

Abstract
Several viruses, including human papillomaviruses, depend on endosomal acidification for successful infection. Hence, the multisubunit enzyme vacuolar ATPase (V-ATPase), which is mainly responsible for endosome acidification in the cell, represents an attractive target for antiviral strategies. In the present study, we show that V-ATPase is required for human papillomavirus (HPV) infection and that uncoating/disassembly but not endocytosis is affected by V-ATPase inhibition. The infection inhibitory potencies of saliphenylhalamide, a proven V-ATPase inhibitor, and its derivatives, as well as those of other V-ATPase inhibitors, were analyzed on different HPV types in relevant cell lines. Variation in the selectivity indices among V-ATPase inhibitors was high, while variation for the same inhibitor against different HPV subtypes was low, indicating that broad-spectrum anti-HPV activity can be provided.
INTRODUCTION
Papillomaviruses are nonenveloped double-stranded DNA (dsDNA) viruses that cause benign or malign neoplasias in epithelial tissues. A leading type of cancer caused by the high-risk group of human papillomaviruses (HPV) is cervical cancer. Approximately half a million new cases and nearly 250,000 deaths among women are observed each year (1). The majority of those cases are associated with one or several oncogenic HPV subtypes, including HPV16, -18, -31, -33, and -45 (2). High-risk HPVs can also cause cancers of the vulva, vagina, penis, anus, and perianal region, and they cause about 20% of head and neck cancers (3). Low-risk HPV subtypes can cause benign skin alterations, e.g., genital warts, which are associated with HPV subtypes 6 and 11 in 90% of cases (4).
Currently, two vaccines are available for effective prevention of infection with high-risk HPV16 and -18. Some cross-reactivity of the vaccines against other subtypes has been observed but is insufficient to provide full cross-protection against all oncogenic subtypes (5). In developing countries where cervical cancer is most frequent, vaccinations are unlikely to resolve the situation because of high vaccine cost and the requirement of multiple injections at certain time points to confer full protection. Also, the long-term effectiveness of these prophylactic vaccines in the vaccinated population remains unclear (5–7). Altogether, this indicates that even in the era of HPV vaccination, effective early stage inhibitors of HPV infection are required. The interruption of an early step in the viral replication cycle, for instance, attachment, endocytosis, or uncoating of virus, represents a promising strategy.
Primary attachment of papillomavirus particles to the cell surface is mediated through heparan sulfate proteoglycans (8, 9). After conformational changes in both capsid proteins L1 and L2 (9–11), viral particles are transferred to a non-heparin sulfate proteoglycan receptor complex (12–15), which triggers a clathrin-independent internalization of HPV (13, 16, 17). After endocytosis, intracellular trafficking through the endosomal compartment with acidification of the endocytic vesicles is a prerequisite for viral uncoating and release of the L2-HPV genome complex (18–21). Disassembly/uncoating of HPV virions can be blocked by inhibitors of endosomal acidification (16, 18–22). As we showed earlier for influenza A virus, which also requires endosomal acidification for cytosol entry, inhibition of endosomal acidification through vacuolar ATPase (V-ATPase) inhibitors can be used for successful treatment in vitro and in vivo (23, 24).
Here, we show that HPV infection can be blocked by V-ATPase inhibition, compare various V-ATPase inhibitors regarding their half-maximal effective concentrations (EC50s), and compare those values to their toxicities, expressed as half-maximal cytotoxic concentrations (CC50s), to give selectivity indices (SIs) as the ratio of CC50/EC50.
MATERIALS AND METHODS
Cell culture.
The human cervix adenocarcinoma cell line HeLa was obtained from the German Resource Centre for Biological Material (DSMZ, Braunschweig, Germany), and the human keratinocyte cell line HaCaT from Cell Lines Services (CLS, Eppelheim, Germany). The human embryonic kidney cell line 293TT was kindly provided by Chris Buck, Bethesda, MD (25). Cells were grown at 37°C in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS), 1% Glutamax I (Invitrogen, United States), 1% modified Eagle's medium with nonessential amino acids and antibiotics. Normal human epidermal keratinocytes (NHEK) were obtained from PromoCell (Heidelberg, Germany) and were cultivated according to the manufacturer's instructions. Given the combination of ease of handling and a significant pseudovirion (PsV) infectivity readout, we chose HeLa cells for the initial V-ATPase inhibitor/HPV infectivity screens. The infectivity inhibition by the most potent inhibitors was then reproduced in the more relevant cell line HaCaT and, finally, in primary keratinocytes.
HPV pseudovirions.
HPV pseudovirions were prepared as previously described (25). Briefly, expression plasmids carrying codon-optimized HPV L1 and L2 cDNA were cotransfected with a pCMV-GLuc control (New England Bioscience, United States) or pcDNA3.1/luciferase reporter plasmid into 293TT cells (26). Forty-eight hours posttransfection, the cells were processed by lysis and nuclease digestion. After maturation, the pseudovirions were purified from the cell lysates by Optiprep gradient centrifugation. Codon-optimized L1 and L2 expression vectors for HPV16, HPV6, and HPV11 were kindly provided by Martin Müller, Heidelberg, Germany, for HPV18 by Chris Buck, Bethesda, MD, and for HPV31 by Tadahito Kanda, Tokyo, Japan (27–31).
V-ATPase inhibitors.
