

Abstract
Antibiotics can play dual roles in Clostridium difficile infection (CDI); antibiotic treatment increases the risk of CDI, and antibiotics are used to treat CDI. The glycylcycline antibiotic tigecycline has broad antimicrobial activity, yet it is rarely associated with the development of CDI, presumably due to its activity against C. difficile. In this study, we investigated how tigecycline treatment affects the structure of the gut microbiota and susceptibility to CDI by treating mice with tigecycline (n = 20) or saline (n = 8) for 10 days. A sequence analysis of the bacterial 16S rRNA gene amplicons was used to monitor changes in the fecal microbiota. A subset of the mice was followed for 5 weeks after the end of treatment. The remaining mice were challenged with C. difficile strain VPI 10463 spores 2 days after the tigecycline treatment ended. Tigecycline treatment resulted in major shifts in the gut microbiota, including large decreases in Bacteroidetes levels and large increases in Proteobacteria levels. Mice with tigecycline-altered microbial communities were susceptible to challenge with C. difficile spores and developed clinical signs of severe CDI. Five weeks after the cessation of tigecycline treatment, the recovery of the bacterial community was incomplete and diversity was lower than in the untreated controls. Antibiotics with intrinsic activity against C. difficile can still alter the microbiota in a way that leads to susceptibility to CDI after discontinuation of the drug. These results indicate that microbiotic dynamics are key in the development of CDI, and a better understanding of these dynamics may lead to better strategies to prevent and treat this disease.
INTRODUCTION
The toxin-producing bacterium Clostridium difficile causes >350,000 cases of diarrhea and colitis per year in the United States (1). A major risk factor for C. difficile infection (CDI) is antibiotic administration (2). An elevated risk has been associated with the use of clindamycin, quinolones, cephalosporins, and aminopenicillins, although virtually all antibiotics have been implicated as potential risk factors for developing CDI (3–5). The mechanism by which antibiotics predispose patients to CDI is thought to be through the alteration of the community structure of the indigenous gut microbiota, which results in a loss of colonization resistance against C. difficile (6–8). While most antibiotics have been implicated as risk factors for CDI, the broad-spectrum glycylcycline antibiotic, tigecycline, is associated with lower rates of CDI (9).
The antibiotic tigecycline was approved in 2005 for the treatment of complicated skin and soft tissue infections and complicated intra-abdominal infections (10). It was subsequently approved for the treatment of community-acquired pneumonia (11). Tigecycline is a broad-spectrum glycylcycline antibiotic that is potent against both Gram-positive and Gram-negative facultative and obligate anaerobes, including C. difficile (12–16). Tigecycline has a low MIC against C. difficile in vitro (17). Recent evidence suggests that administration of the drug is associated with low risk of subsequent CDI (9). It is not known if this is due to the potent anti-C. difficile activity of tigecycline or if the drug is able to spare members of the gut microbiota that are important in maintaining colonization resistance.
Recent studies in humans and animals have shown that tigecycline does not significantly alter the anaerobic bacterial population in the gastrointestinal tract, including Bacteroides spp. (18, 19). It should be noted that this relative sparing of the indigenous microbiota was based on standard microbiologic culture and thus only addressed a subset of the entire microbiota. Although more studies are needed to determine the absolute risk of developing CDI following tigecycline administration, case reports suggest it may be effective, particularly in combination with other antibiotics, for treating severe refractory CDI without relapse (13, 14).
Due to the broad-spectrum activity of tigecycline, we hypothesized that tigecycline administration would alter the murine gut microbiota, but it was not known whether this would render mice susceptible to C. difficile infection. In this study, mice treated with tigecycline for 10 days showed significant changes to the murine gut microbiota. Mice challenged with C. difficile spores after tigecycline treatment were susceptible to CDI.
MATERIALS AND METHODS
Ethics statement.
The University Committee on the Care and Use of Animals (UCUCA) at the University of Michigan approved this study. The University of Michigan laboratory animal care policies follow the Public Health Service policy on Humane Care and Use of Laboratory Animals. The animals were assessed twice daily for physical condition and behavior, and those assessed as moribund were humanely euthanized by CO2 asphyxiation. Trained animal technicians performed animal husbandry in an Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC)-accredited facility.
Animals and housing.
Wild-type C57BL/6 mice (male or female) were obtained from a breeding colony that was established using animals purchased from Jackson Laboratories. The mice were approximately 5 weeks old at the beginning of the study. The mice were housed with autoclaved food, bedding, and water. Cage changes were performed in a laminar flow hood. Mice had a cycle of 12 h of light and 12 h of darkness.
C. difficile spore preparation.
C. difficile VPI 10463 (ATCC 43255) spores were prepared as previously described (20). Briefly, C. difficile VPI 10463 was grown overnight from a single colony in a 2-ml culture of Columbia broth at 37°C, under anaerobic conditions. The next day, the inoculum was added to 40 ml of Clospore medium (21). The culture was incubated at 37°C for 5 to 7 days under anaerobic conditions. The spores were harvested by centrifugation and washed with cold water at least three times. The spore stocks were stored at 4°C in sterile water. C. difficile spores were heat treated for 20 min at 65°C to ensure that any remaining vegetative bacilli were killed prior to gavaging the animals. Viable spores were enumerated by plating for CFU/ml on TCCFA (taurocholate, cefoxitin, cycloserine, and fructose agar).
Antibiotic administration and challenge with C. difficile spores.
