

Abstract
The noncanonical alcohol dehydrogenase AlkJ is encoded on the alkane-metabolizing alk operon of the mesophilic bacterium Pseudomonas putida GPo1. To gain insight into the enzymology of AlkJ, we have produced the recombinant protein in Escherichia coli and purified it to homogeneity using His6 tag affinity and size exclusion chromatography (SEC). Despite synthesis in the cytoplasm, AlkJ was associated with the bacterial cell membrane, and solubilization with n-dodecyl-β-d-maltoside was necessary to liberate the enzyme. SEC and spectrophotometric analysis revealed a dimeric quaternary structure with stoichiometrically bound reduced flavin adenine dinucleotide (FADH2). The holoenzyme showed thermal denaturation at moderate temperatures around 35°C, according to both activity assay and temperature-dependent circular dichroism spectroscopy. The tightly bound coenzyme was released only upon denaturation with SDS or treatment with urea-KBr and, after air oxidation, exhibited the characteristic absorption spectrum of FAD. The enzymatic activity of purified AlkJ for 1-butanol, 1-hexanol, and 1-octanol as well as the n-alkanol derivative ω-hydroxy lauric acid methyl ester (HLAMe) was quantified in the presence of the artificial electron acceptors phenazine methosulfate (PMS) and 2,6-dichlorophenolindophenol (DCPIP), indicating broad substrate specificity with the lowest activity on the shortest alcohol, 1-butanol. Furthermore, AlkJ was able to accept as cosubstrates/oxidants the ubiquinone derivatives Q0 and Q1, also in conjunction with cytochrome c, which suggests coupling to the bacterial respiratory chain of this membrane-associated enzyme in its physiological environment. Using gas chromatographic analysis, we demonstrated specific biocatalytic conversion by AlkJ of the substrate HLAMe to the industrially relevant aldehyde, thus enabling the biotechnological production of 12-amino lauric acid methyl ester via subsequent enzymatic transamination.
INTRODUCTION
The alkane-metabolizing alk operon of Pseudomonas putida GPo1 (formerly known as P. oleovorans) has attracted considerable interest in industrial biotechnology (1–3). This plasmid-encoded degradation system consists of up to 10 enzymes as well as auxiliary proteins that enable full conversion of bare hydrocarbons into fatty acids, thus allowing the bacterium to generate acetyl coenzyme A (acetyl-CoA) from nonactivated alkanes (4). Several mechanistic studies have focused on the oxy-functionalization of alkane substrates, which is followed by catabolic degradation in the microbes (5–8). Furthermore, approaches to utilize parts of the Alk system in biosynthetic applications have recently emerged (9–11). So far, most investigations have dealt with those alk genes that are responsible for the initial introduction of the ω-hydroxy function, i.e., the AlkBGT system, comprising a monooxygenase (AlkB) that becomes charged with electrons by a rubredoxin-electron shuttle (AlkG), which in turn receives electrons from the general cell metabolism via a NADH-dependent rubredoxin reductase (AlkT) (12).
Efforts to employ the AlkBGT system in combination with a transaminase for the selective ω-amino functionalization of fatty acids have reached advanced stage, allowing, for example, the generation of ω-amino lauric acid methyl ester, which serves as precursor for high-performance polymers (13). However, during initial conversion of the substrate lauric acid methyl ester (LAMe) into the alcohol hydroxy-LAMe (HLAMe) by AlkBGT, considerable overoxidation to the aldehyde oxo-LAMe (OLAMe) and even to dodecanedioic acid monomethyl ester (DDAMe) was observed, which impairs the overall process yield (10) (Fig. 1). This side activity was assigned to AlkB, which apparently can further oxidize the enzyme-attached initial alcohol product to the aldehyde and even to the (di)carboxylic acid.
FIG 1.
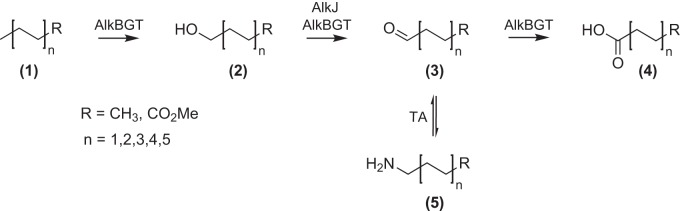
Reaction cascade for the enzymatic ω-oxidation/functionalization of long-chain aliphatic alcohols or their derivatives, such as LAMe. Initially, the hydrocarbon substrate (1) is hydroxylated via the AlkBGT system, resulting in the alcohol (2), which can be further oxidized by either AlkBGT or AlkJ to yield the aldehyde (3). Finally, overoxidation by AlkBGT may lead to the carboxylic acid (4), which normally constitutes an undesired by-product. Using an ω-transaminase (TA), the aldehyde (3) can be diverted to a primary amine (5), thus yielding a valuable building block for industrial biotechnology, for example, 12-amino lauric acid methyl ester (ALAMe).
Apart from AlkB, the alcohol dehydrogenase AlkJ encoded on the alk operon is also capable of oxidizing the primary alcohol to the aldehyde (14), possibly providing a better-defined substrate and product specificity. On the basis of its amino acid sequence, AlkJ has been classified as member of the glucose-methanol-choline (GMC) oxidoreductase family (15). These enzymes share a conserved flavin adenine dinucleotide (FAD)-binding site and also include several NAD-independent oxidases, lyases, and dehydrogenases. Some of them are under consideration for industrial biotransformations (16) or are already in practical application, such as in the case of glucose oxidase (17) or cholesterol oxidase (18).
Unlike most other enzymes of the alk operon, little is known about the biochemical properties of AlkJ. Benson and Shapiro (14) were first able to assign an alcohol dehydrogenase (ADH) function to AlkJ. Later, van Beilen et al. (19) assayed AlkJ-containing membranes of transformed P. putida and Escherichia coli, thus demonstrating the fundamental substrate specificity of AlkJ for mid- to long-chain alkanols. In contrast to other GMC enzymes, AlkJ was described as a membrane-bound enzyme, which usually poses a burden for solubilization and purification in a functional state (20).
In the present study, we describe the first purification to electrophoretic homogeneity of the membrane-associated AlkJ enzyme in its physiologically active state after heterologous expression in E. coli. The enzyme was biochemically characterized with regard to the nature of its prosthetic group and its putative physiological electron acceptor. Furthermore, we have elucidated the enzyme kinetics of oxidation for three aliphatic alcohols as well as for the industrially relevant fatty acid methyl ester alcohol HLAMe.
MATERIALS AND METHODS
Chemicals, plasmids, and strains.
Unless otherwise stated, all chemicals were from Sigma-Aldrich (Munich, Germany), Applichem (Darmstadt, Germany), or VWR (Darmstadt, Germany) in the highest purity available. 2,6-Dichlorophenolindophenol (DCPIP) and n-dodecyl-β-d-maltoside (DDM) were from Carl Roth (Karlsruhe, Germany), and n-alkanol substrates as well as gel filtration standards were from Sigma-Aldrich. Anhydrotetracycline (aTc) was purchased from Acros Organics (Geel, Belgium), while HLAMe, OLAMe, and DDAMe were kindly provided by Evonik Industries (Marl, Germany). Primers and enzymes for DNA manipulation were from Thermo Scientific (Schwerte, Germany), Agilent Technologies (Boeblingen, Germany), and NEB (Frankfurt am Main, Germany). The coding region of alkJ was obtained from Evonik Industries cloned on the pACYCDuet-1 vector (Merck, Darmstadt, Germany). Expression plasmids constructed in this study were based on the pASK-IBA vector series (IBA, Göttingen, Germany). Cloning was performed using E. coli XL1-Blue (21), whereas E. coli BL21 (22) was used for recombinant gene expression.
Gene cloning.
The alkJ gene was amplified from pACYC-AlkJ using Herculase II Fusion DNA polymerase with the primers 5′-GCC TAC GAC TAT ATA ATC GTT GGT GCT G-3′ and 5′-CCC CCT AAG CTT ACA TGC ACA CGG CGA TCA TGG CCA ACT CTA GCT CTG-3′, which carry recognition sites (underlined) for EheI (only one half) and HindIII, respectively. After digestion with HindIII and phosphorylation with polynucleotide kinase, the product was ligated with the backbones of the vectors pASK-IBA5+ and pASK-IBA35+, providing an N-terminal His6 or Strep-tag II, respectively (23, 24), which had been cut with both restriction enzymes. The integrity of the resulting expression plasmids was checked by analytical restriction digestion and automated dideoxy sequencing using an ABI-Prism Genetic Analyzer (Perkin-Elmer, Weiterstadt, Germany) with the BigDye Terminator kit.
