

Abstract
Protein engineering is commonly used to improve the robustness of enzymes for activity and stability at high temperatures. In this study, we identified four residues expected to affect the thermostability of Streptomyces sp. strain S9 xylanase XynAS9 through multiple-sequence analysis (MSA) and molecular dynamic simulations (MDS). Site-directed mutagenesis was employed to construct five mutants by replacing these residues with proline or glutamic acid (V81P, G82E, V81P/G82E, D185P/S186E, and V81P/G82E/D185P/S186E), and the mutant and wild-type enzymes were expressed in Pichia pastoris. Compared to the wild-type XynAS9, all five mutant enzymes showed improved thermal properties. The activity and stability assays, including circular dichroism and differential scanning calorimetry, showed that the mutations at positions 81 and 82 increased the thermal performance more than the mutations at positions 185 and 186. The mutants with combined substitutions (V81P/G82E and V81P/G82E/D185P/S186E) showed the most pronounced shifts in temperature optima, about 17°C upward, and their half-lives for thermal inactivation at 70°C and melting temperatures were increased by >9 times and approximately 7.0°C, respectively. The mutation combination of V81P and G82E in adjacent positions more than doubled the effect of single mutations. Both mutation regions were at the end of long secondary-structure elements and probably rigidified the local structure. MDS indicated that a long loop region after positions 81 and 82 located at the end of the inner β-barrel was prone to unfold. The rigidified main chain and filling of a groove by the mutations on the bottom of the active site canyon may stabilize the mutants and thus improve their thermostability.
INTRODUCTION
Hemicellulose is the second most abundant natural polysaccharide after cellulose. Xylan is the main carbohydrate found in hemicellulose, which constitutes 30 to 35% of lignocellulosic biomass (1). Efficient utilization of xylan is essential for conversion of hemicellulosic materials to other value-added products. Due to the complex structure of xylan, it requires an enzyme system of several hydrolytic enzymes for complete degradation. Among them, xylanase plays a crucial role in xylan hydrolysis, as it catalyzes the hydrolysis of 1,4-β-d-xylosidic linkages in xylan to short xylooligosaccharides. Currently, xylanase has been attracting much attention due to its wide biotechnological applications in the food, animal feed, pulp and paper, and textile industries and in the deconstruction of lignocellulose for biofuel production (2–6). On the basis of the catalytic domains, xylanases are mainly classified into glycoside hydrolase (GH) families 5, 8, 10, 11, 30, and 43 (7); GH10 and GH11 xylanases are the best studied.
High-temperature-active or -stable enzymes are more favorable than mesophilic counterparts, since high temperatures enhance the mass transfer rate, reduce the substrate viscosity, and reduce the risk of contamination (8, 9). For example, thermophilic xylanases are applied in bioconversion processes and pulp bleaching, during which a variety of high-temperature treatments are used prior to or simultaneously with enzyme treatment. In preparing animal feed, thermostable xylanases are added into the feed before the pelleting process (typically carried out at 70 to 95°C). To obtain thermostable xylanases, one strategy is to search for novel xylanases from extremophiles. A number of thermostable GH10 xylanases have been reported for thermophilic Thermotoga maritima MSB8 (90°C) (10), Thermotoga petrophila RKU-1 (90°C) (11), and Thermobifida alba UL JB1 (80°C) (12) and for acidomesophilic Bispora sp. strain MEY-1 (85°C) (13). The available sequences and structures of these thermophilic xylanases (i.e., 1VBU and 3NIY [10, 11]) provide valuable information for better understanding of the structural and functional characters of thermostable enzymes.
Improving the thermal properties of mesophilic enzymes by protein engineering is the other important strategy to obtain thermostable enzymes. The approaches generally used include but are not limited to replacing the N terminus, introducing disulfide bridges, and increasing the number of salt bridges or hydrogen bonds (14). Many successful protein engineering examples for thermostability improvement have been published. Wintrode et al. (15) employed DNA shuffling to develop a protease mutant with increased melting temperature (Tm) of 25°C and increased half-life at 60°C (1,200-fold). In combination with computer-aided prediction and rational and random design, this method has been proved to be more efficient. Fenel et al. (16) and Wang et al. (17) specifically introduced a disulfide bridge into the xylanases of Trichoderma reesei and Thermomyces lanuginosus, shifting their temperature optima at least 10°C higher. Joo et al. (18) improved the Tm of Bacillus circulans xylanase by 4.2°C based on thermal fluctuation analysis.
XynAS9, a GH10 xylanase from Streptomyces sp. strain S9, has been expressed in Escherichia coli and showed superior properties, such as broad ranges of pH adaptation and stability (19). However, its thermostability is poor at temperatures above 65°C, the most common industrial temperature. To widen its application spectrum in industries, it is necessary to improve the thermal properties of XynAS9. Recombinant XynAS9 produced in Pichia pastoris showed an increased temperature optimum (73°C) but was still liable at 65°C and above (data not shown). The objective of the present study was to develop thermostable XynAS9 by employing bioinformatics-driven, rational engineering. By replacing some key residues, the thermostability of XynAS9 was expected to be improved significantly.
MATERIALS AND METHODS
Strains, plasmids, media, and chemicals.
Streptomyces sp. S9 was the donor of the GH10 xylanase gene xynAS9 (GenBank accession number EU153378) (19). Escherichia coli Trans I-T1 (Transgen, Beijing, China) was used for plasmid amplification. P. pastoris GS115 (Invitrogen, Carlsbad, CA) was used as the expression host. Plasmids pEASY-T3 (Tiangen, Beijing, China) and pPIC9 (Invitrogen) were used for cloning and expression, respectively. Pfu DNA polymerase and restriction endonucleases and T4 DNA ligase were from Tiangen and New England BioLabs (Hitchin, United Kingdom), respectively. The low-molecular-weight calibration kit was from GE Healthcare (Pittsburgh, PA).
The substrate birchwood xylan was purchased from Sigma-Aldrich (St. Louis, MO). Minimal dextrose medium or minimal methanol medium for transformant selection and buffered minimal glycerol complex medium and buffered methanol complex medium for P. pastoris growth and induction, respectively, were prepared according to the manual for the Pichia expression kit (Invitrogen). All other chemicals were of analytical grade.
Identification of key residues.
Multiple-sequence alignment (MSA) of XynAS9 and six GH10 xylanases with different temperature optima (from 73°C to 90°C; see Fig. 1) was performed by using ClustalW. The residues that might be related to their thermophilic properties were identified. The three-dimensional structures of XynAS9 and its five mutants (V81P, G82E, V81P/G82E, D185P/S186E, and V81P/G82E/D185P/S186E mutants) were modeled using Discovery studio 2.5.5 software (Accelrys, San Diego, CA) and the crystal structures of chain A (3CUF) (20) and chain E (3NDY) (21) as the templates.