Wild-type C57BL/6 mice (males or females) were given either tigecycline (6.25 mg/kg of body weight) (n = 20) or saline (n = 8) by subcutaneous injection twice a day for a total of 10 days. This dose of tigecycline was selected for the mice because it reaches the maximum concentration of drug in serum (Cmax) of 1.17 μg/ml, which is similar to a Cmax of 0.93 μg/ml, which correlates to a dose of 100 mg every 12 hours in humans (16). The mice were approximately 5 weeks old at the beginning of the study. Two days after the end of treatment, mice (n = 10 from tigecycline group and n = 3 from the saline group) were challenged with 450 C. difficile VPI 10463 spores by oral gavage. As a positive control for susceptibility to experimental CDI, 5 mice were treated with cefoperazone in drinking water (0.5 mg/ml) prior to challenge, as previously described (20). The animals were monitored for clinical signs of severe CDI, including inappetence, diarrhea, and hunched posture. All C. difficile-challenged animals were euthanized by CO2 asphyxiation at 2 days postchallenge, because the tigecycline- and cefoperazone-treated mice met their experimental endpoint (loss of 20% of initial baseline weight or the development of severe clinical disease). At the time of necropsy (2 days postchallenge), bacterial enumeration of C. difficile in cecal content was done on TCCFA selective agar. The cecal content and tissue were collected at the time of necropsy and flash frozen for microbiome analysis.
Another set of mice from each group (n = 5 from the tigecycline group and n = 5 from the saline group) were not challenged with C. difficile and were followed for 5 weeks after the end of tigecycline treatment to monitor long-term changes in the gut microbiota following the administration of this antibiotic. The remaining tigecycline-treated mice (n = 5) were used to measure fecal levels of tigecycline.
Microbiotic sequencing.
DNA was isolated from murine fecal pellets using a PowerSoil-htp 96-well soil DNA isolation kit (Mo Bio Laboratories, Inc.). The library construction of V5V3 16S rRNA gene amplicons was based on a Human Microbiome Project (HMP) protocol (see http://www.hmpdacc.org/doc/16S_Sequencing_SOP_4.2.2.pdf). Each 20-μl PCR mixture contained 2 μl AccuPrime PCR buffer II (Life Technologies), 0.15 μl AccuPrime Taq DNA polymerase high fidelity (Life Technologies), 0.2 μM primer A (CCATCTCATCCCTGCGTGTCTCCGACTCAGXXXXXCCGTCAATTCMTTTRAGT), 0.2 μM primer B (CCTATCCCCTGTGTGCCTTGGCAGTCTCAGCCTACGGGAGGCAGCAG), and 1 μl DNA (bold portions of primer A and primer B are 926R and 357F, respectively). The region of primer A represented by XXXXX is the nucleotides 5 to 10 barcode sequence. The remainder of primer A and primer B are the A adapter sequence and the B adapter sequence, respectively, required for emulsion PCR (emPCR) and 454 sequencing. The PCRs were run for 2 min at 95°C, followed by 30 cycles of 95°C for 20 s, 50°C for 30 s, and 72°C for 5 min. The PCR products were purified with AMPure XP (Agencourt) according to the manufacturer's instructions, except 0.6× the amplicon volume (10.8 μl) of beads was used rather than 1.2×, in order to remove more of the small products. The purified PCR products were quantified with a Quant-iT PicoGreen double-stranded DNA (dsDNA) kit (Invitrogen), according to the manufacturer's instructions, and combined into a pool with equal amounts of each amplicon. To accommodate all of the samples, 2 pools were made. Each pool was then purified with AMPure XP (Agencourt) according to the manufacturer's instructions, except the volume of beads was 0.6× the pool volume. The pools were quantified with a library quantification kit for Roche 454 GS Titanium (KAPA) sequencing. Large-volume Lib-L emPCRs (Roche 454) were performed, and 454 sequencing was done using the GS FLX Titanium platform (Roche), according to the manufacturer's instructions.
Sequence analysis.
The sequences were processed with mothur version 1.27.0 according to the Schloss standard operating procedures (SOP) of August 2012 and March to May 2013 (22, 23). In summary, each .sff file was input into sffinfo, and the sequencing error was reduced by running the version of PyroNoise (24) within mothur. All sequences with >2 mismatches to the 926R region of the primer, >1 mismatch to the barcode region of the primer, a homopolymer of >8 nucleotides, or ≤200 nucleotides were discarded. The fasta, name, and group files (4 of each, 2 from each sequencing run) were concatenated into one fasta, name, and group file, respectively. The sequences were aligned to the Silva reference alignment (25, 26). In order to compare sequences over the same region of the alignment, we set the end position at 27659 and chose a start position that was met by 95% of the sequences. Sequences within 2 nucleotides were merged by using the “pre.cluster” command. Chimeras were identified with chimera.uchime (27) and removed. The sequences were classified by the Wang method using a modified form of RDP training set version 9 (trainset9_032012.pds.tax and trainset9_032012.pds.fasta), with an 80% minimum bootstrap value (28, 29). Sequences classified as chloroplast, mitochondria, Archaea, Eukaryota, or an unknown kingdom were removed. A total of 3,572 sequences were subsampled from each sample. Samples with <3,572 sequences were discarded from the sequence analysis. A distance matrix made with dist.seqs was used with the average neighbor algorithm to group sequences into operational taxonomic units (OTUs) with the cluster command. OTUs that were 3% different were used for further analysis. The make.shared command was used to produce a table (shared file) of the number of sequence reads assigned to each OTU in each sample. The shared file was used to calculate the θYC distance (1 − θYC) between bacterial communities in all of the samples, and the results were formatted into a distance matrix. θYC is a similarity index that takes into account the relative abundances of both shared and nonshared OTUs (30). Principal coordinates analysis (PCoA) was used to visualize the θYC distance matrix, and an analysis of molecular variance (AMOVA) was used to test the statistical significance of the differences between the bacterial communities of different groups.