Recombinant gene expression and protein purification.
AlkJ was expressed in 5-liter shake flasks containing 2 liters of TB medium (25) supplemented with 100 mg/liter ampicillin. After inoculation, the cells were grown at 22°C to an optical density at 550 nm (OD550) of 0.8, and then gene expression was induced by adding 0.2 mg/liter aTc. After further shaking for 15 h at 22°C, the bacteria were harvested by centrifugation (5,000 × g for 10 min at 4°C). Fifteen grams of wet cells was resuspended in 25 ml of buffer X (50 mM NaPi [pH 8.0], 150 mM NaCl, 15% [vol/vol] glycerol) and lysed by four passages through a French pressure cell (1,000 lb/in2; SLM Aminco, Urbana, IL). Intact cells and aggregates were removed by a short centrifugation (10,000 × g for 10 min at 4°C), and the supernatant was recovered. The cell membranes were then sedimented by ultracentrifugation (125,000 × g for 2 h at 4°C). The pellet was resuspended in buffer X and solubilized by adding of 0.5% (wt/vol) DDM (as a solid). Unsolubilized protein and aggregates were removed by a second ultracentrifugation (125,000 × g for 1 h at 4°C) prior to performance of immobilized-metal affinity chromatography (IMAC).
To this end, the solubilized protein was immediately applied to an Ni-Sepharose High Performance column (GE Healthcare, Freiburg, Germany) equilibrated with buffer X. Weakly bound proteins were washed away with buffer Z (buffer X supplemented with 0.025% [wt/vol] DDM) containing 20 mM imidazole-HCl (pH 8.0). AlkJ was then eluted by applying a concentration gradient up to 300 mM imidazole-HCl (pH 8.0) in buffer Z. The elution fractions containing apparently pure AlkJ, as judged from SDS-PAGE analysis, were dialyzed twice against buffer Z and subjected to rechromatography. This time, AlkJ was eluted in a step with 300 mM imidazole-HCl (pH 8.0) in buffer Z and directly applied to size exclusion chromatography (SEC) in the presence of buffer Z on a HiLoad Superdex 16/60 200 column (GE Healthcare) using an Äkta purifier system (GE Healthcare). For estimation of the molecular size, the column was calibrated with blue dextran, β-amylase, alcohol dehydrogenase, bovine serum albumin, ovalbumin, carbonic anhydrase, and cytochrome c.
The purified enzyme was stored at 4°C and used for biochemical characterization within 5 days. Protein purity was checked using SDS-PAGE with a high-molarity Tris buffer system (26). Gel band intensity was densitometrically quantified on a Perfection V700 photo scanner (Epson, Meerbusch, Germany) with TotalLabQuant software (TotalLab, Newcastle upon Tyne, United Kingdom). Protein concentration was measured via UV spectrophotometry using a molar extinction coefficient of 57,340 M−1 cm−1, which had been calculated with the ProtParam tool on the ExPASy server (27).
Spectroscopic measurements and identification of the prosthetic group.
Absorption spectra of recombinant AlkJ were recorded in a 1-cm-path-length quartz cuvette (Hellma, Müllheim, Germany) using either an S-3100 photodiode array photometer (Scinco, Seoul, South Korea) or an Ultrospec 2100 pro UV-visible spectrophotometer (GE Healthcare).
For spectroscopic analysis of the prosthetic group itself, IMAC-purified AlkJ was denatured by adding 0.2% (wt/vol) SDS, and the spectrum from 190 to 600 nm was recorded. To elucidate whether the prosthetic group is bound covalently or by noncovalent interactions, AlkJ was immobilized via the His6 tag on Ni-Sepharose and treated with chaotropic reagents by washing with a mixture of 2 M urea and 2 M KBr in the presence of 50 mM NaPi (pH 8.0) according to the procedure described by Hefti et al. (28). Subsequently, AlkJ was eluted in a step with 300 mM imidazole-HCl (pH 8.0) in buffer Z. This enzyme preparation was treated with SDS and subjected to spectroscopic analysis as described above.
To precisely determine the proportion of bound FAD, freshly prepared AlkJ was precipitated from buffer Z by adding (NH4)2SO4 from a saturated stock solution to a final concentration of 70% (wt/vol), and the pelleted enzyme was resuspended in 50 mM NaPi (pH 8.0), 8 M urea, and 2 M KBr. After incubation for 2 h at room temperature, AlkJ was precipitated with 10% (wt/vol) trichloroacetic acid (TCA) and pelleted by centrifugation (15,000 × g for 30 min at 4°C). The supernatant was collected, and the protein pellet was washed with acetone and redissolved in buffer Z. The concentration of recovered apo-AlkJ was determined according to its protein extinction coefficient (see above), while FAD in the supernatant was quantified using a molar extinction coefficient of 9,300 M−1 cm−1 at 450 nm (29).
Near-UV circular dichroism (CD) measurements were carried out in a Jasco J-810 spectropolarimeter (Jasco, Tokyo, Japan) at 25°C using a 0.1-mm-path-length quartz cuvette (Hellma). A 10 μM AlkJ solution was used, which had been dialyzed, after tandem IMAC, against CD buffer (20 mM KPi [pH 7.5], 50 mM K2SO4) using 12- to 16-kDa molecular-mass-cutoff (MWCO) Spectra/Por dialysis tubing (Spectrum Laboratories, Los Angeles, CA). Up to 6 spectra were accumulated in the wavelength range of 260 to 190 nm (response = 1 s, bandwidth = 1 nm, data pitch = 0.1 nm, and scan speed = 50 nm/min).
Enzyme assays.
The catalytic activity of AlkJ was assessed by using the artificial electron acceptors 2,6-dichlorophenolindophenol (DCPIP) and phenazine methosulfate (PMS) according to a published procedure (14). The reaction is fueled by the alcohol substrate, whose electrons are passed on via PMS to DCPIP, which loses its intense blue color upon reduction. Reactions were performed in triplicate using multiwell plates (Mikrotest plate 96-well F; Sarstedt, Nümbrecht, Germany) in a total reaction volume of 250 μl. The reaction mixture contained 50 mM NaPi (pH 7.5) with 0.1 mM DCPIP, 0.2 mM PMS, 0.2 μM AlkJ, and various amounts of different alcohol substrates dissolved in tetrahydrofuran, whose final concentration in the assay was 0.5% (vol/vol) throughout. The mixture of all reaction substrates was allowed to equilibrate for 5 min at 30°C before the assay was started by addition of the enzyme.
Monitoring of the initial reaction velocity was performed by measuring the absorption at 600 nm in a Synergy 2 microplate reader (BioTek Instruments, Bad Friedrichshall, Germany). DCPIP concentrations at different time points were deduced from a standard curve of this substance. As this assay is known for strong background reaction (30) as well as light bleaching of the DCPIP dye (31), appropriate control assays without the alcohol substrate were carried out, and the corresponding slope of time-dependent absorption was subtracted from the actual measurements. The reaction rate of the control reaction accounted for 10 to 15% of the maximally observed initial reaction velocity. The corrected initial reaction velocities were analyzed using the enzyme kinetics module of SigmaPlot (version 12.0; Systat Software, San Jose, CA) by applying Michaelis-Menten kinetics, considering uncompetitive substrate inhibition where appropriate with V = Vmax/(1 + Km/S + S/Ki).
To investigate the role of ubiquinone as a potential electron acceptor of AlkJ, the kinetic constants for the water soluble short-chain analogue 2,3-dimethoxy-5-methyl-6-(3-methyl-2-butenyl)-1,4-benzoquinone (Q1) were determined. Thus, Q1 replaced PMS as an intermediate electron acceptor in the assay described above, this time using a fixed concentration of 0.1 mM 1-octanol in the presence of 0.1 mM DCPIP and 0.2 μM AlkJ while varying the concentration of Q1 in the range from 0 to 200 μM. Again, the reaction was monitored via decolorization of DCPIP. Similarly, reduction of cytochrome c as a terminal electron acceptor was assessed by applying 5 μM Q1 and 10 μM cytochrome c from Equus caballus, replacing both PMS and DCPIP, respectively. In this case, the redox state of cytochrome c was monitored using its change in absorption at 550 nm (32).