FIG 1.

MSA of GH 10 xylanases with various temperature optima using the ClustalW program. TmxB, the xylanase from Thermotoga maritima; TpXyl10B, xylanase 10b from Thermotoga petrophila RKU-1; XYL10C, the endo-β-1,4-xylanase from Bispora sp. MEY-1; XylA, the β-1,4-endoxylanase from Thermobifida alba; XylE, the endo-1,4-xylanase from Penicillium canescens; XynC, the xylanase from Phanerochaete chrysosporium; and XynAS9, the xylanase from Streptomyces sp. S9. The catalytic residues are indicated with asterisks. The mutation sites are gray.
MDS.
The modeled structures of wild-type XynAS9 and its mutants were optimized using NAMD 2.7 and Gromacs 4.4.5, to investigate the possible mechanism of thermal stability increase. NAMD is a unique strategy for fast parallel molecular dynamic simulation (MDS) using the CHARMM27 force field (22–25). The box size was 63.17 Å by 95.22 Å by 111.34 Å, which was large enough to equilibrate the protein within it. Transferable intermolecular potential 3P (TIP3P) water was used as the aqueous solution (26). Na+ was used as counterion to neutralize the negative charges of proteins, and the system was subjected to a steepest-descent energy minimization with 10,000 steps. Each system was equilibrated in an NPT ensemble with a constant number of particles (N), constant system pressure (P), and constant temperature (T) at 1 atm pressure. The structure of XynAS9 was optimized and simulated at 20 ns. The average conformation of the wild-type protein in the simulation was used as the template to construct variations with Discovery Studio 2.5.5. The wild-type and mutant enzymes were simulated for 20 ns with NAMD at 300 K and 400 K, respectively. For all simulations, the time step was defined as low as 2 fs, and cutoffs were 12 Å (22). The data were calculated at a 5-ns timescale from 15 ns to 20 ns. The analysis for root mean square deviation (RMSD) and root mean square fluctuation (RMSF) was performed using standard NAMD tools, and the hydrogen bonds and salt bridges were analyzed by VMD (http://www.ks.uiuc.edu/Research/vmd/). Analysis with Gromacs followed the protocol of NAMD.
Generation, expression, and purification of wild-type XynAS9 and its mutants.
By combining the results from MSA and MDS, the key residues related to XynAS9 thermostability were determined. Site-directed mutagenesis to replace the specific residue(s) identified in the analyses described above was performed by overlap extension PCR (27). Mutations were introduced into the oligonucleotide primers (see Table S1 in the supplemental material), and the overlapping ends of the fragments were annealed to each other. Final products were purified using the gel extraction kit (Tiangen), ligated into the pEASY-T3 vector, and sequenced. The gene fragments encoding the wild-type XynAS9 and mutant enzymes were inserted into the expression vector pPIC9 at the EcoRI and NotI sites, respectively. The recombinant plasmids were linearized with BglII and then individually transformed into P. pastoris GS115 competent cells by electroporation. Recombinant expression was performed as described in the manual for the Pichia expression kit (Invitrogen).
To purify recombinant XynAS9 and its five mutants, the supernatants (∼1 liter) of the induced cultures were collected by centrifugation at 4°C. The clear supernatants were concentrated to 300 ml using the hollow fiber, followed by further concentration to 40 ml using Vivaflow 50 ultrafiltration membranes with a 5-kDa molecular mass cutoff (Vivascience, Hannover, Germany). The xylanases in the supernatants were each precipitated by the addition of 80 ml of acetone. The precipitates were collected by centrifugation for 10 min at 12,000 × g and 4°C and resuspended in 20 mM Tris-HCl (pH 8.0). Each suspension was loaded onto a HiTrap desalting column (GE Healthcare, Uppsala, Sweden) for desalination and then applied to a HiTrap Q Sepharose XL 5-ml fast-performance liquid chromatography (FPLC) column (GE Healthcare). Proteins were eluted using a linear NaCl gradient (0 to 0.6 M) in the same buffer. Fractions showing xylanase activities were pooled, concentrated, and stored at 4°C.
The concentrations of the purified proteins were determined using the Bio-Rad protein assay kit (Boston, MA). To remove N-glycosylation, purified enzymes were treated with 250 U of endo-β-N-acetylglucosaminidase H (endo H) for 2 h at 37°C according to the manufacturer's instructions (New England BioLabs, Ipswich, MA). The deglycosylated and untreated enzymes were analyzed by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) (28).
Xylanase activity assay.
The enzymatic activity of xylanase was determined by measuring the release of reducing sugar from birchwood xylan with 3,5-dinitrosalicylic acid (DNS) (29). The standard reaction mixture consisting of 100 μl of appropriately diluted enzyme and 900 μl of McIlvaine buffer (200 mM Na2HPO4, 100 mM citric acid [pH 6.5]) containing 1% (wt/vol) xylan substrate was incubated at 70°C for 10 min, and then the reaction was stopped with 1.5 ml of DNS reagent. One unit of xylanase activity was defined as the amount of enzyme that produced 1 μmol of reducing sugar per minute. Each reaction and its controls were run in triplicate.
Determination of kinetic parameters of XynAS9 and its mutants.
The kinetic parameters Km, kcat, and maximum rate of metabolism (Vmax) were determined in McIlvaine buffer (pH 6.5) containing 0.5 to 10.0 mg/ml of birchwood xylan for 5 min. The kinetic values were determined by fitting the Lineweaver-Burk plot.
Measurement of optimal pH for activity and stability.
The pH optimum of each enzyme was determined by measuring the xylanase activity at 70°C for 10 min in the pH range of 3.0 to 12.0. The pH stability of each enzyme was determined by measuring the residual activities under optimal conditions (pH 6.0 or 6.5, 73 to 90°C, 10 min) after preincubation at 37°C and pH 3.0 to 12.0 for 1 h. The buffers used were McIlvaine buffer (pH 2.0 to 8.0) as mentioned above and 50 mM glycine-NaOH (pH 8.0 to 12.0).
Optimal temperature and thermal stability studies.
The optimal temperatures of XynAS9 and its five mutants were determined in the range of 30 to 95°C at optimal pH (6.0 or 6.5) with a 10-min incubation time. Thermal stability was determined by measuring the half-life of enzyme inactivation (t1/2) at 65 to 80°C and optimum pH. All purified enzymes were diluted to 100 μg/ml in McIlvaine buffer (pH 6.5) and incubated for different durations. The residual enzyme activities were measured under standard conditions.
