

Abstract
Knowledge of the trophisms that underpin bowel microbiota composition is required in order to understand its complex phylogeny and function. Stable-isotope (13C)-labeled inulin was added to the diet of rats on a single occasion in order to detect utilization of inulin-derived substrates by particular members of the cecal microbiota. Cecal digesta from Fibruline-inulin-fed rats was collected prior to (0 h) and at 6, 12, 18 and 24 h following provision of the [13C]inulin diet. RNA was extracted from these cecal specimens and fractionated in isopycnic buoyant density gradients in order to detect 13C-labeled nucleic acid originating in bacterial cells that had metabolized the labeled dietary constituent. RNA extracted from specimens collected after provision of the labeled diet was more dense than 0-h RNA. Sequencing of 16S rRNA genes amplified from cDNA obtained from these fractions showed that Bacteroides uniformis, Blautia glucerasea, Clostridium indolis, and Bifidobacterium animalis were the main users of the 13C-labeled substrate. Culture-based studies of strains of these bacterial species enabled trophisms associated with inulin and its hydrolysis products to be identified. B. uniformis utilized Fibruline-inulin for growth, whereas the other species used fructo-oligosaccharide and monosaccharides. Thus, RNA–stable-isotope probing (RNA-SIP) provided new information about the use of carbon from inulin in microbiota metabolism.
INTRODUCTION
The large bowel of vertebrates harbors a diverse community, largely composed of bacteria, which is often referred to as the microbiota (1). Polymeric substances originating in plant structures present in the diet pass undigested to the large bowel, where they become growth substrates for members of the microbiota. Analogous to the rumen ecosystem, it is probable that hydrolysis of plant polymers yields fermentable carbohydrate molecules that are used by both degraders and nondegraders as carbon and energy sources (2). In monogastric animals, however, details of these trophic phenomena are scarce. Knowledge of the trophisms that underpin bowel microbiota composition is necessary in order to understand its complex phylogeny and self-regulation. The presence of numerous species in a community indicates the existence of a wide range of ecological niches (3). Identification of the ecological niches of bowel inhabitants is important because it may lead to the ability to predict desirable or undesirable outcomes for the host resulting from alterations to the microbiota due to dietary modifications.
The use of stable-isotope-labeled substrates to probe the microbial utilization of specific substances has gained attention in environmental microbial ecology and, more recently, in bowel microbial ecology (4–7). Stable-isotope probing (SIP) can provide insights into both microbial phylogeny and metabolic activity by tracking stable-isotope-labeled atoms from substrates into microbial cellular components such as nucleic acids. RNA has higher turnover rates in the cell than does DNA and is thus more responsive to changes in metabolic conditions. The faster turnover of RNA leads to faster incorporation of the isotope so that the times required to reach a measurable amount of stable isotope in RNA are shorter than those for DNA (6). Small-subunit rRNA sequences (16S rRNA genes) provide the cornerstone of bacterial phylogeny, identification, and detection (8). Therefore, RNA-SIP has been used in environmental science to explore plant-microorganism interactions and to identify bacteria responsible for the degradation or utilization of particular substrates (9–12). Pioneering use of RNA-SIP in bowel ecological studies has used in vitro systems and substrates such as [13C]glucose, 13C-galacto-oligosaccharides, or [13C]urea (13–15). Starch has been used in RNA-SIP in an in vitro system representing the human colon (16). Investigations using natural bacterial communities inhabiting living animals have not yet been reported.
We report here the use of stable-isotope-labeled inulin ([13C]inulin) to detect carbon utilization from inulin by bacteria inhabiting the rat cecum. Bacterial species shown in this way to be utilizers of inulin-derived carbon were then used in culture-based studies to confirm hydrolysis of inulin and/or use of its hydrolytic products.
MATERIALS AND METHODS
Inulins.
The chicory inulins used in the diets were Fibruline XL (Cosucra, Warcoing, Belgium), which has a minimum degree of polymerization (DP) of 20, and [U-13C]inulin from IsoLife bv, Wageningen, The Netherlands (uniformly labeled, >97 atom% 13C, 97% DP of >3). Therefore, while Fibruline could be considered a polymer, the [13C]inulin was likely to be a mixture of fructo-oligosaccharides of various DPs. This provided a labeled substrate to detect utilization of inulin-derived substrates by the cecal microbiota.
Rat experiment.
The experimental protocol was approved by the Crown Research Institute Animal Ethics Committee, Palmerston North, New Zealand, according to the NZ Animal Welfare Act 1999 (approval number 12462). Weanling male Sprague-Dawley rats (45- to 50-g live weight), which had not been exposed to any diet other than that of the dams, were randomly assigned to groups for sampling at different times. The rats were held individually in metabolism cages and fed a Fibruline-inulin diet (Table 1; 10 g per day per rat, which is effectively ad libitum; available for 24 h). The animal room was maintained at a temperature of 22 ± 1°C and humidity of 60% ± 5%, with air exchange 12 times/hour and a 12-h light/dark cycle. The Fibruline-inulin diet was fed for 3 days, and then 10 g of 13C-labeled inulin diet (0.5 g of [13C]inulin) was provided on day 4. Animals were euthanized by carbon dioxide anesthesia, and samples of cecal digesta were collected from 3 rats at each of the following times: before the [13C]inulin dose (0 h) and 6, 12, 18, and 24 h after the [13C]inulin dose. A temporal study was necessary because the 13C-labeled diet was consumed during a 24-h period. The cecal samples were stored at −80°C prior to extraction of RNA.