Biocatalysis of HLAMe oxidation.
To study the biocatalytic conversion of the industrially relevant alcohol derivative HLAMe by AlkJ, an excess of 2,3-dimethoxy-5-methyl-1,4-benzoquinone (Q0) was applied as a terminal oxidant. The reaction mixture contained 0.6 mM Q0 and 0.5 mM HLAMe in 50 mM NaPi (pH 7.5) with the addition of 1% (vol/vol) tetrahydrofuran (to mediate substrate solubility) in a total volume of 5 ml. The reaction was started by addition of 0.8 μM AlkJ, previously purified by tandem IMAC, and was continued at ambient temperature for 60 min. At various time points, small samples were collected and the educts/products were extracted with ethyl acetate supplemented with 0.5 mM decane as internal standard. Product analysis was performed on a Clarus 500 gas chromatograph (Perkin-Elmer, Rodgau, Germany) equipped with a flame ionization detector using an Optima 5 HT column (Macherey-Nagel, Düren, Germany). The initial oven temperature was 90°C, followed by a ramp of 15°C/min to 280°C. The temperature was then raised at 45°C/min to 300°C and held for 3 min. Peaks were analyzed and quantified by comparison with authentic standards using the instrument software.
Measurement of enzyme thermal stability.
To determine the half-life of irreversible thermal denaturation, 1 μM solutions of the purified AlkJ holoenzyme in buffer Z were incubated in a thermocycler (Mastercycler gradient; Eppendorf, Hamburg, Germany) at constant temperatures in the range of 20 to 60°C for 1 h. After cooling to 4°C, the residual enzyme activity was measured using the assay described further above by applying a fixed substrate concentration of 0.1 mM 1-octanol in the presence of 0.2 mM PMS and 0.1 mM DCPIP at 30°C.
Quasireversible thermal denaturation of AlkJ was recorded in a Jasco J-810 spectropolarimeter equipped with a Peltier element (ramp = 60 K/h, data pitch = 0.1 K) at a wavelength of 215 nm, where the most pronounced spectral difference upon denaturation was observed. To this end, a 5 μM protein solution in CD buffer (see above) was applied in a 1-mm-path-length quartz cuvette (Hellma). The ellipticity data were fitted assuming a two-state unfolding model, employing KaleidaGraph software (Synergy, Reading, PA), as previously described (33).
RESULTS
E. coli expression, solubilization, and purification of AlkJ.
The coding sequence for AlkJ from P. putida (UniProt ID Q00593) was cloned in parallel on the vectors pASK-IBA5+ and pASK-IBA35+ with two different N-terminal affinity tags, the Strep-tag II (23) and the His6 tag (24), respectively. In both cases, cytoplasmic expression was under the control of the tetracycline promoter/operator (34). Initial attempts to isolate AlkJ from the total cell lysate of E. coli (in the absence of detergents), using either tag, remained unsuccessful, as AlkJ always appeared in the flowthrough of the affinity chromatography. Eventually, solubilization of the recombinant protein from the bacterial membrane fraction with nonionic detergents enabled binding of AlkJ to both Strep-Tactin and IMAC resins. A concentration of 0.5% (wt/vol) DDM turned out to offer the best compromise between efficient solubilization and preserving the biologically active state, as assessed from SDS-PAGE densitometry and from enzyme assays (see below). Under these conditions, up to approximately 75% native AlkJ was recovered from the bacterial membrane fraction.
Since the recombinant protein equipped with the amino-terminal His6 tag resulted in higher expression yields, this version was used for all further experiments. While solubilization of AlkJ from the total cell homogenate was possible, preparation of the cell membrane fraction via ultracentrifugation, followed by resuspension in the presence of DDM, allowed the essentially complete removal of contaminating E. coli host proteins in one subsequent IMAC step (Fig. 2). However, efforts to concentrate the affinity-purified AlkJ prior to SEC via ultrafiltration (MWCO, 30 kDa) led to the formation of aggregates starting at a detergent concentration of approximately 0.1% (wt/vol). This phenomenon was most likely due to the rising concentration of DDM, which forms large micelles of 50 to 70 kDa and, thus, also becomes concentrated during ultrafiltration (35). This problem was overcome by dialysis of the protein obtained from IMAC (to remove imidazole from the elution buffer) and then subjecting it to a second IMAC, this time applying step elution. This procedure resulted in about 80% recovery of concentrated AlkJ (∼5 mg/ml) in a small volume of 2 ml, which could be directly used for SEC on a preparative column.
FIG 2.
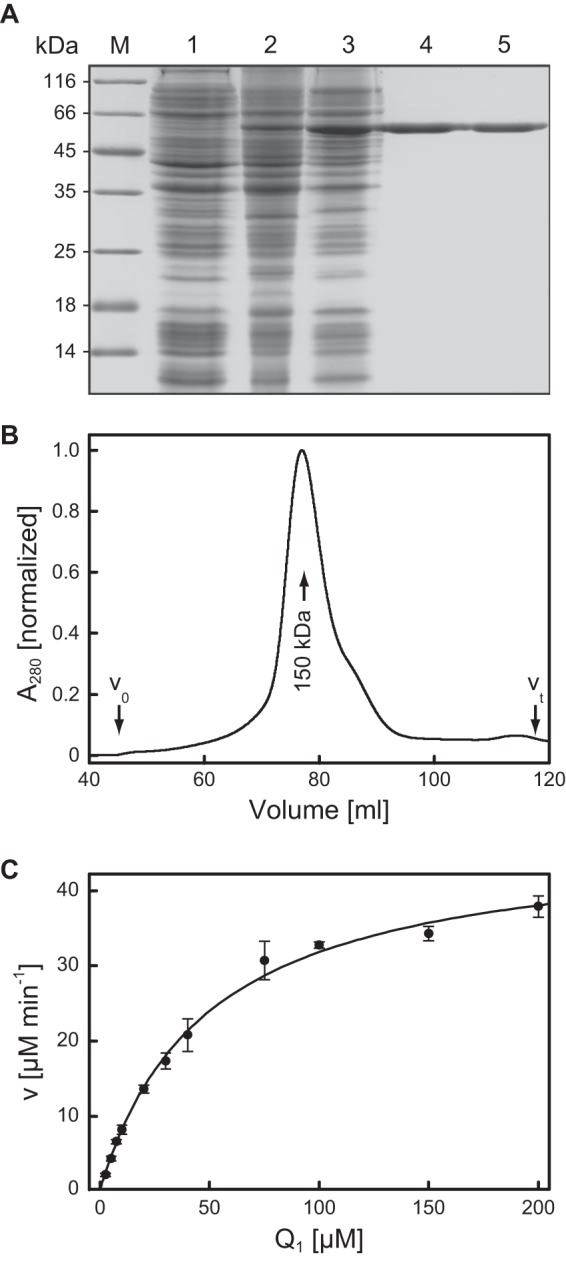
Purification of AlkJ produced in E. coli and enzymatic analysis of the ubiquinone analogue Q1 as a cosubstrate. (A) Overview of the production and purification of recombinant AlkJ analyzed via reducing SDS-PAGE. Lanes: M, molecular size marker; 1, total protein lysate of E. coli strain BL21 harboring pASK-IBA35(+)-AlkJ prior to induction; 2, total cell lysate after 15 h of recombinant gene expression at harvest; 3, solubilized proteins from the cell membrane fraction after removal of aggregates; 4, pooled elution fractions after IMAC of the detergent-solubilized membrane protein fraction; 5, purified AlkJ after SEC. (B) SEC profile of AlkJ after IMAC purification. AlkJ elutes at a volume corresponding to a molecular size of 150 kDa, which is equivalent to a protein dimer with approximately 43 DDM and 2 FADH2 molecules associated. The slight tailing of the peak is due to a small proportion of monomeric AlkJ, as checked by SDS-PAGE. The fractions containing the dimer were isolated and used for the kinetic measurements. (C) For determination of catalytic activity with Q1 as an electron acceptor, the reaction was performed at 30°C in 50 mM NaPi (pH 7.5) using 0.2 μM recombinant AlkJ in the presence of 0.1 mM 1-octanol and monitored via decolorization of the terminal electron acceptor reagent DCPIP at 600 nm. The initial velocities with various Q1 concentrations were fitted according to the Michaelis-Menten equation, yielding Km = 41 μM and kcat = 3.5 s−1.