To determine their thermal tolerance (temperature at which 50% of activity is lost [T50]), wild-type XynAS9 and mutant enzymes were diluted to 50 μg/ml in McIlvaine buffer (pH 6.5) and then heated for 30 min at each temperature point within the range of 50 to 90 at intervals of 5°C. After heating, the enzymes were immediately placed on ice for 10 min; the residual xylanase activity was then measured using the assay described above.
CD for protein structure analysis.
Circular dichroism (CD) measurements of wild-type XynAS9 and its mutants were performed on a Chirascan spectropolarimeter (Applied Photophysics, Surrey, United Kingdom) (30) at 25°C. The proteins were diluted to 200 μg/ml (4.2 μM) in 10 mM phosphate-buffered saline (PBS). Spectra were continuously recorded from 190 nm to 250 nm using a 1-mm cell and a bandwidth of 1 nm over 3 scans at a scan rate of 120 nm/min. The results were analyzed by CDNN processing software.
DSC.
Differential scanning calorimetry (DSC) was performed on a Nano-DSC (TA Instruments, New Castle, DE) at a heating rate of 1°C/min and a scanning rate of 1°C/min. The proteins, 350 μg of each sample, were dissolved in 1 ml of 10 mM PBS (pH 7.4). Before measurements, the samples were degassed in an evacuated chamber for 15 min at an atmospheric pressure of 51 to 85 kPa and then immediately loaded into the calorimeter cell, a 0.3-ml sample cell against a 0.3-ml reference cell. The same amount of buffer was measured as the baseline, which was subtracted from each measured scan. A constant pressure of 3 atm was maintained during all DSC experiments to prevent possible degassing of the solution on heating. The melting temperature, Tm, obtained from the DSC experiments corresponded to the maximum of the transition peak, and the test was repeated at least twice at temperatures between 25°C and 90°C.
Substrate docking analysis.
AutoDock 4.0 (http://autodock.scripps.edu/) was used as an appropriate docking tool to carry out the docking simulations of substrate xylohexaose and XynAS9 or the V81P/G82E and V81P/G82E/D185P/S186E mutants. The output from AutoDock and all modeling studies as well as images was generated with PyMOL 0.99 (http://pymol.en.uptodown.com). Hydrophobic and hydrogen bonding interactions of docked molecules were compared using the LigPlot program (31).
RESULTS
Identification of key residues.
Based on the MSA results (Fig. 1), a regular pattern was identified from the consensus sequences. Compared with mesophilic counterparts, thermophilic xylanases with a temperature optimum of 80°C or higher have proline at sites 81 and 185 and glutamic acid at sites 82 and 186 with a higher frequency, corresponding to valine and aspartic acid and glycine and serine in XynAS9, respectively. By replacing the corresponding residues in XynAS9 with proline and glutamic acid, the modeled V81P/G82E/D185P/S186E structure mutant (Fig. 2) indicated that all mutation sites are at the end of a long secondary-structure element; i.e., the mutations V81P and G82E are located on the internal β-strand 2 of the (α/β)8 domain and D185P and S186E on the surface region of the barrel at the N terminus of the fourth α-helix. MDS analysis with NAMD showed that these mutations decreased the conformational flexibility of XynAS9 at a temperature of 400 K with low RMSD values (see Fig. S1 in the supplemental material), suggesting that the V81P/G82E/D185P/S186E mutant may have a more stable structure. Thus, the crucial role of proline and glutamic acid substitutions in thermostability was supported by MDS results.
FIG 2.
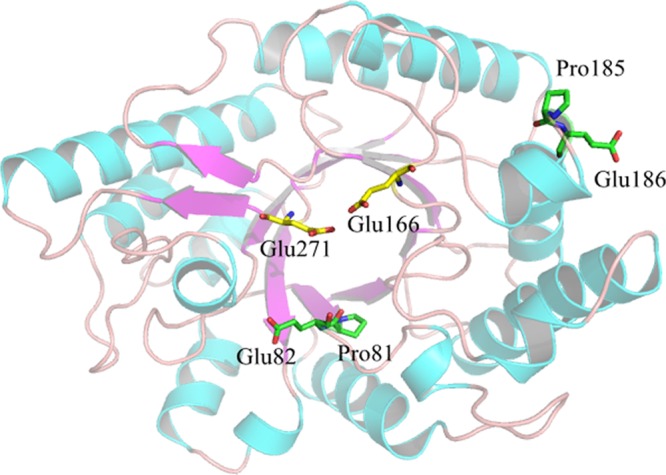
Three-dimensional structure model of the V81P/G82E/D185P/S186E mutant. Glu166 and Glu271 are putative catalytic residues.
Generation, expression, and purification of wild-type XynAS9 and its mutants.
The wild-type XynAS9 and its mutants were generated and expressed in Pichia and purified to electrophoretic homogeneity as described above. SDS-PAGE revealed that all recombinant enzymes had apparent molecular masses of ∼45 to 55 kDa, which were higher than their theoretical molecular masses (46 kDa). After treatment with endo H, all enzymes showed a single band corresponding to the theoretical mass (see Fig. S2 in the supplemental material).
Determination of kinetic parameters of XynAS9 and its mutants.
The specific activities and kinetic values of wild-type XynAS9 and its mutants were determined with birchwood xylan as the substrate (Table 1). Compared with the high specific activity of XynAS9 (630.3 U/mg), all mutants showed decreased specific activities, ranging from 78.5 to 206.9 U/mg. Moreover, these residue substitutions had profound impacts on the kinetics of all mutants. Except for the varying Km values from 0.60 to 4.02 mg/ml, all mutants were found to have decreased kcat, kcat/Km, and Vmax values. The results indicated that both residue substitutions V81P/G82E and substitutions far from them (D185P/S186E) decreased the catalytic efficiency of these mutant enzymes. Although the Km value of G82E for birchwood xylan decreased, which means an increased affinity to a substrate, its low catalytic efficiency (low specific activity and kcat/Km value) suggests that it might be inefficient in product releases.
TABLE 1.