TABLE 1.
Compositions (g/kg) of inulin diets
| Ingredient | Amt (g/kg of diet) |
|
|---|---|---|
| Inulin (5%) diet | [13C]inulin diet | |
| Lactic caseina | 120 | 120 |
| Vitamin mixb | 50 | 50 |
| Salt mixc | 50 | 50 |
| Corn oild | 120 | 120 |
| Starche | 540 | 540 |
| Sucrosef | 45 | 45 |
| Celluloseg | 25 | 25 |
| Inulinh | 50 | |
| [13C]inulini | 50 | |
Alacid 80 mesh; New Zealand Milk Products, Wellington, New Zealand.
The mixture contains the following components: in mg/kg diet, retinol acetate, 5.0; dl-α-tocopheryl acetate, 100.0; menadione, 3.0; thiamine hydrochloride, 5.0; riboflavin, 7.0; pyridoxine hydrochloride, 8.0; d-pantothenic acid, 20.0; folic acid, 2.0; nicotinic acid, 20.0; d-biotin, 1.0; myoinositol, 200.0; choline chloride, 1,500.0; in μg/kg diet, ergocalciferol, 25.0; cyanocobalamin, 50.0.
The mixture contains the following components: in g/kg diet, Ca, 6.29; Cl, 7.79; Mg, 1.06; P, 4.86; K, 5.24; Na, 1.97; in mg/kg diet, Cr, 1.97; Cu, 10.7; Fe, 424.0; Mn, 78.0; Zn, 48.2; in μg/kg diet, Co, 29.0; I, 151.0; Mo, 152.0; Se, 151.0.
Davis Trading Company, Palmerston North, New Zealand.
Wheaten corn flour from Starch Australasia, Goodman Fielder Group, Tamworth, NSW, Australia.
Chelsea Sugar Refinery, Auckland, New Zealand.
Avicel PH102; Commercial Minerals Ltd., Auckland, New Zealand.
Fibruline XL; Cosucra, Warcoing, Belgium.
IsoLife bv, Wageningen, The Netherlands.
Extraction of RNA from cecal digesta.
A 1/10 (wt/vol) cecal digesta homogenate was prepared in sterile phosphate-buffered saline (pH 7.0). A 500-microliter aliquot of homogenate was brought to 1.0 ml with sterile phosphate-buffered saline and centrifuged at 150 × g for 5 min at 5°C. The supernatant was transferred to a microcentrifuge tube and centrifuged at 5,000 × g for 5 min at 5°C. The pellet was suspended in 200 microliters of lysis buffer (20 mg lysozyme in 80 microliters of 10 mM Tris-HCl–10 mM EDTA) and incubated at room temperature for 30 min. Fifty microliters of 20% (wt/vol) sodium dodecyl sulfate (SDS) solution was added together with 300 microliters of 50 mM sodium acetate-10 mM EDTA (pH 5.1) solution. The preparation was transferred to a bead beater tube, and 300 microliters of phenol saturated with 50 mM sodium acetate-10 mM EDTA buffer (pH 5.1) was added to the tubes. The sample was shaken at 5,000 rpm for 2 min in a bead beater. After centrifugation at 14,000 × g for 10 min at 4°C, the supernatant was transferred to a microcentrifuge tube, and 600 microliters of phenol saturated with sodium acetate-EDTA buffer (pH 5.1) was added. Samples were mixed by vortexing for 1 min and centrifuged under the conditions described above. Then, 600 microliters of phenol-chloroform-isoamyl alcohol (25:24:1) was added to the supernatant, and the mixture was vortexed for 1 min and centrifuged. This step was repeated once. Next, 600 microliters of chloroform-iso-amyl alcohol (24:1) was added to the supernatants, which were vortexed for 1 min and centrifuged for 5 min at 14,000 × g. This step was repeated once. Nucleic acids were precipitated in 1 ml of isopropanol overnight at −20°C. The precipitated nucleic acids were obtained by centrifugation at 14,000 × g for 20 min at 4°C. They were washed with 1 ml of 80% ethanol and centrifuged at 14,000 × g for 10 min, the supernatant was discarded, and the pellet was dried in air at 37°C. Further purification of RNA was achieved using the Qiagen-AllPrep DNA/RNA minikit and treatment with the Turbo DNA-free kit (Ambion Inc., Austin, TX, USA) according to the manufacturers' instructions. The concentration and quality of the RNA were measured with a Nanodrop 1000 spectrometer.
RNA-SIP methodology.