Tigecycline detection in stool by LC-MS analysis.
Methanol with an internal standard was used to extract tigecycline from mouse fecal samples. A volume of 400 μl of extraction solvent was added to fecal samples in an Eppendorf tube and homogenized by a probe sonicator. The homogenized samples were vortexed briefly, allowed to sit on ice for 5 min, vortexed again, and finally centrifuged at 15,000 × g for 10 min. The supernatants were dried using an N2 dryer at room temperature. The samples were reconstituted in 100 μl of 50:50 acetonitrile to mobile phase (0.1% formic acid in water) and transferred to autosampler vials for liquid chromatography-mass spectrometry (LC-MS) analysis.
An Agilent 1200 liquid chromatography-Agilent 6420 series triple quadrupole mass spectrometer with electrospray ionization (ESI), which was operated in positive mode, was used for sample analysis. A Waters XBridge BEH C18 column was used for chromatographic separation. The following transition was used to identify and quantify tigecycline: m/z 586.3→m/z 513.2. The data were processed with the MassHunter workstation software, version B.06.
The concentrations of tigecycline in the mouse stool samples were analyzed by LC-MS. The tigecycline-treated mice were analyzed from day 10 (n = 5) and 1 week after stopping the antibiotic (n = 5).
Statistical analysis.
Prism 5 (GraphPad Software, Inc.) was used for statistical analysis. Significance was determined by a nonparametric Kruskal-Wallis one-way analysis of variance (ANOVA) test followed by Dunn's posttest. Differences were considered significant at a P value of <0.05.
RESULTS
Tigecycline treatment alters the murine gut microbiota.
Three days after the initiation of tigecycline treatment, the fecal bacterial community had shifted dramatically and continued to change over the 10-day course of tigecycline treatment and the first 3 weeks of recovery (Fig. 1). The 3% OTU-based θYC distances between bacterial communities at consecutive time points were statistically different through the 3rd week of recovery (AMOVA, P < 0.05 for all; see Table S1 in the supplemental material). No further statistically significant changes in θYC distances occurred between the 3rd and 5th weeks of recovery. However, the community failed to return to baseline. The community structure after 5 weeks of recovery from tigecycline was significantly different from the community structure observed prior to tigecycline treatment (AMOVA, P < 0.001). The bacterial communities from untreated mice (prior to saline or tigecycline treatment) and from saline-treated (during and after saline treatment) mice were similar and clustered together (Fig. 1).
FIG 1.

Tigecycline treatment alters the murine gut microbiota. Principal coordinates analysis (PCoA) illustrates the θYC distances (1 − θYC) between the bacterial communities of mouse fecal samples. The θYC distances were calculated using a subsample of 3,572 sequences per sample and a 3% OTU definition.
Prior to treatment, Bacteroidetes was the major phylum of the fecal bacterial community, with over 80% of 16S rRNA sequences on average (Fig. 2A). The Bacteroidetes organisms consisted mainly of Porphyromonadaceae, Rikenellaceae, Bacteroidaceae, and some unclassified Bacteroidales species (Fig. 2A; see also Table S2 in the supplemental material). After 3 days of tigecycline treatment, Bacteroidetes levels decreased to an average of about 20% of the bacterial 16S rRNA sequences and consisted almost entirely of Porphyromonadaceae species. The decrease in Bacteroidetes levels was accompanied by a decrease in Firmicutes levels and corresponding increases in Enterobacteriaceae and Verrucomicrobiaceae levels (Fig. 2A).
FIG 2.

Average bacterial community composition changes during tigecycline treatment and recovery. The bar graphs depict the mean percent abundances of the top bacterial families (≥1% relative abundance at a minimum of one time point) prior to and during tigecycline treatment (A) or saline treatment (B) and recovery. The family-level percent abundances were based on the classification of a subsample of 3,572 bacterial 16S rRNA gene sequences per sample. The respective phyla are listed to the right of the family names.
After tigecycline treatment stopped, the fecal bacterial community continued to change. By 5 weeks of recovery, the Porphyromonadaceae and unclassified Bacteroidales species, prominent members of the Bacteroidetes, had increased to levels similar to those of the baseline, while the Enterobacteriaceae and Verrucomicrobiaceae levels had decreased and were nearly undetectable (Fig. 2A; see also Table S2 in the supplemental material). Other members of the Bacteroidetes (unclassified Bacteroidetes, Rikenellaceae, and Bacteroidaceae) remained at much lower levels than on day 0. Within the Firmicutes, all of the families present prior to tigecycline treatment returned by week 5 to relative abundances at least as high as on day 0. Most were at very similar relative abundances compared with day 0 (within 1.10%), with the exception of the Erysipelotrichaceae, which started at <1% on day 0 and reached 12.68% ± 4.36% (mean ± standard deviation [SD]) after 10 days of tigecycline treatment and 5 weeks of recovery. The Erysipelotrichaceae level increased similarly in the saline-treated mice between day 0 (2.12% ± 2.02%) and week 5 of recovery (12.58% ± 4.29%), indicating that this change was independent of tigecycline treatment (Fig. 2B).
