

Abstract
Pseudomonas syringae pv. actinidiae is a reemerging pathogen which causes bacterial canker of kiwifruit (Actinidia sp.). Since 2008, a global outbreak of P. syringae pv. actinidiae has occurred, and in 2010 this pathogen was detected in New Zealand. The economic impact and the development of resistance in P. syringae pv. actinidiae and other pathovars against antibiotics and copper sprays have led to a search for alternative management strategies. We isolated 275 phages, 258 of which were active against P. syringae pv. actinidiae. Extensive host range testing on P. syringae pv. actinidiae, other pseudomonads, and bacteria isolated from kiwifruit orchards showed that most phages have a narrow host range. Twenty-four were analyzed by electron microscopy, pulse-field gel electrophoresis, and restriction digestion. Their suitability for biocontrol was tested by assessing stability and the absence of lysogeny and transduction. A detailed host range was performed, phage-resistant bacteria were isolated, and resistance to other phages was examined. The phages belonged to the Caudovirales and were analyzed based on morphology and genome size, which showed them to be representatives of Myoviridae, Podoviridae, and Siphoviridae. Twenty-one Myoviridae members have similar morphologies and genome sizes yet differ in restriction patterns, host range, and resistance, indicating a closely related group. Nine of these Myoviridae members were sequenced, and each was unique. The most closely related sequenced phages were a group infecting Pseudomonas aeruginosa and characterized by phages JG004 and PAK_P1. In summary, this study reports the isolation and characterization of P. syringae pv. actinidiae phages and provides a framework for the intelligent formulation of phage biocontrol agents against kiwifruit bacterial canker.
INTRODUCTION
In November 2010, the New Zealand kiwifruit industry first identified the phytopathogen Pseudomonas syringae pv. actinidiae on Actinidia chinensis Hort16A (1). P. syringae pv. actinidiae was subsequently detected in over 2,300 orchards on both Actinidia deliciosa cv. Hayward and A. chinensis, which represent >70% of the total area of kiwifruit orchards in New Zealand. This outbreak led to the removal of approximately 2,000 ha of kiwifruit orchards in New Zealand, resulting in significant economic loss (2).
P. syringae pv. actinidiae was first discovered as the causative agent of bacterial canker of kiwifruit in Japan in 1989 (3) although later reports indicate its presence in China in 1984 (see reference 4). Subsequently, the pathogen was detected in Italy, South Korea, and China (5–8). Although the disease was destructive, antibiotics or good orchard management was sufficient to control the disease and spread of the pathogen.
Between 2008 and 2011, P. syringae pv. actinidiae reemerged throughout the kiwifruit-growing regions of the world and was detected in Italy (9), South Korea (10), France (11), Portugal (12), Spain (13, 14), Turkey (15), Chile (16), and New Zealand. Many of these outbreak strains proved highly aggressive compared with those previously identified, resulting in entire orchards being uprooted in Italy and elsewhere due to the high susceptibility of the A. chinensis germplasm cultivated (Hort16A, Jin Tao, and Soreli) (9, 17, 18).
P. syringae pv. actinidiae enters the plant through natural openings and lesions (4). The initial symptoms of the bacterium on both green flesh (A. deliciosa) and yellow flesh (A. chinensis) kiwifruit are brown leaf spotting with yellow haloes and necrosis. In plants infected with more virulent P. syringae pv. actinidiae, the bacterium is also found in lenticels and phloem, with biofilm formation occurring both inside and outside plants (4). Other symptoms include brown discoloration to buds and flowers, twig, leader and cane dieback, reddening of lenticels, canker formation on the trunk and vines, and red or white exudates that can include high levels of bacteria (4, 19, 20).
The reemergence of P. syringae pv. actinidiae and the traits that make this pathogen successful have been reviewed by Scortichini et al. (20). In particular, genome sequencing and multilocus sequence typing have led to a greater understanding of the origins, evolution, and biology of this pathogen (19, 21–24). It has been proposed that the Italian, Chilean, and New Zealand strains originated independently from China (21). The current pandemic strains differ in only a few single nucleotide polymorphisms (SNPs) in their core genomes, with variable genomic island content accounting for any major differences (19, 21, 23). These strains also differ significantly from the less virulent strains previously identified. For example, the current, aggressive population displays four putative effector genes, namely, hopA1, hopAA1-2, hopH1, and hopZ2-like, which are not present in the strains previously isolated in Japan and Italy prior to these recent outbreaks. In New Zealand, P. syringae pv. actinidiae strains were termed virulent and less virulent. It has recently been shown that the less virulent strains are distinct from virulent P. syringae pv. actinidiae (21, 24).
In New Zealand, management programs have been developed to limit the spread of P. syringae pv. actinidiae (2). These strategies involve regular monitoring, the removal of infected plant material, spraying with streptomycin and/or copper, and the use of biological control agents (BCAs) and host resistance elicitors. However, streptomycin is not a viable control option in many countries; for example, the use of streptomycin to control plant diseases has been banned in the European Union (25). Furthermore, streptomycin resistance genes have been detected in P. syringae pv. actinidiae (26). Copper-based agrichemicals are currently the mainstay protective spray used against P. syringae pv. actinidiae in New Zealand (2). Continual use of copper can lead to a toxic buildup of copper residues within the environment (27, 28), and bacterial resistance to copper has been documented (29–31).
An alternative strategy for the control of phytopathogens is the use of naturally occurring bacteriophages (phages) (reviewed in references 32 to 34). Phages are viruses that specifically infect bacteria, and their replication results in the lysis and killing of the host cell and the release of more viral particles. Trials using phages for plant disease control against a variety of phytopathogens, including “Dickeya solani” (35), Erwinia amylovora (36–38), Pectobacterium carotovorum (39), and Ralstonia solanacearum (40, 41), have been conducted. An example of the commercial application of phages in agriculture is the production of AgriPhage by OmniLytics for the control of the tomato pathogens Pseudomonas syringae pv. tomato and Xanthomonas campestris pv. vesicatoria (42–46). Here, we present the first report of the isolation and characterization of a large bank of potential biocontrol phages that infect strains of P. syringae pv. actinidiae, the causative agent of kiwifruit canker.
MATERIALS AND METHODS
Materials, bacterial strains, and culture conditions.
The bacterial strains used in this study are listed in Table S1 in the supplemental material and include Pseudomonas isolates from New Zealand, Japan, South Korea, Italy, the United Kingdom, Spain, Chile, China, France, the United States, South Africa, Brazil, and Ethiopia. All strains were grown in nutrient broth (NB; 5 g liter−1 peptone, 3 g liter−1 yeast extract, and 5 g liter−1 NaCl) at 25°C or on solid NB medium containing 1.5% (wt/vol) agar. Soft medium (overlay) agar (0.35%, wt/vol) was used for bacteriophage host range assays. Phage buffer was composed of 10 mM Tris-HCl, pH 7.4, 10 mM MgSO4, and 0.01% (wt/vol) gelatin. The chloroform used in this study was saturated with sodium hydrogen carbonate.
Phage isolation.
Soil, water, and leaf litter samples were collected from infected kiwifruit orchards in the Te Puke region (July 2011) and from areas around Dunedin, Otago, New Zealand (January 2012). Wastewater inflow was collected from the Tahuna Wastewater Treatment Plant, Dunedin, New Zealand (August 2011). Phages were isolated directly from samples or following an enrichment step. For direct isolation, 2 g of the sample was mixed with 5 ml of NB and gently mixed at room temperature for 1 to 2 h to wash any phages off the sample. The mixture was centrifuged (120 × g for 10 min) to pellet the sample, and 100 μl of the supernatant was mixed with an equal volume of a culture of a host bacterium that had been grown overnight. The phage-host mixture was then plated using a soft-agar overlay. Water samples (100 μl) were used directly in the soft-agar overlay. Five milliliters of wastewater was vortexed with 500 μl of chloroform to kill any bacteria before being used in the soft-agar overlay. For isolation of phages using enrichment, 5 g or 5 ml of sample and 2 ml of bacterial culture were added to 45 ml of NB. Flasks were incubated at 25°C with gentle agitation for 18 h. Cells and the sample debris were removed by centrifugation (2,442 × g for 10 min at room temperature), and the supernatant was used in the soft-agar overlay. Plaques on soft-agar overlays were picked using a pipette tip and resuspended in 1 ml of phage buffer. For phages ϕPsa1 to ϕPsa191, phage lysates were prepared and used for host range testing. The remaining phages were plaque purified into 1 ml of phage buffer, and host range testing was performed on these small-scale samples.
Initially, P. syringae pv. actinidiae strains from Japan (International Collection of Microorganisms from Plants [ICMP] 9853), New Zealand (ICMP 18708, 18800, 18804, and 18806), and Italy (ICMP 18744) and Pseudomonas fluorescens strains 46A and SBW25 and ICMP strains 3636, 7279, and 11288 were used for phage isolation in direct and enrichment approaches. During enrichment, samples were mixed with either single strains or a mixture of strains in an attempt to isolate phages with broad host ranges. Since the New Zealand P. syringae pv. actinidiae isolates have arisen from a recent clonal expansion (21, 24), the remaining phages were plaque purified following plating on ICMP 18800 to increase throughput.
Phage lysates were prepared by serial dilution in phage buffer and incorporation into soft agar with host bacteria. Next, the soft agar containing the phage and bacteria was poured onto agar plates and incubated overnight. Phage lysates were prepared from agar plates with semiconfluent lysis. Briefly, the soft-agar overlay was removed using a sterile glass slide, and 3 ml of phage buffer was washed over the agar plate and then added to the soft agar. Chloroform was mixed with the agar vigorously, the agar mix was centrifuged (2,442 × g for 30 min at 4°C), and the supernatant was retained. The phage lysates were stored over 100 μl of chloroform at 4°C.
Host range analysis.
A total of 59 bacteria isolated from orchards were used in host range studies and were identified as follows. Genomic DNA from bacterial isolates was purified using a Qiagen DNeasy Blood and Tissue DNA extraction kit, according to the manufacturer's instructions for Gram-negative bacteria. Isolates were identified by amplification of a 1.5-kb partial 16S rRNA sequence by PCR using the genomic DNA as the template and the universal primer pair U16A and U16B (47). Each PCR was carried out in a total volume of 25 μl containing 1 μl of each primer (5 μM), 2 μl of the deoxynucleoside triphosphates (dNTPs; 2 mM), 2.5 μl of 10× buffer, 1 μl of 50 mM MgCl2, 0.10 μl of Taq polymerase (5 U μl−1; Invitrogen) and 1 μl of template DNA (20 to 50 ng μl−1) in sterile water. PCR amplification was performed in a GeneAmp PCR system 9700 (Applied Biosystems) thermocycler using the following conditions: 2 min for initial denaturation at 94°C, followed by 30 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 1 min, with a final extension step of 72°C for 7 min. PCR products were separated in a 1% agarose gel and visualized using a Multi Doc-It Digital Imaging System (UVP/Bio-Strategy). PCR products were then purified using a Qiagen PCR purification kit according to the manufacturer's instructions.