Inhibitors of the salicylihalamide type were synthesized in the laboratory of J. K. De Brabander (32–35, 44). If common names for inhibitors were unavailable, internal laboratory nomenclature was used to refer to the specific substance. ApiA, ApiOpen, PalmA, and decarbamoylated PalmA were from the laboratory of Martin E. Maier. The control compound 543229 was obtained from Hit2Lead, United States.
siRNA experiments.
Transfections of HeLa cells were performed with RNAiMAX (Invitrogen) and 30 nM small interfering RNA (siRNA) according to the manufacturer's recommendations. Subsequent experiments (infectivity assays, Western blotting) were done 48 h after siRNA transfection, as we observed that this was the time point of maximal knockdown by siRNA. siRNAs targeting the V-ATPase subunit D1 (ATP6V0D1) and the Allstars negative-control siRNA were purchased from Qiagen. The ATP6V0D1 siRNA target sequences were as follows: siNRA 1, CTGGCTCGGGCTGACGACTAT; siNRA 2, TTACAGTTACATGATCGACAA; siNRA 3, CAAGAACGTGGCCGATTACTA; and siNRA 4, CTACCTGGAGTCCTTCTACAA.
Knockdown of ATP6V0D1 was confirmed 48 h after siRNA transfection by Western blotting using a specific monoclonal antibody (sc-81887; Santa Cruz). Tubulin was probed as a loading control, using the monoclonal antibody B-5-1-2 (Sigma-Aldrich).
Infectivity assays.
Cells were infected with approximately 1,000 pseudovirions per cell. For reverse infections, cells were trypsinized, resuspended in medium, infected in suspension, and seeded on culture plates. ATP6V0D1 siRNA transfections were done 48 h before infection, while V-ATPase inhibitors were added to the cells immediately after infection. Infectivity rates were assessed 24 to 48 h postinfection (p.i.). Experiments were conducted in triplicates. Infectivity assays with the firefly luciferase reporter plasmid were quantified using the cell culture lysis reagent and luciferase assay system (Promega); Infectivity assays with the Gaussia luciferase reporter plasmid were performed in the cell culture supernatant using the Gaussia Juice kit (PJK). Measurements were done with the Tristar LB 941 luminometer (Berthold Technologies).
Cell viability and cytotoxicity.
The potential cytotoxicities of inhibitors and siRNAs were assessed with an XTT cell proliferation kit (AppliChem) according to the manufacturer's instructions. The same culture conditions and treatment parameters were used for infectivity and XTT viability/cytotoxicity assays. Determination of CC50 by XTT in HeLa cells was done after 5 days of incubation with inhibitors. The infectivity values in siRNA assays were normalized by the XTT measurements.
Disassembly/uncoating analysis by immunofluorescence.
HeLa cells were grown on glass coverslips and infected either after siRNA transfection or prior to inhibitor addition. At various time points after infection as indicated below, cells were washed with phosphate-buffered saline (PBS) and fixed/permeabilized with methanol (−20°C). Fixed cells were washed and blocked with 1% bovine serum albumin (BSA)-PBS. Coverslips were incubated for 1 h at 37°C with primary antibodies (indicated below) diluted in BSA-PBS and then washed and blocked with BSA-PBS. Incubation with secondary, Alexa Fluor 488-conjugated antibodies diluted in BSA-PBS was performed for 45 min at 37°C. DNA was stained with Hoechst 33342 (Sigma-Aldrich). After BSA-PBS and PBS washing, coverslips were mounted on slides using Fluoprep mounting medium (bioMérieux). Fluorescence microscopy was done with a Zeiss Axiovert 200 M microscope equipped with a Plan-Apochromat 100× (1.4 numerical aperture) objective, a Zeiss Axiocam digital camera, and Axiovision software, version 4.6. For quantification of L1-7 signals, 10 random images per experiment were taken with identical settings (fixed exposure time and focal plane); the pictures were not deconvoluted and no modifications regarding contrast and brightness were made. Pixel-by-pixel analysis was performed using the Axiovision colocalization module with fixed thresholds for all images. The total number of L1-7 pixels corrected by the total number of DNA (cell nuclei) pixels (L1-7/DNA) was set as the measure for uncoating. Uncoating in control experiments (control siRNA and dimethyl sulfoxide [DMSO]) was set to 100%.
Endocytosis assay.
HeLa cells were infected either 48 h after siRNA transfection or before the addition of inhibitor. For immunostaining of cell surface-bound pseudovirions, cells were incubated with anti-L1 (K75) antibody (36) in 0.5% fetal calf serum (FCS)-PBS for 45 min at 4°C. Background staining was determined using noninfected control cells. Subsequently, cells were washed with FCS-PBS and incubated with anti-rabbit IgG-Alexa Fluor 488 (1:250 in FCS-PBS) for 30 min at 4°C. Cells were washed with FCS-PBS and resuspended in PBS, and the mean fluorescence intensity of extracellular virions was quantified by flow cytometry. Cell surface-bound virions were quantified at 1 and 20 h postinfection. The difference in bound extracellular virions between 1 h and 20 h in the controls (control siRNA and DMSO) corresponds to the amount of internalized virions and was set to 100% endocytosis activity. All flow cytometry experiments were done on a FACScan (Becton, Dickinson).
Data analysis.
