

Abstract
Genotype 6 (GT6) hepatitis C virus (HCV) is prevalent in Southeast Asia and southern China, where it can constitute up to 50% of HCV infections. Despite this, no direct-acting antivirals are approved to treat GT6 HCV infection, and no cell culture systems have been described. In this study, we aimed to develop a GT6 HCV subgenomic replicon to facilitate the identification and development of new HCV therapies with pan-genotype activity. A subgenomic replicon cDNA encoding a GT6a consensus sequence plus an NS5A amino acid substitution (S232I) was synthesized. Electroporation of RNA encoding the GT6a replicon into Huh-7-derived cells consistently yielded 20 to 100 stable replicon colonies. Genotypic analyses of individual replicon colonies revealed new adaptive mutations across multiple viral nonstructural proteins. The E30V and K272R mutations in NS3 and the K34R mutation in NS4A were observed most frequently and were confirmed to enhance GT6a replicon replication in the presence of the NS5A amino acid substitution S232I. These new adaptive mutations allowed establishment of robust luciferase-encoding GT6a replicons for reproducible quantification of HCV replication, and the luciferase-encoding replicons enabled efficient determinations of antiviral activity for HCV inhibitors in a 384-well assay format. While nucleoside/nucleotide NS5B inhibitors and cyclophilin A inhibitors had similar antiviral activities against both GT6a and GT1b replicons, some nonnucleoside NS5B inhibitors, NS3 protease inhibitors, and NS5A inhibitors had less antiviral activity against GT6a replicons. In conjunction with other genotype replicons, this robust GT6a replicon system will aid in the development of pan-genotypic HCV regimens.
INTRODUCTION
Chronic hepatitis C virus (HCV) infection affects an estimated 170 million people worldwide and represents a significant global health burden (1, 2). Until recently, the standard of care was 24- to 48-week courses of pegylated alpha interferon (PegIFN) plus ribavirin (RBV) (3). Due to the partial efficacy and poor tolerability of this regimen, the discovery and development of new antiviral agents have been pursued intensely. These efforts have culminated in the recent FDA approval of two NS3 protease inhibitors (boceprevir and telaprevir) for use in combination with PegIFN and RBV for the treatment of chronic genotype 1 (GT1) HCV infection (4).
HCV is a positive-strand RNA virus that exhibits extraordinary genetic diversity. Six major genotypes (genotypes 1 to 6) and multiple subtypes (e.g., genotypes 1a and 1b) have been reported (5). Genotypes 1, 2, and 3 are common throughout the world (6–8). However, GT6 is prevalent in Southeast Asia and southern China and can constitute up to 50% of HCV infections in many of these areas (9, 10). Despite its limited geographical presence, GT6 represents a significant portion of the global unmet medical need associated with chronic HCV infection, due to the high HCV disease burdens in Southeast Asia and southern China (with more than 32 million people infected). Furthermore, in contrast to the case in North America and Europe, the occurrence of new incidences of HCV infection also remains high in these regions due to a higher risk of exposure to contaminated blood products and intravenous drug use (9, 10).
Currently, the standard treatment for GT6 HCV patients remains PegIFN and RBV for 24 to 48 weeks (10). Although GT6 infection is more responsive to PegIFN-RBV than GT1 infection is (sustained virologic responses of 86% and 52%, respectively) (11), this treatment is still partially efficacious and contraindicated in many patients. No direct-acting antivirals (DAAs) have been approved to treat GT6 HCV infection (4). Many HCV DAAs are in advanced clinical development, but few are being developed to treat GT6 infections. Thus, there is an urgent need to develop novel therapeutic agents for the treatment of chronic GT6 HCV infection. This need also aligns with the tremendous interest in developing “pan-genotypic” drugs that are active against all HCV genotypes to simplify the treatment of HCV (12, 13).