DNA sequencing of the 16S rRNA PCR products was performed by Macrogen, Inc. (Seoul, South Korea). Sequencing reactions were initially performed using primers U16A and U16B, and subsequently PCR for gap filling was performed with primers PSFwd1 (5′-ACCCTGGTAGTCCACGCCGT-3′) and PSRev2 (5′-GATGCAGTTCCCAGGTTGA-3′) to ensure that at least two reads per sequence were obtained across the 16S rRNA fragment amplified. Resulting DNA sequences were trimmed to 1,343 bp to remove primer binding sites using Geneious Pro, version 5.5.6 (48), and homology searches of the nonredundant (nr) database of the NCBI (National Centre for Biotechnology Information, Bethesda, MD) were conducted using BLASTN (49). The identification for each strain using the 16S rRNA gene sequence is provided in Table S1 in the supplemental material. The identity of all P. syringae pv. actinidiae strains (virulent and less virulent) were confirmed using quantitative real-time PCR (unpublished data).
Host range studies were performed by pouring a soft-agar overlay containing 100 μl of the test bacterial strain onto nutrient agar plates and allowing the agar to set. Phages were aliquoted into 96-well plates, and a 48-pin replicator was used to transfer approximately 3 μl onto the agar plate surface. Each plate was incubated overnight, and phages that were able to cause an area of local clearing were noted. For a subset of phages, permissive hosts were confirmed by titration to single plaques to rule out effects caused by lysis from without (50).
Bacteriophage-insensitive mutant (BIM) generation, efficiency of plating (EOP), and lysogeny assays.
Spontaneous mutants of P. syringae pv. actinidiae that were resistant to phage infection were isolated by combining 100 μl of overnight culture with 100 μl of high-titer phage lysate (∼107 to 1011 PFU ml−1) in a soft-agar overlay. The plates were incubated at 25°C for approximately 2 days until colonies appeared. Colonies were isolated and streaked to single colonies at least twice to ensure that a single strain was selected and to remove any phage particles.
The effect of the spontaneous mutations on phage infection was measured by the efficiency of plating (EOP). Phage lysates were serially diluted, and 3-μl drops were applied to soft agar containing either the original P. syringae pv. actinidiae host or the BIM. The plates were incubated overnight. The EOP was determined semiquantitatively as the inverse of the dilution where single plaques were visible on the BIM divided by the dilution where single plaques were visible on the original host.
Tests for lysogeny were performed as described previously (51). Supernatants from overnight cultures of BIMs were collected and tested for the spontaneous release of phage by spotting 5-μl drops onto soft agar containing P. syringae pv. actinidiae ICMP 18800. The supernatant from P. syringae pv. actinidiae ICMP 18800 was used as a negative control, and phage lysates were used as the positive control for plaque formation.
Transduction assays.
Transduction assays were performed as previously described (51), with several modifications for P. syringae pv. actinidiae. Briefly, phages were tested for the ability to transfer the kanamycin resistance cassette from P. syringae pv. actinidiae ICMP 18800 TnKm12 (Tn-DS1028lacZKm [52]) to wild-type P. syringae pv. actinidiae ICMP 18800. A transposon mutant was generated as follows. Transfer of Tn-DS1028lacZKm from Escherichia coli BW20767 pKRCPN1 into P. syringae pv. actinidiae ICMP 18800 by conjugation was performed by pelleting 20 μl of overnight culture of each strain by centrifugation, resuspending the cultures in 20 μl NB, and combining the cultures. The conjugation mix was spotted onto a NB agar plate and incubated at 25°C overnight. The mixture was suspended in 1 ml of NB, serially diluted, and plated onto NB agar plates containing kanamycin (50 μg ml−1). The presence of the transposon was tested by PCR using primers PF332 (5′-TTTACTAGTCTGATCCTTCAACTCAGC-3′) and PF333 (5′-TTTACTAGTCTCTGCCAGTGTTACAACC-3′). P. syringae pv. actinidiae ICMP 18800 TnKm12 was used for the transduction assays. Transduction was measured by the production of kanamycin-resistant colonies and confirmed by amplification of the kanamycin resistance cassette by PCR. A high-titer lysate of each phage was prepared using P. syringae pv. actinidiae ICMP 18800 TnKm12 as the host. One hundred microliters of lysate was mixed with 3 ml of a wild-type P. syringae pv. actinidiae ICMP 18800 overnight culture and incubated at room temperature without agitation for 45 min. The samples were then incubated at 28°C at 160 rpm for 30 min. Following incubation, the cells were collected by centrifugation and resuspended in 300 μl of NB; 150 μl of the suspension was spread onto each of two nutrient agar plates containing kanamycin. Controls for spontaneous resistance to kanamycin and lysate contamination were also included. Plates were incubated for 48 h at 25°C.
Phage DNA extraction and restriction digestion.
Phage DNA was isolated using the cetyltrimethylammonium bromide (CTAB) method adapted from Manfioletti and Schneider (53). Briefly, 2.5 ml of high-titer phage lysate was mixed with 100 ng of RNase A and 100 U of DNase I and incubated at 37°C for 30 min to remove any bacterial nucleic acids. The nucleases were inactivated by the addition of EDTA (pH 8.0) to a final concentration of 40 mM. Phage particles were lysed by the addition of 0.5 mg of proteinase K and incubation at 45°C for 15 min. Next, 220 μl of 10% CTAB (wt/vol) in 4% NaCl (wt/vol) (at 55°C) was added, gently mixed, and cooled on ice for 15 min. The CTAB-DNA complex was collected by centrifugation, and the pellet was resuspended in 1.2 M NaCl. The DNA was then precipitated with isopropanol, washed with 75% ethanol, and resuspended in ultrapure water. The concentration was determined using a Nanodrop ND1000. Nucleases used for restriction digests of phage DNA were used as recommended by the manufacturer (Roche). Restriction digests contained ∼1 μg of DNA and 20 U of enzyme and were incubated at 37°C for 16 to 18 h. DNA was separated on 1% agarose gels by running at 40 V for 18 h, stained in ethidium bromide (0.5 μg ml−1) for 20 min, then destained in distilled H2O for 30 min with agitation, and visualized under UV light.
PFGE.
To estimate genome size, phage DNA was loaded into a 1% pulsed-field gel electrophoresis (PFGE)-certified agarose gel, and the wells were sealed with agarose. A Chef DR III system (Bio-Rad) was used to perform the electrophoresis in 0.5× Tris-borate-EDTA buffer at 6 V cm−1 for 18 h at 12°C. Conditions for electrophoresis were as follows: included angle of 120°, initial switch time of 5 s, and final switch time of 15 s. Midrange PFG Marker I (NEB) was used as the ladder. DNA was visualized as described above.
TEM.
The titer and purity of phage lysates were increased for transmission electron microscopy (TEM) by ultracentrifugation (109,760 × g for 30 min at 4°C) of 5 ml of phage lysate. The phage pellets were resuspended in ultrapure water overnight and were then dialyzed against water for 4 days to remove any residual salt. Carbon-coated copper grids were plasma glowed, and 10 μl of high-titer phage lysate was placed on the grid and left for 1 min. The grid was blotted dry and negatively stained with 10 μl of 1% phosphotungstic acid (pH 6.8). If necessary, the samples on the grids were washed five times in ultrapure water prior to staining. Grids were blotted dry immediately and air dried before examination in a Philips CM100 transmission electron microscope. All images were taken at the same magnification.
Phage genome sequencing.
DNA of nine Myoviridae phages (ϕPsa267, ϕPsa300, ϕPsa315, ϕPsa347, ϕPsa374, ϕPsa381, ϕPsa397, ϕPsa410, and ϕPsa440) was isolated as described above for restriction digestion and PFGE; the DNA was further purified and eluted using components of a Qiagen DNeasy Blood and Tissue kit. Sequencing was performed by New Zealand Genomics, Ltd. (NZGL). The sequencing libraries were prepared using an Illumina TruSeq DNA Sample Preparation, version 2, kit. The libraries were quantitated using a Bioanalyzer 2100 DNA 1000 chip (Aligent, Santa Clara, CA, USA) and a Qubit Fluorometer using a dsDNABR kit (Life Technologies, Burlington, ON, Canada). MiSeq 150-bp paired-end (PE) sequencing was performed and demultiplexed using the ea-utils suite of tools (54).
Phage genome assembly, annotation, and mapping.
ϕPsa374 was assembled using Geneious, version 6.1.6 (48) (de novo assembly using default settings with increased sensitivity). The assembly generated two large contigs. The contig ends overlapped by approximately 90 and 850 bp, allowing a single circular phage scaffold assembly to be generated. The reads were mapped back to the assembly for validation. An automated annotation of ϕPsa374 was generated using RAST (55), and tRNAs were identified using tRNAscan-SE (56) and ARAGORN (57). A pairwise comparison of ϕPsa374 and a related Myoviridae phage, JG004 (58), was visualized using Easyfig (59). The assembled genome of ϕPsa374 was used as a reference to compare the sequencing reads of the remaining eight phages with a mapping approach in Geneious. BLAST Ring Image Generator (BRIG) (60) was used to visualize the results.
Nucleotide sequence accession numbers.
The sequencing reads from the nine phages have been deposited in the NCBI SRA with the BioProject number PRJNA236447. The ϕPsa374 genome has been deposited in the NCBI GenBank database with the accession number KJ409772.
RESULTS
Generation of a repository of P. syringae pv. actinidiae phages.
To develop phage-based approaches for the control and/or detection of P. syringae pv. actinidiae, a bank of phages was collected that infect P. syringae pv. actinidiae. The phages were isolated from a range of leaf and vine samples as well as from soil obtained from orchards in New Zealand that are infected with P. syringae pv. actinidiae, in addition to locations where kiwifruit are not grown (see Materials and Methods). In total, 275 phages were isolated from 25 independent samples and locations. Detailed information of the phages isolated is provided in Data Set S1 in the supplemental material.
Most P. syringae pv. actinidiae phages have a narrow host range.