From SEC analysis, the apparent size of solubilized AlkJ was determined as 150 kDa (Fig. 2B), which corresponds to the mass of an AlkJ homodimer associated with approximately 43 DDM molecules as well as 2 FADH2 molecules (see below). After detergent solubilization and tandem IMAC, approximately 5 mg essentially monodisperse AlkJ was obtained from 1 liter of E. coli culture, finally yielding 3.5 mg AlkJ after preparative SEC (if required). The enzyme was stored in buffer Z (optionally containing 300 mM imidazole-HCl if used without SEC) for up to 5 days at 4°C while retaining full activity.
Spectroscopic properties of AlkJ.
Although AlkJ was shown in previous investigations (14) to be independent of nicotinamide cosubstrates, there has been no experimental evidence for FAD as its redox-active prosthetic group to date. UV-visible spectroscopy of the SEC-purified recombinant protein did not reveal the two characteristic peaks for oxidized FAD at 372 and 450 nm that have been observed for other flavoenzymes (29). A minor shoulder of the peak arising from absorption of the aromatic amino acid side chains at 280 nm, stretching to 355 nm, provided a hint of the presence of fully reduced FADH2 (36). Therefore, denaturation of the purified AlkJ with SDS was investigated, resulting in the quick appearance of two peaks in the UV-visible spectrum at 377 and 449 nm in the course of 25 s (Fig. 3). This phenomenon is most likely explained by the release of enzyme-bound FADH2, which was subsequently oxidized by dissolved air oxygen in the assay solution.
FIG 3.

Spectroscopic characterization of the recombinant AlkJ holoenzyme and thermal stability analysis. (A) Time-dependent cofactor release from AlkJ upon enzyme denaturation. Reoxidation of the liberated FADH2 from a 10 μM solution of IMAC-purified AlkJ after addition of 0.2% (wt/vol) SDS was monitored at different time points using a diode array spectrophotometer. During the progress of this reaction, two absorption maxima emerged, at around 370 and 450 nm, presumably reflecting the oxidation of the released coenzyme FADH2 by dissolved oxygen. The inset depicts the UV-visible spectrum of the initial IMAC-purified AlkJ, which lacks the characteristic peaks for oxidized FAD but reveals the typical prominent absorption maximum at 280 nm for the aromatic amino acid side chains of the enzyme. This peak is neighbored by a broad shoulder extending to longer wavelengths, which is indicative of FADH2. (B) Far-UV circular dichroism (CD) spectrum of IMAC-purified recombinant AlkJ. The presence of both α-helical and β-sheet secondary structure can be deduced from the shape of the broad negative band in the region of 210 to 220 nm. Estimation of the secondary structure content was performed with the K2D algorithm available at the Dichroweb server (66). (C) Quasireversible thermal denaturation at pH 7.5 was monitored in a CD spectropolarimeter at 215 nm while heating at a constant rate of 60 K/h. Refolding could not be measured in this assay due to the formation of protein aggregates upon denaturation. (D) Thermally induced irreversible denaturation at pH 8.0 was assessed after 60 min of incubation at various temperatures by determining the residual enzymatic activity toward 0.1 mM 1-octanol (with 0.2 mM PMS and 0.1 mM DCPIP) at 30°C.
Furthermore, it was possible to deplete the enzyme-bound FADH2 by washing AlkJ, after immobilization to Ni-Sepharose, with urea and KBr, confirming that this coenzyme is not covalently bound. To elucidate the molar ratio between AlkJ and its prosthetic group, enzyme-bound FADH2 was liberated by denaturing the purified protein with urea-KBr. After precipitating AlkJ with TCA, the yellow colored supernatant containing the coenzyme was separated while the enzyme was redissolved in buffer Z, and both fractions were used for individual UV-visible spectroscopic quantification. Using the appropriate molar absorption coefficients (see Materials and Methods), the FADH2/AlkJ ratio was determined to be 1.04.
Finally, the secondary structure composition of the purified AlkJ holoenzyme was investigated by circular dichroism (CD) spectroscopy. Deconvolution of the far-UV spectrum revealed 37% α-helix, 26% β-sheet, and 37% random coil (Fig. 3B). This is in line with the secondary structure content of the most closely related enzyme (31% amino acid identity) with known three-dimensional structure, choline oxidase (CHOX) from Arthrobacter globiformis (PDB entry 2JBV) (37), which features 23% β-sheet and 25% α-helix as calculated from the crystal structure using the Stride web server (38).
Identification of the in vivo electron acceptor.
In enzyme activity assays of holo-AlkJ with 0.1 mM 1-octanol and the artificial electron acceptors PMS and DCPIP (39), supplementation of up to 2 mM NAD+ or NADP+ did not influence the reaction velocity. This proved that, in contrast to conventional ADHs, AlkJ cannot transfer electrons from alcohol oxidation to a nicotinamide cosubstrate, in line with the likely unfavorable redox potential if it is assumed that the enzyme-bound FAD acts as the primary electron acceptor. Notably, supplementation with excess FAD (up to 25 μM) did not boost the reaction either, confirming that AlkJ isolated from E. coli via the detergent solubilization and affinity purification procedure described above is fully charged with this prosthetic group.
In light of the association of AlkJ with the bacterial membrane, a mechanistic linkage of its redox catalytic activity to the pseudomonal respiratory chain via ubiquinone was previously suggested by van Beilen et al. (19). Thus, we applied Q1, a functional ubiquinone analogue with a shortened terpenoid side chain, as the electron acceptor in an enzyme assay. By measuring the redox state of the terminal electron acceptor DCPIP at 600 nm, we could trace the AlkJ-catalyzed transfer of electrons from 1-octanol to Q1 in a concentration-dependent manner (Fig. 2C). Indeed, a Michaelis-Menten-type enzyme kinetic behavior was observed for Q1, with a kcat value of 3.5 s−1 and a remarkably low Km value of 41 μM. From this experiment it appears plausible that under the physiological conditions in P. putida, ubiquinone accepts electrons from AlkJ and distributes them into the respiratory chain, where they finally reduce O2. In a further experiment, electron transport from AlkJ to cytochrome c in the presence of 5 μM Q1 was also observed (not shown), whereas cytochrome c alone was not reduced by AlkJ.
Substrate specificity and kinetic measurements.
The aliphatic alcohol substrate specificity of AlkJ was examined by applying PMS and DCPIP as artificial electron acceptors, constituting a well established colorimetric redox system that has been frequently applied for the study of FAD-dependent enzymes (14, 39). To this end, a series of three n-alkanols, 1-butanol, 1-hexanol, and 1-octanol, was compared with the industrially relevant long-chain alcohol derivative HLAMe. AlkJ revealed a classical Michaelis-Menten kinetic behavior (40) for 1-butanol, whereas 1-hexanol, 1-octanol, and HLAMe showed a decreased initial reaction velocity at higher substrate concentrations (Fig. 4). In these cases, the data could be fitted under the assumption of uncompetitive substrate inhibition (41, 42). The resulting kinetic parameters and inhibitory constants are summarized in Table 1.
FIG 4.

Catalytic activity of AlkJ on different alcohol substrates. The kinetic features of the purified recombinant enzyme toward the aliphatic alcohols 1-butanol, 1-hexanol, and 1-octanol as well as HLAMe, an industrially relevant long-chain alcohol derivative, were investigated using a 0.2 μM solution of the enzyme in 50 mM NaPi (pH 7.5) and PMS/DCPIP as oxidants in microwell plates. The redox state of the terminal electron acceptor DCPIP was monitored in situ at 600 nm. The initial reaction velocities measured with various alcohol concentrations were subjected to curve fitting according to Michaelis-Menten kinetics, taking into account uncompetitive substrate inhibition for the three alcohols with longer hydrocarbon chains.
TABLE 1.