Specific activities and kinetics of wild-type XynAS9 and its mutants with birchwood xylan as the substrate
| Enzyme | Sp act (U/mg)a | Km (mg/ml)a | kcat (/s)a | kcat/Km (ml/s · mg) | Vmax (U/mg)a |
|---|---|---|---|---|---|
| XynAS9 | 630.3 ± 12.5 | 1.03 ± 0.21 | 790.2 ± 14.3 | 760.3 | 950.8 ± 23.12 |
| V81P mutant | 155.2 ± 2.1 | 3.19 ± 0.91 | 218.4 ± 3.8 | 68.2 | 268.4 ± 8.52 |
| G82E mutant | 78.5 ± 1.4 | 0.60 ± 0.01 | 75.7 ± 3.1 | 121.4 | 90.3 ± 3.14 |
| V81P/G82E mutant | 206.9 ± 9.6 | 1.05 ± 0.15 | 212.5 ± 7.4 | 202.5 | 255.6 ± 8.33 |
| D185P/S186E mutant | 90.7 ± 3.4 | 4.02 ± 1.32 | 184.4 ± 6.4 | 45.8 | 221.9 ± 7.30 |
| V81P/G82E/D185P/S186E mutant | 160.4 ± 5.3 | 2.21 ± 0.79 | 235.6 ± 6.8 | 106.5 | 283.5 ± 7.97 |
Values are means ± standard deviations.
pH optima and stability studies.
The pH activity profiles of wild-type XynAS9 and its mutants were essentially quite similar and showed two activity peaks at pH 6.0 or 6.5 (maximum) and pH 8.0 or 9.0 (Fig. 3A), respectively. The enzymes retained >55% of maximal activities at pH 5.0 to 9.0, and the V81P/G82E and V81P/G82E/D185P/S186E mutants remained active even at pH 10.0 (>20% activity). All enzymes were stable at pH 4.0 to 11.0, and some enzymes retained activity after 1 h of incubation under extremely acidic (pH 3.0; G82E, V81P/G82E, and V81P/G82E/D185P/S186E mutants) and alkaline (pH 12.0; wild-type XynAS9 and V81P and D185P/S186E mutants) conditions (Fig. 3B).
FIG 3.

Enzymatic properties of wild-type XynAS9 and its mutants. (A) pH-dependent activity profiles; (B) pH stability; (C) temperature-dependent activity profiles; (D) enzyme inactivation at different temperatures. Symbols: □, wild-type XynAS9; ●, V81P mutant; ○, G82E mutant; -, V81P/G82E mutant; ■, D185P/S186E mutant; ▲,V81P/G82E/D185P/S186E mutant.
Temperature optima and thermostability studies.
Five mutants showed improved activity at high temperatures (Fig. 3C). Among them, the V81P, G82E, and D185P/S186E mutants had temperature optima of 75°C, 80°C, and 80°C, respectively, 2 to 7°C higher than that of wild-type XynAS9 (73°C). The V81P/G82E and V81P/G82E/D185P/S186E mutants showed the greatest shift in temperature optimum, from ∼73°C to 90°C. All these mutants showed 50 to 100% of the maximal activity at 85°C, much higher than that of wild-type XynAS9 (20% of the maximal activity). The temperature optimum increase (7°C) of the G82E mutant was more significant than that of the V81P mutant (2°C), and the sum of their increases (∼9°C) was less than the gain in the V81P/G82E double mutant (∼17°C). The V81P/G82E/D185P/S186E quadruple mutant showed the same increase in the apparent temperature optimum (∼17°C). The results indicated that mutations at the positions identified by MSA had a cumulative effect on enzyme activity at elevated temperatures. Thermostabilities of wild-type XynAS9 and its mutants were assessed by their half-lives (t1/2s) (Table 2). The V81P and D185P/S186E mutants had a marginally increased t1/2 value (1- to 2-fold). The G82E mutant was much more thermostable, with a t1/2 increased up to 3-fold at 65°C. And the V81P/G82E and V81P/G82E/D185P/S186E mutants showed the greatest improvement in thermostability, with t1/2 increased up to 16-fold at 65°C, 10-fold at 70°C, and 4-fold at 75°C and 80°C. The strongest effect on thermostability was therefore observed for the V81P/G82E double mutant, exactly in the same way as the highest increase in temperature optimum.
TABLE 2.
Half-lives of wild-type XynAS9 and its mutants for thermal inactivationa
| Enzyme |
t1/2 (min) |
|||
|---|---|---|---|---|
| 65°C | 70°C | 75°C | 80°C | |
| XynAS9 | 16 | 9 | 8 | 4 |
| V81P mutant | 27 | 12 | 7 | 4 |
| G82E mutant | 45 | 12 | 9 | 6 |
| V81P/G82E mutant | 255 | 94 | 33 | 15 |
| D185P/S186E mutant | 35 | 15 | 8 | 5 |
| V81P/G82E/D185P/S186E mutant | 228 | 81 | 33 | 18 |
The enzyme activity was assayed at pH 6.5 and 70°C for 10 min.
Thermal tolerance (T50) of wild-type XynAS9 and its mutants was determined at the temperature range of 50°C to 90°C (Fig. 3D). The T50 value of XynAS9 was determined to be 62.0°C, and the V81P, G82E, V81P/G82E, D185P/S186E, and V81P/G82E/D185P/S186E mutants showed increased values of 1.7°C, 6.2°C, 6.9°C, 2.3°C, and 11.2°C, respectively. It was found that single mutation at position 82 with glutamic acid played a key role in the improvement of thermal tolerance and had an additive effect when combined with V81P. The V81P/G82E/D185P/S186E quadruple mutant was most striking in thermal tolerance improvement.
CD measurements.
CD analysis was conducted to check whether the improved thermal properties of mutants were caused by the secondary-structure changes of proteins. As shown in Fig. 4A, the far-UV CD spectrum of XynAS9 exhibited a pronounced maximum and minimum at 195 nm and 222 nm, respectively, which are characteristics of β-sheet and α-helix structures in aqueous solution, respectively (32). Thus, XynAS9 is an autonomous structural protein that contains both α-helix and β-sheet secondary structures as predicted from homology modeling. Residue substitutions did not change the secondary structure of the G82E and D185P/S186E mutants, which had a far-UV CD spectrum similar to that of XynAS9. In contrast, the V81P, V81P/G82E, and V81P/G82E/D185P/S186E mutants yielded different far-UV CD spectra with deep minima near 200 nm, which is typical of irregular structure (32). The results may explain the significant contribution of residue substitution at site 81 with proline to improved thermal properties. Moreover, TmxB from T. maritima (1VBU) (10) and XynAS9 had different residues at positions 81 and 82 (Fig. 1), but their local structures in these positions are similar (modeled XynAS9 with 3CUF and 3NDY as the templates). This result indicates that the improved thermostability is still based on minor structural changes.
FIG 4.