For biocontrol use, it is important to understand the host range of the selected phages. For example, we sought to identify phages that infected the pathogen but were unable to infect other bacteria present in the environment of kiwifruit orchards. The host range of the first 40 phages was tested against a range of 60 Gram-negative bacteria (∼2,400 combinations) including a selection of Pseudomonas species (see Data Set S2 in the supplemental material). The phages did not infect more distantly related bacteria including Pseudomonas aeruginosa, Citrobacter rodentium, Pectobacterium atrosepticum, E. coli, and Serratia sp. strain ATCC 39006. However, some phages infected Pseudomonas pathogens more closely related to P. syringae pv. actinidiae, including the following P. syringae pathovars: morsprunorum, syringae, phaseolicola, and tomato. Strains of Pseudomonas corrugata, Pseudomonas viridiflava, and other poorly characterized Pseudomonas syringae and Pseudomonas strains were also lysed by some phages. It is worth noting that P. viridiflava causes blossom blight on Actinidia species (61) and that several of the phages isolated have potential applications for the control of this kiwifruit pathogen.
Next, host range screening of the entire repository of 275 phages was carried out against a selection of environmental strains specifically isolated from kiwifruit orchards in New Zealand (see Data Set S3 in the supplemental material). The identities of these isolates were determined using 16S data (see Table S1). In addition, select representative P. syringae pv. actinidiae strains from Japan, Italy, and New Zealand were tested. In total, ∼20,000 phage-host combinations were examined and showed clear differences in the phage profiles of P. syringae pv. actinidiae strains from distinct geographic locations. There are examples of phages that infect only the Japanese ICMP 9853 isolate (e.g., ϕPsa259) or the New Zealand isolates (e.g., ϕPsa271). In addition, the majority of phages that infect the New Zealand P. syringae pv. actinidiae less virulent strains do not infect P. syringae pv. actinidiae virulent strains, consistent with the data that these strains are distinct (21, 22, 24). The 1992 Italian P. syringae pv. actinidiae ICMP 19090 isolate showed a distinct phage profile compared with the strains responsible for the more recent disease outbreaks in Italy and New Zealand (see Data Set S2 in the supplemental material). However, there are phages that infect combinations of the different P. syringae pv. actinidiae isolates from various geographical locations. As expected, many of these host range differences reflect the strains used for initial phage isolation, and, as planned, our phage bank predominantly contains phages that target P. syringae pv. actinidiae from New Zealand (see Data Sets S2 and S3). The variation in phage profiles for each strain raises the possibility of establishing a diagnostic phage-typing assay using a selection of phages from our set.
Analysis of infection of the other bacterial isolates from the kiwifruit phyllosphere indicated that the majority of these strains were not sensitive to the P. syringae pv. actinidiae phages (see Data Set S3 in the supplemental material). However, several phages that infected P. syringae pv. actinidiae, P. viridiflava, or P. syringae also generated plaques on a small number of other bacterial isolates. For example, the ABAC10 isolate, putatively identified as P. syringae and confirmed not to be a P. syringae pv. actinidiae virulent or less virulent strain using a recently developed PCR diagnostic (Andersen et al., submitted), was infected by a large number of phages, suggesting that it is closely related to P. syringae pv. actinidiae. Several phages, for example, ϕPsa17, ϕPsa369, ϕPsa410, and ϕPsa411, were able to infect P. fluorescens ABAC62 but not the other P. fluorescens strains tested. Pseudomonas sp. strain ABAC61 was also infected by phages, including ϕPsa8, ϕPsa315, and ϕPsa323.
From the high-throughput host range screening of ∼20,000 phage-host combinations, 24 phages were selected for further investigation. The phages were chosen to include both those with a broader host range and other more host-specific isolates. Due to the possibility of false positives because of lysis from without, high-titer phage lysates were prepared for the 24 phages, and quantitative efficiency of plating (EOP) assays were used to confirm the host range data (Table 1; see also Data Set S4 in the supplemental material).
TABLE 1.
Host ranges of phages against P. syringae pv. actinidiae and related strains
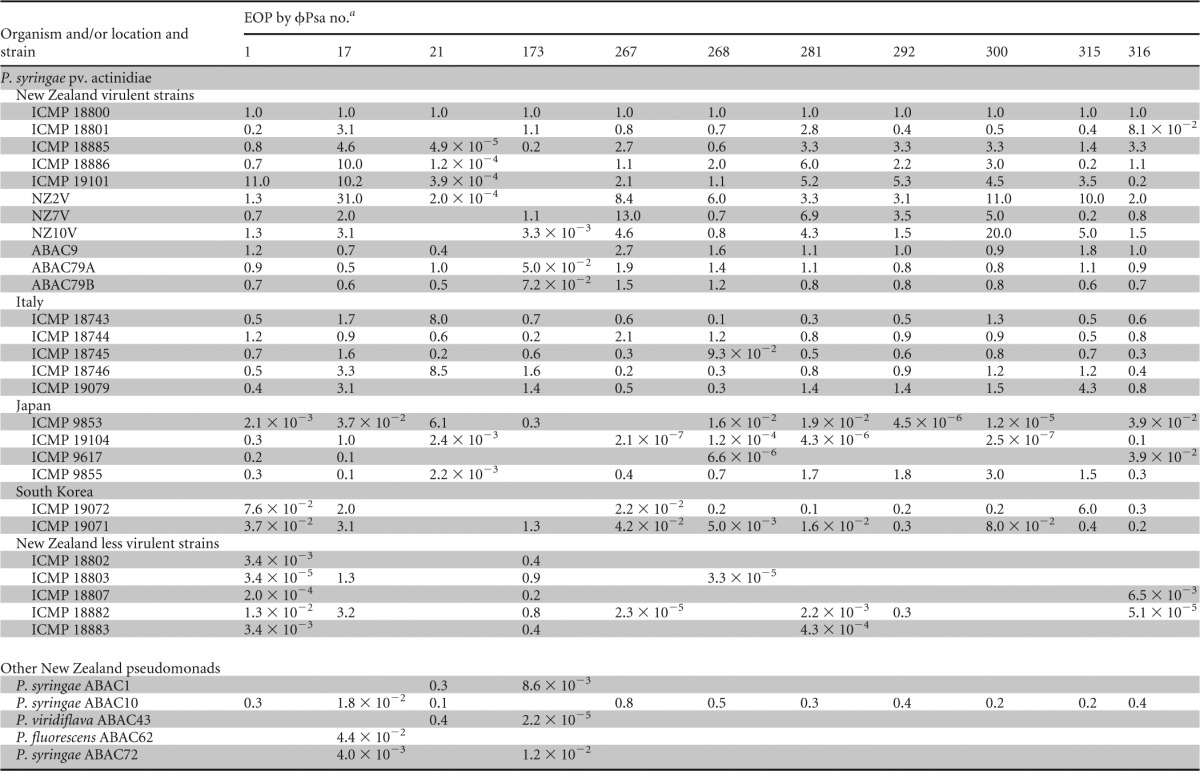
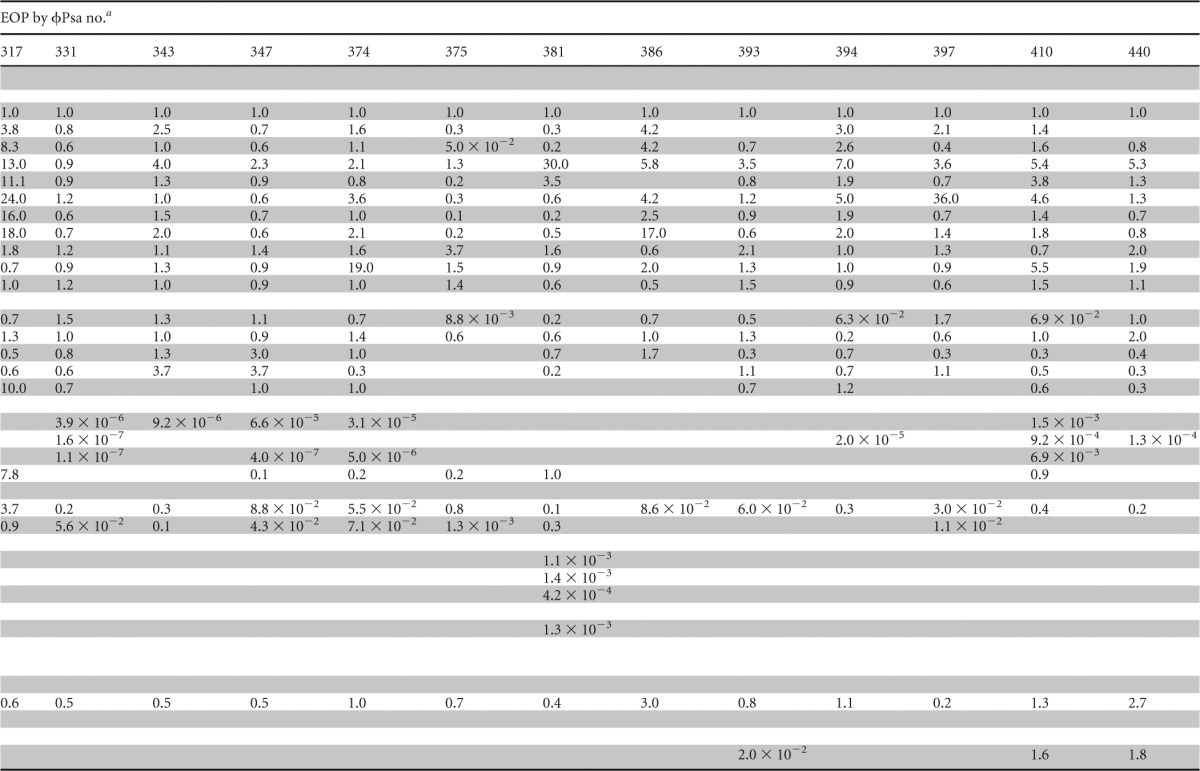
EOP was calculated as the titer (PFU ml−1) on the test strain/titer (PFU ml−1) on P. syringae pv. actinidiae ICMP 18800. Raw phage titers are provided in Data Set S4 in the supplemental material. Blank cells indicate that no plaques were detected.