The response values (absorbance for XTT and luminescence for luciferase experiments) of triplicates were curve fitted to their respective inhibitor concentration values in Sigmaplot 9.0 (Systat Software) to obtain a Hill curve. The half-maximal effective concentration values (CC50 for toxicity and EC50 for anti-HPV effect) were derived from Hill curves. Statistical significance tests were performed with Student's t test (*, P < 0.05 compared to control).
RESULTS
V-ATPase is required for HPV infection.
Acidification of endosomes is an essential step for successful HPV infection. An enrichment of V-ATPase subunits in endosomes of HPV-infected cells was observed in a mass spectrometry-based screen, indicating a role of V-ATPase in HPV infection (37; also L. Florin and K. D. Scheffer, unpublished data). To analyze the functional relevance of V-ATPase for HPV, infectivity studies were performed in HeLa cells after V-ATPase D knockdown using 4 individual, specific siRNAs (Fig. 1). HPV16 infectivity was significantly reduced by the V-ATPase D-specific siRNAs compared to the results for the control siRNA. This demonstrates that a functional V-ATPase is required for successful HPV infection.
FIG 1.

Depletion of V-ATPase subunit D1 inhibits HPV16 infectivity. (a) HeLa cells were transfected for 48 h with different siRNAs targeting the V-ATPase subunit D1 or a control siRNA and subsequently infected for 24 h with HPV16 pseudovirions. Depicted are the means ± SD relative to the control siRNA (n = three independent experiments). (b) At 48 h after siRNA transfection, HeLa cells were analyzed by Western blotting for the levels of V-ATPase D1 and, as a loading control, tubulin.
V-ATPase is required for uncoating/disassembly of HPV.
Next, we analyzed which step of HPV infection is affected by V-ATPase inhibition. To examine the role of V-ATPase in viral endocytosis, control and V-ATPase D-depleted cells were incubated with HPV16 pseudovirions (PsVs) for 1 or 20 h, and surface-associated virions were quantified by flow cytometry using the polyclonal anti-L1 antibody K75 (Fig. 2a). The decrease of surface-bound particles between 1 and 20 h of infection represents HPV endocytosis. We found that endocytosis of HPV16 particles was not affected in V-ATPase subunit D1 knockdown cells (Fig. 2b). To analyze the role of V-ATPase in HPV uncoating/disassembly during infection, we examined the amounts of disassembled capsids in endosomes of control and V-ATPase siRNA-transfected cells using the specific anti-L1 monoclonal antibody L1-7, which recognizes an epitope that is located on the inner side of intact capsids and is only accessible after internalization and disassembly of capsids (Fig. 2c) (10, 13, 38, 39). Quantitative analysis of fluorescence micrographs revealed a significant inhibition of HPV16 uncoating/disassembly in cells with V-ATPase subunit D1 knockdown (Fig. 2d).
FIG 2.

Depletion of V-ATPase D1 inhibits HPV16 uncoating but not endocytosis. (a) HeLa cells were transfected with control or V-ATPase D1 siRNA 1 (si #1) for 48 h and subsequently incubated with HPV16 PsVs for 1 or 20 h as indicated. The amount of surface-associated PsVs was assessed by flow cytometry using anti-L1 polyclonal antibody (K75). (b) The difference in mean fluorescence intensities between 1 and 20 h of infection with control siRNA was set to 100% endocytosis. Bars represent compiled results of three independent experiments ± SD. (c) HeLa cells were transfected as described for panel a and infected with HPV16 PsVs for 8 h. Virus disassembly/uncoating was analyzed by immunofluorescence with monoclonal antibody L1-7 (green), recognizing an intraluminal L1 epitope of the capsid. DNA was stained with Hoechst 33342 (blue). (d) Virus uncoating/disassembly in control siRNA-treated cells was set to 100%. Depicted are the means of three independent experiments ± SD.
Saliphenylhalamide inhibits HPV infection.
To investigate whether HPV infection can be blocked by a small molecule inhibitor of V-ATPase, we quantified the effects of saliphenylhalamide (SaliPhe), a specific V-ATPase inhibitor, against low-risk HPV6 and -11, as well as high-risk HPV16, -18, and -31 (Fig. 3). The results show that SaliPhe is effective against all HPV subtypes tested, with EC50s ranging from 91 nM for HPV31 to 226 nM for HPV11. Considering a CC50 value of 411 nM for the 5-day toxicity of SaliPhe on HeLa cells, selectivity indices (SIs [CC50/EC50]) of 1.8 for HPV11 and 3.3 for HPV6 were obtained. For the high-risk subtypes, the SI values were 2.6 for HPV16, 3.7 for HPV18, and 4.5 for HPV31. Similar to siRNA knockdown, inhibition of V-ATPase with SaliPhe did not impair endocytosis of HPV16 PsVs but it significantly reduced uncoating of HPV16 PsVs (Fig. 4).
FIG 3.

Cytotoxic and antiviral effects of SaliPhe in HeLa cells. (a) HeLa cells were incubated with serial dilutions of SaliPhe for 5 days and subsequently stained with XTT. Raw data of triplicates were fitted to a Hill curve to calculate the CC50. (b to f) HeLa cells were infected with the indicated HPV PsVs for 24 h in the presence of serial dilutions of SaliPhe. Raw data of triplicates were fitted to Hill curves to calculate the indicated EC50s. Depicted are the means of three independent experiments ± SD.
FIG 4.