RNA was extracted from 3 rats per sampling time. The RNA was used to prepare preparations from individual rats for isopycnic density gradient centrifugation. The preparations contained, per gradient, 4.08 ml of cesium trifluoroacetate (GE Healthcare UK Ltd., Little Chalfont, England), 153 microliters of formamide (Sigma-Aldrich, St. Louis, MO, USA), 160 nanograms of RNA, and 865 microliters of ultrapure water (Gibco, Life Technologies, Auckland, New Zealand) minus the volume of RNA solution. The gradients were prepared in ultracentrifuge tubes (Beckman Coulter Optiseal [1/2 in.]; Beckman Coulter Australia Pty Ltd., Gladesville, Australia) which were centrifuged in an upright position (Beckman Coulter VTi 65.2 rotor) for 67 h at 45,000 rpm at 15°C in a Beckman Coulter Optima L-80XP ultracentrifuge. The density gradients were fractionated in 200-microliter volumes per minute using a Beckman Coulter fraction recovery system. Fifteen fractions per gradient were collected with flushing of the recovery system with 1 ml of 0.1 M sodium hydroxide followed by 1 ml of absolute ethanol between gradients. RNA was precipitated from the fractions by the addition of 600 microliters of cold isopropanol and then held at −20°C for 24 h. The fractions were centrifuged for 20 min at −10°C at 14,000 × g, the isopropanol was decanted, and the pelleted RNA was washed once using 1 ml of cold isopropanol. The pellets were dried, and then RNA from the respective fractions from three rats per sampling time was pooled in 30 μl of ultrapure water. The pooling was to ensure that an adequate amount of RNA (cDNA) was available per fraction for phylogenetic analysis. By repetition of experiments, results from three replicate density gradients per sampling time were obtained. Figure 1 provides a schema to assist with comprehension of the experimental protocol.
FIG 1.
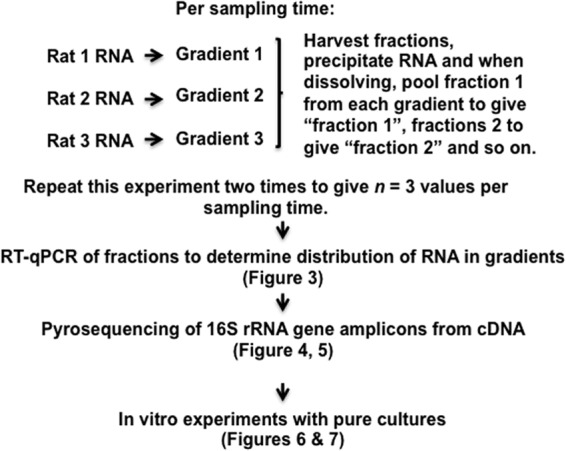
Schema summarizing the experimental protocol.
Three density gradients without added RNA were also prepared to determine that satisfactory gradients had been obtained. After centrifugation and fractionation, each fraction was weighed and the refraction was measured with a refractometer (Reichert AR200 digital refractometer; Reichert Inc., Depew, NY, USA).
Detection and identification of 13C-labeled bacterial populations.
RNA from density gradient fractions was converted to cDNA using an Invitrogen Superscript III First-Strand Synthesis Supermix kit (Life Technologies) according to the manufacturer's protocol. The relative amounts of bacterial 16S rRNA gene sequences in cDNA from each fraction were then determined by real-time quantitative PCR (qPCR) using the universal bacterial primers (targeting the 16S rRNA genes of all bacteria) 8fAll (GRGTTYGATYMTGGCTCAG) and 340R (ACTGCTGCCTCCCGTAGGAGT) and the following protocol. qPCR was carried out using an ABI 7500 Fast system in MicroAmp Fast Optical 96-well plates with optical adhesive film (Applied Biosystems, Foster City, CA). All reactions were carried out in a final volume of 20 microliters containing 1× Fast SYBR Green PCR Mastermix (Applied Biosystems), 300 nM (each) primer, and 2 microliters of template cDNA. The thermocycling program consisted of an initial activation of the polymerase at 95°C for 30 s, followed by 40 cycles of 95°C for 10 s and 60°C for 30 s. Fluorescence levels were measured after the 60°C annealing/extension step. A melt curve was generated to analyze product specificity. All reactions were carried out in duplicate and were run twice on separate plates. No-template controls were also included on each plate.
To determine which bacterial groups were represented in density gradient fractions, 16S rRNA gene sequences in cDNA were amplified by PCR, pyrosequenced, and analyzed for phylogenetic composition. Specifically, a region comprising the V1 to V3 regions of the bacterial 16S rRNA gene was amplified using a two-step protocol similar to that described by Dowd et al. (17). First-round PCR was carried out for 15 cycles using the 8fAll (GRGTTYGATYMTGGCTCAG)/HDA2 (GTATTACCGCGGCTGCTGGCAC) primer set under the following conditions: 94°C for 1 min, 57°C for 1 min, and 72°C for 1 min, with a final extension step of 72°C for 5 min. This product was diluted 1/5 with PCR-grade water, and 1 microliter was used as the template in a 20-microliter secondary PCR. The secondary PCR was carried out for 30 cycles using the 8fAll primer with the 454 sequencing Lib-A adapter sequence A (CGTATCGCCTCCCTCGCGCCATCAGGRGTTYGATYMTGGCTCAG) and the HDA2 primer with the 454 sequencing Lib-A adapter sequence B plus a 10-base barcode, shown as N′s (CTATGCGCCTTGCCAGCCCGCTCAGNNNNNNNNNNGTATTACCGCGGCTGCTGGCAC), using conditions identical to those for the primary PCR. Products were cleaned using Qiagen PCR cleanup columns (Qiagen, Hilden, Germany) and quantified using a Nanodrop 1000 spectrometer. Equivalent quantities of PCR product from each sample were pooled, and the pooled DNA was recleaned through a Qiagen PCR cleanup column, quantified, and sent to Macrogen (South Korea) for unidirectional sequencing from the reverse primer using the Roche-454 genome sequencer with titanium chemistry. Sequences were processed using a combination of methods from the QIIME v.1.3 software package (18). Sequences were excluded from analysis if they were <250 or >550 bases in length, had an average quality score of <25, contained one or more ambiguous bases, had >1 mismatch with the sequencing primer, or had a homopolymer run of >6. Following splitting into barcoded samples and initial quality filtering, the sequences were passed through the QIIME pipeline using default parameters, including chimera checking. Species-level taxonomy was obtained by filtering operational taxonomic unit (OTU) tables, containing taxonomic data generated using the RDP classifier, at a genus level; extracting representative sequences; and using BLASTn to identify species-level matches within the NCBI database.