This restricted group of 24 phages was subsequently assayed (by EOP) against a wider selection of P. syringae pv. actinidiae isolates from New Zealand, Italy, Japan, and South Korea (Table 1). There were clear differences in the phage sensitivity profiles of these isolates and in the ability of some phages to infect particular strains. For example, the P. syringae pv. actinidiae less virulent strains (ICMP 18802, 18803, 18807, 18882, and 18883) had significantly reduced EOPs for most phages except ϕPsa173, which is consistent with their designation as distinct from P. syringae pv. actinidiae virulent strains (21, 22). In general, the remaining P. syringae pv. actinidiae isolates from New Zealand showed similar EOPs to ICMP 18800, which was used for most phage isolations. The host range of ϕPsa21 was difficult to determine since it had tiny plaques (<1 mm) but could infect quite different hosts. Indeed, it was the only phage able to infect P. viridiflava ABAC43. Determining the host range of ϕPsa173 was also difficult due to the turbid plaques it formed on virulent strains of P. syringae pv. actinidiae. In summary, the host range information collected in this study demonstrates that appropriate phage selection should have little, if any, effect on nonpathogenic bacteria present in kiwifruit orchards and will assist in the informed design of phage cocktails that target desired P. syringae pv. actinidiae strains.
The P. syringae pv. actinidiae phages are stable.
To investigate stability, which is important for phage storage and application, phage lysates were stored at either 4°C or 25°C and sampled after 1, 3, and 6 months and 1 year (see Fig. S1 in the supplemental material). Storage at 25°C gives an indication of the stability of the phages at higher temperatures that might be encountered in an orchard setting. However, other factors, particularly UV light, affect phage stability in the environment (33). When the phages were stored at 4°C, the PFU count ml−1 of all the phages did not change dramatically after 6 months, with a decrease in ϕPsa21, ϕPsa173, ϕPsa317, and ϕPsa386 after 1 year. The majority of phages were also stable when stored at 25°C; the PFU count ml−1 of ϕPsa17 decreased over 6 months and was not detected at 1 year, and ϕPsa21 and ϕPsa173 had decreased below the level of detection after 3 months. These results demonstrate that most phages can be stored successfully at either 4°C or ambient temperature (25°C) for extended periods of time.
Extensive resistance profiling indicates varied infection strategies.
Consideration of phage resistance is important in designing a phage biocontrol strategy (33). For example, resistance profiles have the potential to aid in the design of phage cocktails to minimize the emergence of phage-resistant strains of the pathogen. Spontaneous bacteriophage insensitive mutants (BIMs) of P. syringae pv. actinidiae ICMP 18800 were isolated for 22 out of the 24 phages. We could not isolate BIMs for ϕPsa21 and ϕPsa173 using the same method, which is probably due to the difficulty of generating completely lysed bacterial lawns with these phages. The ability of each phage to infect every spontaneous BIM was determined by semiquantitatively measuring the EOP for all 524 combinations (Fig. 1; see also Data Set S5 in the supplemental material). In one case, resistance against ϕPsa316 resulted in resistance to all other phages. Interestingly, ϕPsa316 could still infect most of the other BIMs isolated against other phages. BIMs isolated following challenge with phages ϕPsa1, ϕPsa17, ϕPsa267, ϕPsa300, ϕPsa315, ϕPsa316, ϕPsa317, and ϕPsa410 resulted in resistance against most, but not all, phages. Phages ϕPsa17, ϕPsa21, and ϕPsa173 generally could infect the majority of BIMs, which might reflect their distinct morphologies and possible alternative receptors (Fig. 2). Unfortunately, since we did not have BIMs against ϕPsa21 and ϕPsa173, we could not check for their resistance to other phages. Phages ϕPsa268, ϕPsa281, and ϕPsa381 could infect many of the BIMs, but some displayed a reduced EOP. Overall, these single spontaneous BIMs provide one snapshot of the possible phage resistance profiles. In addition, different mutations will enable resistance, and by chance some will have more severe consequences and therefore affect sensitivity to a wider range of phages. For example, mutations in the inner core of lipopolysaccharide will have greater effects than mutations that alter the O antigen. Determining the receptors used by these phages will require further work. However, these results indicate that resistance profiling might be a useful tool to aid in the careful selection of phages to reduce the emergence of resistance.
FIG 1.

Phage resistance profiles calculated as the EOP of each phage on each BIM compared with the wild-type ICMP 18800 strain. Raw phage titers are provided in Data Set S5 in the supplemental material. WT, wild type; ND, not determined; <10−7, complete resistance; 10−2 to 10−7, partial resistance; 0.1 to 10, infection similar to that of the WT. All raw phage titers are provided in Data Set S5 in the supplemental material.
FIG 2.
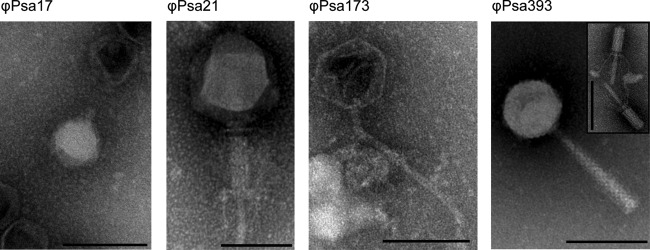
Four representative morphologies of 24 phages active against P. syringae pv. actinidiae. Phage lysates were stained with 1% phosphotungstic acid (pH 6.8) and viewed using a Philips CM100 TEM. Scale bars represent 100 nm. The insets represent alternative tail morphologies that were observed (extended or contracted). Details of the measurements and classifications are provided in Table 2, and TEM images of all other phages are available in Fig. S2 in the supplemental material.
The P. syringae pv. actinidiae phages are not detectably lysogenic or transducing.
Phages used for biocontrol should not genetically modify the pathogen through lysogenic conversion (62). Consequently, the 24 phages were tested to establish if they were temperate and capable of lysogeny. Lysogens contain a prophage and are typically resistant to reinfection by the same phage, which results in turbid plaques via superinfection immunity. We tested the supernatants of all the BIMs for the spontaneous release of phage particles, and no plaquing was observed when they were tested against P. syringae pv. actinidiae ICMP 18800. However, since we did not isolate ϕPsa21- or ϕPsa173-resistant mutants, we did not test these for lysogeny. In addition, all of the phage isolates, except ϕPsa173, produced clear plaques, indicative of virulent phages. Interestingly, ϕPsa173 infected the less virulent P. syringae pv. actinidiae ICMP 18804 strain and gave larger clear plaques than P. syringae pv. actinidiae ICMP 18800, where turbid plaques were observed. Turbid plaque formation by ϕPsa173 indicates that this phage might be temperate, but this requires further analysis since turbid plaques can result from other properties, such as the exopolysaccharides produced by the bacterial host (63).
The DNA packaging mechanisms of some phages have a lower fidelity, resulting in the rare inclusion of bacterial DNA during phage assembly (62). As a result, transduction can lead to the movement of host DNA, such as resistance genes and virulence determinants, between bacterial species, which is undesirable for a BCA. None of the 24 phages were able to transfer the kanamycin resistance cassette from a P. syringae pv. actinidiae ICMP 18800 transposon mutant to the unmarked wild-type P. syringae pv. actinidiae ICMP 18800 strain (Table 2). Together, the results from the lysogeny and transduction assays show that all 24 phages are incapable of generalized transduction under the conditions tested and, therefore, might be suitable as BCAs.
TABLE 2.
Summary of phages characterized in this study
| Phage name | Source (source name) and location | Method of isolation | Date isolated (mo/day/yr) | Plaque morphology (0.35% NA) | Classification | Size of phage head (nm [±SD]) | Total length of phage (nm [±SD]) | Genome size (kb)a |
|---|---|---|---|---|---|---|---|---|
| ϕPsa1 | Soil (S1), Te Puke | Direct | 8/3/2011 | Clear, fuzzy edge, 2 mm | Myoviridae | 76.8 (±1.8) | 206.9 (±3.6) | 95 |
| ϕPsa17 | Wastewater (W1), Tahuna | Direct | 8/10/2011 | Clear center, 3–4 mm; turbid ring, 1 mm; total diam, 4–5 mm | Podoviridae | 55.9 (±1.9) | 55.9 (±1.9) | 30 |
| ϕPsa21 | Leaf litter (L1), Te Puke | Direct | 8/15/2011 | Clear, <1 mm | Myoviridae | 126.1 (±6.6) | 293.1 (±8.4) | >300 |
| ϕPsa173 | Leaf litter (L1), Te Puke | Direct | 8/22/2011 | Turbid, 2 mm | Siphoviridae | 77.5 (±3.9) | 252.8 (±8.3) | 110 |
| ϕPsa267 | Soil (KH), Dunedin | Direct | 1/23/2012 | Clear, 1–2 mm | Myoviridae | 71.6 (±2.9) | 186.6 (±11.5) | 95 |
| ϕPsa268 | Soil (KH), Dunedin | Direct | 1/23/2012 | Clear, 2–3 mm | Myoviridae | 67.5 (±2.8) | 181.4 (±1.6) | 95 |
| ϕPsa281 | Soil (KH), Dunedin | Direct | 1/23/2012 | Clear, 2 mm | Myoviridae | 70.9 (±2.3) | 179.2 (±7.2) | 95 |
| ϕPsa292 | Compost (CT1), Dunedin | Direct | 1/23/2012 | Clear, 2 mm | Myoviridae | 77.3 (±3.1) | 204.7 (±2.9) | 95 |
| ϕPsa300 | Compost (CT1), Dunedin | Direct | 1/23/2012 | Clear, 2 mm | Myoviridae | 70.8 (±3.4) | 201.7 (±6.2) | 95 |
| ϕPsa315 | Soil (CR), Dunedin | Direct | 1/23/2012 | Clear, fuzzy edge, 1 mm | Myoviridae | 67.1 (±3.1) | 181.0 (±18.5) | 95 |
| ϕPsa316 | Soil (CR), Dunedin | Direct | 1/23/2012 | Clear, 2 mm | Myoviridae | 68.7 (±2.8) | 184.3 (±15.1) | 95 |
| ϕPsa317 | Soil (CR), Dunedin | Direct | 1/23/2012 | Clear, 1 mm | Myoviridae | 69.5 (±3.4) | 173.5 (±23.5) | 95 |
| ϕPsa331 | Soil (TP), Dunedin | Direct | 1/23/2012 | Clear, fuzzy edge, 1 mm | Myoviridae | 73.2 (±3.3) | 202.6 (±6.3) | 95 |
| ϕPsa343 | Soil (TP), Dunedin | Direct | 1/23/2012 | Clear, fuzzy edge, 1–2 mm | Myoviridae | 74.6 (±7.4) | 194.1 (±14.5) | 95 |
| ϕPsa347 | Soil (CT2), Dunedin | Enrichment | 1/23/2012 | Clear, 1–2 mm | Myoviridae | 75.3 (±3.7) | 202.3 (±5.0) | 95 |
| ϕPsa374 | Soil (HB), Dunedin | Direct | 1/31//2012 | Clear, fuzzy edge, 1–2 mm | Myoviridae | 75.0 (±1.9) | 204.6 (±3.3) | 97.8 |
| ϕPsa375 | Soil (HB), Dunedin | Direct | 1/31//2012 | Clear, 1–2 mm | Myoviridae | 68.3 (±4.3) | 167.7 (±14.2) | 95 |
| ϕPsa381 | Compost (KM5), Dunedin | Direct | 2/1/2012 | Clear, 1–2 mm | Myoviridae | 72.7 (±2.2) | 194.2 (±7.1) | 95 |
| ϕPsa386 | Compost (KM5), Dunedin | Direct | 2/1/2012 | Clear, fuzzy edge, 1 mm | Myoviridae | 73.0 (±2.3) | 168.2 (±19.3) | 95 |
| ϕPsa393 | Compost (KM6), Dunedin | Direct | 2/1/2012 | Clear, fuzzy edge, 1 mm | Myoviridae | 70.1 (±2.2) | 202.2 (±4.3) | 95 |
| ϕPsa394 | Compost (KM6), Dunedin | Direct | 2/1/2012 | Clear, fuzzy edge, 1 mm | Myoviridae | 71.8 (±6.1) | 198.1 (±10.4) | 95 |
| ϕPsa397 | Compost (KM6), Dunedin | Direct | 2/1/2012 | Clear, 1 mm | Myoviridae | 72.5 (±2.4) | 169.8 (±12.1) | 95 |
| ϕPsa410 | Soil (KM8), Dunedin | Direct | 2/2/2012 | Clear, fuzzy edge, 1.5 mm | Myoviridae | 76.8 (±2.9) | 208.4 (±6.9) | 95 |
| ϕPsa440 | Compost (JR2), Dunedin | Direct | 2/3/2012 | Clear, fuzzy edge, 1 mm | Myoviridae | 75.2 (±3.8) | 208.2 (±5.8) | 95 |
Genome size was estimated from PFGE except for the data for ϕPsa21 (draft genome sequence, unpublished data) and ϕPsa374 (assembled genome).