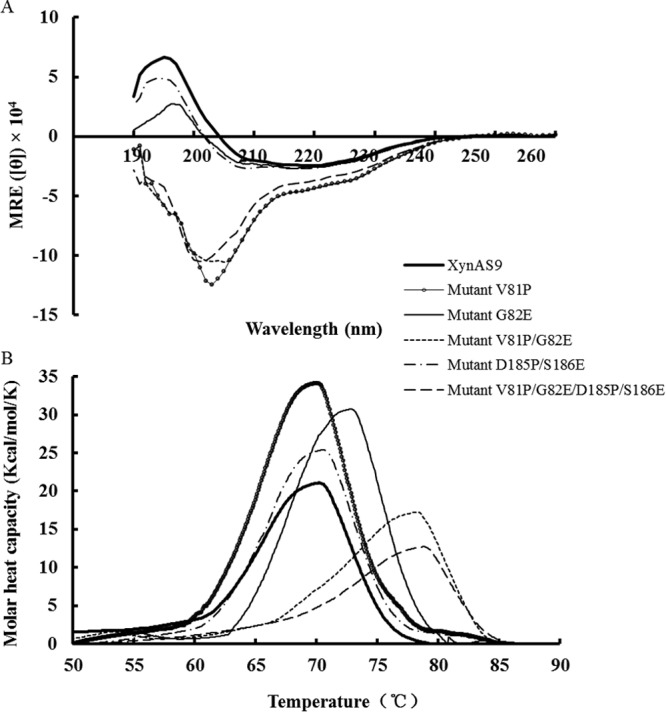
(A) Far-UV CD spectra of wild-type XynAS9 and its mutants in 10 mM PBS (pH 7.4); (B) thermograms, measured using DSC. The calorimetric recordings for XynAS9 and its mutants were scanned at 1°C/min in 10 mM PBS (pH 7.4) with the protein concentration of 350 μg/ml, respectively.
DSC analysis.
DSC was performed to determine the Tm values of wild-type XynAS9 and its mutants over the temperature range of 25°C to 90°C (Fig. 4B). The Tm values of the V81P, G82E, V81P/G82E, D185P/S186E, and V81P/G82E/D185P/S186E mutants were 69.1°C, 71.8°C, 75.1°C, 69.4°C, and 75.2°C, respectively, which were 0.9 to 7.0°C higher than that of wild-type XynAS9 (68.2°C). This is in agreement with the results above that residue substitutions at sites 81 and 82 make the most remarkable contribution to the thermal stability.
Structural changes during MDS.
MDS at 400 K was used to interpret the effect of mutations on structural changes. During the course of simulation, the inner β-barrel stayed quite stable, but the loops and α-helix layer started to experience changes (see Fig. S3 in the supplemental material). When the residues at four sites were mutated, several α-helices were unfolded and changed to long loops, especially around positions 81 and 82. A long loop located after 81 and 82 (region 83 to 86 and 91 to 98) changed completely its conformation. The local structures immediately before and after positions 185 and 186 did not essentially change, but there were changes in other main-chain regions nearby. Amphiphilic α-helices are important in the stabilization of the whole enzyme structure. The large changes from α-helices to long loops after positions 81 and 82 could be relevant for the interpretation of the effects of mutations at positions 81 and 82. According to MDS, the eventual decrease of flexibility was observed in large areas of the protein with residue replacement (see Fig. S1 in the supplemental material).
VMD analysis of hydrogen bonds and salt bridges.
In the modeled structure of XynAS9, Val81 does not form any hydrogen bond with other residues, whereas there is a putative hydrogen bond between the main-chain nitrogen of Gly82 and the main-chain oxygen of Thr58. Within the distance of 6 Å, there are other four putative hydrogen bonds around Val81 and Gly82 (see Fig. S4A in the supplemental material), i.e., Asn83 to Met85 or Lys86 and Thr80 to the carboxyl or amino group of Thr58, and five around Asp185 and Ser186 (Fig. S4C), i.e., Arg174 to Asp185 or Gln180 and Ser186 to Ala189 or Glu190. When these residues are replaced with proline and glutamic acid, one or two additional putative hydrogen bonds are predicted to be formed between Glu82 and Asn83 (see Fig. S4B and S5) and one additional bond is predicted between Glu186 and Tyr187 (see Fig. S4D). Dihedral rotation of Ser186 does not make any hydrogen bonds, whereas rotated Glu186 can form several hydrogen bonds to the residues mentioned above, thus possibly strengthening the local structure. On the other hand, Glu186 is fully exposed to solution, whereas Glu82 appears to have favorable hydrophobic interactions in the hydrophobic part of the side chain with Trp311 and a strong polar environment for the carboxyl group of Glu82, including ND2 of Asn83 and NE of Trp311 and the side active site. Therefore, Glu82 fills very well the groove on the bottom of the active site and is packed well against W311 and N83 (see Fig. S5). The space filling with hydrophobic and polar interactions is likely to stabilize the local area around the cavity.
The protein structure changed considerably during the course of MDS, and new interactions were formed. The distances between Glu82 and Asp59 and between Glu186 and Lys217 of the V81P/G82E/D185P/S186E mutant became short enough to form hydrogen bonds and even a salt bridge between Glu186 and Lys217. The same phenomenon was also observed in the distance change of Glu82 and Lys316. The putative salt bridge between Glu82 and Lys316 may stabilize the structures of the big loop from Trp311 to Trp319 and the β-strand in close proximity. Asp59 and Asp312 are separated from Glu82 at the distances of 3.2 and 4.9 Å, respectively; within this distance a force of mutual electrostatic repulsion might be produced. VMD analysis indicates that unfolded protein may create new interactions between natively distant positions and thus in some degree stabilize the unfolded structure.
Docking of xylohexaose into XynAS9 and the V81P/G82E mutant.
Docking simulation with AutoDock 4.0 (see Fig. S6 in the supplemental material) showed that xylohexaose was docked into the substrate binding pocket of XynAS9 by residues Asn83, Trp123, Glu166, Glu170, Asn210, His243, Glu271, Trp311, and Trp319. The schematic diagrams of protein-ligand interactions by Ligplot showed that the catalytic residue Glu271 formed a strong intermolecular hydrogen bond with xylohexaose–O-25. Gly82 formed a hydrophobic interaction with xylohexaose. One potential hydrogen bond was formed between Asn83 and xylohexaose–O-18. When xylohexaose was docked with the modeled structure of the V81P/G82E mutant, the filling of the bottom of the catalytic canyon by Glu82 forced the substrate to occupy a higher position at this end of the canyon, therefore changing the sites of interaction with the enzyme. The catalytic Glu271 may form one additional hydrogen bond with xylohexaose–O-23, and Glu82 may form three potential hydrogen bonds, two with xylohexaose–O-11 and one with xylohexaose–O-12. Glu82 and Tyr317 may compete to form a hydrogen bond with xylohexaose–O-11. Moreover, Asn83 in the V81P/G82E mutant may form two hydrogen bonds with xylohexaose–O-13/O-14 instead of one with xylohexaose–O-18 in XynAS9. The docking result for the V81P/G82E/D185P/S186E mutant was the same as that for the V81P/G82E mutant (data not shown). This result suggests that residue substitution with glutamic acid at site 82 may form new hydrogen bonds, increase the stability of enzyme-substrate complex, and consequently improve the enzyme activity at high temperatures.