Utilization of inulin, fructo-oligosaccharide, glucose, and fructose by bacterial cultures.
Based on the results of phylogenetic analysis of bacteria represented in density gradient fractions, isolates of Bifidobacterium animalis subsp. animalis, Bacteroides uniformis, Blautia glucerasea, and Clostridium indolis (Table 2) were tested in triplicate for their ability to ferment Fibruline-inulin, raftilose (a fructo-oligosaccharide), glucose, and fructose (each used at 0.5%, wt/vol) in PY medium as described by Holdeman and Moore (19) (per 100 ml: peptone, 1 g; yeast extract, 1 g; Tween 80, 100 microliters; vitamin K-hemin solution [0.005% menadione, 0.05% hemin], 1 ml; salts solution [per liter: calcium chloride, 0.2 g; magnesium sulfate, 0.2 g; dipotassium phosphate, 1.0 g; monopotassium phosphate, 1.0 g; sodium bicarbonate, 10.0 g; sodium chloride, 2.0 g], 4 ml; sodium sulfide–l-cysteine solution [per 100 ml: 1.25 g of each chemical], 2 ml). The pHs of cultures were measured after anaerobic incubation at 37°C for 48 h and compared to that of uninoculated medium (pH 7.2). As defined by Holdeman and Moore, a change (reduction) in pH of ≥0.9 pH unit was considered to indicate fermentation of the substrate (19).
TABLE 2.
Bacterial strains used in fermentation experiments
| Bacterial species | Source |
|---|---|
| Bifidobacterium animalis DSM 20104T | American Type Culture Collection |
| Bifidobacterium animalis R12 | Rat cecal isolate; this study |
| Bacteroides uniformis ATCC 8492T | American Type Culture Collection |
| Bacteroides uniformis 201 | Rat cecal isolate; this study |
| Blautia glucerasea DSM 22028T | German Culture Collection |
| Clostridium indolis DSM 755T | German Culture Collection |
HPAEC to show hydrolysis of Fibruline by B. uniformis.
The hydrolysis of Fibruline-inulin by the bacteria was monitored by high-performance anion-exchange chromatography (HPAEC) of cell-free culture supernatant, harvested after 24 h of incubation, and compared with uninoculated controls. Duplicate samples of cell-free medium diluted 20-fold (20 μl) were analyzed on a CarboPac PA-100 (4- by 250-mm) column equilibrated in 150 mM sodium hydroxide, using a Dionex ICS 3000 chromatographic system (Dionex Corp., Sunnyvale, CA, USA), and eluted with a linear gradient of sodium acetate (0 to 500 mM) from 1 to 60 min after injection. The eluent was monitored by pulsed amperometric detection. The concentration of free fructose in the medium was determined by calibration with standard fructose (0 to 0.05 mg ml−1), and the DP of individual oligosaccharides was determined by comparison with oligosaccharides extracted from tubers of Helianthus tuberosus as described by Sims et al. (20).
RESULTS
Density gradients.
The preparation and examination of preparations without the addition of RNA showed that gradients of an appropriate density spectrum, as indicated by reference to the work of others (16), were obtained (Fig. 2).
FIG 2.
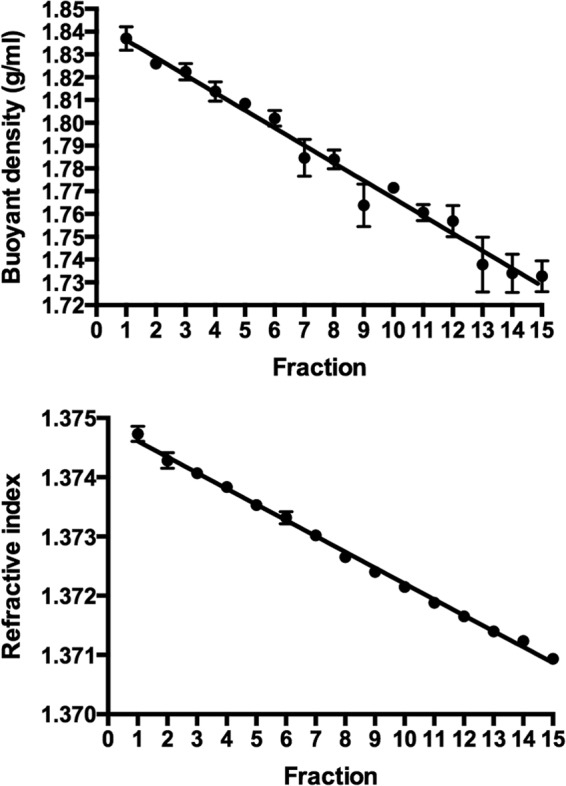
Buoyant density and refraction curves prepared from density gradients. Means and standard errors of three gradients are shown.
