as the samples tested were different. Further studies, such as studies with larger patient populations from different locations, are needed to confirm these findings to further evaluate the global role of IL-23 in the immunopathogenesis of CDI.
The average concentrations of lactoferrin and calprotectin in healthy adults have been found to range from 1.45 to 4.6 μg/ml (24, 25) and to be ≤10 μg/ml (26), respectively. These are consistent with the concentrations obtained in our study: 6.8 ± 0.85 μg/ml for lactoferrin and 10.2 ± 0.92 μg/ml for calprotectin. Elevated levels of lactoferrin and calprotectin in stools are associated with colonic inflammation (27). Our data show that 88% and 80% of the CDI-positive stools had average lactoferrin and calprotectin concentrations higher than the average concentrations in the stools of the hospitalized patients without diarrhea, respectively. These results agree with those of Shastri et al. (28), in which 85.1% and 82.8% of CDI patient stool samples had higher levels of lactoferrin and calprotectin, respectively, than stool samples from healthy adults. Lactoferrin and calprotectin serve as part of the innate inflammatory response, and so their overexpression during CDI may provide insight into the extent of inflammation associated with this infection.
It is important to note that a proportion of CDI-positive stool samples had levels of either lactoferrin (12%) or calprotectin (20%) below the average levels in stools of the hospitalized patients without diarrhea. This underscores the complex nature of CDI immunopathogenesis, as evidenced by the wide range of clinical and symptomatic phenotypes observed in CDI patients (29). Subjects with relatively low lactoferrin and calprotectin concentrations may represent patients diagnosed with CDI who are simply colonized by C. difficile but have diarrhea due to another cause. The more sensitive methods of detecting the C. difficile toxins do not always differentiate between C. difficile carriage and C. difficile-associated diarrhea. Combining the presence of an inflammatory biomarker in diarrheal stools together with a positive assay result for C. difficile toxin may move us closer to a “gold standard” for CDI diagnosis.
The mechanisms and factors that contribute to antibiotic-associated diarrhea may also trigger inflammation in patients that tested negative for CDI. In fact, our study shows that 44% and 30% of the CDI-negative stools had average lactoferrin and calprotectin concentrations higher than the average concentrations in the stools of the hospitalized patients without diarrhea, respectively. This offers insight into the colonic inflammatory nature of a subset of patients with non-CDI hospital-associated diarrhea. Some of the proinflammatory cytokines and inflammatory-associated proteins that were found in the CDI-positive stools were also detected in the CDI-negative stools. As a result, confirmation of the extent of colonic inflammation by testing colonic mucosal tissues would have been useful, although that was beyond the scope of this study.
We also note that secretory forms of diarrhea can theoretically dilute intraluminal contents. If the concentration of mucosal cytokines was the same in patients with secretory diarrhea as in healthy controls, the patients with diarrhea would likely have lower levels of detectable fecal cytokines. This could be a factor in the study. However, we also compared cytokine levels in CDI-negative patients with diarrhea. Theoretically, fecal cytokines from both diarrheal groups should be diluted, but the levels of some of the cytokines detected, such as IL-8, were higher in CDI-positive diarrheal stools than in the nondiarrheal stools.
The differentiation of naive CD4+ helper T cells into either Th1 or Th2 cells is critical in the development of the adaptive immune response (30). A Th1 inflammatory response usually induces IFN-γ production, leading to activation of phagocytes, whereas a Th2 response results in humoral immunity and allergic inflammation and stimulates host resistance to intracellular infections or agents (31, 32). The patterns of Th1- and Th2-associated cytokines found in the CDI-positive stools were not distinct from those found in the CDI-negative stools. However, the concentrations of TNF-α and IL-13 in the stools of CDI-positive patients were significantly higher than those in the stools of the CDI-negative patients and the hospitalized patients without diarrhea. This suggests a mixed Th1/Th2 response during CDI and infers that the host response to CDI is complex and proinflammatory and encompasses both the innate and the adaptive arms of the immune system. These results may also denote a probable intracellular response to the toxins and an extracellular response to the bacterium.
ACKNOWLEDGMENTS
This work was supported in part by discretionary funds from The University of Texas School of Public Health and Public Health Service grant DK56338, which funds the Texas Medical Center Digestive Disease Center and the Roderick D. MacDonald Research Fund at St. Luke's Episcopal Hospital.
We have no conflicts to report.