DISCUSSION
XynAS9 from Streptomyces sp. S9 contains an (α/β)8 barrel catalytic domain and has weak thermostability (19). Quite a lot of knowledge exists about the stabilizing factors in (α/β)8-barrel enzymes, including efficient packing of the hydrophobic core, the presence of prolines at the N termini of α-helices, and cavity filling and stabilization of loops and N- and C-terminal regions (33, 34). In this study, we developed several highly thermostable mutants of XynAS9 by combining MSA, MDS, and site-directed mutagenesis techniques. MSA of various GH10 xylanases with different temperature optima provides the most direct and readily available information related to enzyme properties. Computational design methods provide more precise guidance for enzyme engineering and make mutation more efficient. According to the results of MSA and MDS, four key residues probably involved in enzyme activity and stability at high temperatures were identified, and five mutants were then constructed. Compared with wild-type XynAS9, all five mutants—the V81P, G82E, V81P/G82E, D185P/S186E, and V81P/G82E/D185P/S186E mutants—showed enhanced resistance to high temperatures, and the V81P/G82E and V81P/G82E/D185P/S186E double and quadruple mutants showed the greatest shift, 17°C, in temperature optima and the best thermostability at high temperatures (i.e., 85°C).
Glutamic acid has an anionic carboxylate that easily forms hydrogen bonds and salt bridges and has helix-forming propensity, which contributes to thermostability (35, 36). For example, replacement of aspartic acid with glutamic acid made mesophilic Aspergillus awamori glucoamylase thermostable up to 70°C (36), glutamic acid substitution increased the Tm of T4 lysozyme by 1.5°C by forming a salt bridge (37), and the modified xylanase A of Streptomyces lividans with glutamic acid substitution showed a longer half-life at 60°C than its wild-type counterpart (38). In this study, we also introduced glutamic acid residues into XynAS9, which may produce additional putative salt bridges, hydrogen bonds, and hydrophobic interactions (see Fig. S4 and S5 in the supplemental material). The mutant glutamate side chain of G82E fills a groove on the bottom of the active site canyon with some favorable side chain interactions like hydrogen bonds, and therefore, the improved packing and filling of the groove may play a central role in thermostability. The same space filling can be seen in T. maritima xylanase 10B (1VBU) and Cellulomonas fimi XYN10A (3CUF) by the side chain of glutamic acid (data shown in the Protein Data Bank[PDB] database). Besides these molecular interactions, the mutation S186E at the end of the helix might increase the length of the helix, thus creating higher rigidity in the region. In both regions there are mainly acidic amino acids near the introduced glutamic acid (plus one lysine close to S186E); the roles of these ionic interactions remain unclear, however. These interactions in combination contributed to the more stable conformation and improved the enzyme robustness at high temperatures.
Besides glutamic acid, proline is highly prevalent in thermophilic proteins because it has a side chain of distinctive cyclic structure that locks its backbone and leads to an exceptional conformational rigidity in the turns and loops. Introduction of proline residues at certain positions has been used successfully to improve the thermostabilities of many enzymes (39–44). In the present study, we introduced proline at sites 81 and 185, which are located at the second position of the β-turn and the N-cap of the α-helix (Fig. 2), respectively. Proline substitution at site 81 probably affected the local structure with lower RMSD values (as shown in Fig. S7 in the supplemental material). After position 81, there is a curvy loop (starting from 82) that is probably stabilized by the rigid proline and enables the enzyme to perform its catalytic function at higher temperature. Thus, we can conclude that proline substitutions also are one of the key factors responsible for the improved thermal properties of the mutated XynAS9.
Considering the increases in temperature optima and Tm values of mutants G82E and V81P (7°C and 3.6°C versus 2°C and 0.9°C), the effect of glutamic acid is clearly more significant than that of proline. The combination of the two mutations in adjacent positions with apparently differing stabilization mechanisms was additive in the improvement of temperature optimum. Therefore, both improved rigidity of the main chain and improved side chain interactions contributed to the final effect in the combination mutant. These results showed that the enzyme had a very weak point on the bottom of the active-site canyon that was largely repaired by only two mutations.
It is known that substrate itself increases the thermostability of xylanases (34). In the presence of substrate, hydrogen bonds were formed between xylohexaose and catalytic residues and Glu82 of the G82E/V81P mutant, respectively, as shown in the corresponding glutamic acid of C. fimi xylanase (3CUJ) and Bacillus subtilis xylanase (2B46) that forms two hydrogen bonds to the substrate (data shown in PDB databases). These interactions may increase the robustness of the xylanase-xylohexaose complex, which benefits the adaptability and stability of XynAS9 over high temperatures. Experimental results showing that the increases in apparent temperature optima of the G82E and V81P mutants were greater than that in melting temperature further confirm the role of the substrate in the stabilization of enzymes at high temperatures. These improved interactions between enzyme and substrate can remarkably improve the tolerance of enzymes to high temperatures.
Modified XynAS9 with double to quadruple mutations showed significant improvement in enzyme thermostability and activity under alkaline conditions. These characteristics make them favorable for wide industrial application, especially in the bleaching of kraft pulp. However, the price of these stabilizing mutations could be a lowered catalytic activity. While the mutations improved both apparent temperature optimum and thermostability, there was a clear trend to decreased catalytic efficiency. It indicates how sensitive the balance between activity and stability can be, and the increase of enzymatic performance at higher temperatures may require a compromise of the catalytic efficiency. This phenomenon can be interpreted by the stability-function hypothesis that enzyme residues involved in function are not optimized for stability and vice versa (45). The consequence could then be that the increased rigidity leads to lowered activity, as it was observed in our molecular dynamic simulations that the mutations decreased structural flexibility.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National High-Tech Research and Development Program (863 Program, grant no. 2012AA022208), the National Science Fund for Distinguished Young Scholars (grant no. 31225026), and the National Natural Science Foundation of China (grant no. 31101742).
Footnotes
Published ahead of print 24 January 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03458-13.