Stable-isotope-labeled bacterial populations.
cDNA from RNA extracted from the ceca of rats that had not been dosed with [13C]inulin (0 h) was detected by qPCR in fractions 13 to 15 of density gradients (Fig. 3). In contrast, most of the cDNA was detected in fractions 7 to 12 in the case of rats sampled after consumption of the 13C-diet (Fig. 3). These results provided evidence of bacterial utilization of the [13C]inulin and consequent incorporation of [13C]carbon into bacterial RNA. It was notable that most of the RNA obtained from gradients at 6 to 24 h was present in the denser fractions. In other words, there was little [12C]RNA in the preparations at these sampling times. This was likely to have been due to enrichment of bacterial populations by feeding long-chain inulin (Fibruline) to the rats for 3 days prior to pulsing with [13C]inulin. This was supported by the results of phylogenetic analysis of bacterial 16S RNA gene sequences in fractions 13 to 15 in 0-h gradients in which 16S rRNA gene sequences from 19 bacterial families were detected. However, Bifidobacteriaceae, Lachnospiraceae, and Bacteroidaceae 16S rRNA gene sequences predominated (Fig. 4). Members of these families also predominated in fractions containing [13C]RNA (Fig. 4). While it seems possible to obtain relatively well separated [12C] and [13C]RNA fractions using pure cultures of bacteria (6), the reality was a gradation of [13C]RNA probably due to the variable amount of labeling between RNA molecules that occurred in cells in vivo (21). Thus, the [13C]RNA formed a “smear” in the gradient rather than a single band of [13C]RNA (Fig. 5).
FIG 3.

Relative amounts of RNA, determined by qPCR of cDNA, in fractions collected from density gradients at 0 (before) and 6, 12, 18, and 24 h after provision of the [13C]inulin diet. Means and standard errors of three gradients per sampling time are shown.
FIG 4.

Results from pyrosequencing data (cDNA) showing abundances of 16S rRNA gene sequences from bacterial groups in pooled fractions before (fractions 13 to 15, 0 h) and after provision of the [13C]inulin diet (fractions 7 to 12). Means and standard errors of the means are shown.
FIG 5.
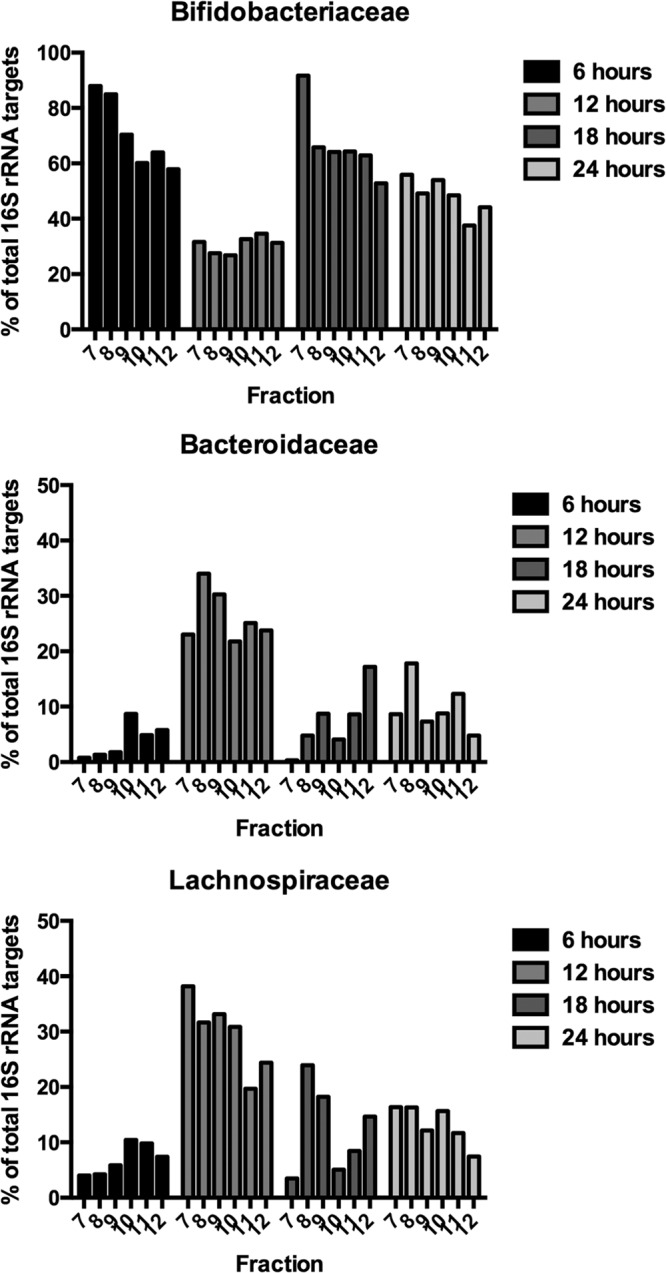
Results from pyrosequencing data (cDNA) of individual fractions 7 to 12 of representative gradients. Abundances of 16S rRNA gene sequences from bacterial families at 6, 12, 18, and 24 h after provision of the [13C]inulin diet are shown.