Kinetic properties of AlkJ with four different alcohol substrates measured using a photometric assay with PMS and DCPIP as electron acceptors
| Substrate | Km (μM) | kcat (s−1) | Ki (μM) | kcat/Km (mM−1 s−1) | Activity (U mg−1) |
|---|---|---|---|---|---|
| 1-Butanol | 194 | 0.24 | 1.2 | 0.23 | |
| 1-Hexanol | 37.5 | 0.38 | 399 | 10.0 | 0.36 |
| 1-Octanol | 39.0 | 0.39 | 95 | 9.9 | 0.37 |
| HLAMe | 20.9 | 0.51 | 125 | 24.5 | 0.49 |
AlkJ showed a markedly lower substrate affinity for 1-butanol, with a Km value of around 200 μM, than for 1-hexanol, 1-octanol, and HLAMe (Table 1), indicating broad substrate specificity for alkanols having carbon chain lengths of ≥6, with Km = 20 to 40 μM. Likewise, AlkJ exhibited a higher (and almost uniform) catalytic efficiency for 1-hexanol, 1-octanol, and HLAMe than for 1-butanol. Specific activities of AlkJ for the different n-alkanol substrates ranged from 0.23 U mg−1 for 1-butanol to 0.49 U mg−1 for HLAMe when using the electron acceptors PMS and DCPIP, as published previously (19). Finally, substrate inhibition was more pronounced for 1-octanol (Ki = 95 μM) and HLAMe (Ki = 125 μM) than for 1-hexanol (Ki = 399 μM).
Thermal stability.
With regard to industrial application, the thermal stability of the purified recombinant AlkJ was studied using two different experimental setups (Fig. 3). In an assay of irreversible thermal denaturation, comprising 1 h of incubation at a given temperature in 50 mM NaPi (pH 8.0), 150 mM NaCl, 15% (vol/vol) glycerol, and 0.025% (wt/vol) DDM followed by measurement of the residual enzyme activity, the highest activity was measured after incubation around room temperature up to 26°C. Tstab, the temperature where the enzyme showed 50% residual activity, was determined as ∼34°C (Fig. 3D). At incubation temperatures above 40°C, AlkJ exhibited less than 5% residual activity. These findings were supported by thermally induced denaturation experiments with AlkJ as determined by far-UV CD spectroscopy, where 50% of the enzyme adopted the unfolded state at a melting temperature (Tm) of ∼35°C (Fig. 3C). Since AlkJ formed aggregates during heating in the CD buffer (20 mM KPi [pH 7.5], 50 mM K2SO4), denaturation turned out to be irreversible under these conditions.
Biocatalytic application of AlkJ for the oxidation of the industrially relevant substrate HLAMe.
The AlkJ-catalyzed oxidation of HLAMe to OLAMe was further investigated by substrate/product analysis via gas chromatography (Fig. 5). To this end, 0.5 mM HLAMe was oxidized in the presence of Q0 (as a physiologically more relevant kind of electron acceptor than PMS/DCPIP, especially with regard to application as a whole-cell biocatalyst) using a 0.8 μM concentration of the purified recombinant enzyme. After 10 min, a maximal amount of the primary reaction product OLAMe was detected, corresponding to a specific enzyme activity of 1.46 U mg−1. For comparison, AlkJ activity under the same conditions toward 1-octanol was 1.42 U mg−1. However, the reaction did not stop at this stage, since the emerging aldehyde OLAMe was further oxidized by AlkJ to the (di)carboxylic acid DDAMe, albeit with 9-fold-lower velocity (0.16 U mg−1).
FIG 5.
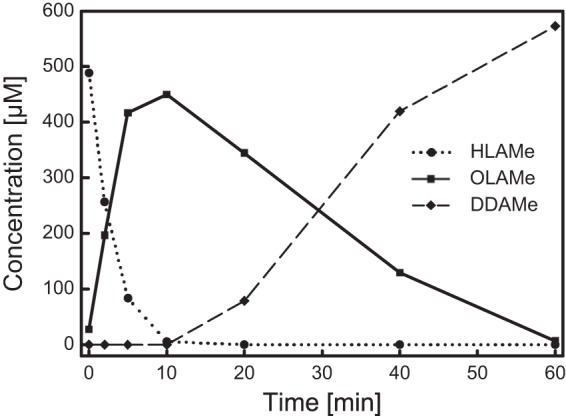
Product analysis during biocatalytic application of AlkJ to the oxidation of HLAMe, an industrially relevant intermediate. Oxidation of 0.5 mM HLAMe (circles) to the aldehyde OLAMe (squares), followed by overoxidation to the (di)acid DDAMe (diamonds), was monitored over 60 min, using 0.8 μM AlkJ with Q0 as the electron acceptor. Samples were collected at different time points, and the substrates/products were quantified by gas chromatography analysis after extraction from the aqueous phase.
DISCUSSION
Here we report the first biochemical analysis of AlkJ as a purified enzyme, whereas previous studies have relied on crude extracts or membrane fractions of P. putida and E. coli (14, 19). Using the isolated recombinant protein, we were able to prove that AlkJ is a flavoenzyme with tightly bound FADH2 (even after IMAC and SEC purification). Furthermore, we could show that this ADH can utilize ubiquinone as a cosubstrate, which presumably connects the catalyzed redox reaction to the bacterial respiratory chain. Kinetic characterization of the enzyme with four different alcohol substrates indicated that AlkJ shows broad substrate recognition of long-chain n-alkanols. Unexpectedly, beyond conversion into the corresponding aldehydes, AlkJ can also catalyze further oxidation to the corresponding carboxylic acid, though with clearly lower velocity, thus resulting in two distinct reaction phases.
AlkJ exhibits pronounced affinity to the bacterial cell membrane, which necessitated solubilization with the detergent DDM to extract the recombinant enzyme from E. coli. In fact, attempts to dissociate AlkJ from the bacterial membrane with high concentrations of NaCl, a procedure commonly used for disrupting the electrostatic interaction of peripheral membrane proteins, remained unsuccessful (not shown). This behavior distinguishes AlkJ from CHOX, the closest homologue with known X-ray structure (37). Although AlkJ and CHOX are both grouped within the class of GMC oxidoreductases and have a similar fold, the subcellular localizations of the two enzymes appear to be markedly different, since the latter was described as a soluble protein (43). Despite structural similarity, the GMC family contains both membrane-associated enzymes, such as polyethylene glycol dehydrogenase (PEGDH) from Sphingopyxis terrae (44) or glucose dehydrogenase from Pseudomonas fluorescens (45), and cytoplasmic enzymes such as formate oxidase from Aspergillus oryzae (46). This suggests that the membrane-interacting region of AlkJ is not part of the conserved catalytic or cofactor-binding domains.
During SEC analysis, which was performed after the IMAC purification step, AlkJ eluted predominantly as a dimer, presumably with detergent molecules attached. Thus, solubilization with the mild nonionic detergent DDM has obviously retained the quaternary structure. Notably, the estimated number of 43 DDM molecules per AlkJ dimer is small compared to those for other, integral membrane proteins (47), which is in agreement with the absence of a distinct hydrophobic transmembrane segment, thus indicating a peripheral membrane association of AlkJ. Nevertheless, it seems that the membrane localization of AlkJ is relevant for its stability, considering that after 15 h of production in E. coli, at least 90% of the folded protein can be detected in the bacterial membrane fraction by SDS-PAGE.
After solubilization from the pelleted cell membrane, AlkJ showed a thermal stability of about 35°C both in enzyme activity assays and in CD measurements. While this value seems to represent the thermal stability of the purified enzyme quite fairly, it probably underestimates the robustness of AlkJ in vivo. In fact, it is well known that solubilization of a membrane-associated protein with detergent can cause loss of stabilizing interactions compared with the original lipid environment (48).
One of the conserved features among GMC oxidoreductases is their common Rossmann fold (49), which inter alia allows identification of the nucleotide-binding motif at the amino acid sequence level (50). Indeed, the Rossmann motif was also identified in the AlkJ primary structure, which initially contributed to its classification as a member of the GMC oxidoreductase family (51). However, experimental evidence for the association of the coenzyme FAD(H2) with AlkJ was still missing. The recorded absorption spectrum of the purified recombinant enzyme was devoid of the characteristic peaks for (oxidized) FAD at 372 and 450 nm. Nevertheless, a plateau in the range of 325 to 355 nm was observed in the UV-visible spectrum, with decaying absorption to baseline at about 500 nm.