OTU-level diversity remained low even as family-level community composition recovered.
After tigecycline treatment stopped, the family-level composition of the fecal microbiota appeared to return to the baseline composition by 2 weeks after the end of treatment (Fig. 2A). However, an analysis of the microbiota at a finer level, with sequences assigned to 3% OTUs, revealed a failure to return to the baseline community structure after 5 weeks of recovery (Fig. 1). To quantitatively compare dynamics at the family level versus the OTU level, we calculated the average θYC distances between the baseline samples and samples from the other time points based on family designations and OTU designations (Fig. 3). By the third day of tigecycline treatment, the average distance from the baseline microbiota increased greatly compared to the average distance between all of the baseline microbiota samples in both the family-based and OTU-based analyses (Fig. 3). However, in the family-based analysis, the distance from baseline started decreasing after day 3 of tigecycline treatment, and by 2 weeks after tigecycline treatment, the distance from baseline was not statistically different from the distance between the baseline samples (Fig. 3A). In the OTU-based analysis, the distance from baseline remained relatively high throughout the 5 weeks of recovery (Fig. 3B). Only minimal changes to the fecal bacterial community were observed in mice that were treated with saline (see Fig. S1 in the supplemental material).
FIG 3.

Mean θYC distances from baseline community during tigecycline treatment and recovery, based on family-level (A) and 3% OTU-level (B) sequence assignments. *, P < 0.05, which indicates a significant difference from the mean distance between all communities at day 0 (day 0 to day 0) by a nonparametric Kruskal-Wallis test, followed by Dunn's multiple comparison test. The error bars represent the SD.
The return to baseline at the family level but not at the OTU level is further illustrated by the fate of the 10 most abundant OTUs in the Porphyromonadaceae family during and after tigecycline treatment (Fig. 4). The Porphyromonadaceae family consisted of 10 OTUs present at >1% relative abundance on day 0. After 3 days of tigecycline treatment, only 2 Porphyromonadaceae OTUs remained at levels of >1% of the total bacterial 16S rRNA gene sequences, on average. After 5 weeks of recovery, there were still only 2 Porphyromonadaceae OTUs present at >1%, although the relative abundance of the Porphyromonadaceae family as a whole had returned to a level similar to that at day 0.
FIG 4.
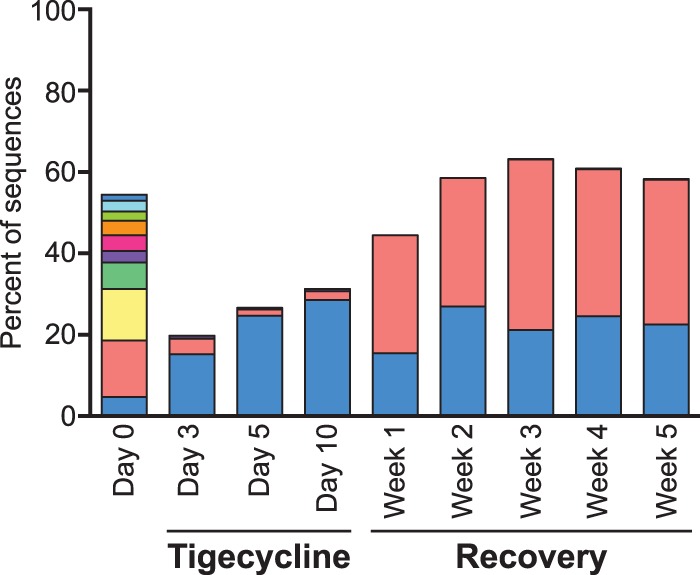
Loss of Porphyromonadaceae OTUs with tigecycline treatment. The relative abundances are shown for the top Porphyromonadaceae OTUs (all OTUs classified as Porphyromonadaceae with a relative abundance of ≥1% at a minimum of one time point) prior to and during tigecycline treatment and recovery. Each of the top Porphyromonadaceae OTUs is represented by a different color. The relative abundances are based on a subsample of 3,572 bacterial 16S rRNA gene sequences per sample with a 3% OTU definition.
Consistent with these results, overall diversity as measured by the OTU-based inverse Simpson diversity index decreased with tigecycline treatment (Fig. 5A). Although the diversity of the fecal microbiota increased during recovery, it remained less diverse than prior to tigecycline treatment (Fig. 5A). In contrast, the diversity of the fecal microbiota of saline-treated mice did not decrease over the course of treatment and recovery (Fig. 5B). Although there were reproducible changes to the gut microbiota of mice throughout tigecycline treatment, the levels of tigecycline detected in the stools of mice on the last day of treatment and 1 week later were below the limit of detection, or <0.2 μg/g of feces.
FIG 5.