Identification of 13C-labeled bacterial populations.
cDNA from cecal digesta prior to provision of the [13C]inulin diet was in gradient fractions above 12, whereas cDNA from rats administered [13C]inulin was predominantly in gradient fractions below 12. Therefore, pyrosequencing results from fractions 7 to 12 were analyzed to determine the kinds of bacteria that had been labeled. Three bacterial families predominated in the fractions: Bifidobacteriaceae, Bacteroidaceae, and Lachnospiraceae. Further BLASTn alignments of 16S rRNA gene sequences in fractions 7 to 12 showed that four bacterial species predominated in the fractions: Bifidobacterium animalis, Bacteroides uniformis, Blautia glucerasea, and Clostridium indolis (Table 3).
TABLE 3.
Bifidobacterial, Bacteroides, and lachnospiral groups detected in gradient fractions 7 to 12 at abundances of >1%
| Bacterial family and species | Relative abundance (%)a |
|---|---|
| Bifidobacteriaceae | |
| Bifidobacterium animalis | 94.79 |
| Bifidobacterium bifidum | 1.99 |
| Bifidobacterium pseudocatenulatum | 4.29 |
| Bacteroidaceae | |
| Bacteroides acidifaciens | 8.21 |
| Bacteroides uniformis | 50.73 |
| Bacteroides xylanisolvens | 8.74 |
| Lachnospiraceae | |
| Anaerostipes sp. | 0.89 |
| Blautia glucerasea | 77.90 |
| Blautia luti | 15.78 |
| Blautia producta | 4.52 |
| Clostridium indolis | 80.79 |
Percentage of bifidobacterial, Bacteroides, or lachnospiral 16S rRNA sequences.
Utilization of inulin, fructo-oligosaccharide, glucose, and fructose by bacterial cultures.
Inulin is composed of fructose chains with glucose end residues (22). Figure 6 shows the results of tests to determine the fermentation of Fibruline-inulin and possible products of its hydrolysis (fructo-oligosaccharide, glucose, and fructose) by these bacteria. B. uniformis was the only species of the four that could ferment Fibruline-inulin, whereas all species could ferment fructo-oligosaccharide and glucose. B. animalis did not ferment fructose, but the other species were able to do so. Thus, these experiments confirmed that the particular species could grow using inulin and/or its hydrolytic products and associated the species with specific trophisms.
FIG 6.

Fermentation of inulin and derivatives by bacterial species in PY medium. A change of 0.9 pH unit (dashed line) or more relative to an uninoculated control indicates fermentation of a substrate. Means of triplicate cultures are shown. Standard error of the mean, ≤0.05.
Inulin hydrolysis by B. uniformis.
Analysis of inulin consumption by HPAEC showed that the amplitude of the peaks corresponding to oligosaccharides with a DP of >15 had decreased in spent medium compared with uninoculated controls (∼90% to 55% peak area). Peaks corresponding to oligosaccharides with DPs of 3 to 10 remained little changed, or were increased in size, compared with uninoculated controls (Fig. 7). Free fructose was present in the spent medium at 24 h (0.27 mg ml−1 medium, or ∼1.5 μmol ml−1 medium) but was present at only trace levels in uninoculated medium. These data are consistent with enzymatic hydrolysis of Fibruline by B. uniformis, resulting in a shift from longer to shorter oligosaccharide chain lengths and release of fructose into the medium.
FIG 7.

Hydrolysis of inulin (Fibruline, red line) by Bacteroides uniformis strain 201, compared with an uninoculated control (black line), analyzed by HPAEC on a CarboPac PA100 column. The right-hand panel is magnified 10 times to show oligosaccharide profiles clearly. The elution positions of fructose (F) and oligosaccharides with increasing degrees of polymerization (numbers) are shown. PAD, pulsed amperometric detection.
DISCUSSION
In prebiotic (functional foods) experimentation, detection of a bacterial bloom (increase in abundance of a particular bacterial population) is generally taken as evidence of utilization of a nondigestible food component as a carbon and energy source for the bacteria (23). However, whether bacterial blooms can be considered a normal phenomenon, or indeed desirable, in the large bowel ecosystem is arguable. We do not really know whether such major disturbances, apart from effects produced by antibiotics (24), occur in the day-to-day life of humans. Where temporal comparisons of fecal microbiota compositions have been made, phylogenetic stability seems to be the norm. However, based on limited data, changes in the relative abundances of core populations have been noted. Whether these can be considered to be blooms due to dietary intake is so far unclear (25). In any case, generation of bacterial blooms by adding relatively large amounts of nondigestible polymers to the diet to obtain evidence of bacterial utilization may be a crude approach. RNA-SIP, on the other hand, provides a potentially subtle method of detecting utilization of a particular substrate by specific bowel bacteria. As a result of administration of a 13C-labeled substance in the diet, bacterial populations that have used the substrate for growth contain cells whose components were synthesized from 13C originating, directly or indirectly, from that particular substrate.