Thus, the absorption spectrum of AlkJ resembled that of fully reduced FADH2 in the GMC enzyme glucose oxidase (52). Since the pronounced peak at 280 nm due to the aromatic side chains of the protein partially overlapped with the low and broad absorption of FADH2, spectroscopic characterization of the enzyme-bound reduced cofactor was not further pursued. Instead, the coenzyme was liberated from AlkJ by protein denaturation with SDS and spectrometrically quantified according to the specific absorption of the oxidized FAD at 450 nm, in line with common practice (29). These findings suggest that AlkJ provides a protein microenvironment that stabilizes FADH2 in the cofactor-binding pocket against air oxidation, somewhat similar to the case for CHOX, where the covalently bound FAD was detected as a mixture of oxidized and anionic semiquinone states (43).
Quantification after separation of the prosthetic group from the recombinant protein indicated a roughly equimolar FADH2/AlkJ ratio, demonstrating that the enzyme is efficiently charged with FADH2 during production in E. coli and does not release its cofactor during detergent solubilization or in the course of various chromatographic steps. The coenzyme is tightly though not covalently bound to AlkJ, which was confirmed by its release during SDS-mediated protein denaturation as well as by the more specific removal of FADH2 upon application of urea and KBr during IMAC. Whereas AlkJ shares this type of strong noncovalent FAD binding with other members of the GMC oxidoreductase family (50, 53, 54), the related enzyme CHOX features a covalent linkage to this prosthetic group (43).
Contrasting with nicotinamide cosubstrates, which typically diffuse from the enzyme after hydride ion transfer, FAD(H2) is usually bound very tightly, in accordance with its susceptibility to autoxidation when freely dissolved (55). This implies that the abstracted electrons from the substrate have to be passed on to another electron acceptor in the so-called oxidation half-reaction in order to regenerate the prosthetic group (56). In some members of the GMC oxidoreductase family this is achieved via direct reduction of molecular oxygen, as reported, e.g., for aryl alcohol oxidase (57), cholesterol oxidase (53), or CHOX (58). For other GMC family members, such as cellobiose dehydrogenase, the physiological electron acceptor is still elusive (59), while for the closely related PEGDH, which shares 42% amino acid sequence identity with AlkJ, ubiquinone has been identified as the physiological electron acceptor (51).
Likewise, ubiquinone was proposed as electron acceptor for AlkJ (19), which has been experimentally proven in the present study by enzyme kinetic measurements using the water-soluble short-chain derivative Q1 instead of Q9 or Q10. Indeed, oxidation of the substrate 1-octanol was clearly dependent on Q1 as an intermediate electron acceptor because the reaction did not proceed at all or was markedly slowed down, respectively, if merely cytochrome c or DCPIP was present. The measured AlkJ Km value of 41 μM for Q1 is just slightly higher than the cosubstrate affinity described for PEGDH with Q10 (11 μM) (51), while it is in a range similar to the Km value for Q1 (70 μM) determined for fumarate reductase (60). Since Pseudomonas bacteria have Q9 in their respiratory chains (61, 62), it seems very likely that in vivo AlkJ funnels electrons via this cosubstrate into the respiratory chain, where they are further transferred to a cytochrome (as also demonstrated in our study) and eventually to O2.
The ability of the bacterial strain P. putida GPo1 to convert aliphatic alkanes into corresponding aldehydes was anticipated since the first studies on its endogenous OCT plasmid (63). Subsequently, the dehydrogenase activity on aliphatic alcohols detectable in isolated P. putida cell membranes was mapped to the alkJ cistron as part of the alk operon (14). The goal of our study was to obtain deeper insight into the role of AlkJ-assisted catalysis for alkane metabolization on the basis of a comprehensive kinetic characterization of the purified recombinant enzyme. As result, AlkJ exhibits conventional Michaelis-Menten kinetics for the biocatalytic oxidation of 1-butanol, while in the case of other linear alcohols with 6 or more carbon atoms enzyme kinetics was complicated by substrate inhibition (Table 1). This is in part reflected by the stronger substrate affinity of AlkJ for 1-hexanol, 1-octanol, and HLAMe, showing a Km range of 20 to 40 μM, than for 1-butanol, where the Km value is almost 10-fold higher.
Moreover, we observed that the purified AlkJ showed a markedly lower specific activity for 1-octanol than was measured in previous studies (0.37 versus 3.84 U mg−1) using enriched P. putida membrane fractions in the same enzyme assay (14). It is likely that AlkJ is more active in its physiological membrane environment than in a DDM micelle after solubilization. The presence of a native lipid membrane might also lead to an elevated effective concentration by enrichment of the hydrophobic substrate within the lipid bilayer that surrounds the enzyme, resulting in a higher apparent enzyme activity. Furthermore, electron transport should be considerably accelerated by the presence of natural electron acceptors, such as ubiquinones, that are also associated with the native bacterial cell membrane.
Indeed, the use of Q0 as a physiological homologue of ubiquinone instead of the artificial electron acceptor pair PMS/DCPIP led to an approximately 3-fold-increased specific activity of AlkJ toward HLAMe and 1-octanol: the activity for HLAMe was raised from 0.49 U mg−1 to about 1.46 U mg−1 and that for 1-octanol from 0.37 U mg−1 to 1.42 U mg−1. AlkJ shares this cosubstrate preference with other FAD/ubiquinone-dependent enzymes such as caffeine dehydrogenase (64), which was shown to have 1.6-fold activity if Q0 is used in place of the artificial electron acceptors PMS/DCPIP.
As demonstrated in this study, AlkJ is able to catalyze the oxidation of n-alkyl alcohol substrates to the corresponding aldehydes, which constitute useful starting materials for numerous applications in the pharmaceutical, cosmetic, and chemical industries, especially with regard to subsequent reductive transamination to primary amines (65). In particular, AlkJ, with its proven substrate specificity, seems to be predestined for the biocatalytic synthesis of OLAMe from HLAMe. OLAMe in turn can be enzymatically transaminated to 12-amino lauric acid methyl ester (ALAMe), a valuable building block for high-performance polymers (13).
Unexpectedly, AlkJ can further oxidize the initial product OLAMe to the undesired (di)carboxylic acid. Nevertheless, this catalytic activity is about 9 times lower than that for conversion of HLAMe into the aldehyde. This selective oxidation of the alcohol substrate by AlkJ should offer an opportunity to reduce the known overoxidation during the biocatalytic ω-functionalization of LAMe with the P. putida AlkBGT system, which was shown to yield only approximately equimolar amounts of aldehyde and (di)carboxylic acid from LAMe (13). By sequestering the initial biocatalytic reaction product of AlkBGT, HLAMe, and accelerating its selective oxidation to OLAMe via AlkJ, the overall yield of aldehyde may be boosted, thus providing a more efficient substrate supply for the subsequent transamination to the industrially relevant ALAMe (Fig. 1).
ACKNOWLEDGMENTS
This work was financially supported by the German Bundesministerium für Bildung und Forschung in the framework of the project “BISON” (grant no. 0316044).
We are grateful to Evonik Industries AG, Marl, Germany, for providing materials and equipment.