Footnotes
Published ahead of print 29 January 2014
REFERENCES
- 1.Loo VG, Poirier L, Miller MA, Oughton M, Libman MD, Michaud S, Bourgault AM, Nguyen T, Frenette C, Kelly M, Vibien A, Brassard P, Fenn S, Dewar K, Hudson TJ, Horn R, Rene P, Monczak Y, Dascal A. 2005. A predominantly clonal multi-institutional outbreak of Clostridium difficile-associated diarrhea with high morbidity and mortality. N. Engl. J. Med. 353:2442–2449. 10.1056/NEJMoa051639 [DOI] [PubMed] [Google Scholar]
- 2.DuPont HL. 2011. The search for effective treatment of Clostridium difficile infection. N. Engl. J. Med. 364:473–475. 10.1056/NEJMe1013236 [DOI] [PubMed] [Google Scholar]
- 3.Voth DE, Ballard JD. 2005. Clostridium difficile toxins: mechanism of action and role in disease. Clin. Microbiol. Rev. 18:247–263. 10.1128/CMR.18.2.247-263.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thelestam M, Florin I. 1994. Assay of cytopathogenic toxins in cultured cells. Methods Enzymol. 235:679–690. 10.1016/0076-6879(94)35181-3 [DOI] [PubMed] [Google Scholar]
- 5.Von Eichel-Streiber C, Boquet P, Sauerborn M, Thelestam M. 1996. Large clostridial cytotoxins—a family of glycosyltransferases modifying small GTP-binding proteins. Trends Microbiol. 4:375–382. 10.1016/0966-842X(96)10061-5 [DOI] [PubMed] [Google Scholar]
- 6.Steiner TS, Flores CA, Pizarro TT, Guerrant RL. 1997. Fecal lactoferrin, interleukin-1beta, and interleukin-8 are elevated in patients with severe Clostridium difficile colitis. Clin. Diagn. Lab. Immunol. 4:719–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Torres JF, Lyerly DM, Hill JE, Monath TP. 1995. Evaluation of formalin-inactivated Clostridium difficile vaccines administered by parenteral and mucosal routes of immunization in hamsters. Infect. Immun. 63:4619–4627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manabe YC, Vinetz JM, Moore RD, Merz C, Charache P, Bartlett JG. 1995. Clostridium difficile colitis: an efficient clinical approach to diagnosis. Ann. Intern. Med. 123:835–840. 10.7326/0003-4819-123-11-199512010-00004 [DOI] [PubMed] [Google Scholar]
- 9.Desai D, Faubion WA, Sandborn WJ. 2007. Review article: biological activity markers in inflammatory bowel disease. Aliment. Pharmacol. Ther. 25:247–255. 10.1111/j.1365-2036.2006.03184.x [DOI] [PubMed] [Google Scholar]
- 10.Schroder O, Naumann M, Shastri Y, Povse N, Stein J. 2007. Prospective evaluation of faecal neutrophil-derived proteins in identifying intestinal inflammation: combination of parameters does not improve diagnostic accuracy of calprotectin. Aliment. Pharmacol. Ther. 26:1035–1042. 10.1111/j.1365-2036.2007.03457.x [DOI] [PubMed] [Google Scholar]
- 11.Guerrant RL, Araujo V, Soares E, Kotloff K, Lima AA, Cooper WH, Lee AG. 1992. Measurement of fecal lactoferrin as a marker of fecal leukocytes. J. Clin. Microbiol. 30:1238–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bjerke K, Halstensen TS, Jahnsen F, Pulford K, Brandtzaeg P. 1993. Distribution of macrophages and granulocytes expressing L1 protein (calprotectin) in human Peyer's patches compared with normal ileal lamina propria and mesenteric lymph nodes. Gut 34:1357–1363. 10.1136/gut.34.10.1357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roseth AG, Schmidt PN, Fagerhol MK. 1999. Correlation between faecal excretion of indium-111-labelled granulocytes and calprotectin, a granulocyte marker protein, in patients with inflammatory bowel disease. Scand. J. Gastroenterol. 34:50–54. 10.1080/00365529950172835 [DOI] [PubMed] [Google Scholar]
- 14.Garey KW, Jiang ZD, Ghantoji S, Tam VH, Arora V, Dupont HL. 2010. A common polymorphism in the interleukin-8 gene promoter is associated with an increased risk for recurrent Clostridium difficile infection. Clin. Infect. Dis. 51:1406–1410. 10.1086/657398 [DOI] [PubMed] [Google Scholar]
- 15.Darkoh C, DuPont HL, Kaplan HB. 2011. Novel one-step method for detection and isolation of active-toxin-producing Clostridium difficile strains directly from stool samples. J. Clin. Microbiol. 49:4219–4224. 10.1128/JCM.01033-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tixier E, Lalanne F, Just I, Galmiche JP, Neunlist M. 2005. Human mucosa/submucosa interactions during intestinal inflammation: involvement of the enteric nervous system in interleukin-8 secretion. Cell. Microbiol. 7:1798–1810. 10.1111/j.1462-5822.2005.00596.x [DOI] [PubMed] [Google Scholar]