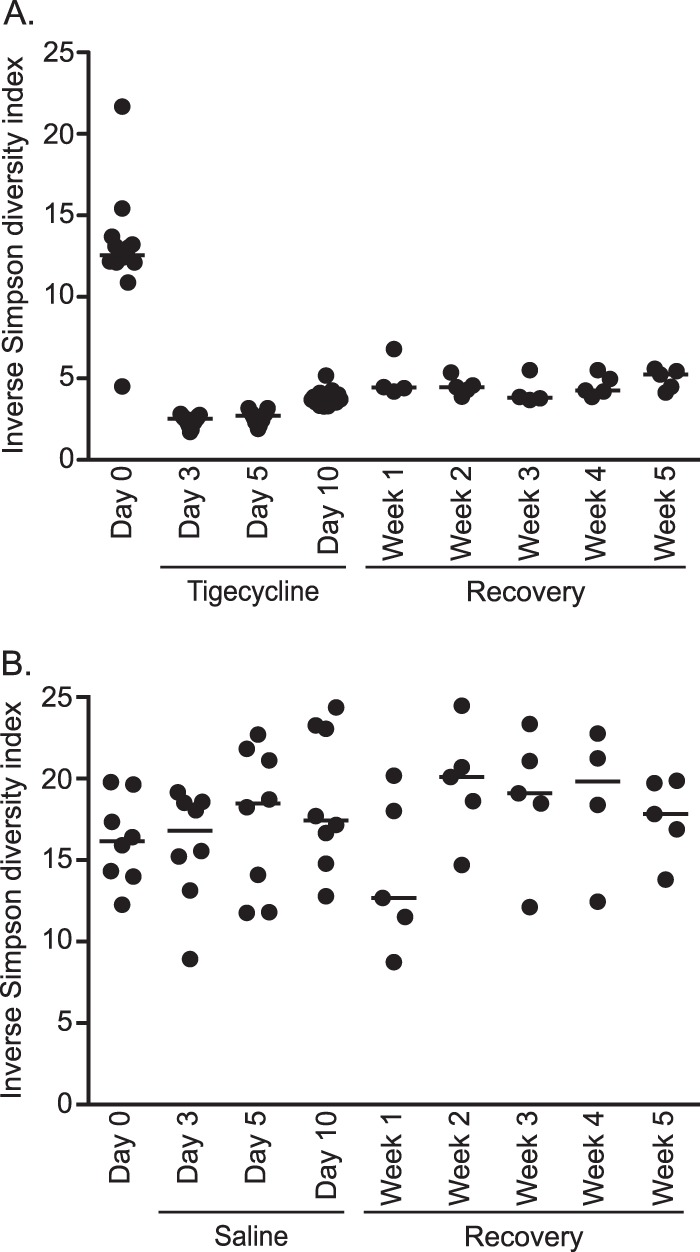
Bacterial diversity decreases during tigecycline treatment and remains low during recovery. Shown are the inverse Simpson index values of the gut microbiota prior to and during tigecycline treatment (A) and saline treatment (B) and recovery. Dots (•) represent values for each individual sample and horizontal bars represent the median values for each time point. The inverse Simpson diversity index was calculated from a subsample of 3,572 bacterial 16S rRNA gene sequences per sample with a 3% OTU definition.
Mice are susceptible to C. difficile infection after tigecycline treatment.
Mice were challenged with VPI 10463 spores 2 days after completing saline, cefoperazone, or tigecycline treatment (Fig. 6). On the day of challenge, the fecal microbiota of the saline-treated mice consisted mainly of Bacteroidetes and Firmicutes (Fig. 6). As seen in previous studies (31), the microbiota of cefoperazone-treated mice was dominated by Lactobacillaceae (Fig. 6). On the day of challenge, the tigecycline-treated mice had high levels of Enterobacteriaceae and Verrucomicrobiaceae and decreased levels of Bacteroidetes and Firmicutes relative to those of the saline-treated mice (Fig. 6).
FIG 6.

Susceptibility to C. difficile after tigecycline, saline, and cefoperazone treatment. Mice were subcutaneously injected with saline (negative control) every 12 h, given cefoperazone in their drinking water (positive control), or subcutaneously injected with tigecycline every 12 h for 10 days (upper left). All mice were allowed to recover for 2 days without treatment and were then challenged with C. difficile spores to assess susceptibility to colonization and infection. The bacterial community composition on the day of challenge with C. difficile spores is depicted by bar graphs (top) of the mean percent abundances of 16S rRNA sequences of the top bacterial families (≥1% relative abundance in a minimum of one sample) for each treatment group. The respective phyla are listed to the right of the family names. The family-level percent abundances are based on the classification of a subsample of 3,572 bacterial 16S rRNA gene sequences per sample. Samples with <3,572 sequences were not included in sequence analysis; therefore, for the bacterial community composition bar graph, n = 3 for saline, n = 9 for tigecycline, and n = 4 for cefoperazone. The mean number of C. difficile CFUs isolated and enumerated per gram of cecal contents of mice 2 days postchallenge (tigecycline versus saline, P < 0.05; tigecycline versus cefoperazone, nonsignificant [NS]) is shown (upper right). Error bars represent the SD. The mean percentage of the baseline weight for animals in each group is shown (bottom) from the day of challenge to 2 days postchallenge (saline versus tigecycline, P < 0.05; saline versus cefoperazone, P < 0.05). *, P <0.05.
Consistent with our previous studies (20, 31), C. difficile-challenged cefoperazone-treated mice were colonized with high levels of C. difficile (mean, 8.9 × 108 CFU/g of cecal content) and exhibited signs of clinically severe CDI, including significant weight loss and histopathological changes to the murine cecum (Fig. 6 and data not shown). Similar to cefoperazone-treated mice, tigecycline-treated mice were also colonized with C. difficile (mean, 3.7 × 108 CFU/g of cecal content), had a significant loss in weight by day 2 postchallenge, and had severe cecal histopathology (Fig. 6 and data not shown). As expected, at 2 days postchallenge, the mice treated with saline were not colonized with detectable levels of C. difficile and did not show signs of disease, including weight loss and cecal histopathology (Fig. 6 and data not shown).