Footnotes
Published ahead of print 7 February 2014
REFERENCES
- 1.van Beilen JB, Kingma J, Witholt B. 1994. Substrate specificity of the alkane hydroxylase system of Pseudomonas oleovorans GPo1. Enzyme Microb. Technol. 16:904–911. 10.1016/0141-0229(94)90066-3 [DOI] [Google Scholar]
- 2.Bosetti A, van Beilen JB, Preusting H, Lageveen RG, Witholt B. 1992. Production of primary aliphatic alcohols with a recombinant Pseudomonas strain, encoding the alkane hydroxylase enzyme system. Enzyme Microb. Technol. 14:702–708. 10.1016/0141-0229(92)90109-2 [DOI] [Google Scholar]
- 3.Koch DJ, Chen MM, van Beilen JB, Arnold FH. 2009. In vivo evolution of butane oxidation by terminal alkane hydroxylases AlkB and CYP153A6. Appl. Environ. Microbiol. 75:337–344. 10.1128/AEM.01758-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Beilen JB, Panke S, Lucchini S, Franchini AG, Röthlisberger M, Witholt B. 2001. Analysis of Pseudomonas putida alkane-degradation gene clusters and flanking insertion sequences: evolution and regulation of the alk genes. Microbiology 147:1621–1630 [DOI] [PubMed] [Google Scholar]
- 5.van Beilen JB, Penninga D, Witholt B. 1992. Topology of the membrane-bound alkane hydroxylase of Pseudomonas oleovorans. J. Biol. Chem. 267:9194–9201 [PubMed] [Google Scholar]
- 6.Shanklin J, Whittle E, Fox BG. 1994. Eight histidine residues are catalytically essential in a membrane-associated iron enzyme, stearoyl-CoA desaturase, and are conserved in alkane hydroxylase and xylene monooxygenase. Biochemistry 33:12787–12794. 10.1021/bi00209a009 [DOI] [PubMed] [Google Scholar]
- 7.Alonso H, Roujeinikova A. 2012. Characterization and two-dimensional crystallization of membrane component AlkB of the medium-chain alkane hydroxylase system from Pseudomonas putida GPo1. Appl. Environ. Microbiol. 78:7946–7953. 10.1128/AEM.02053-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cooper HL, Mishra G, Huang X, Pender-Cudlip M, Austin RN, Shanklin J, Groves JT. 2012. Parallel and competitive pathways for substrate desaturation, hydroxylation, and radical rearrangement by the non-heme diiron hydroxylase AlkB. J. Am. Chem. Soc. 134:20365–20375. 10.1021/ja3059149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grant C, Woodley JM, Baganz F. 2011. Whole-cell bio-oxidation of n-dodecane using the alkane hydroxylase system of P. putida GPo1 expressed in E. coli. Enzyme Microb. Technol. 48:480–486. 10.1016/j.enzmictec.2011.01.008 [DOI] [PubMed] [Google Scholar]
- 10.Schrewe M, Magnusson AO, Willrodt C, Bühler B, Schmid A. 2011. Kinetic analysis of terminal and unactivated C-H bond oxyfunctionalization in fatty acid methyl esters by monooxygenase-based whole-cell biocatalysis. Adv. Synth. Catal. 353:3485–3495. 10.1002/adsc.201100440 [DOI] [Google Scholar]
- 11.Panke S, Meyer A, Huber CM, Witholt B, Wubbolts MG. 1999. An alkane-responsive expression system for the production of fine chemicals. Appl. Environ. Microbiol. 65:2324–2332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peterson JA, Basu D, Coon MJ. 1966. Enzymatic ω-oxidation. I. Electron carriers in fatty acid and hydrocarbon hydroxylation. J. Biol. Chem. 241:5162–5164 [PubMed] [Google Scholar]
- 13.Schrewe M, Ladkau N, Bühler B, Schmid A. 2013. Direct terminal alkylamino-functionalization via multistep biocatalysis in one recombinant whole-cell catalyst. Adv. Synth. Catal. 355:1693–1697. 10.1002/adsc.201200958 [DOI] [Google Scholar]
- 14.Benson S, Shapiro J. 1976. Plasmid-determined alcohol dehydrogenase activity in alkane-utilizing strains of Pseudomonas putida. J. Bacteriol. 126:794–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cavener DR. 1992. GMC oxidoreductases: a newly defined family of homologous proteins with diverse catalytic activities. J. Mol. Biol. 223:811–814. 10.1016/0022-2836(92)90992-S [DOI] [PubMed] [Google Scholar]
- 16.Goswami P, Chinnadayyala SS, Chakraborty M, Kumar AK, Kakoti A. 2013. An overview on alcohol oxidases and their potential applications. Appl. Microbiol. Biotechnol. 97:4259–4275. 10.1007/s00253-013-4842-9 [DOI] [PubMed] [Google Scholar]
- 17.Bankar SB, Bule MV, Singhal RS, Ananthanarayan L. 2009. Glucose oxidase—an overview. Biotechnol. Adv. 27:489–501. 10.1016/j.biotechadv.2009.04.003 [DOI] [PubMed] [Google Scholar]
- 18.Pollegioni L, Piubelli L, Molla G. 2009. Cholesterol oxidase: biotechnological applications. FEBS J. 276:6857–6870. 10.1111/j.1742-4658.2009.07379.x [DOI] [PubMed] [Google Scholar]
- 19.van Beilen JB, Eggink G, Enequist H, Bos R, Witholt B. 1992. DNA sequence determination and functional characterization of the OCT-plasmid-encoded alkJKL genes of Pseudomonas oleovorans. Mol. Microbiol. 6:3121–3136. 10.1111/j.1365-2958.1992.tb01769.x [DOI] [PubMed] [Google Scholar]
- 20.Tan S, Tan HT, Chung MCM. 2008. Membrane proteins and membrane proteomics. Proteomics 8:3924–3932. 10.1002/pmic.200800597 [DOI] [PubMed] [Google Scholar]
- 21.Bullock WO, Fernandez JM, Short JM. 1987. XL1-Blue: a high efficiency plasmid transforming recA Escherichia coli strain with β-galactosidase selection. Biotechniques 5:376–379 [Google Scholar]
- 22.Studier FW, Moffatt BA. 1986. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189:113–130. 10.1016/0022-2836(86)90385-2 [DOI] [PubMed] [Google Scholar]
- 23.Schmidt TG, Skerra A. 2007. The Strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nat. Protoc. 2:1528–1535. 10.1038/nprot.2007.209 [DOI] [PubMed] [Google Scholar]
- 24.Skerra A, Pfitzinger I, Plückthun A. 1991. The functional expression of antibody Fv fragments in Escherichia coli: improved vectors and a generally applicable purification technique. Biotechnology 9:273–278. 10.1038/nbt0391-273 [DOI] [PubMed] [Google Scholar]
- 25.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 26.Fling SP, Gregerson DS. 1986. Peptide and protein molecular weight determination by electrophoresis using a high-molarity tris buffer system without urea. Anal. Biochem. 155:83–88. 10.1016/0003-2697(86)90228-9 [DOI] [PubMed] [Google Scholar]
- 27.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. 2005. Protein identification and analysis tools on the ExPASy server, p 571–607 In Walker JM. (ed), The proteomics protocols handbook. Humana Press, New York, NY [Google Scholar]
- 28.Hefti MH, Milder FJ, Boeren S, Vervoort J, van Berkel WJ. 2003. A His-tag based immobilization method for the preparation and reconstitution of apoflavoproteins. Biochim. Biophys. Acta 1619:139–143. 10.1016/S0304-4165(02)00474-9 [DOI] [PubMed] [Google Scholar]
- 29.Aliverti A, Curti B, Vanoni M. 1999. Identifying and quantitating FAD and FMN in simple and in iron-sulfur-containing flavoproteins. Methods Mol. Biol. 131:9–23 [DOI] [PubMed] [Google Scholar]
- 30.Engel PC. 1981. Butyryl-CoA dehydrogenase from Megasphaera elsdenii. Methods Enzymol. 71:359–366. 10.1016/0076-6879(81)71045-0 [DOI] [Google Scholar]
- 31.McIntire WS. 1990. Trimethylamine dehydrogenase from bacterium W3A1. Methods Enzymol. 188:250–260. 10.1016/0076-6879(90)88042-9 [DOI] [PubMed] [Google Scholar]
- 32.Margoliash E, Frohwirt N. 1959. Spectrum of horse-heart cytochrome c. Biochem. J. 71:570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schlehuber S, Skerra A. 2002. Tuning ligand affinity, specificity, and folding stability of an engineered lipocalin variant—a so-called ‘anticalin'—using a molecular random approach. Biophys. Chem. 96:213–228. 10.1016/S0301-4622(02)00026-1 [DOI] [PubMed] [Google Scholar]