GT6 is the most genetically diverse HCV genotype, with at least 23 currently known subtypes and new subtypes expected to be identified continuously (14). It is well documented that individual HCV genotypes respond differently to direct-acting antivirals due to high HCV genetic diversity between and within genotypes (3, 15). For example, essentially all HCV NS3 protease inhibitors, although potent against GT1, have significantly reduced antiviral activity against GT3; this is due largely to GT3 polymorphisms at known drug resistance sites within NS3 protease, including residue 168 (16). For NS5A inhibitors, earlier compounds often inhibit the GT2a JFH-1 virus efficiently but have much weaker antiviral activities (>200-fold) against more common GT2 strains carrying the M31 polymorph in NS5A (17). The considerable genetic diversity of GT6, combined with a limited virological characterization of this genotype compared to common GT1 strains, creates significant challenges to DAA development against this genotype. Meeting this challenge will require the establishment of efficient GT6 HCV tools for the identification and development of new therapies.
HCV replicons are self-replicating viral RNAs that have served as workhorses for molecular virology studies and drug discovery (18). These replicons have been crucial in the identification of novel inhibitor classes, the optimization of clinical candidates, and the characterization of clinical resistance. Despite initial successes in generating replicons derived from genotype 1a, 1b, or 2a (19–21), it has proven difficult to generate efficiently replicating RNAs from other genotypes (22, 23). Not until very recently were GT3 and GT4 replicons successfully established, representing a significant expansion of virological tools for HCV research and drug development (24–26).
Here we report the establishment of the first GT6a replicons that efficiently replicate in hepatoma cell lines. We demonstrate that robust replication requires novel adaptive mutations in either HCV NS3 or NS4A, combined with the NS5A amino acid substitution S232I. By incorporating adaptive mutations into luciferase-encoding constructs, we established efficient replication of GT6a HCV RNA in both stably replicating and transiently transfected cultures. The replicons support efficient activity profiling of antiviral compounds and will be valuable tools for molecular virology and drug development applications.
MATERIALS AND METHODS
Cell culture.
Huh-7 Lunet cells were obtained from ReBLikon GmbH (Mainz, Germany) (27). 51C cells were derived by curing a Huh-7 Lunet-based GT1a replicon clone and were described previously (28). 1C cells were derived by curing a GS-5885-resistant GT1a replicon clone derived from 51C cells, and they showed much more permissiveness to GT1a replicon replication (24). All cell lines were propagated in Dulbecco's modified Eagle's medium (DMEM) as described previously (29). Replicon cell lines were selected and maintained in complete DMEM containing 0.25 to 0.5 mg/ml G418 (Geneticin; Invitrogen).
Construction of plasmids encoding GT6a HCV subgenomic replicons.
A plasmid (pGT6aNeoSG) encoding a subgenomic GT6a replicon based on the consensus sequence (see Fig. S1 in the supplemental material) of 16 GT6a genomes available in the European HCV database (see Table S1) was prepared by DNA synthesis and cloning (GenScript, Piscataway, NJ). The synthesized replicon incorporated the following elements (5′ to 3′) (Fig. 1): (i) the GT6a consensus 5′-untranslated region (5′-UTR) (342 nucleotides [nt]) plus the first 48 nt of the core region; (ii) a linker with the nucleotide sequence 5′-GGCGCGCCCA-3′, which introduces the AscI restriction site (underlined); (iii) the neomycin phosphotransferase II (neo) gene; (iv) a linker with the nucleotide sequence 5′-GGCCGGCCA-3′, which introduces the FseI restriction site (underlined); (v) the encephalomyocarditis virus (EMCV) internal ribosome entry site (IRES); (vi) a linker with the nucleotide sequence 5′-ACGCGTATG-3′, which introduces the MluI restriction site (underlined) and an ATG start codon for HCV polyprotein expression; (vii) the NS3-NS5B polyprotein region of the GT6a consensus sequence, including the sequence encoding the NS5A amino acid substitution S232I (equivalent to an S2204I substitution in the GT1 polyprotein); and (viii) the 3′-UTR of GT2a JFH-1 (239 nt; no GT6a 3′-UTR is available). The synthetic DNA fragment encoding the GT6a replicon was inserted into pUC57, between EcoRI and XbaI restriction sites.