The combination of enrichment and 13C probing that we have used revealed new information about bacterial trophisms in relation to inulin and its derivatives. The “enriched” bacterial populations associated with dietary long-chain inulin (Fibruline) contained bacterial species that hydrolyzed inulin and/or used the hydrolysis products for growth. We were not sure of the sensitivity of the RNA-SIP protocol; therefore, we fed the Fibruline diet to the rats prior to the [13C]inulin diet. As a result, bacteria capable of utilizing long-chain inulin or products of inulin hydrolysis when the [13C]inulin was administered dominated the cecal microbiota. Now that the utility of RNA-SIP has been shown in rat experiments, it would be preferable in future experiments to omit this prior enrichment. This would allow the sensitivity of the RNA-SIP methodology to be tested in a nonmanipulated ecosystem.
The trophic structure of a community is the arrangement of groups based on their feeding relationships (26). All organisms in an ecosystem can be placed in trophic levels depending on what energy source they rely on. Thus, the trophic structure controls the passage of energy and nutrients from one group to another in an ecosystem. It is likely that bowel bacteria interact with each other through food webs, based on what they use for growth and what they provide for others to utilize. In our study, Bacteroides uniformis was demonstrated to occupy what could be termed trophic level 1 because it hydrolyzed inulin. The remaining three species that were identified as benefiting from inulin administration in the rat diet could be said to occupy trophic level 2. They did not utilize inulin but fermented the hydrolysis product fructo-oligosaccharide. Trophic level 3, based on the utilization of monosaccharides, could be divided into two groups: those bacteria that fermented glucose (all species) and those that fermented fructose (all except B. animalis). Thus, RNA-SIP can help to identify bacterial species in the large bowel microbiota that have particular trophisms. Our study emphasizes the need for a more thorough understanding of bowel ecology in the development of functional foods aimed at influencing the bowel microbiota. The addition of a nondigestible labeled substance to the diet, as in our example, can influence several phylogenetic groups of bacteria whose metabolism and other properties may have as-yet-unknown influences in the bowel ecosystem and on the vertebrate host.
ACKNOWLEDGMENTS
An introduction to the methodology of RNA-SIP by Maria-Luisa Gutierrez-Zamora and Mike Manefield, University of New South Wales, Australia, was greatly appreciated. We thank Sheridan Martell and Hannah Smith, Plant & Food Research, Palmerston North, New Zealand, for the care of the animals.
This research was supported by Foundation for Research, Science and Technology program contract CO2X0703.
Footnotes
Published ahead of print 31 January 2014
REFERENCES
- 1.Ley RE, Lozupone CA, Hamady M, Knight R, Gordon JI. 2008. Worlds within worlds: evolution of the vertebrate gut microbiota. Nat. Rev. Microbiol. 6:776–788. 10.1038/nrmicro1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flint HJ, Bayer EA, Rincon MT, Lamed R, White BA. 2008. Polysaccharide utilization by gut bacteria: potential for new insights from genomic analysis. Nat. Rev. Microbiol. 6:121–131. 10.1038/nrmicro1817 [DOI] [PubMed] [Google Scholar]
- 3.Tannock GW. 2009. The microbiota of humans, p 129–138 In Henderson B, Curtis MA, Seymour RM, Donos N. (ed), Periodontal medicine and systems biology. Blackwell Publishing Ltd, Chichester, United Kingdom [Google Scholar]
- 4.Radajewski S, Ineson P, Parekh NR, Murrell JC. 2000. Stable-isotope probing as a tool in microbial ecology. Nature 403:646–649. 10.1038/35001054 [DOI] [PubMed] [Google Scholar]
- 5.Manefield M, Whiteley AS, Griffiths RI, Bailey MJ. 2002. RNA stable isotope probing, a novel means of linking microbial community function to phylogeny. Appl. Environ. Microbiol. 68:5367–5373. 10.1128/AEM.68.11.5367-5373.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whiteley AS, Thomson B, Lueders T, Manefield M. 2007. RNA stable-isotope probing. Nat. Protoc. 2:838–843. 10.1038/nprot.2007.115 [DOI] [PubMed] [Google Scholar]
- 7.Egert M, de Graaf AA, Maathius A, de Waard P, Plugge CM, Smidt H, Deutz NEP, Dijkema C, de Vos WM, Venema K. 2007. Identification of glucose-fermenting bacteria present in an in vitro model of the human intestine by RNA-stable isotope probing. FEMS Microbiol. Ecol. 60:126–135. 10.1111/j.1574-6941.2007.00281.x [DOI] [PubMed] [Google Scholar]