- 34.Skerra A. 1994. Use of the tetracycline promoter for the tightly regulated production of a murine antibody fragment in Escherichia coli. Gene 151:131–135. 10.1016/0378-1119(94)90643-2 [DOI] [PubMed] [Google Scholar]
- 35.le Maire M, Champeil P, Moller JV. 2000. Interaction of membrane proteins and lipids with solubilizing detergents. Biochim. Biophys. Acta 1508:86–111. 10.1016/S0304-4157(00)00010-1 [DOI] [PubMed] [Google Scholar]
- 36.Ghisla S, Massey V, Lhoste JM, Mayhew SG. 1974. Fluorescence and optical characteristics of reduced flavines and flavoproteins. Biochemistry 13:589–597. 10.1021/bi00700a029 [DOI] [PubMed] [Google Scholar]
- 37.Quaye O, Lountos GT, Fan F, Orville AM, Gadda G. 2008. Role of Glu312 in binding and positioning of the substrate for the hydride transfer reaction in choline oxidase. Biochemistry 47:243–256. 10.1021/bi7017943 [DOI] [PubMed] [Google Scholar]
- 38.Frishman D, Argos P. 1995. Knowledge-based protein secondary structure assignment. Proteins 23:566–579. 10.1002/prot.340230412 [DOI] [PubMed] [Google Scholar]
- 39.Adachi O, Ano Y, Toyama H, Matsushita K. 2007. Biooxidation with PQQ- and FAD-dependent dehydrogenases, p 1–41 In Schmid RD, Urlacher VB. (ed), Modern biooxidation: enzymes, reactions and applications. Wiley-VCH, Weinheim, Germany [Google Scholar]
- 40.Johnson KA, Goody RS. 2011. The original Michaelis constant: translation of the 1913 Michaelis-Menten paper. Biochemistry 50:8264–8269. 10.1021/bi201284u [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Theissen U, Martin W. 2008. Sulfide:quinone oxidoreductase (SQR) from the lugworm Arenicola marina shows cyanide- and thioredoxin-dependent activity. FEBS J. 275:1131–1139. 10.1111/j.1742-4658.2008.06273.x [DOI] [PubMed] [Google Scholar]
- 42.Shrivastava R, Basu A, Phale P. 2011. Purification and characterization of benzylalcohol- and benzaldehyde-dehydrogenase from Pseudomonas putida CSV86. Arch. Microbiol. 193:553–563. 10.1007/s00203-011-0697-6 [DOI] [PubMed] [Google Scholar]
- 43.Fan F, Ghanem M, Gadda G. 2004. Cloning, sequence analysis, and purification of choline oxidase from Arthrobacter globiformis: a bacterial enzyme involved in osmotic stress tolerance. Arch. Biochem. Biophys. 421:149–158. 10.1016/j.abb.2003.10.003 [DOI] [PubMed] [Google Scholar]
- 44.Kawai F, Kimura T, Tani Y, Yamada H, Kurachi M. 1980. Purification and characterization of polyethylene glycol dehydrogenase involved in the bacterial metabolism of polyethylene glycol. Appl. Environ. Microbiol. 40:701–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matsushita K, Ameyama M. 1982. d-Glucose dehydrogenase from Pseudomonas fluorescens, membrane-bound. Methods Enzymol. 89:149–154. 10.1016/S0076-6879(82)89026-5 [DOI] [PubMed] [Google Scholar]
- 46.Maeda Y, Doubayashi D, Oki M, Nose H, Sakurai A, Isa K, Fujii Y, Uchida H. 2009. Expression in Escherichia coli of an unnamed protein gene from Aspergillus oryzae RIB40 and cofactor analyses of the gene product as formate oxidase. Biosci. Biotechnol. Biochem. 73:2645–2649. 10.1271/bbb.90497 [DOI] [PubMed] [Google Scholar]
- 47.Møller JV, le Maire M. 1993. Detergent binding as a measure of hydrophobic surface area of integral membrane proteins. J. Biol. Chem. 268:18659–18672 [PubMed] [Google Scholar]
- 48.Prive GG. 2007. Detergents for the stabilization and crystallization of membrane proteins. Methods 41:388–397. 10.1016/j.ymeth.2007.01.007 [DOI] [PubMed] [Google Scholar]
- 49.Rossmann MG, Moras D, Olsen KW. 1974. Chemical and biological evolution of a nucleotide-binding protein. Nature 250:194–199. 10.1038/250194a0 [DOI] [PubMed] [Google Scholar]
- 50.Kiess M, Hecht H-J, Kalisz HM. 1998. Glucose oxidase from Penicillium amagasakiense. Eur. J. Biochem. 252:90–99. 10.1046/j.1432-1327.1998.2520090.x [DOI] [PubMed] [Google Scholar]
- 51.Ohta T, Kawabata T, Nishikawa K, Tani A, Kimbara K, Kawai F. 2006. Analysis of amino acid residues involved in catalysis of polyethylene glycol dehydrogenase from Sphingopyxis terrae, using three-dimensional molecular modeling-based kinetic characterization of mutants. Appl. Environ. Microbiol. 72:4388–4396. 10.1128/AEM.02174-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Massey V, Palmer G. 1966. On the existence of spectrally distinct classes of flavoprotein semiquinones. A new method for the quantitative production of flavoprotein semiquinones. Biochemistry 5:3181–3189. 10.1021/bi00874a016 [DOI] [PubMed] [Google Scholar]
- 53.Vrielink A, Ghisla S. 2009. Cholesterol oxidase: biochemistry and structural features. FEBS J. 276:6826–6843. 10.1111/j.1742-4658.2009.07377.x [DOI] [PubMed] [Google Scholar]
- 54.Ruiz-Duenas FJ, Ferreira P, Martinez MJ, Martinez AT. 2006. In vitro activation, purification, and characterization of Escherichia coli expressed aryl-alcohol oxidase, a unique H2O2-producing enzyme. Protein Expr. Purif. 45:191–199. 10.1016/j.pep.2005.06.003 [DOI] [PubMed] [Google Scholar]
- 55.Walsh CT, Wencewicz TA. 2013. Flavoenzymes: versatile catalysts in biosynthetic pathways. Nat. Prod. Rep. 30:175–200. 10.1039/c2np20069d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ghisla S, Massey V. 1989. Mechanisms of flavoprotein-catalyzed reactions. Eur. J. Biochem. 181:1–17. 10.1111/j.1432-1033.1989.tb14688.x [DOI] [PubMed] [Google Scholar]
- 57.Hernandez-Ortega A, Lucas F, Ferreira P, Medina M, Guallar V, Martinez AT. 2012. Role of active site histidines in the two half-reactions of the aryl-alcohol oxidase catalytic cycle. Biochemistry 51:6595–6608. 10.1021/bi300505z [DOI] [PubMed] [Google Scholar]
- 58.Gadda G. 2003. Kinetic mechanism of choline oxidase from Arthrobacter globiformis. Biochim. Biophys. Acta 1646:112–118. 10.1016/S1570-9639(03)00003-7 [DOI] [PubMed] [Google Scholar]
- 59.Henriksson G, Johansson G, Pettersson G. 2000. A critical review of cellobiose dehydrogenases. J. Biotechnol. 78:93–113. 10.1016/S0168-1656(00)00206-6 [DOI] [PubMed] [Google Scholar]
- 60.Maklashina E, Cecchini G. 1999. Comparison of catalytic activity and inhibitors of quinone reactions of succinate dehydrogenase (succinate-ubiquinone oxidoreductase) and fumarate reductase (menaquinol-fumarate oxidoreductase) from Escherichia coli. Arch. Biochem. Biophys. 369:223–232. 10.1006/abbi.1999.1359 [DOI] [PubMed] [Google Scholar]
- 61.Zannoni DD. 1989. The respiratory chains of pathogenic pseudomonads. Biochim. Biophys. Acta 975:299–316. 10.1016/S0005-2728(89)80337-8 [DOI] [PubMed] [Google Scholar]
- 62.Matsushita K, Yamada M, Shinagawa E, Adachi O, Ameyama M. 1980. Function of ubiquinone in the electron transport system of Pseudomonas aeruginosa grown aerobically. J. Biochem. 88:757–764 [DOI] [PubMed] [Google Scholar]
- 63.Grund A, Shapiro J, Fennewald M, Bacha P, Leahy J, Markbreiter K, Nieder M, Toepfer M. 1975. Regulation of alkane oxidation in Pseudomonas putida. J. Bacteriol. 123:546–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yu CL, Kale Y, Gopishetty S, Louie TM, Subramanian M. 2008. A novel caffeine dehydrogenase in Pseudomonas sp. strain CBB1 oxidizes caffeine to trimethyluric acid. J. Bacteriol. 190:772–776. 10.1128/JB.01390-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rausch C, Lerchner A, Schiefner A, Skerra A. 2013. Crystal structure of the ω-aminotransferase from Paracoccus denitrificans and its phylogenetic relationship with other Class III amino-transferases that have biotechnological potential. Proteins 81:774–787. 10.1002/prot.24233 [DOI] [PubMed] [Google Scholar]
- 66.Whitmore L, Wallace BA. 2004. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 32:W668–W673. 10.1093/nar/gkh371 [DOI] [PMC free article] [PubMed] [Google Scholar]