- 17.Jiang ZD, Garey KW, Price M, Graham G, Okhuysen P, Dao-Tran T, LaRocco M, DuPont HL. 2007. Association of interleukin-8 polymorphism and immunoglobulin G anti-toxin A in patients with Clostridium difficile-associated diarrhea. Clin. Gastroenterol. Hepatol. 5:964–968. 10.1016/j.cgh.2007.04.018 [DOI] [PubMed] [Google Scholar]
- 18.Iwakura Y, Ishigame H. 2006. The IL-23/IL-17 axis in inflammation. J. Clin. Invest. 116:1218–1222. 10.1172/JCI28508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, Kleinschek MA, Owyang A, Mattson J, Blumenschein W, Murphy E, Sathe M, Cua DJ, Kastelein RA, Rennick D. 2006. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J. Clin. Invest. 116:1310–1316. 10.1172/JCI21404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, Vega F, Yu N, Wang J, Singh K, Zonin F, Vaisberg E, Churakova T, Liu M, Gorman D, Wagner J, Zurawski S, Liu Y, Abrams JS, Moore KW, Rennick D, de Waal-Malefyt R, Hannum C, Bazan JF, Kastelein RA. 2000. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 13:715–725. 10.1016/S1074-7613(00)00070-4 [DOI] [PubMed] [Google Scholar]
- 21.Parham C, Chirica M, Timans J, Vaisberg E, Travis M, Cheung J, Pflanz S, Zhang R, Singh KP, Vega F, To W, Wagner J, O'Farrell AM, McClanahan T, Zurawski S, Hannum C, Gorman D, Rennick DM, Kastelein RA, de Waal Malefyt R, Moore KW. 2002. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J. Immunol. 168:5699–5708 [DOI] [PubMed] [Google Scholar]
- 22.Lankford CS, Frucht DM. 2003. A unique role for IL-23 in promoting cellular immunity. J. Leukoc. Biol. 73:49–56. 10.1189/jlb.0602326 [DOI] [PubMed] [Google Scholar]
- 23.Buonomo EL, Madan R, Pramoonjago P, Li L, Okusa MD, Petri WA., Jr 2013. Role of interleukin 23 signaling in Clostridium difficile colitis. J. Infect. Dis. 208:917–920. 10.1093/infdis/jit277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kane SV, Sandborn WJ, Rufo PA, Zholudev A, Boone J, Lyerly D, Camilleri M, Hanauer SB. 2003. Fecal lactoferrin is a sensitive and specific marker in identifying intestinal inflammation. Am. J. Gastroenterol. 98:1309–1314. 10.1111/j.1572-0241.2003.07458.x [DOI] [PubMed] [Google Scholar]
- 25.Joshi S, Lewis SJ, Creanor S, Ayling RM. 2010. Age-related faecal calprotectin, lactoferrin and tumour M2-PK concentrations in healthy volunteers. Ann. Clin. Biochem. 47:259–263. 10.1258/acb.2009.009061 [DOI] [PubMed] [Google Scholar]
- 26.Dolwani S, Metzner M, Wassell JJ, Yong A, Hawthorne AB. 2004. Diagnostic accuracy of faecal calprotectin estimation in prediction of abnormal small bowel radiology. Aliment Pharmacol. Ther. 20:615–621. 10.1111/j.1365-2036.2004.02128.x [DOI] [PubMed] [Google Scholar]
- 27.Konikoff MR, Denson LA. 2006. Role of fecal calprotectin as a biomarker of intestinal inflammation in inflammatory bowel disease. Inflamm. Bowel Dis. 12:524–534. 10.1097/00054725-200606000-00013 [DOI] [PubMed] [Google Scholar]
- 28.Shastri YM, Bergis D, Povse N, Schafer V, Shastri S, Weindel M, Ackermann H, Stein J. 2008. Prospective multicenter study evaluating fecal calprotectin in adult acute bacterial diarrhea. Am. J. Med. 121:1099–1106. 10.1016/j.amjmed.2008.06.034 [DOI] [PubMed] [Google Scholar]
- 29.Eckert C, Barbut F. 2010. Clostridium-difficile-associated infections. Med. Sci. (Paris) 26:153–158 (In French.) 10.1051/medsci/2010262153 [DOI] [PubMed] [Google Scholar]
- 30.Murphy KM, Reiner SL. 2002. The lineage decisions of helper T cells. Nat. Rev. Immunol. 2:933–944. 10.1038/nri954 [DOI] [PubMed] [Google Scholar]
- 31.Abbas AK, Murphy KM, Sher A. 1996. Functional diversity of helper T lymphocytes. Nature 383:787–793. 10.1038/383787a0 [DOI] [PubMed] [Google Scholar]
- 32.Finkelman FD, Shea-Donohue T, Morris SC, Gildea L, Strait R, Madden KB, Schopf L, Urban JF., Jr 2004. Interleukin-4- and interleukin-13-mediated host protection against intestinal nematode parasites. Immunol. Rev. 201:139–155. 10.1111/j.0105-2896.2004.00192.x [DOI] [PubMed] [Google Scholar]