FIG 1.

Schematic diagrams of GT6a subgenomic replicons used in this study. Elements engineered into the replicons included the neomycin phosphotransferase II gene (Neo) (A), a renilla luciferase (Rluc)-neomycin phosphotransferase II gene fusion reporter (Rluc-Neo) (B), and a poliovirus IRES-driven Rluc reporter (Pi-Rluc) (C). All replicons carried the GT6a 5′-UTR; an EMCV IRES controlling translation of the NS3-NS5B polyprotein region of the GT6a consensus sequence, including an NS5A amino acid substitution (S232I); and the 3′-UTR of GT2a JFH-1 virus. Black bars indicate the GT6a core N-terminal sequence. Gray bars indicate the HCV polyprotein sequence. Neo and Rluc-Neo translation was controlled by an HCV IRES (A and B), whereas Rluc translation was controlled by a Pi-IRES (C).
A second plasmid (pGT6aRlucNeoSG), encoding a subgenomic replicon that incorporated the humanized renilla luciferase (hRluc) reporter gene, was generated as follows. The pGT6aNeoSG plasmid (described above) was cut using the AscI and MluI restriction enzymes (to remove the neo gene) and then gel purified using a commercial kit (Qiagen, Valencia, CA). A gene fragment carrying the hRluc gene fused with the neo gene, along with the EMCV region from the phRlucNeoSG2a plasmid (described below), was PCR amplified using Accuprime supermix I (Invitrogen) and the following primers: 2aRlucNeoAscIFor (5′-AAC ACC ATC GGC GCG CCC ATG GCT TCC AAG GTG TAC GAC-3′; the AscI site was introduced by the primer and is underlined) and 2aEMCVIRESMluIRev (5′-TCG GGG CCA TAC GCG TAT CGT GTT TTT CAA AGG-3′; the MluI site is underlined). The subsequent PCR fragment was cut with AscI and MluI and gel purified using a commercial kit (Qiagen). The vector and insert pieces were ligated using the LigaFast Rapid DNA ligation system per the manufacturer's protocol (Promega, Madison, WI). The resulting vector, pGT6aRlucNeoSG, was sequenced to confirm the correct orientation and sequence of the hRluc-Neo cassette. The phRlucNeoSG2a plasmid was constructed by replacing the Luc-Neo fragment in the pLucNeoSG2a plasmid with the hRluc-Neo gene cassette amplified from the plasmid hRluc-Neo Flexi (Promega), as previously described (28, 29).
A third plasmid (pGT6aPiRlucSG), encoding a bicistronic replicon with the hRluc reporter gene downstream of the poliovirus IRES (Pi-IRES) and the GT6a consensus HCV nonstructural genes (NS3 to NS5B) downstream of the EMCV IRES, was used for transient-transfection studies. The plasmid was generated as follows. The pGT6aRlucNeoSG plasmid (described above) was cut using the AscI and MluI restriction enzymes (to remove the Rluc-Neo gene cassette) and then gel purified using a commercial kit (Qiagen). A gene fragment carrying the Pi-IRES, hRluc gene, and EMCV IRES regions was PCR amplified from the GT1b Pi-Rluc plasmid by using Accuprime supermix I (Invitrogen) and the following primers: 6aPiRlucAscIFor (5′-AAC ACC ATC GGC GCG CCA AAC CAA GTT CAA TAG-3′; the AscI site was introduced by the primer and is underlined) and 1bEMCVIRESMluIRev (5′-TCG GGG CCA TAC GCG TAT CGT GTT TTT CAA AGG-3′; the MluI site is underlined). The subsequent PCR fragment was cloned and ligated as described above to generate the resulting vector, pGT6aPiRlucSG.
Construction of mutant replicons.