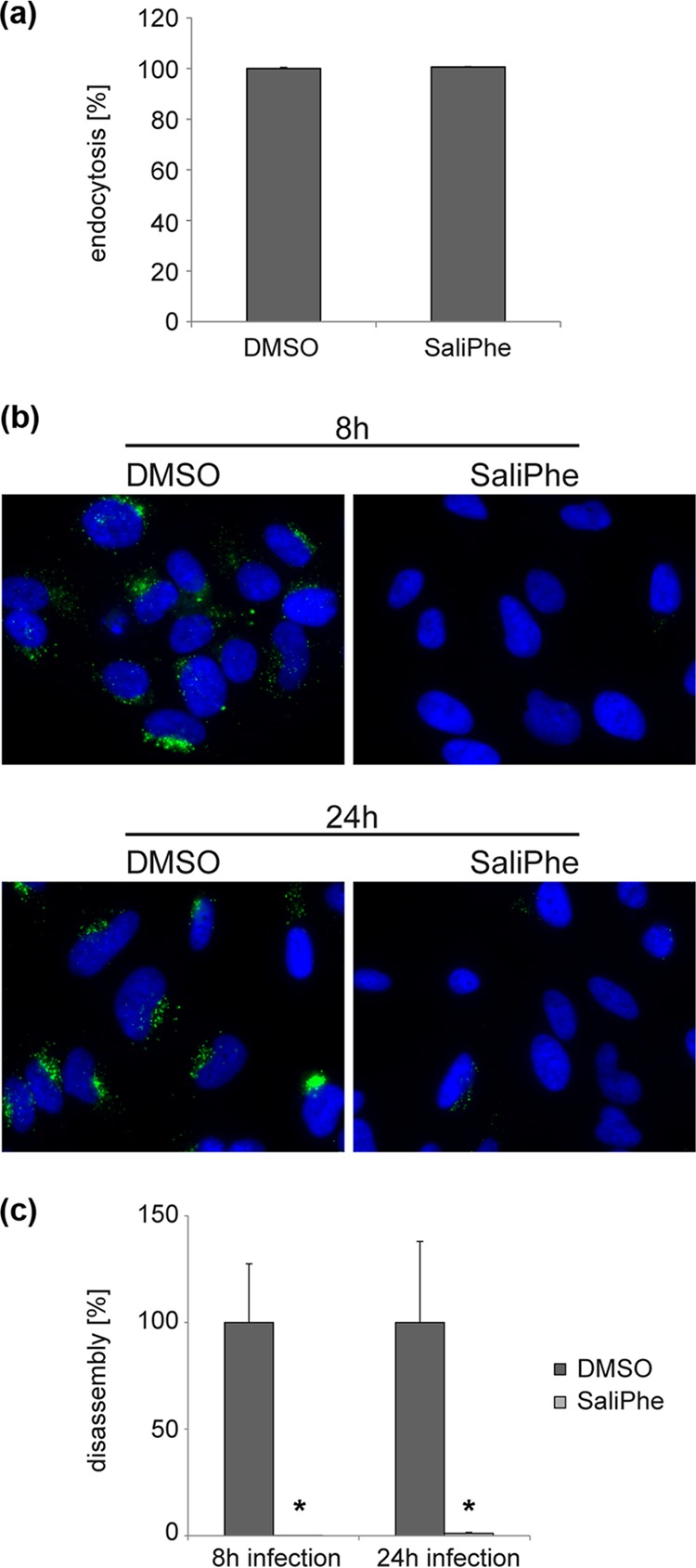
SaliPhe inhibits uncoating/disassembly of HPV16. (a) HeLa cells were infected for 1 or 20 h with HPV16 in the presence of 1.5 μM SaliPhe or DMSO as control and subsequently processed as described in the legend to Fig. 2a and b to determine the rate of endocytosis. Depicted are the means of three independent experiments ± SD. (b, c) HeLa cells were infected for 8 or 24 h with HPV16 in the presence of 1.5 μM SaliPhe or DMSO as control and subsequently processed as described in the legend to Fig. 2c and d to determine the rate of capsid uncoating/disassembly. Depicted are the means of three independent experiments ± SD.
Effects of other V-ATPase inhibitors on HPV infection.
Derivatives of salicylihalamide A and other specific V-ATPase inhibitors were screened against high-risk HPV subtypes 16, 18, and 31 to analyze whether small modifications on the side chain or on the backbone could improve selectivity or reveal pharmacological differences between the three subtypes (see Tables S1 and S2 in the supplemental material). Compound 543229, which shares similarities with enamides but does not contain a functional enamide side chain, was included as a negative control. As expected, 543229 did not show an anti-HPV or cytotoxic effect at concentrations up to 50 μM. The lowest EC50s were observed for concanamycin A against HPV31 (0.138 nM), while SL-2073 was ineffective against all HPV subtypes at concentrations up to 50 μM. The EC50s determined for the different compounds were similar for the three HPV types. The CC50 values, as a read-out of cytotoxicity, were measured after a 5-day incubation of cells with inhibitors. The 5-day cytotoxicity is particularly strict to also account for longer treatment durations. The lowest CC50 value was obtained for concanamycin A with 0.19 nM, and the highest for SL-2036 with 18.39 μM. The EC50 and CC50 values of macrolactone V-ATPase inhibitors were in the subnanomolar range, except for ApiOpen, which had EC50s of around 30 nM, indicating that the transannular oxygen that is absent in ApiOpen compared to ApiA has a role in the antiviral and cytotoxic effects.