- 8.Woese CR. 1987. Bacterial evolution. Microbiol. Rev. 51:221–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prosser JI, Rangel-Castro JI, Killham K. 2006. Studying plant-microbe interactions using stable isotope technologies. Curr. Opin. Biotechnol. 17:98–102. 10.1016/j.copbio.2006.01.001 [DOI] [PubMed] [Google Scholar]
- 10.Boschker HTS, Nold SC, Wellsbury P, Bos D, de Graaf W, Pel R, Parkes RJ, Cappenberg TE. 1998. Direct linking of microbial populations to specific biogeochemical processes by 13C-labelling of biomarkers. Nature 392:801–805. 10.1038/33900 [DOI] [Google Scholar]
- 11.Kalyuzhnaya MG, Lapidus A, Ivanova N, Copeland AC, McHardy AC, Szeto E, Salamov A, Grigoriev IV, Suciu D, Levine SR, Markowitz VM, Rigoutsos I, Tringe SG, Bruce DC, Richardson PM, Lidstrom ME, Chistoserdova L. 2008. High-resolution metagenomics targets specific functional types in complex microbial communities. Nat. Biotechnol. 26:1029–1034. 10.1038/nbt.1488 [DOI] [PubMed] [Google Scholar]
- 12.Miyatake T, MacGregor BJ, Boschker HTS. 2009. Linking microbial community function to phylogeny of sulfate-reducing Deltaproteobacteria in marine sediments by combining stable isotope probing with magnetic-bead capture hybridization of 16S rRNA. Appl. Environ. Microbiol. 75:4927–4935. 10.1128/AEM.00652-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Graaf AA, Maathuis A, de Waard P, Deutz NE, Dijkema C, de Vos WM, Venema K. 2010. Profiling human gut bacterial metabolism and its kinetic using (U-13C)glucose and NMR. NMR Biomed. 23:2–12. 10.1002/nbm.1418 [DOI] [PubMed] [Google Scholar]
- 14.Reichardt N, Barclay AR, Weaver LT, Morrison DJ. 2011. Use of stable isotopes to measure the metabolic activity of the human intestinal microbiota. Appl. Environ. Microbiol. 77:8009–8014. 10.1128/AEM.05573-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maathuis AJ, van den Heuvel EG, Schoterman MH, Venema K. 2012. Galacto-oligosaccharides have prebiotic activity in a dynamic in vitro colon model using a (13)C-labeling technique. J. Nutr. 142:1205–1212. 10.3945/jn.111.157420 [DOI] [PubMed] [Google Scholar]
- 16.Kovatcheva-Datchary P, Ergert M, Maathuis A, Rajilic-Stojanovic M, de Graaf AA, Smidt H, de Vos W, Venema K. 2009. Linking phylogenetic identities of bacteria to starch fermentation in an in vitro model of the large intestine by RNA-based stable isotope probing. Environ. Microbiol. 11:914–926. 10.1111/j.1462-2920.2008.01815.x [DOI] [PubMed] [Google Scholar]
- 17.Dowd SE, Callaway TR, Wolcott RD, Sun Y, McKeehan T, Hagevoort RG, Edrington TS. 2008. Evaluation of the bacterial diversity in the feces of cattle using 16S rDNA bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP). BMC Microbiol. 8:125–132. 10.1186/1471-2180-8-125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7:335–336. 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holdeman LV, Moore WEC. 1973. Anaerobe laboratory manual, 2nd ed. VPI Anaerobe Laboratory, Virginia Polytechnic Institute and State University, Blacksburg, VA [Google Scholar]
- 20.Sims IM, Smouter H, Pollock CJ, Simpson RJ. 1991. The separation of complex mixtures of fructo-oligosaccharides from plants. Plant Physiol. Biochem. 29:257–267 [Google Scholar]
- 21.Dumont MG, Pommerenke B, Casper P, Conrad R. 2011. DNA-, rRNA-, and mRNA-based stable isotope probing of aerobic methanotrophs in lake sediment. Environ. Microbiol. 13:1153–1167. 10.1111/j.1462-2920.2010.02415.x [DOI] [PubMed] [Google Scholar]
- 22.Niness K. 1999. Inulin and oligofructose: what are they? J. Nutr. 129:1402S–1406S [DOI] [PubMed] [Google Scholar]
- 23.Walker AW, Ince J, Duncan SH, Webster LM, Holtrop G, Ze X, Brown D, Stares MD, Scott P, Bergerat A, Louis P, McIntosh F, Johnstone AM, Lobley GE, Parkhill J, Flint HJ. 2011. Dominant and diet-responsive groups of bacteria within the human colonic microbiota. ISME J. 5:220–230. 10.1038/ismej.2010.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tannock GW, Munro K, Taylor C, Lawley B, Young W, Byrne B, Emery J, Louie T. 2010. A new macrocyclic antibiotic, fidaxomicin (OPT-80) causes less alteration to the bowel microbiota of Clostridium difficile-infected patients than does vancomycin. Microbiology 156:3354–3359. 10.1099/mic.0.042010-0 [DOI] [PubMed] [Google Scholar]
- 25.Rajilic-Stojanovic M, Heilig HG, Tims S, Zoetendal EG, de Vos WM. 2013. Long-term monitoring of the human intestinal microbiota composition. Environ. Microbiol. 15:1146–1159. 10.1111/1462-2920.12023 [DOI] [PubMed] [Google Scholar]
- 26.Pimm SL, Lawton JH. 1978. On feeding on more than one trophic level. Nature 275:542–544. 10.1038/275542a0 [DOI] [Google Scholar]