Adaptive mutations were introduced into the pGT6aNeoSG, pGT6aRlucNeoSG, or pGT6aPiRlucSG replicon by site-directed mutagenesis using a QuikChange Lightning kit (Stratagene, La Jolla, CA). A replication-defective NS5B polymerase GND mutant was constructed by introducing a point substitution in the nucleotide sequence that encodes amino acid 318 in NS5B, changing the sequence from GAT to AAT, and therefore altering the GDD motif of the NS5B polymerase active site to GND and abolishing the polymerase activity (GenScript) (20). All substitutions were confirmed by DNA sequencing by Elim Biopharm (Hayward, CA).
RNA transcription.
Plasmids encoding GT6a subgenomic HCV replicons were linearized with XbaI and purified using a PCR purification kit (Qiagen). RNAs were synthesized and purified with T7 MEGAScript (Ambion, Austin, TX) and RNeasy (Qiagen) kits, respectively, according to the manufacturers' instructions. RNA concentrations were measured using the optical density at 260 nm and were confirmed by 0.8% agarose gel electrophoresis (Invitrogen).
RNA transfection and isolation of stable replicon cell lines.
Ten micrograms of in vitro-transcribed RNA was transfected into Huh-7 Lunet or 1C cells by electroporation as previously described (28). Briefly, cells were collected by trypsinization and centrifugation and then washed twice with ice-cold phosphate-buffered saline (PBS) and resuspended in Opti-MEM medium (Invitrogen) at a concentration of 107 cells/ml. Replicon RNA was added to 400 μl of cell suspension in a Gene Pulser (Bio-Rad, Hercules, CA) cuvette (0.4-cm gap). Cells were electroporated at 270 V and 960 μF, incubated at room temperature for 10 min, resuspended in 30 ml complete DMEM, and then plated in two 100-mm-diameter dishes. Forty-eight hours after plating, the medium was replaced with complete DMEM supplemented with 0.25 mg/ml G418, which was refreshed twice per week. After 3 weeks, cell clones were isolated, expanded with 0.5 mg/ml G418, and cryopreserved at early passages.
Replicon colony formation assays.
To determine the efficiency of replicon colony formation, cells were electroporated with the indicated amounts of replicon RNA or extracted cellular RNA and then plated at multiple densities, ranging from 2 × 105 to 2 × 106 cells/100-mm dish. Forty-eight hours after plating, the medium was replaced with complete DMEM supplemented with 0.5 mg/ml G418, which was refreshed twice per week. Three weeks later, colony plates were used for cell expansion or G418-resistant foci were fixed with 4% formaldehyde and stained with 0.05% crystal violet.
Extraction, amplification, and genotypic analysis of HCV RNA.
HCV RNA isolation, reverse transcription-PCR (RT-PCR), and population sequencing were performed as described previously (29). Briefly, HCV replicon cellular RNAs were extracted and purified using an RNeasy kit (Qiagen) according to the manufacturer's protocol. RT-PCR was performed using the SuperScript III first-strand synthesis system (Invitrogen), and PCR products were subsequently sequenced by Elim Biopharm (Hayward, CA).
Replicon cell NS3-4A protease assay.
Replicon cells were seeded in 96-well plates at a concentration of 1 × 104 cells per well. The cells were incubated for 24 h, after which the culture medium was removed. Replicon cells were then lysed with 90 μl of 1× Promega luciferase lysis buffer supplemented with 150 mM NaCl at room temperature for 20 min on a plate shaker. Ten microliters of 1 μM europium-labeled NS3-4A substrate diluted in the above lysis buffer was added to each well. Protease activity data were collected and analyzed as previously described (29).
Detection of NS5A protein by indirect immunofluorescence.
Replicon cells were plated in 96-well plates at a density of 1 × 104 cells per well. After incubation for 24 h, cells were then stained for NS5A protein as described previously (29). Briefly, cells were fixed in 4% paraformaldehyde for 20 min. Cells were then washed three times with PBS and blocked with 3% bovine serum albumin, 0.5% Triton X-100, and 10% fetal bovine serum (FBS). NS5A protein staining was performed using a 1:10,000 dilution of mouse monoclonal antibody 9E10 (Apath, Brooklyn, NY). After washing in PBS three times, a secondary anti-mouse antibody conjugated to Alexa Fluor 555 was used to detect anti-NS5A antibody-labeled cells (Invitrogen). Nuclei were stained with 1 μg/ml Hoechst 33342 dye (Invitrogen). Cells were washed with PBS and imaged with a Zeiss fluorescence microscope (Zeiss, Thornwood, NY).