To compare all compounds with respect to cytotoxicity and anti-HPV effect, SIs are shown in Table S1 in the supplemental material. SI values above 1 indicate an antiviral effect distinct from cytotoxic effects. For the compounds tested, the SI values ranged from 0.002 for SL-3177 to 11 for SaliPhe. While the SI values differed substantially between compounds, the SI values between the three HPV types were comparable for a given compound. LX-1231, SL-2072, and SL-3175 had SI values below 1 and were ineffective against HPV. ArchB had an SI value of about 1. All other compounds were considered active.
Effects of selected V-ATPase inhibitors on HPV in HaCaT and NHEK cells.
To validate our results in more relevant cell types, we tested some selected inhibitors in HaCaT cells, an immortalized human keratinocyte cell line and, more importantly, in normal human epithelial (NHEK) cells, primary keratinocytes which constitute the natural target cells of HPV infection (Fig. 5).
FIG 5.

Cytotoxic and antiviral effects of selected V-ATPase inhibitors in HaCaT and NHEK cells. (a to c) HaCaT cells were infected for 24 h with HPV16 PsVs in the presence of serial dilutions of the indicated V-ATPase inhibitors. In addition, XTT was measured in HaCaT cells after 24 h of incubation with serial dilutions of the indicated inhibitors. Raw data of triplicates were fitted to Hill curves, and CC50 and EC50 values deduced. XTT resp., XTT response. (d) NHEK cells were infected with HPV16 PsVs in the presence of SaliPhe serial dilutions. After 48 h, infection rates and XTT responses were determined. Raw data of triplicates were fitted to a Hill curve, and CC50 and EC50 values deduced. Depicted are the means of three independent experiments ± SD.
We observed that the EC50s, as well as the CC50s, were increased in HaCaT cells compared to the values in HeLa cells (Fig. 5a to c). The EC50s of SaliPhe, SL-1295, and SL-2036 were 0.96 μM, 3.17 μM, and 7.5 μM for HPV16 infection of HaCaT cells. The CC50 toxicity values of those compounds were 13.2 μM, 5.6 μM, and 17.5 μM, respectively. This results in calculated SIs of 13.75 for SaliPhe, 1.8 for SL-1295, and 2.3 for SL-2036.
SaliPhe, which had the best SI in HaCaT cells, was also tested in NHEK cells (Fig. 5d). Therefore, NHEK cells were infected for 48 h with HPV16 PsVs in the presence of serial dilutions of SaliPhe. The resulting EC50 and CC50 were 0.12 μM and 7.2 μM, respectively. This gives an SI of 60, indicative of a strong HPV16 infection inhibitory potency at low cytotoxicity for SaliPhe in NHEK cells.
DISCUSSION
We report here the functional requirement of V-ATPase for HPV infection and provide a comparison of different V-ATPase inhibitors with respect to their anti-HPV activities. We show that endocytosis of HPV particles was not affected by V-ATPase inhibition but that capsid disassembly/uncoating within the endosomal compartment was strongly dependent on V-ATPase activity. This finding can be pharmacologically exploited to prevent HPV infection by using small molecule V-ATPase inhibitors. Interestingly, not all V-ATPase inhibitors tested in this study were pharmacologically active (SI < 1); however, the majority of inhibitors that proved to be active were equally effective against different HPV subtypes. This is indicative of broad-spectrum anti-HPV activity of this group of compounds. Although the SI seems low, V-ATPase inhibitors may nevertheless have some merit when combined with other inhibitors applied, for instance, as lubricants or vaginal gels to build up a cascade of several steps for prevention of sexually transmitted viral infections, including HPV.
Antiviral compounds that inhibit the early steps of HPV infection represent an attractive alternative for HPV prevention, as vaccines do not yet confer full protection against all high-risk HPV types and might be too expensive for general use in some countries. Hence, chemoprevention of HPV infection might be an interesting prophylactic approach to prevent sexual transmission of HPVs. Besides V-ATPase inhibitors, several other HPV antivirals inhibiting different steps of early HPV infection have already been described. Primary attachment of HPV particles to target cells can be inhibited by carrageenans (40), which opsonize the virions by covering the surface and thus inhibit attachment to target cells, or by cationic polymers, such as polyethylenimine (41), which block the primary receptors on the cell surface. DSTP27, a derivative of dispirotripiperazine, also inhibits primary HPV-cell interaction when administered prior to infection. When administered postattachment, this molecule directs virions into a noninfectious internalization pathway (12). A recent screen of a small-compound library has identified new antiviral molecules that inhibit a not-yet-defined postentry step of HPV infection (42). In the same line, lately a genome-wide siRNA screen has identified the retromer as a cellular factor required for HPV infection and shown that Retro-2, a small molecule inhibitor of retrograde transport, inhibits HPV16 infection (43). Interestingly, in the same screen, V-ATPase was also identified as a cellular factor necessary for HPV infection, confirming our report. As the HPV inhibitors described above act on different steps of infection, combinations of these molecules could be used in formulations, such as lubricants, for application in the anogenital area to confer multiple layers of protection against HPV.