REFERENCES
- 1.Collins T, Gerday C, Feller G. 2005. Xylanases, xylanase families and extremophilic xylanases. FEMS Microbiol. Rev. 29:3–23. 10.1016/j.femsre.2004.06.005 [DOI] [PubMed] [Google Scholar]
- 2.Li X, Jiang Z, Li L, Yang S, Feng W, Fan J, Kusakabe I. 2005. Characterization of a cellulase-free, neutral xylanase from Thermomyces lanuginosus CBS 288.54 and its biobleaching effect on wheat straw pulp. Bioresour. Technol. 96:1370–1379. 10.1016/j.biortech.2004.11.006 [DOI] [PubMed] [Google Scholar]
- 3.Polizeli M, Rizzatti A, Monti R, Terenzi H, Jorge J, Amorim D. 2005. Xylanases from fungi: properties and industrial applications. Appl. Microbiol. Biotechnol. 67:577–591. 10.1007/s00253-005-1904-7 [DOI] [PubMed] [Google Scholar]
- 4.Xiong H, von Weymarn N, Turunen O, Leisola M, Pastinen O. 2005. Xylanase production by Trichoderma reesei Rut C-30 grown on l-arabinose-rich plant hydrolysates. Bioresour. Technol. 96:753–759. 10.1016/j.biortech.2004.08.007 [DOI] [PubMed] [Google Scholar]
- 5.Huang Y, Chen Y, Chen C, Chen W, Ciou Y, Liu W, Yang C. 2011. Production of ferulic acid from lignocellulolytic agricultural biomass by Thermobifida fusca thermostable esterase produced in Yarrowia lipolytica transformant. Bioresour. Technol. 102:8117–8122. 10.1016/j.biortech.2011.05.062 [DOI] [PubMed] [Google Scholar]
- 6.Zhang J, Tuomainen P, Siika-Aho M, Viikari L. 2011. Comparison of the synergistic action of two thermostable xylanases from GH families 10 and 11 with thermostable cellulases in lignocellulose hydrolysis. Bioresour. Technol. 102:9090–9095. 10.1016/j.biortech.2011.06.085 [DOI] [PubMed] [Google Scholar]
- 7.Henrissat B. 1991. A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem. J. 280:309–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Badieyan S, Bevan DR, Zhang C. 2012. Study and design of stability in GH5 cellulases. Biotechnol. Bioeng. 109:31–44. 10.1002/bit.23280 [DOI] [PubMed] [Google Scholar]
- 9.Turner P, Mamo G, Karlsson EN. 2007. Potential and utilization of thermophiles and thermostable enzymes in biorefining. Microb. Cell Fact. 6:9. 10.1186/1475-2859-6-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ihsanawati , Kumasaka T, Kaneko T, Morokuma C, Yatsunami R, Sato T, Nakamura S, Tanaka N. 2005. Structural basis of the substrate subsite and the highly thermal stability of xylanase 10B from Thermotoga maritima MSB8. Proteins 61:999–1009. 10.1002/prot.20700 [DOI] [PubMed] [Google Scholar]
- 11.Santos CR, Meza AN, Hoffmam ZB, Silva JC, Alvarez TM, Ruller R, Giesel GM, Verli H, Squina FM, Prade RA. 2010. Thermal-induced conformational changes in the product release area drive the enzymatic activity of xylanases 10B: crystal structure, conformational stability and functional characterization of the xylanase 10B from Thermotoga petrophila RKU-1. Biochem. Biophys. Res. Commun. 403:214–219. 10.1016/j.bbrc.2010.11.010 [DOI] [PubMed] [Google Scholar]
- 12.Blanco J, Coque J, Velasco J, Martin J. 1997. Cloning, expression in Streptomyces lividans and biochemical characterization of a thermostable endo-β-1,4-xylanase of Thermomonospora alba UL JB1 with cellulose-binding ability. Appl. Microbiol. Biotechnol. 48:208–217. 10.1007/s002530051040 [DOI] [PubMed] [Google Scholar]
- 13.Luo H, Li J, Yang J, Wang H, Yang Y, Huang H, Shi P, Yuan T, Fan Y, Yao B. 2009. A thermophilic and acid stable family-10 xylanase from the acidophilic fungus Bispora sp. MEY-1. Extremophiles 13:849–857. 10.1007/s00792-009-0272-0 [DOI] [PubMed] [Google Scholar]
- 14.Li W, Zhou X, Lu P. 2005. Structural features of thermozymes. Biotechnol. Adv. 23:271–281. 10.1016/j.biotechadv.2005.01.002 [DOI] [PubMed] [Google Scholar]
- 15.Wintrode PL, Miyazaki K, Arnold FH. 2001. Patterns of adaptation in a laboratory evolved thermophilic enzyme. Biochim. Biophys. Acta 1549:1–8. 10.1016/S0167-4838(01)00226-6 [DOI] [PubMed] [Google Scholar]
- 16.Fenel F, Leisola M, Janis J, Turunen O. 2004. A de novo designed N-terminal disulphide bridge stabilizes the Trichoderma reesei endo-1,4-β-xylanase II. J. Biotechnol. 108:137–143. 10.1016/j.jbiotec.2003.11.002 [DOI] [PubMed] [Google Scholar]
- 17.Wang Y, Fu Z, Huang H, Zhang H, Yao B, Xiong H, Turunen O. 2012. Improved thermal performance of Thermomyces lanuginosus GH11 xylanase by engineering of an N-terminal disulfide bridge. Bioresour. Technol. 112:275–279. 10.1016/j.biortech.2012.02.092 [DOI] [PubMed] [Google Scholar]
- 18.Joo JC, Pack SP, Kim YH, Yoo YJ. 2011. Thermostabilization of Bacillus circulans xylanase: computational optimization of unstable residues based on thermal fluctuation analysis. J. Biotechnol. 151:56–65. 10.1016/j.jbiotec.2010.10.002 [DOI] [PubMed] [Google Scholar]
- 19.Li N, Meng K, Wang Y, Shi P, Luo H, Bai Y, Yang P, Yao B. 2008. Cloning, expression, and characterization of a new xylanase with broad temperature adaptability from Streptomyces sp. S9. Appl. Microbiol. Biotechnol. 80:231–240. 10.1007/s00253-008-1533-z [DOI] [PubMed] [Google Scholar]
- 20.Bedarkar S, Gilkes NR, Kilburn DG, Kwan E, Rose DR, Miller RC, Jr, Warren RA, Withers SG. 1992. Crystallization and preliminary X-ray diffraction analysis of the catalytic domain of Cex, an exo-β-1,4-glucanase and β-1,4-xylanase from the bacterium Cellulomonas fimi. Appl. Microbiol. Biotechnol. 228:693–695 [DOI] [PubMed] [Google Scholar]