DISCUSSION
The broad-spectrum glycylcycline antibiotic, tigecycline, significantly altered the composition of the murine gut microbiota and rendered mice susceptible to CDI. Several tigecycline-induced changes to the microbiota, including increased Proteobacteria and Verrucomicrobia levels and the loss of certain Bacteroidetes species, may contribute to CDI susceptibility.
Many antibiotics, including third-generation cephalosporins and clindamycin, are associated with a decrease in colonization resistance to C. difficile (2). Previous studies by our laboratory and others have found that the effects of these antibiotics on the murine gut microbiota increase susceptibility to C. difficile colonization and disease (31). These antibiotics alter the gut microbiota in different ways and result in multiple structures that allow for C. difficile colonization (6, 7, 32, 33). In this study, tigecycline-treated mice demonstrated CDI susceptibility similar to that of cefoperazone-treated mice, which have a dramatically different Lactobacillaceae-dominated microbiota. Murine gut microbial structures that allow for C. difficile colonization are commonly associated with an increase in members from the Proteobacteria phylum (Enterobacteriaceae family) and a decrease in overall diversity (6, 7, 33). This is consistent with human studies in which high levels of Proteobacteria and low levels of Bacteroidetes in the gut microbiota were associated with C. difficile colonization (34–39).
While some aspects of the tigecycline-altered bacterial community recovered within 5 weeks of stopping treatment, others persisted. The bacterial compositions at the phylum and family levels were returning to baseline (resembling saline controls). However, at the 3% OTU level, many members of the community failed to return, and the diversity remained low. Although the gut microbiota did not fully recover after antibiotics, it did return to a new stable state. Like many antibiotics, tigecycline has the potential to cause long-lasting effects on the murine gut microbiota (6, 32, 40). Even though the composition of the microbiota did not fully recover, it is unknown if the metabolic function of the community recovered. Distinct communities can have similar metabolic functions and resistances to CDI (31).
Previous culture-based studies looking at the effects of tigecycline on the murine gut microbiota have shown a relative sparing of the anaerobic bacteria and the Bacteroidetes population (18). Another culture-based study looking at how tigecycline affects the human intestinal microbiota reported a reduction of the Lactobacillus and Bifidobacterium populations, although again with no effect on Bacteroides (19). However, when tigecycline was added to a three-stage chemostat gut model, it showed marked decreases in Bacteroides and Bifidobacterium levels and lesser effects on the facultative anaerobes (41). Until now, there has not been a comprehensive culture-independent study detailing the effects of tigecycline on indigenous gut microbiota. In our study, the use of culture-independent techniques provided a comprehensive view of the gut bacterial community and revealed that tigecycline significantly altered the murine gut microbiota, including many members of the phylum Bacteroidetes, even though fecal levels of tigecycline were lower than those of humans (19).
Additional culture-independent studies are needed to determine if tigecycline affects the human gut microbiota, allowing for increased risk of CDI. There is some evidence that tigecycline exposure does not increase the risk of CDI in humans. In four phase III clinical studies, the overall rates of CDI after tigecycline exposure in humans were low but were not significantly different than those of the comparator agents (imipenem, vancomycin, aztreonam, levofloxacin, ciprofloxacin, and linezolid) (9). Furthermore, as an intravenously administered antibiotic reserved for complicated infections, tigecycline use is relatively limited compared to the use of other broad-spectrum antibiotics. Therefore, studies conducted more broadly in humans are needed to define the risk of CDI with tigecycline treatment.
If the risk of CDI with tigecycline treatment is truly low in humans, it is likely due to the anti-C. difficile activity of tigecycline. Tigecycline has a low MIC for C. difficile in vitro and is currently being used in Europe to treat severe refractory CDI in humans (13, 17). There are multiple reports that tigecycline can successfully treat severe CDI in humans, with a low incidence of relapse (13–15). It still remains to be seen if a drug that has potent anti-C. difficile activity is also one that can increase the risk for CDI. The interaction between tigecycline, the gut microbiota, and the pathogen C. difficile will be important in the future for evaluating the role of tigecycline as a treatment for patients with CDI.
Supplementary Material
ACKNOWLEDGMENTS
We thank Kathy Wozniak, Charlie Koumpouras, Gabrielle Hatton, and the University of Michigan ULAM staff for help with the mouse work. We also thank Sue Foltin for the 454 pyrosequencing and Anna Seekatz for assistance with data analysis.
The work on this paper utilized the Metabolomics Core Services supported by grant U24 DK097153 of NIH Common Funds Project to the University of Michigan. Funding for this project comes from Pfizer (Advancing Science through Pfizer-Investigator Research Exchange [ASPIRE], grant 673305) and NIH grant U19AI090871. C.M.T. is supported by the NIH Metabolomics Common Fund and the National Institute of General Medical Science under award K01GM109236.
Footnotes
Published ahead of print 3 March 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02262-13.