In conclusion, we show for the first time that the vacuolar ATPase is involved in the viral uncoating step during papillomavirus infection. V-ATPase inhibitors seem to have broad-spectrum anti-HPV activity, providing new pharmacological opportunities.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the generous gift of ArchB from Dirk Trauner (Munich). We thank Fatima Boukhallouk (Mainz) for excellent technical assistance.
This work was supported by grants to L.F. (SFB490/D2) from the German Research Foundation and the intern-funding program of the Johannes Gutenberg University Mainz and a grant to K.H.M. from the Fonds National de la Recherche Luxembourg (FNR). J.K.D.B. holds the Julie and Louis Beecherl, Jr., Chair in Medical Science, and wishes to thank the Robert A. Welch Foundation for support (grant I-1422).
The funders have not played any decision-making role in the research.
Footnotes
Published ahead of print 10 March 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02284-13.
REFERENCES
- 1.Arbyn M, Castellsague X, de Sanjose S, Bruni L, Saraiya M, Bray F, Ferlay J. 2011. Worldwide burden of cervical cancer in 2008. Ann. Oncol. 22:2675–2686. 10.1093/annonc/mdr015 [DOI] [PubMed] [Google Scholar]
- 2.Cogliano V, Baan R, Straif K, Grosse Y, Secretan B, El Ghissassi F, WHO International Agency for Research on Cancer 2005. Carcinogenicity of human papillomaviruses. Lancet Oncol. 6:204. 10.1016/S1470-2045(05)70086-3 [DOI] [PubMed] [Google Scholar]
- 3.zur Hausen H. 2009. Papillomaviruses in the causation of human cancers—a brief historical account. Virology 384:260–265. 10.1016/j.virol.2008.11.046 [DOI] [PubMed] [Google Scholar]
- 4.Greer CE, Wheeler CM, Ladner MB, Beutner K, Coyne MY, Liang H, Langenberg A, Yen TS, Ralston R. 1995. Human papillomavirus (HPV) type distribution and serological response to HPV type 6 virus-like particles in patients with genital warts. J. Clin. Microbiol. 33:2058–2063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paavonen J, Jenkins D, Bosch FX, Naud P, Salmeron J, Wheeler CM, Chow SN, Apter DL, Kitchener HC, Castellsague X, de Carvalho NS, Skinner SR, Harper DM, Hedrick JA, Jaisamrarn U, Limson GA, Dionne M, Quint W, Spiessens B, Peeters P, Struyf F, Wieting SL, Lehtinen MO, Dubin G, HPV PATRICIA study group 2007. Efficacy of a prophylactic adjuvanted bivalent L1 virus-like-particle vaccine against infection with human papillomavirus types 16 and 18 in young women: an interim analysis of a phase III double-blind, randomised controlled trial. Lancet 369:2161–2170. 10.1016/S0140-6736(07)60946-5 [DOI] [PubMed] [Google Scholar]
- 6.Garland SM, Hernandez-Avila M, Wheeler CM, Perez G, Harper DM, Leodolter S, Tang GW, Ferris DG, Steben M, Bryan J, Taddeo FJ, Railkar R, Esser MT, Sings HL, Nelson M, Boslego J, Sattler C, Barr E, Koutsky LA, Females United to Unilaterally Reduce Endo/Ectocervical Disease II 2007. Quadrivalent vaccine against human papillomavirus to prevent anogenital diseases. N. Engl. J. Med. 356:1928–1943. 10.1056/NEJMoa061760 [DOI] [PubMed] [Google Scholar]
- 7.Villa LL, Costa RL, Petta CA, Andrade RP, Ault KA, Giuliano AR, Wheeler CM, Koutsky LA, Malm C, Lehtinen M, Skjeldestad FE, Olsson SE, Steinwall M, Brown DR, Kurman RJ, Ronnett BM, Stoler MH, Ferenczy A, Harper DM, Tamms GM, Yu J, Lupinacci L, Railkar R, Taddeo FJ, Jansen KU, Esser MT, Sings HL, Saah AJ, Barr E. 2005. Prophylactic quadrivalent human papillomavirus (types 6, 11, 16, and 18) L1 virus-like particle vaccine in young women: a randomised double-blind placebo-controlled multicentre phase II efficacy trial. Lancet Oncol. 6:271–278. 10.1016/S1470-2045(05)70101-7 [DOI] [PubMed] [Google Scholar]
- 8.Joyce JG, Tung JS, Przysiecki CT, Cook JC, Lehman ED, Sands JA, Jansen KU, Keller PM. 1999. The L1 major capsid protein of human papillomavirus type 11 recombinant virus-like particles interacts with heparin and cell-surface glycosaminoglycans on human keratinocytes. J. Biol. Chem. 274:5810–5822. 10.1074/jbc.274.9.5810 [DOI] [PubMed] [Google Scholar]