- 21.Privett HK, Kiss G, Lee TM, Blomberg R, Chica RA, Thomas LM, Hilvert D, Houk KN, Mayo SL. 2012. Iterative approach to computational enzyme design. Proc. Natl. Acad. Sci. U. S. A. 109:3790–3795. 10.1073/pnas.1118082108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Darden T, York D, Pedersen L. 1993. Particle mesh Ewald: an N · log(N) method for Ewald sums in large systems. J. Chem. Phys. 98:10089–10096. 10.1063/1.464397 [DOI] [Google Scholar]
- 23.Wilson GV, Lu P. 1996. Parallel programming using C Plus Plus. MIT Press, Cambridge, MA [Google Scholar]
- 24.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. 2005. Scalable molecular dynamics with NAMD. J. Comp. Chem. 26:1781–1802. 10.1002/jcc.20289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.MacKerell AD, Bashford D, Bellott M, Dunbrack R, Evanseck J, Field MJ, Fischer S, Gao J, Guo H, Ha S. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102:3586–3616. 10.1021/jp973084f [DOI] [PubMed] [Google Scholar]
- 26.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. 10.1063/1.445869 [DOI] [Google Scholar]
- 27.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77:51–59. 10.1016/0378-1119(89)90358-2 [DOI] [PubMed] [Google Scholar]
- 28.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. 10.1038/227680a0 [DOI] [PubMed] [Google Scholar]
- 29.Miller GL. 1959. Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal. Chem. 31:426–428. 10.1021/ac60147a030 [DOI] [Google Scholar]
- 30.Maheut G, Castaings A, Pécaut J, Lawson Daku LM, Pescitelli G, Di Bari L, Marchon JC. 2006. Chiroptical and computational studies of a bridled chiroporphyrin and of its nickel (II), copper (II), and zinc (II) complexes. J. Am. Chem. Soc. 128:6347–6356. 10.1021/ja054926o [DOI] [PubMed] [Google Scholar]
- 31.Wallace AC, Laskowski RA, Thornton JM. 1995. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 8:127–134. 10.1093/protein/8.2.127 [DOI] [PubMed] [Google Scholar]
- 32.Kelly SM, Jess TJ, Price NC. 2005. How to study proteins by circular dichroism. Biochim. Biophys. Acta 1751:119–139. 10.1016/j.bbapap.2005.06.005 [DOI] [PubMed] [Google Scholar]
- 33.Lo Leggio L, Kalogiannis S, Bhat M, Pickersgill R. 1999. High resolution structure and sequence of T. aurantiacus xylanase I: implications for the evolution of thermostability in family 10 xylanases and enzymes with βα-barrel architecture. Proteins 36:295–306. [DOI] [PubMed] [Google Scholar]
- 34.Anbarasan S, Jänis Paloheimo JM, Laitaoja M, Vuolanto M, Karimäki J, Vainiotalo P, Leisola M, Turunen O. 2010. Effect of pH, glycosylation and additional domains on the thermostability of family 10 xylanase of Thermopolyspora flexuosa. Appl. Environ. Microbiol. 76:356–360. 10.1128/AEM.00357-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dougherty DA. 2006. Modern physical organic chemistry. University Science Books, Sausalito, CA [Google Scholar]
- 36.Chen H, Ford C, Reilly PJ. 1995. Identification and elimination by site-directed mutagenesis of thermolabile aspartyl bonds in Aspergillus awamori glucoamylase. Protein Eng. 8:575–582. 10.1093/protein/8.6.575 [DOI] [PubMed] [Google Scholar]
- 37.Sun D, Sauer U, Nicholson H, Matthews BW. 1991. Contributions of engineered surface salt bridges to the stability of T4 lysozyme determined by directed mutagenesis. Biochemistry 30:7142–7153. 10.1021/bi00243a015 [DOI] [PubMed] [Google Scholar]
- 38.Moreau A, Shareck F, Kluepfel D, Morosoli R. 1994. Increase in catalytic activity and thermostability of the xylanase A of Streptomyces lividans 1326 by site-specific mutagenesis. Enzyme Microb. Technol. 16:420–424. 10.1016/0141-0229(94)90158-9 [DOI] [PubMed] [Google Scholar]
- 39.Tian J, Wang P, Gao S, Chu X, Wu N, Fan Y. 2010. Enhanced thermostability of methyl parathion hydrolase from Ochrobactrum sp. M231 by rational engineering of a glycine to proline mutation. FEBS J. 277:4901–4908. 10.1111/j.1742-4658.2010.07895.x [DOI] [PubMed] [Google Scholar]
- 40.Suzuki Y, Hatagaki K, Oda H. 1991. A hyperthermostable pullulanase produced by an extreme thermophile, Bacillus flavocaldarius KP 1228, and evidence for the proline theory of increasing protein thermostability. Appl. Microbiol. Biotechnol. 34:707–714 [DOI] [PubMed] [Google Scholar]
- 41.Watanabe K, Masuda T, Ohashi H, Mihara H, Suzuki Y. 1994. Multiple proline substitutions cumulatively thermostabilize Bacillus cereus ATCC 7064 oligo-1,6-glucosidase. Eur. J. Biochem. 226:277–283. 10.1111/j.1432-1033.1994.tb20051.x [DOI] [PubMed] [Google Scholar]
- 42.Allen MJ, Coutinho PM, Ford CF. 1998. Stabilization of Aspergillus awamori glucoamylase by proline substitution and combining stabilizing mutations. Protein Eng. 11:783–788. 10.1093/protein/11.9.783 [DOI] [PubMed] [Google Scholar]
- 43.Zhu G, Xu C, Teng M, Tao L, Zhu X, Wu C, Hang J, Niu L, Wang YZ. 1999. Increasing the thermostability of d-xylose isomerase by introduction of a proline into the turn of a random coil. Protein Eng. 12:635–638. 10.1093/protein/12.8.635 [DOI] [PubMed] [Google Scholar]
- 44.Goihberg E, Dym O, Telr S, Levin I, Peretz M, Burstein Y. 2007. A single proline substitution is critical for the thermostabilization of Clostridium beijerinckii alcohol dehydrogenase. Proteins 66:196–204. 10.1002/prot.21170 [DOI] [PubMed] [Google Scholar]
- 45.Shoichet BK, Baase WA, Kuroki R, Matthews BW. 1995. A relationship between protein stability and protein function. Proc. Natl. Acad. Sci. U. S. A. 92:452–456. 10.1073/pnas.92.2.452 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.