P. syringae pv. actinidiae phages are Caudovirales.
The morphology of the 24 P. syringae pv. actinidiae phages was examined by transmission electron microscopy (TEM) (Fig. 2; see also Fig. S2 in the supplemental material). The phages belong to the Myoviridae (22 phages), Podoviridae (ϕPsa17), or Siphoviridae (ϕPsa173) families of the order Caudovirales. For the Myoviridae, where possible, images of both contracted and extended tails are shown (Fig. 2; see also Fig. S2). The dimensions of the icosahedral phage heads and the entire phage, as measured from at least four particles, are provided in Table 2. All members of the Myoviridae, with the exception of the large ϕPsa21, appeared very similar in size and might be closely related. However, they are unlikely to be identical due to their different host range profiles (Table 1). In summary, from this morphological characterization, it was possible to clearly distinguish four morphotypes of the P. syringae pv. actinidiae phages.
To further differentiate between the 24 phages, genomic DNA was extracted, and the approximate size and restriction patterns were analyzed. Estimates of genome size by PFGE indicated that the ϕPsa173 genome size was ∼110 kb, ϕPsa17 was the smallest at ∼30 kb, and 21 phages had genome sizes of ∼95 kb (Table 2; see also Fig. S3 in the supplemental material). The ϕPsa21 genome could not be accurately resolved by PFGE despite multiple attempts. A preliminary draft assembly of ϕPsa21 genome sequence data suggests this is a “giant” or “jumbo” phage with a genome of >300 kb (unpublished data). The ∼95-kb estimated size of the 21 Myoviridae genomes supports the morphological data and infers they are closely related.
To investigate more subtle differences in phage sequences, the DNA was also analyzed by restriction digestion. NheI and SphI cut the majority of the phage genomes (Fig. 3). Two groups of phages showed identical restriction patterns, suggesting that they were identical or very closely related (group 1, ϕPsa292 and ϕPsa300; group 2, ϕPsa331, ϕPsa343, and ϕPsa347). Indeed, the resistance profiles of each of these two phage groups were very similar.
FIG 3.
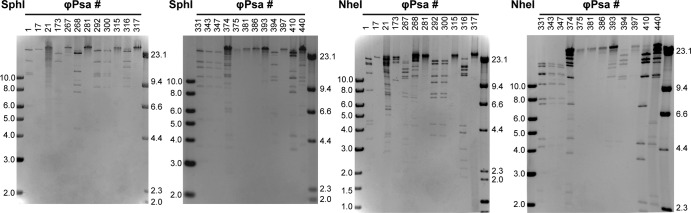
Restriction digest profiles of 24 P. syringae pv. actinidiae phages. Phage genomic DNA was digested with either SphI or NheI and separated on agarose gels.
Myoviridae genome sequences.
To investigate how closely related the group of Myoviridae phages were, the genomes of nine Myoviridae phages (ϕPsa267, ϕPsa300, ϕPsa315, ϕPsa347, ϕPsa374, ϕPsa381, ϕPsa397, ϕPsa410, and ϕPsa440) were sequenced using the Illumina MiSeq platform. An average of 1.3 × 106 reads per phage were obtained to produce an average depth of coverage of 2,000 times, based on the estimated 95-kb genome size. The sequencing reads of ϕPsa374 were assembled into a single, circular genome consisting of 97,761 bp with a GC content of 47.4%. This assembly correlated well with the PFGE data and demonstrates the usefulness of PFGE to aid and verify genome assembly. Eleven tRNA genes and 173 coding sequences (CDS) were identified in ϕPsa374. Comparison of ϕPsa374 to JG004 (58), a member of the Myoviridae that infects P. aeruginosa, shows that the genome organization is similar even though the level of sequence similarity is low (Fig. 4). Only 46 regions longer than 55 bp in ϕPsa374 and JG004 were found to have greater than 65% sequence similarity. Mapping the ϕPsa374 reads back to the assembled ϕPsa374 genome showed that there was even coverage over the genome, except where coverage is increased at one point (Fig. 5, inner ring). This spike in coverage is likely due to terminal redundancy at the genome ends (64), suggesting a linear genome. Indeed, the genome of the related Pseudomonas phage, PaP1, contains 1,190 bp of terminal redundancy (65).
FIG 4.
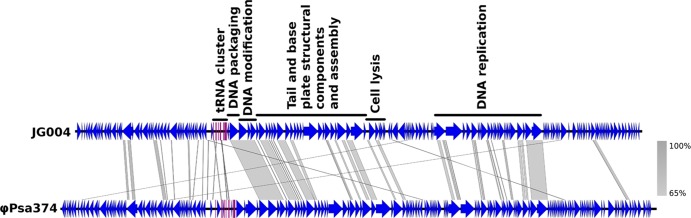
BLASTn comparison of JG004 with ϕPsa374. ϕPsa374 was compared to a similar-sized Myoviridae phage that infects P. aeruginosa, JG004, using Easyfig (59). The annotation of JG004 functional assignments is taken from Garbe et al. (58). The comparison was performed with BLAST 2.2.28+, and the regions of similarity greater than 65% are shown by gray shading. CDS are in blue, and predicted tRNA genes are in fuchsia.
FIG 5.

Comparison of nine related Myoviridae genomes. The inner circle portrays the nonredundant reference genome of ϕPsa374, and the second inner-most ring shows the coverage of ϕPsa374 sequencing reads mapped to the assembled genome (shown as a red graph). The next nine colored rings (identified at right), representing the nine query phage genomes, indicate the sequence similarity (based on BLASTn) between ϕPsa374 and ϕPsa267, ϕPsa300, ϕPsa315, ϕPsa347, ϕPsa374 (included as a control), ϕPsa381, ϕPsa397, ϕPsa410, and ϕPsa440. Regions where the sequence similarity is less than 90% between the phages are represented as white gaps in the rings. The RAST-predicted CDS annotated in the ϕPsa374 genome are shown as arrows in the outer ring along with the predicted tRNA genes (in fuchsia). CDS predicted to be associated with tellurite resistance and the tape measure protein, as mentioned in the text, are highlighted in teal. The figure was prepared using the BLAST Ring Image Generator (BRIG) (60).
As the remaining eight genomes were likely to be highly similar to the genome of ϕPsa374, the ϕPsa374 assembly was used as a reference to which the reads from the other phages were mapped, as shown in Fig. 5. The eight genomes are very similar to the genome of ϕPsa374, with >90% nucleotide identity over almost the entire length of the reference genome. However, they all have clear differences compared with the reference genome and each other, as demonstrated by both the breaks in sequence similarity, shown as white gaps in the colored rings, and by different percent nucleotide identities (Fig. 5).
Typical for phage genomes, the majority (169 out of 173) of the CDS identified using automated RAST annotation in the ϕPsa374 genome were of unknown function. However, of particular interest, we found that all nine phage genomes contain a cluster of four genes related to tellurium resistance, highlighted in the outer ring of Fig. 5 in teal at 4 to 6 kbp. The first three genes are similar to terD (50.5% similarity over 198 amino acids), terC (27.1% similarity over 373 amino acids), and terF (24.7% similarity over 417 amino acids) from the terZABCDEF system of plasmid R478 originally isolated from Serratia marcescens (66) (GenBank accession number U59239.2), and the fourth gene shares similarity with telA (37.8% similarity over 418 amino acids) from the telAB system of plasmid RK2Te-r from Klebsiella aerogenes (67) (DDBJ accession number M62846.1). The terZABCDEF and telAB operons confer resistance to tellurium (66, 67). The terZABCDEF operon also mediates resistance to phages and colicins (66). Tellurite has been used in selective medium for the isolation of pathogens such as Shigella and E. coli, but the role of tellurium resistance genes is unknown (68). Genes related to tellurium resistance (terB) have also been found in phages that infect Salmonella (69) and Cronobacter (GenBank accession number NC_019402). The importance of this cluster of genes in the nine Myoviridae phages is unknown as they do not comprise a full tellurium resistance system, and the function of the individual genes is not known.
One gene of interest (Fig. 5, outer ring 44 to 46 kbp) is the putative tape measure protein, which varies between some of the phages. The predicted function of this gene was determined by comparison to JG004. The length of these genes correlates to the length of the phage tail, but these proteins are also thought to play other roles during infection (70, 71). It is possible that the observed variation in the tape measure gene could contribute to the differences in host range and resistance profiles.
Based on genome size and morphology, we conclude that there are four major phage groups (represented by ϕPsa374, ϕPsa17, ϕPsa21, and ϕPsa173) with at least 21 different phage sequence types in this collection of 24 phages. Genome sequencing has shown that the dominant Myoviridae type contains a closely related sequence family. The details of the isolation and characterization of all the 24 P. syringae pv. actinidiae phages are summarized in Table 2.