- 9.Giroglou T, Florin L, Schafer F, Streeck RE, Sapp M. 2001. Human papillomavirus infection requires cell surface heparan sulfate. J. Virol. 75:1565–1570. 10.1128/JVI.75.3.1565-1570.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bienkowska-Haba M, Patel HD, Sapp M. 2009. Target cell cyclophilins facilitate human papillomavirus type 16 infection. PLoS Pathog. 5:e1000524. 10.1371/journal.ppat.1000524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Selinka HC, Giroglou T, Nowak T, Christensen ND, Sapp M. 2003. Further evidence that papillomavirus capsids exist in two distinct conformations. J. Virol. 77:12961–12967. 10.1128/JVI.77.24.12961-12967.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Selinka HC, Florin L, Patel HD, Freitag K, Schmidtke M, Makarov VA, Sapp M. 2007. Inhibition of transfer to secondary receptors by heparan sulfate-binding drug or antibody induces noninfectious uptake of human papillomavirus. J. Virol. 81:10970–10980. 10.1128/JVI.00998-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spoden G, Freitag K, Husmann M, Boller K, Sapp M, Lambert C, Florin L. 2008. Clathrin- and caveolin-independent entry of human papillomavirus type 16–involvement of tetraspanin-enriched microdomains (TEMs). PLoS One 3:e3313. 10.1371/journal.pone.0003313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Surviladze Z, Dziduszko A, Ozbun MA. 2012. Essential roles for soluble virion-associated heparan sulfonated proteoglycans and growth factors in human papillomavirus infections. PLoS Pathog. 8:e1002519. 10.1371/journal.ppat.1002519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scheffer KD, Gawlitza A, Spoden GA, Zhang XA, Lambert C, Berditchevski F, Florin L. 2013. Tetraspanin CD151 mediates papillomavirus type 16 endocytosis. J. Virol. 87:3435–3446. 10.1128/JVI.02906-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schelhaas M, Shah B, Holzer M, Blattmann P, Kuhling L, Day PM, Schiller JT, Helenius A. 2012. Entry of human papillomavirus type 16 by actin-dependent, clathrin- and lipid raft-independent endocytosis. PLoS Pathog. 8:e1002657. 10.1371/journal.ppat.1002657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spoden G, Kuhling L, Cordes N, Frenzel B, Sapp M, Boller K, Florin L, Schelhaas M. 2013. Human papillomaviruses type 16, 18, and 31 share similar endocytic requirements for entry. J. Virol. 87:7765–7773. 10.1128/JVI.00370-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Selinka HC, Giroglou T, Sapp M. 2002. Analysis of the infectious entry pathway of human papillomavirus type 33 pseudovirions. Virology 299:279–287. 10.1006/viro.2001.1493 [DOI] [PubMed] [Google Scholar]
- 19.Dabydeen SA, Meneses PI. 2009. The role of NH4Cl and cysteine proteases in human papillomavirus type 16 infection. Virol. J. 6:109. 10.1186/1743-422X-6-109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith JL, Campos SK, Wandinger-Ness A, Ozbun MA. 2008. Caveolin-1-dependent infectious entry of human papillomavirus type 31 in human keratinocytes proceeds to the endosomal pathway for pH-dependent uncoating. J. Virol. 82:9505–9512. 10.1128/JVI.01014-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bienkowska-Haba M, Williams C, Kim SM, Garcea RL, Sapp M. 2012. Cyclophilins facilitate dissociation of the human papillomavirus type 16 capsid protein L1 from the L2/DNA complex following virus entry. J. Virol. 86:9875–9887. 10.1128/JVI.00980-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Day PM, Lowy DR, Schiller JT. 2003. Papillomaviruses infect cells via a clathrin-dependent pathway. Virology 307:1–11. 10.1016/S0042-6822(02)00143-5 [DOI] [PubMed] [Google Scholar]
- 23.Müller KH, Kainov DE, El Bakkouri K, Saelens X, De Brabander JK, Kittel C, Samm E, Muller CP. 2011. The proton translocation domain of cellular vacuolar ATPase provides a target for the treatment of influenza A virus infections. Br. J. Pharmacol. 164:344–357. 10.1111/j.1476-5381.2011.01346.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Müller KH, Kakkola L, Nagaraj AS, Cheltsov AV, Anastasina M, Kainov DE. 2012. Emerging cellular targets for influenza antiviral agents. Trends Pharmacol. Sci. 33:89–99. 10.1016/j.tips.2011.10.004 [DOI] [PubMed] [Google Scholar]
- 25.Buck CB, Pastrana DV, Lowy DR, Schiller JT. 2004. Efficient intracellular assembly of papillomaviral vectors. J. Virol. 78:751–757. 10.1128/JVI.78.2.751-757.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schneider MA, Spoden GA, Florin L, Lambert C. 2011. Identification of the dynein light chains required for human papillomavirus infection. Cell. Microbiol. 13:32–46. 10.1111/j.1462-5822.2010.01515.x [DOI] [PubMed] [Google Scholar]
- 27.Kondo K, Ishii Y, Ochi H, Matsumoto T, Yoshikawa H, Kanda T. 2007. Neutralization of HPV16, 18, 31, and 58 pseudovirions with antisera induced by immunizing rabbits with synthetic peptides representing segments of the HPV16 minor capsid protein L2 surface region. Virology 358:266–272. 10.1016/j.virol.2006.08.037 [DOI] [PubMed] [Google Scholar]