REFERENCES
- 1.McDonald LC, Lessa F, Sievert D, Wise M, Herrera R, Gould C, Malpiedi P, Dudeck M, Srinivasan A, Fridkin S, Cardo D. 2012. Vital signs: preventing Clostridium difficile infections. MMWR Morb. Mortal. Wkly. Rep. 61:157–162 [PubMed] [Google Scholar]
- 2.Freeman J, Wilcox MH. 1999. Antibiotics and Clostridium difficile. Microbes Infect. 1:377–384. 10.1016/S1286-4579(99)80054-9 [DOI] [PubMed] [Google Scholar]
- 3.Pépin J, Saheb N, Coulombe MA, Alary ME, Corriveau MP, Authier S, Leblanc M, Rivard G, Bettez M, Primeau V, Nguyen M, Jacob CE, Lanthier L. 2005. Emergence of fluoroquinolones as the predominant risk factor for Clostridium difficile-associated diarrhea: a cohort study during an epidemic in Quebec. Clin. Infect. Dis. 41:1254–1260. 10.1086/496986 [DOI] [PubMed] [Google Scholar]
- 4.Baxter R, Ray GT, Fireman BH. 2008. Case-control study of antibiotic use and subsequent Clostridium difficile-associated diarrhea in hospitalized patients. Infect. Control Hosp. Epidemiol. 29:44–50. 10.1086/524320 [DOI] [PubMed] [Google Scholar]
- 5.Gifford AH, Kirkland KB. 2006. Risk factors for Clostridium difficile-associated diarrhea on an adult hematology-oncology ward. Eur. J. Clin. Microbiol. Infect. Dis. 25:751–755. 10.1007/s10096-006-0220-1 [DOI] [PubMed] [Google Scholar]
- 6.Reeves AE, Theriot CM, Bergin IL, Huffnagle GB, Schloss PD, Young VB. 2011. The interplay between microbiome dynamics and pathogen dynamics in a murine model of Clostridium difficile infection. Gut Microbes 2:145–158. 10.4161/gmic.2.3.16333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buffie CG, Jarchum I, Equinda M, Lipuma L, Gobourne A, Viale A, Ubeda C, Xavier J, Pamer EG. 2012. Profound alterations of intestinal microbiota following a single dose of clindamycin results in sustained susceptibility to Clostridium difficile-induced colitis. Infect. Immun. 80:62–73. 10.1128/IAI.05496-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang JY, Antonopoulos DA, Kalra A, Tonelli A, Khalife WT, Schmidt TM, Young VB. 2008. Decreased diversity of the fecal microbiome in recurrent Clostridium difficile-associated diarrhea. J. Infect. Dis. 197:435–438. 10.1086/525047 [DOI] [PubMed] [Google Scholar]
- 9.Wilcox MH. 2007. Evidence for low risk of Clostridium difficile infection associated with tigecycline. Clin. Microbiol. Infect. 13:949–952. 10.1111/j.1469-0691.2007.01792.x [DOI] [PubMed] [Google Scholar]
- 10.Wilcox MH. 2005. Efficacy of tigecycline in complicated skin and skin structure infections and complicated intra-abdominal infections. J. Chemother. 17(Suppl 1):23–29. 10.1179/joc.2005.17.Supplement-1.23 [DOI] [PubMed] [Google Scholar]
- 11.Townsend ML, Pound MW, Drew RH. 2011. Potential role of tigecycline in the treatment of community-acquired bacterial pneumonia. Infect. Drug Resist. 4:77–86. 10.2147/IDR.S6030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Petersen PJ, Bradford PA, Weiss WJ, Murphy TM, Sum PE, Projan SJ. 2002. In vitro and in vivo activities of tigecycline (GAR-936), daptomycin, and comparative antimicrobial agents against glycopeptide-intermediate Staphylococcus aureus and other resistant Gram-positive pathogens. Antimicrob. Agents Chemother. 46:2595–2601. 10.1128/AAC.46.8.2595-2601.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu CL, Liu CY, Liao CH, Huang YT, Wang HP, Hsueh PR. 2010. Severe and refractory Clostridium difficile infection successfully treated with tigecycline and metronidazole. Int. J. Antimicrob. Agents. 35:311–312. 10.1016/j.ijantimicag.2009.11.008 [DOI] [PubMed] [Google Scholar]
- 14.Herpers BL, Vlaminckx B, Burkhardt O, Blom H, Biemond-Moeniralam HS, Hornef M, Welte T, Kuijper EJ. 2009. Intravenous tigecycline as adjunctive or alternative therapy for severe refractory Clostridium difficile infection. Clin. Infect. Dis. 48:1732–1735. 10.1086/599224 [DOI] [PubMed] [Google Scholar]
- 15.Hawser SP. 2010. Activity of tigecycline against recent European clinical isolates of Clostridium difficile. Int. J. Antimicrob. Agents. 35:97–98. 10.1016/j.ijantimicag.2009.09.009 [DOI] [PubMed] [Google Scholar]
- 16.Tang HJ, Ko WC, Chen CC, Chen PL, Toh HS, Weng TC, Yu WL, Chiang SR, Chuang YC. 2011. In vitro and in vivo intracellular killing effects of tigecycline against clinical nontyphoid Salmonella isolates using ceftriaxone as a comparator. Antimicrob. Agents Chemother. 55:2755–2759. 10.1128/AAC.01807-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hecht DW, Galang MA, Sambol SP, Osmolski JR, Johnson S, Gerding DN. 2007. In vitro activities of 15 antimicrobial agents against 110 toxigenic Clostridium difficile clinical isolates collected from 1983 to 2004. Antimicrob. Agents Chemother. 51:2716–2719. 10.1128/AAC.01623-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jump RL, Li Y, Pultz MJ, Kypriotakis G, Donskey CJ. 2011. Tigecycline exhibits inhibitory activity against Clostridium difficile in the colon of mice and does not promote growth or toxin production. Antimicrob. Agents Chemother. 55:546–549. 10.1128/AAC.00839-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nord CE, Sillerström E, Wahlund E. 2006. Effect of tigecycline on normal oropharyngeal and intestinal microflora. Antimicrob. Agents Chemother. 50:3375–3380. 10.1128/AAC.00373-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Theriot CM, Koumpouras CC, Carlson PE, Bergin II, Aronoff DM, Young VB. 2011. Cefoperazone-treated mice as an experimental platform to assess differential virulence of Clostridium difficile strains. Gut Microbes 2:326–334. 10.4161/gmic.19142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perez J, Springthorpe VS, Sattar SA. 2011. Clospore: a liquid medium for producing high titers of semi-purified spores of Clostridium difficile. J. AOAC Int. 94:618–626 [PubMed] [Google Scholar]