Identification of a possible phage-carrier bacterium combination.
One aim of this study was to identify a nonpathogenic bacterium to enable phage amplification in the laboratory and in the environment. Use of such a “carrier” bacterium has been investigated previously with Pantoea agglomerans as the carrier for phage to treat fire blight in apples and pears caused by Erwinia amylovora (36–38, 63, 72, 73). P. agglomerans is proposed to enhance the survival of phages in the environment by supporting their replication, but there may also be protective biocontrol effects of the carrier bacterium alone (reviewed in reference 33). Bacteria were isolated from several commercially available biocontrol products, Bio Sol B Sub Plus (Biological Solutions, Ltd.), SoilFx (AgTurf Products), Superzyme (JH Biotech, Inc.), Blossom Bless (Grow-Chem NZ, Ltd.), and BlightBan A506 (Nufarm Americas, Inc.), and screened against the bank of phages (data not shown; see Data Set S3 in the supplemental material). None of these commercial biocontrol agents provided a suitable carrier.
From the host range tests of our bacterial isolates from kiwifruit, a single putative phage-nonpathogenic carrier strain combination was identified. ϕPsa17 infected strain P. fluorescens ABAC62, albeit with a >10-fold decrease in titer, relative to P. syringae pv. actinidiae (Table 1). Passage of ϕPsa17 on ABAC62 did not increase the EOP relative to that of P. syringae pv. actinidiae, suggesting that restriction modification was not involved (data not shown). Therefore, the potential exists to use P. fluorescens ABAC62 with ϕPsa17 as a phage-carrier combination for phage biocontrol of P. syringae pv. actinidiae.
DISCUSSION
P. syringae pv. actinidiae has reemerged as a major pathogen of kiwifruit, causing severe outbreaks of bacterial canker throughout many parts of the world (20). Given that this disease causes significant economic impacts on fruit production and can lead to the destruction of entire orchards, approaches for control and prevention are actively sought. In this study, we isolated and characterized a suite of phages that could infect this phytopathogen, with the longer-term goal of testing and developing phage-based biocontrol solutions. In total, high-throughput isolation and host range screening resulted in a set of 275 phages which infected P. syringae pv. actinidiae virulent strains or other related bacteria from kiwifruit orchards, including P. syringae pv. actinidiae less virulent strains or P. agglomerans (ϕPan363). The host range of individual phages is narrow, suggesting that the development of a cocktail of phages with activity against P. syringae pv. actinidiae V or other related pathogens would have minimal impact on the remaining microflora.
The phages analyzed all belonged to the Caudovirales order with members of all three families: Siphoviridae, Podoviridae, and Myoviridae. Based on genome size and morphology, four clearly distinct phage types were identified with a siphovirus, podovirus, a large myovirus, and 21 similar members of the Myoviridae (Fig. 2; see also Fig. S2 in the supplemental material). These 21 Myoviridae phages are indistinguishable based on morphology and approximate genome size. Numerous studies have examined phages that infect Pseudomonas species, and more than 97% observed by electron microscopy belong to the Caudovirales (74). In our current study, examples from all three families that comprise this order were isolated, consistent with a worldwide survey of phages that infect P. aeruginosa (75). Our phage isolation procedure selected for chloroform-resistant phages and those that form visible plaques on 0.35% agar. Indeed, a small number of original plaques were unstable, and titers for these could not be redetermined. Therefore, we have detected only a subset of P. syringae pv. actinidiae phages present in New Zealand.
Genome sizes of Pseudomonas phages typically range from 37 to 93 kb and from 200 to >300 kb for giant Myoviridae phages (74). Interestingly, preliminary sequence data indicate that ϕPsa21 has a large, >300-kb, genome (unpublished data). The smaller range also corresponds to the genome sizes for the nine phages characterized by DNA sequencing (i.e., ϕPsa374). JG004, vB_PaeM_c2-10_Ab1, PaP1, and PAK_P1 are highly similar phages, with ∼93-kb genomes, which infect P. aeruginosa (58, 65, 76, 77). Genome comparison of ϕPsa374 to JG004 revealed a low level of similarity in several genomic regions, suggesting that ϕPsa374 and the other eight sequenced phages are more distantly related to JG004 than other members of this related group of Myoviridae that infect P. aeruginosa.
Further analysis of the 21 Myoviridae phages using a combination of host range, restriction digestion, and genome sequencing demonstrated that they are very similar but not identical. The sequencing of more phage genomes will be essential, however, to determine whether the other phages in our collection are appropriate BCAs. For example, we identified the presence of genes related to tellurium resistance. However, due to the shuffled gene organization and paucity of knowledge about the mechanism of tellurite resistance, it is difficult to draw conclusions about the function of these genes and whether they are a concern for use in a BCA. Tellurium resistance genes may prove important for bacterial virulence since they have previously been identified in a range of bacterial pathogens (68).
Various features are desirable for phage-based BCAs, including stability and a lack of lysogeny and transduction. We did not detect lysogeny, suggesting that the phages are unlikely to transfer genetic information to P. syringae pv. actinidiae. However, this does not preclude lysogenization of untested bacteria or under alternative conditions. Furthermore, phages ϕPsa21 and ϕPsa173 were not examined, and ϕPsa173 might be temperate since it forms turbid plaques. We did not observe any transduction by the phages, but transduction could occur at a rate below our limit of detection. Finally, most phages were stable for 6 months at either 4°C or 25°C. Therefore, these phages display the basic properties required for use in phytopathogen biocontrol.
There are a number of challenges in the implementation of phage therapy in agriculture (32–34, 38). For example, phage resistance needs to be considered. To enable a more rational design of a phage cocktail that reduces the likelihood of phage resistance developing, we isolated phage-resistant bacterial mutants and utilized an extensive resistance assay. We are not aware of other studies using this approach, and, although it only provides a snapshot of resistance to the phages, it can identify clear differences in host susceptibility to particular phages (Fig. 1). This technique will aid the informed design of cocktails of phages that are likely to have different receptors or infection strategies. The mechanisms of phage resistance are diverse and include the modification of the cell surface (i.e., receptor mutation or receptor masking), injection blocking, restriction modification, abortive infection, and clustered regularly interspaced short palindromic repeat (CRISPR)-Cas systems (78, 79). From our analysis, the mechanism of resistance was not determined, but spontaneous mutants are most likely to have undergone a loss of function, such as receptor mutation.
Due to the systemic nature of bacterial canker of kiwifruit (4, 20), phage therapy is best suited as a preventative strategy. However, the natural environment, e.g., water, wind, rain, and sunlight, influences the success of phage treatment. One major factor affecting phage longevity in the phyllosphere is UV light. Trials have been performed with protective formulations (42) and carrier bacteria (38, 72) to improve viral survival. Here, we had limited success with the identification of a carrier bacterium isolated from kiwifruit orchards, but a possible P. fluorescens strain was identified. It will be necessary to test if this strain is completely nonpathogenic and if it will influence any phage treatments.
Finally, in addition to characterizing a bank of phages for use as possible BCAs, we showed that phage typing might be suitable for the differentiation of strains of P. syringae pv. actinidiae and related bacteria. The host range data clearly demonstrate that the P. syringae pv. actinidiae isolates from different geographical sources are distinct. In particular, Japanese and South Korean P. syringae pv. actinidiae isolates are quite different from those from both New Zealand and Italy. This is consistent with the genetic data on the origins and relatedness of these strains, which showed that the New Zealand and Italian strains are very closely related, with only a few SNPs and differences in horizontally acquired islands (21, 22, 24). It is reassuring that most phages have the ability to infect both New Zealand and Italian P. syringae pv. actinidiae strains, demonstrating that development of a phage-based BCA using our repository might be applicable both in New Zealand and Europe.
The host range screening also confirmed that P. syringae pv. actinidiae isolates within each country are not entirely clonal. In particular, New Zealand strains originally considered P. syringae pv. actinidiae less virulent showed a significantly different profile of phage susceptibility than the highly aggressive P. syringae pv. actinidiae virulent strains that have proven so damaging since their emergence in New Zealand. The aggressive strains are thought to have spread largely through clonal expansion, yet our host range data indicate that there are detectable differences in phage infection between some isolates (Table 1). A further detailed comparison of multiple New Zealand strains is required to determine if these differences involve receptor alterations, or other resistance mechanisms. P. syringae pv. actinidiae virulent strains are predicted to have entered New Zealand very recently and have not been detected in the South Island (22). However, we have isolated many phages from the South Island that infect P. syringae pv. actinidiae virulent strains, which might indicate that their native host is a closely related pseudomonad already present in the country.
In summary, we have reported the first isolation and characterization of an extensive set of P. syringae pv. actinidiae phages. We have determined that the majority of phages have a very specific host range for P. syringae pv. actinidiae and do not affect any nonpathogenic bacteria isolated from kiwifruit plants. Thus, they have the potential to act as BCAs in the fight against this major horticultural pathogen.
Supplementary Material
ACKNOWLEDGMENTS
Funding for this work was provided by Zespri International, Ltd., by a Rutherford Discovery Fellowship from the Royal Society of New Zealand to P.C.F., and by the New Zealand Ministry of Business, Innovation and Employment, Better Border Biosecurity program.
We acknowledge the facilities as well as scientific and technical assistance from staff at the Otago Centre for Electron Microscopy at the University of Otago. We thank John Tagg, Iain Lamont, Joel Vanneste, Janet Yu, Matt Templeton, Mark Andersen, and Robert Taylor for generously providing some strains and helping with strain information used in this study, members of the Department of Microbiology and Immunology and Tahuna wastewater treatment plant for providing samples for phage isolation, and Bryan Parkes for useful discussions.
Footnotes
Published ahead of print 31 January 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00062-14.