- 28.Leder C, Kleinschmidt JA, Wiethe C, Muller M. 2001. Enhancement of capsid gene expression: preparing the human papillomavirus type 16 major structural gene L1 for DNA vaccination purposes. J. Virol. 75:9201–9209. 10.1128/JVI.75.19.9201-9209.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pastrana DV, Buck CB, Pang YY, Thompson CD, Castle PE, FitzGerald PC, Kruger Kjaer S, Lowy DR, Schiller JT. 2004. Reactivity of human sera in a sensitive, high-throughput pseudovirus-based papillomavirus neutralization assay for HPV16 and HPV18. Virology 321:205–216. 10.1016/j.virol.2003.12.027 [DOI] [PubMed] [Google Scholar]
- 30.Pastrana DV, Gambhira R, Buck CB, Pang YY, Thompson CD, Culp TD, Christensen ND, Lowy DR, Schiller JT, Roden RB. 2005. Cross-neutralization of cutaneous and mucosal Papillomavirus types with anti-sera to the amino terminus of L2. Virology 337:365–372. 10.1016/j.virol.2005.04.011 [DOI] [PubMed] [Google Scholar]
- 31.Mossadegh N, Gissmann L, Muller M, Zentgraf H, Alonso A, Tomakidi P. 2004. Codon optimization of the human papillomavirus 11 (HPV 11) L1 gene leads to increased gene expression and formation of virus-like particles in mammalian epithelial cells. Virology 326:57–66. 10.1016/j.virol.2004.04.050 [DOI] [PubMed] [Google Scholar]
- 32.Lebreton S, Xie X-S, Ferguson D, De Brabander JK. 2004. Ring-closing metathesis: a powerful tool for the synthesis of simplified salicylihalamide-based V-ATPase inhibitors. Tetrahedron 60:9635–9647. 10.1016/j.tet.2004.06.146 [DOI] [Google Scholar]
- 33.Wu Y, Liao X, Wang R, Xie XS, De Brabander JK. 2002. Total synthesis and initial structure-function analysis of the potent V-ATPase inhibitors salicylihalamide A and related compounds. J. Am. Chem. Soc. 124:3245–3253. 10.1021/ja0177713 [DOI] [PubMed] [Google Scholar]
- 34.Lebreton S, Jaunbergs J, Roth MG, Ferguson DA, De Brabander JK. 2008. Evaluating the potential of vacuolar ATPase inhibitors as anticancer agents and multigram synthesis of the potent salicylihalamide analog saliphenylhalamide. Bioorg. Med. Chem. Lett. 18:5879–5883. 10.1016/j.bmcl.2008.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xie XS, Padron D, Liao X, Wang J, Roth MG, De Brabander JK. 2004. Salicylihalamide A inhibits the V0 sector of the V-ATPase through a mechanism distinct from bafilomycin A1. J. Biol. Chem. 279:19755–19763. 10.1074/jbc.M313796200 [DOI] [PubMed] [Google Scholar]
- 36.Rommel O, Dillner J, Fligge C, Bergsdorf C, Wang X, Selinka HC, Sapp M. 2005. Heparan sulfate proteoglycans interact exclusively with conformationally intact HPV L1 assemblies: basis for a virus-like particle ELISA. J. Med. Virol. 75:114–121. 10.1002/jmv.20245 [DOI] [PubMed] [Google Scholar]
- 37.Scheffer KD, Popa-Wagner R, Florin L. 2013. Isolation and characterization of pathogen-bearing endosomes enable analysis of endosomal escape and identification of new cellular cofactors of infection. Methods Mol. Biol. 1064:101–113. 10.1007/978-1-62703-601-6_7 [DOI] [PubMed] [Google Scholar]
- 38.Campos SK, Chapman JA, Deymier MJ, Bronnimann MP, Ozbun MA. 2012. Opposing effects of bacitracin on human papillomavirus type 16 infection: enhancement of binding and entry and inhibition of endosomal penetration. J. Virol. 86:4169–4181. 10.1128/JVI.05493-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sapp M, Kraus U, Volpers C, Snijders PJ, Walboomers JM, Streeck RE. 1994. Analysis of type-restricted and cross-reactive epitopes on virus-like particles of human papillomavirus type 33 and in infected tissues using monoclonal antibodies to the major capsid protein. J. Gen. Virol. 75(Pt 12):3375–3383. 10.1099/0022-1317-75-12-3375 [DOI] [PubMed] [Google Scholar]
- 40.Buck CB, Thompson CD, Roberts JN, Muller M, Lowy DR, Schiller JT. 2006. Carrageenan is a potent inhibitor of papillomavirus infection. PLoS Pathog. 2:e69. 10.1371/journal.ppat.0020069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spoden GA, Besold K, Krauter S, Plachter B, Hanik N, Kilbinger AF, Lambert C, Florin L. 2012. Polyethylenimine is a strong inhibitor of human papillomavirus and cytomegalovirus infection. Antimicrob. Agents Chemother. 56:75–82. 10.1128/AAC.05147-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang HS, Pyeon D, Pearce SM, Lank SM, Griffin LM, Ahlquist P, Lambert PF. 2012. Novel antivirals inhibit early steps in HPV infection. Antiviral Res. 93:280–287. 10.1016/j.antiviral.2011.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lipovsky A, Popa A, Pimienta G, Wyler M, Bhan A, Kuruvilla L, Guie MA, Poffenberger AC, Nelson CD, Atwood WJ, Dimaio D. 2013. Genome-wide siRNA screen identifies the retromer as a cellular entry factor for human papillomavirus. Proc. Natl. Acad. Sci. U. S. A. 110:7452–7457. 10.1073/pnas.1302164110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Brabander JK, Wu Y. May 2004. Synthetic salicylihalamides, apicularens and derivatives thereof. US patent 6,734,209 B2
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.