- 22.Schloss PD, Gevers D, Westcott SL. 2011. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS One 6:e27310. 10.1371/journal.pone.0027310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75:7537–7541. 10.1128/AEM.01541-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quince C, Lanzén A, Curtis TP, Davenport RJ, Hall N, Head IM, Read LF, Sloan WT. 2009. Accurate determination of microbial diversity from 454 pyrosequencing data. Nat. Methods 6:639–641. 10.1038/nmeth.1361 [DOI] [PubMed] [Google Scholar]
- 25.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and Web-based tools. Nucleic Acids Res. 41:D590–D596. 10.1093/nar/gks1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schloss PD. 2009. A high-throughput DNA sequence aligner for microbial ecology studies. PLoS One 4:e8230. 10.1371/journal.pone.0008230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. 2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200. 10.1093/bioinformatics/btr381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cole JR, Chai B, Farris RJ, Wang Q, Kulam SA, McGarrell DM, Garrity GM, Tiedje JM. 2005. The Ribosomal Database Project (RDP-II): sequences and tools for high-throughput rRNA analysis. Nucleic Acids Res. 33:D294–D296. 10.1093/nar/gki038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73:5261–5267. 10.1128/AEM.00062-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yue JC, Clayton MK. 2005. A similarity measure based on species proportions. Commun. Statist.-Theor. Method. 34:2123–2131. 10.1080/STA-200066418 [DOI] [Google Scholar]
- 31.Theriot CM, Koenigsknecht MJ, Carlson PE, Jr, Hatton GE, Nelson AM, Li B, Huffnagle GB, Li JZ, Young VB. 2014. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat. Commun. 5:3114. 10.1038/ncomms4114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Antonopoulos DA, Huse SM, Morrison HG, Schmidt TM, Sogin ML, Young VB. 2009. Reproducible community dynamics of the gastrointestinal microbiota following antibiotic perturbation. Infect. Immun. 77:2367–2375. 10.1128/IAI.01520-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lawley TD, Clare S, Walker AW, Stares MD, Connor TR, Raisen C, Goulding D, Rad R, Schreiber F, Brandt C, Deakin LJ, Pickard DJ, Duncan SH, Flint HJ, Clark TG, Parkhill J, Dougan G. 2012. Targeted restoration of the intestinal microbiota with a simple, defined bacteriotherapy resolves relapsing Clostridium difficile disease in mice. PLoS Pathog. 8:e1002995. 10.1371/journal.ppat.1002995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Skraban J, Dzeroski S, Zenko B, Mongus D, Gangl S, Rupnik M. 2013. Gut microbiota patterns associated with colonization of different Clostridium difficile ribotypes. PLoS One 8:e58005. 10.1371/journal.pone.0058005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Manges AR, Labbe A, Loo VG, Atherton JK, Behr MA, Masson L, Tellis PA, Brousseau R. 2010. Comparative metagenomic study of alterations to the intestinal microbiota and risk of nosocomial Clostridum difficile-associated disease. J. Infect. Dis. 202:1877–1884. 10.1086/657319 [DOI] [PubMed] [Google Scholar]
- 36.Rea MC, O'Sullivan O, O'Toole PW, Stanton C, Ross RP, Hill C. 2012. Clostridium difficile carriage in elderly subjects and associated changes in the intestinal microbiota. J. Clin. Microbiol. 50:867–875. 10.1128/JCM.05176-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hopkins MJ, Macfarlane GT. 2002. Changes in predominant bacterial populations in human faeces with age and with Clostridium difficile infection. J. Med. Microbiol. 51:448–454 [DOI] [PubMed] [Google Scholar]
- 38.Hamilton MJ, Weingarden AR, Unno T, Khoruts A, Sadowsky MJ. 2013. High-throughput DNA sequence analysis reveals stable engraftment of gut microbiota following transplantation of previously frozen fecal bacteria. Gut Microbes 4:125–135. 10.4161/gmic.23571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Theriot CM, Young VB. 11 December 2013. Microbial and metabolic interactions between the gastrointestinal tract and infection. Gut Microbes. 10.4161/gmic.27131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robinson CJ, Young VB. 2010. Antibiotic administration alters the community structure of the gastrointestinal micobiota. Gut Microbes 1:279–284. 10.4161/gmic.1.4.12614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baines SD, Saxton K, Freeman J, Wilcox MH. 2006. Tigecycline does not induce proliferation or cytotoxin production by epidemic Clostridium difficile strains in a human gut model. J. Antimicrob. Chemother. 58:1062–1065. 10.1093/jac/dkl364 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.