REFERENCES
- 1.Everett KR, Taylor RK, Romberg MK, Rees-George J, Fullerton RA, Vanneste JL, Manning MA. 2011. First report of Pseudomonas syringae pv. actinidiae causing kiwifruit bacterial canker in New Zealand. Australas. Plant Dis. Notes 6:67–71. 10.1007/s13314-011-0023-9 [DOI] [Google Scholar]
- 2.Kiwifruit Vine Health, Inc. 2013, posting date Kiwifruit vine health. Kiwifruit Vine Health, Inc., Mount Maunganui, New Zealand: http://www.kvh.org.nz Accessed 1 November 2013 [Google Scholar]
- 3.Takikawa Y, Serizawa S, Ichikawa T, Tsuyumu S, Goto M. 1989. Pseudomonas syringae pv. actinidae pv. nov.: the causal bacterium of canker of kiwifruit in Japan. Ann. Phytopathol. Soc. Jpn. 55:437–444. 10.3186/jjphytopath.55.437 [DOI] [Google Scholar]
- 4.Renzi M, Copini P, Taddei AR, Rossetti A, Gallipoli L, Mazzaglia A, Balestra GM. 2012. Bacterial canker on kiwifruit in Italy: anatomical changes in the wood and in the primary infection sites. Phytopathology 102:827–840. 10.1094/PHYTO-02-12-0019-R [DOI] [PubMed] [Google Scholar]
- 5.Wang Z, Tang X, Liu S. 1992. Identification of the pathogenic bacterium for bacterial canker on Actinidia in Sichuan. J. Southwest Agric. Univ. 1992-06. (In Chinese.) http://en.cnki.com.cn/Article_en/CJFDTOTAL-XNND199206007.htm [Google Scholar]
- 6.Koh J, Cha B, Chung H, Lee D. 1994. Outbreak and spread of bacterial canker in kiwifruit. Korean J. Plant Pathol. 10:68–72 [Google Scholar]
- 7.Scortichini M. 1994. Occurrence of Pseudomonas syringae pv. actinidiae on kiwifruit in Italy. Plant Pathol. 43:1035–1038. 10.1111/j.1365-3059.1994.tb01654.x [DOI] [Google Scholar]
- 8.Liang Y, Zhang X, Tian C, Gao A, Wang P. 2000. Pathogenic identification of kiwifruit bacterial canker in Shaanxi. J. Northwest For. Coll. 2000-01. (In Chinese.) http://en.cnki.com.cn/Article_en/CJFDTOTAL-XBLX200001006.htm [Google Scholar]
- 9.Ferrante P, Scortichini M. 2009. Identification of Pseudomonas syringae pv. actinidiae as causal agent of bacterial canker of yellow kiwifruit (Actinidia chinensis Planchon) in central Italy. J. Phytopathol. 157:768–770. 10.1111/j.1439-0434.2009.01550.x [DOI] [Google Scholar]
- 10.Koh Y, Kim G, Jung J, Lee Y, Hur J. 2010. Outbreak of bacterial canker on Hort16A (Actinidia chinensis Planchon) caused by Pseudomonas syringae pv. actinidiae in Korea. N. Z. J. Crop Hortic. Sci. 38:275–282. 10.1080/01140671.2010.512624 [DOI] [Google Scholar]
- 11.Vanneste JL, Poliakoff F, Audusseau C, Cornish DA, Paillard S, Rivoal C, Yu J. 2011. First report of Pseudomonas syringae pv. actinidiae, the causal agent of bacterial canker of kiwifruit in France. Plant Dis. 95:1311.1. 10.1094/PDIS-03-11-0195 [DOI] [PubMed] [Google Scholar]
- 12.Balestra GM, Renzi M, Mazzaglia A. 2010. First report of bacterial canker of Actinidia delicosa caused by Pseudomonas syringae pv. actinidiae in Portugal. New Dis. Rep. 22:10. 10.5197/j.2044-0588.2010.022.010 [DOI] [Google Scholar]
- 13.Balestra GM, Renzi M, Mazzaglia A. 2011. First report of Pseudomonas syringae pv. actinidiae on kiwifruit plants in Spain. New Dis. Rep. 24:10. 10.5197/j.2044-0588.2011.024.010 [DOI] [Google Scholar]
- 14.Abelleira A, López MM, Peñalver J, Aguín O, Mansilla JP, Picoaga A, García MJ. 2011. First report of bacterial canker of kiwifruit caused by Pseudomonas syringae pv. actinidiae in Spain. Plant Dis. 95:1583.1. 10.1094/PDIS-06-11-0537 [DOI] [PubMed] [Google Scholar]
- 15.Bastas KK, Karakaya A. 2012. First report of bacterial canker of kiwifruit caused by Pseudomonas syringae pv. actinidiae in Turkey. Plant Dis. 96:452. 10.1094/PDIS-08-11-0675 [DOI] [PubMed] [Google Scholar]
- 16.ProMED-mail. 25 March 2011. Bacterial canker, kiwifruit—Chile: first report (O'Higgins, Maule). Int. Soc. Infect. Dis. ProMED-mail:20110325.0940 Accessed 1 November 2013 [Google Scholar]
- 17.Ferrante P, Scortichini M. 2010. Molecular and phenotypic features of Pseudomonas syringae pv. actinidiae isolated during recent epidemics of bacterial canker on yellow kiwifruit (Actinidia chinensis) in central Italy. Plant Pathol. 59:954–962. 10.1111/j.1365-3059.2010.02304.x [DOI] [Google Scholar]
- 18.Balestra GM, Mazaglia A, Quattrucci A, Renzi M, Rossetti A. 2009. Occurrence of Pseudomonas syringae pv. actinidiae in Jin Tao kiwi plants in Italy. Phytopathol. Mediterr. 48:299–301 [Google Scholar]
- 19.Marcelletti S, Ferrante P, Petriccione M, Firrao G, Scortichini M. 2011. Pseudomonas syringae pv. actinidiae draft genomes comparison reveal strain-specific features involved in adaptation and virulence to Actinidia species. PLoS One 6:e27297. 10.1371/journal.pone.0027297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scortichini M, Marcelletti S, Ferrante P, Petriccione M, Firrao G. 2012. Pseudomonas syringae pv. actinidiae: a re-emerging, multi-faceted, pandemic pathogen. Mol. Plant Pathol. 13:631–640. 10.1111/j.1364-3703.2012.00788.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Butler MI, Stockwell PA, Black MA, Day RC, Lamont IL, Poulter RTM. 2013. Pseudomonas syringae pv. actinidiae from recent outbreaks of kiwifruit bacterial canker belong to different clones that originated in China. PLoS One 8:e57464. 10.1371/journal.pone.0057464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chapman JR, Taylor RK, Weir BS, Romberg MK, Vanneste JL, Luck J, Alexander BJR. 2012. Phylogenetic relationships among global populations of Pseudomonas syringae pv. actinidiae. Phytopathology 102:1034–1044. 10.1094/PHYTO-03-12-0064-R [DOI] [PubMed] [Google Scholar]
- 23.Mazzaglia A, Studholme DJ, Taratufolo MC, Cai R, Almeida NF, Goodman T, Guttman DS, Vinatzer BA, Balestra GM. 2012. Pseudomonas syringae pv. actinidiae (PSA) isolates from recent bacterial canker of kiwifruit outbreaks belong to the same genetic lineage. PLoS One 7:e36518. 10.1371/journal.pone.0036518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McCann HC, Rikkerink EHA, Bertels F, Fiers M, Lu A, Rees-George J, Andersen MT, Gleave AP, Haubold B, Wohlers MW, Guttman DS, Wang PW, Straub C, Vanneste J, Rainey PB, Templeton MD. 2013. Genomic analysis of the kiwifruit pathogen Pseudomonas syringae pv. actinidiae provides insight into the origins of an emergent plant disease. PLoS Pathog. 9:e1003503. 10.1371/journal.ppat.1003503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.European Economic Commission. 2004. Commission decision of 30 January 2004 concerning the non-inclusion of certain active substances in Annex I to Council Directive 91/414/EEC and the withdrawal of authorisations for plant protection products containing these substances. Official Journal of the European Communities, No. L 37/27. European Economic Commission, Brussels, Belgium [Google Scholar]
- 26.Han HS, Koh YJ, Hur JS, Jung JS. 2004. Occurrence of the strA-strB streptomycin resistance genes in Pseudomonas species isolated from kiwifruit plants. J. Microbiol. 42:365–368 [PubMed] [Google Scholar]
- 27.Hirst JM, Riche HH, Bascomb CL. 1961. Copper accumulation in the soils of apple orchards near Wisbech. Plant Pathol. 10:105–108. 10.1111/j.1365-3059.1961.tb00127.x [DOI] [Google Scholar]
- 28.Pietrzak U, McPhail DC. 2004. Copper accumulation, distribution and fractionation in vineyard soils of Victoria, Australia. Geoderma 122:151–166. 10.1016/j.geoderma.2004.01.005 [DOI] [Google Scholar]
- 29.Cooksey DA. 1994. Molecular mechanisms of copper resistance and accumulation in bacteria. FEMS Microbiol. Rev. 14:381–386. 10.1111/j.1574-6976.1994.tb00112.x [DOI] [PubMed] [Google Scholar]
- 30.Hwang MSH, Morgan RL, Sarkar SF, Wang PW, Guttman DS. 2005. Phylogenetic characterization of virulence and resistance phenotypes of Pseudomonas syringae. Appl. Environ. Microbiol. 71:5182–5191. 10.1128/AEM.71.9.5182-5191.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Behlau F, Canteros BI, Minsavage GV, Jones JB, Graham JH. 2011. Molecular characterization of copper resistance genes from Xanthomonas citri subsp. citri and Xanthomonas alfalfae subsp. citrumelonis. Appl. Environ. Microbiol. 77:4089–4096. 10.1128/AEM.03043-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Balogh B, Jones JB, Iriarte FB, Momol MT. 2010. Phage therapy for plant disease control. Curr. Pharm. Biotechnol. 11:48–57. 10.2174/138920110790725302 [DOI] [PubMed] [Google Scholar]
- 33.Frampton RA, Pitman AR, Fineran PC. 2012. Advances in bacteriophage-mediated control of plant pathogens. Int. J. Microbiol. 2012:326452. 10.1155/2012/326452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jones JB, Jackson LE, Balogh B, Obradovic A, Iriarte FB, Momol MT. 2007. Bacteriophages for plant disease control. Annu. Rev. Phytopathol. 45:245–262. 10.1146/annurev.phyto.45.062806.094411 [DOI] [PubMed] [Google Scholar]
- 35.Adriaenssens EM, Van Vaerenbergh J, Vandenheuvel D, Dunon V, Ceyssens P-J, De Proft M, Kropinski AM, Noben J-P, Maes M, Lavigne R. 2012. T4-related bacteriophage LIMEstone isolates for the control of soft rot on potato caused by “Dickeya solani.” PLoS One 7:e33227. 10.1371/journal.pone.0033227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boulé J, Sholberg PL, Lehman SM, O'gorman DT, Svircev AM. 2011. Isolation and characterization of eight bacteriophages infecting Erwinia amylovora and their potential as biological control agents in British Columbia, Canada. Can. J. Plant Pathol. 33:308–317. 10.1080/07060661.2011.588250 [DOI] [Google Scholar]
- 37.Gill JJ, Svircev AM, Smith R, Castle AJ. 2003. Bacteriophages of Erwinia amylovora. Appl. Environ. Microbiol. 69:2133–2138. 10.1128/AEM.69.4.2133-2138.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Svircev AM, Castle AJ, Lehman SM. 2010. Bacteriophages for control of phytopathogens in food production systems, p 79–102 In Sabour PM, Griffiths MW. (ed), Bacteriophages in the control of food- and waterborne pathogens. ASM Press, Washington, DC [Google Scholar]
- 39.Ravensdale M, Blom TJ, Gracia-Garza JA, Svircev AM, Smith RJ. 2007. Bacteriophages and the control of Erwinia carotovora subsp. carotovora. Can. J. Plant Pathol. 29:121–130. 10.1080/07060660709507448 [DOI] [Google Scholar]
- 40.Fujiwara A, Fujisawa M, Hamasaki R, Kawasaki T, Fujie M, Yamada T. 2011. Biocontrol of Ralstonia solanacearum by treatment with lytic bacteriophages. Appl. Environ. Microbiol. 77:4155–4162. 10.1128/AEM.02847-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanaka H, Negishi H, Maeda H. 1990. Control of tobacco bacterial wilt by an avirulent strain of Pseudomonas solanacearum M4S and its bacteriophage. Ann. Phytopathol. Soc. Jpn. 56:243–246. 10.3186/jjphytopath.56.243 [DOI] [Google Scholar]
- 42.Balogh B, Jones JB, Momol MT, Olson SM, Obradović A, King P, Jackson LE. 2003. Improved efficacy of newly formulated bacteriophages for management of bacterial spot on tomato. Plant Dis. 87:949–954. 10.1094/PDIS.2003.87.8.949 [DOI] [PubMed] [Google Scholar]
- 43.Flaherty JE, Jones JB, Harbaugh BK, Somodi GC, Jackson LE. 2000. Control of bacterial spot on tomato in the greenhouse and field with H-mutant bacteriophages. HortScience 35:882–884 [Google Scholar]
- 44.Iriarte FB, Balogh B, Momol MT, Smith LM, Wilson M, Jones JB. 2007. Factors affecting survival of bacteriophage on tomato leaf surfaces. Appl. Environ. Microbiol. 73:1704–1711. 10.1128/AEM.02118-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Obradović A, Jones JB, Momol MT, Balogh B, Olson SM. 2004. Management of tomato bacterial spot in the field by foliar applications of bacteriophages and SAR inducers. Plant Dis. 88:736–740. 10.1094/PDIS.2004.88.7.736 [DOI] [PubMed] [Google Scholar]
- 46.Obradović A, Jones JB, Momol MT, Olson SM, Jackson LE, Balogh B, Guven K, Iriarte FB. 2005. Integration of biological control agents and systemic acquired resistance inducers against bacterial spot on tomato. Plant Dis. 89:712–716. 10.1094/PD-89-0712 [DOI] [PubMed] [Google Scholar]
- 47.Wang GC, Wang Y. 1996. The frequency of chimeric molecules as a consequence of PCR co-amplification of 16S rRNA genes from different bacterial species. Microbiology 142:1107–1114. 10.1099/13500872-142-5-1107 [DOI] [PubMed] [Google Scholar]
- 48.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. 10.1093/bioinformatics/bts199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402. 10.1093/nar/25.17.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abedon ST. 2011. Lysis from without. Bacteriophage 1:46–49. 10.4161/bact.1.1.13980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Petty NK, Foulds IJ, Pradel E, Ewbank JJ, Salmond GPC. 2006. A generalized transducing phage (phiIF3) for the genomically sequenced Serratia marcescens strain Db11: a tool for functional genomics of an opportunistic human pathogen. Microbiology 152:1701–1708. 10.1099/mic.0.28712-0 [DOI] [PubMed] [Google Scholar]
- 52.Ramsay JP, Williamson NR, Spring DR, Salmond GPC. 2011. A quorum-sensing molecule acts as a morphogen controlling gas vesicle organelle biogenesis and adaptive flotation in an enterobacterium. Proc. Natl. Acad. Sci. U. S. A. 108:14932–14937. 10.1073/pnas.1109169108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Manfioletti G, Schneider C. 1988. A new and fast method for preparing high quality lambda DNA suitable for sequencing. Nucleic Acids Res. 16:2873–2884. 10.1093/nar/16.7.2873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Aronesty E. 2013. Comparison of sequencing utility programs. Open Bioinformatics J. 7:1–8. 10.2174/1875036201307010001 [DOI] [Google Scholar]
- 55.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O. 2008. The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. 10.1186/1471-2164-9-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25:955–964. 10.1093/nar/25.5.0955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Laslett D, Canback B. 2004. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32:11–16. 10.1093/nar/gkh152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Garbe J, Bunk B, Rohde M, Schobert M. 2011. Sequencing and characterization of Pseudomonas aeruginosa phage JG004. BMC Microbiol. 11:102. 10.1186/1471-2180-11-102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sullivan MJ, Petty NK, Beatson SA. 2011. Easyfig: a genome comparison visualizer. Bioinformatics 27:1009–1010. 10.1093/bioinformatics/btr039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alikhan N-F, Petty NK, Ben Zakour NL, Beatson SA. 2011. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. 10.1186/1471-2164-12-402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wilkie JP, Dye DW, Watson DRW. 1973. Further hosts of Pseudomonas viridiflava. N. Z. J. Agric. Res. 16:315–323. 10.1080/00288233.1973.10421110 [DOI] [Google Scholar]
- 62.Fineran PC, Petty NK, Salmond GPC. 2009. Transduction: host DNA transfer by bacteriophages. In Schaechter M. (ed), The Encyclopedia of microbiology, 3rd ed. Academic Press, London, United Kingdom [Google Scholar]
- 63.Roach DR, Sjaarda DR, Castle AJ, Svircev AM. 2013. Host exopolysaccharide quantity and composition impact Erwinia amylovora bacteriophage pathogenesis. Appl. Environ. Microbiol. 79:3249–3256. 10.1128/AEM.00067-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yee LM, Matsumoto T, Yano K, Matsuoka S, Sadaie Y, Yoshikawa H, Asai K. 2011. The genome of Bacillus subtilis phage SP10: a comparative analysis with phage SPO1. Biosci. Biotechnol. Biochem. 75:944–952. 10.1271/bbb.100921 [DOI] [PubMed] [Google Scholar]
- 65.Lu S, Le S, Tan Y, Zhu J, Li M, Rao X, Zou L, Li S, Wang J, Jin X, Huang G, Zhang L, Zhao X, Hu F. 2013. Genomic and proteomic analyses of the terminally redundant genome of the Pseudomonas aeruginosa phage PaP1: establishment of genus PaP1-like phages. PLoS One 8:e62933. 10.1371/journal.pone.0062933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Whelan KF, Colleran E, Taylor DE. 1995. Phage inhibition, colicin resistance, and tellurite resistance are encoded by a single cluster of genes on the IncHI2 plasmid R478. J. Bacteriol. 177:5016–5027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Walter EG, Thomas CM, Ibbotson JP, Taylor DE. 1991. Transcriptional analysis, translational analysis, and sequence of the kilA-tellurite resistance region of plasmid RK2Ter. J. Bacteriol. 173:1111–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Taylor DE. 1999. Bacterial tellurite resistance. Trends Microbiol. 7:111–115. 10.1016/S0966-842X(99)01454-7 [DOI] [PubMed] [Google Scholar]
- 69.Moreno Switt AI, Orsi RH, den Bakker HC, Vongkamjan K, Altier C, Wiedmann M. 2013. Genomic characterization provides new insight into Salmonella phage diversity. BMC Genomics 14:481. 10.1186/1471-2164-14-481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Roessner CA, Ihler GM. 1984. Proteinase sensitivity of bacteriophage lambda tail proteins gpJ and pH in complexes with the lambda receptor. J. Bacteriol. 157:165–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Boulanger P, Jacquot P, Plançon L, Chami M, Engel A, Parquet C, Herbeuval C, Letellier L. 2008. Phage T5 straight tail fiber is a multifunctional protein acting as a tape measure and carrying fusogenic and muralytic activities. J. Biol. Chem. 283:13556–13564. 10.1074/jbc.M800052200 [DOI] [PubMed] [Google Scholar]
- 72.Lehman SM. 2007. Development of a bacteriophage-based biopesticide for fire blight. PhD thesis Brock University, St. Catharines, Ontario, Canada [Google Scholar]
- 73.Lehman SM, Kropinski AM, Castle AJ, Svircev AM. 2009. Complete genome of the broad-host-range Erwinia amylovora phage phiEa21-4 and its relationship to Salmonella phage felix O1. Appl. Environ. Microbiol. 75:2139–2147. 10.1128/AEM.02352-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ceyssens P-J, Lavigne R. 2010. Bacteriophages of Pseudomonas. Future Microbiol. 5:1041–1055. 10.2217/fmb.10.66 [DOI] [PubMed] [Google Scholar]
- 75.Ceyssens P-J, Noben J-P, Ackermann H-W, Verhaegen J, De Vos D, Pirnay J-P, Merabishvili M, Vaneechoutte M, Chibeu A, Volckaert G, Lavigne R. 2009. Survey of Pseudomonas aeruginosa and its phages: de novo peptide sequencing as a novel tool to assess the diversity of worldwide collected viruses. Environ. Microbiol. 11:1303–1313. 10.1111/j.1462-2920.2008.01862.x [DOI] [PubMed] [Google Scholar]
- 76.Debarbieux L, Leduc D, Maura D, Morello E, Criscuolo A, Grossi O, Balloy V, Touqui L. 2010. Bacteriophages can treat and prevent Pseudomonas aeruginosa lung infections. J. Infect. Dis. 201:1096–1104. 10.1086/651135 [DOI] [PubMed] [Google Scholar]
- 77.Essoh C, Blouin Y, Loukou G, Cablanmian A, Lathro S, Kutter E, Thien HV, Vergnaud G, Pourcel C. 2013. The susceptibility of Pseudomonas aeruginosa strains from cystic fibrosis patients to bacteriophages. PLoS One 8:e60575. 10.1371/journal.pone.0060575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Petty NK, Evans TJ, Fineran PC, Salmond GPC. 2007. Biotechnological exploitation of bacteriophage research. Trends Biotechnol. 25:7–15. 10.1016/j.tibtech.2006.11.003 [DOI] [PubMed] [Google Scholar]
- 79.Richter C, Chang JT, Fineran PC. 2012. Function and regulation of clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR associated (Cas) systems. Viruses 4:2291–2311. 10.3390/v4102291 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.