Replicon antiviral assays.
Replicon RNAs were electroporated into 1C cells as described above. After transfection, cells were quickly transferred to 100 ml of prewarmed culture medium, and 90-μl samples were seeded in 384-well plates at a density of 2,000 cells/well. Cells were treated with 3-fold serial drug dilutions for a total of 10 different concentrations. Cell plates were incubated at 37°C for 3 days, after which the culture medium was removed and cells were assayed for luciferase activity as a marker of the replicon level. Luciferase expression was quantified using a commercial luciferase assay (Promega). Luciferase levels were converted into percentages relative to the levels in the untreated controls (defined as 100%), and data were fitted to the logistic dose-response equation y = a/[1 + (x/b)c] by using XLFit4 software (IDBS, Emeryville, CA).
Antiviral compounds.
Telaprevir, boceprevir, and 2-C-methyladenosine (2-CMeA) were purchased from Acme Bioscience (Belmont, CA). Cyclosporine (CsA) was purchased from Sigma-Aldrich (St. Louis, MO). The Wyeth HCV NS5B site IV inhibitor HCV-796 was synthesized by Curragh Chemistries (Cleveland, OH). Ledipasvir (GS-5885) (30), tegobuvir (GS-9190) (31), GS-9451 (32), GS-9669 (33), sofosbuvir (GS-7977) (34), the Pfizer NS5B thumb site II inhibitor filibuvir, the Merck NS5B thumb site I inhibitor MK-3281 and protease inhibitor MK-5172, and the Bristol-Myers Squibb NS5A inhibitor daclatasvir (BMS-790052) were synthesized by Gilead Sciences.
RESULTS
Construction of a GT6a subgenomic replicon.
The consensus sequence from 16 GT6a genomes available in the European HCV database was used to construct a GT6a subgenomic replicon (GT6a-Neo) as previously described by Lohmann et al. (22) (Fig. 1A). Due to the lack of a complete GT6a 3′-UTR sequence in the database, the 3′-UTR sequence from the highly replicative GT2a HCV isolate JFH-1 was used to construct the GT6a replicon. In addition, the sequence encoding the NS5A amino acid substitution S232I, which was previously shown to enhance the replication of HCV replicons for multiple genotypes, was included to determine if it could enhance the basal replication of the GT6a replicon (23, 24, 26).
Selection of stable GT6a subgenomic replicon colonies.
In vitro-transcribed GT6a-Neo replicon RNAs, with or without the NS5A amino acid substitution S232I, were electroporated into Huh-7-derived Lunet or 1C cells and subjected to G418 selection until stable colonies emerged. In the absence of the S232I substitution, no G418-resistant colonies emerged in either cell background (Fig. 2A). In contrast, in the presence of the S232I substitution, a modest number of G418-resistant colonies were selected in both Lunet cells (average of 35 colonies per transfection) and 1C cells (average of 75 colonies per transfection) (Fig. 2A). These results indicate that the NS5A amino acid substitution S232I enhanced the replication of the parental GT6a replicon and was vital to the selection of stable replicon colonies. Colony formation efficiency was consistently 2-fold greater in 1C cells than in Lunet cells, suggesting that 1C cells provided a slightly more permissive environment for GT6a replicon replication.
FIG 2.

Establishment of stable GT6a replicons. (A) Selection of stable GT6a replicon colonies. In vitro-transcribed RNAs encoding GT6a-Neo replicons, with and without the NS5A S232I substitution, were transfected into Lunet or 1C cells and subjected to G418 selection (0.5 mg/ml). G418-resistant colonies emerged after approximately 4 weeks of treatment and were counted. The values represent the means for three independent assays, with standard errors. (B) Intracellular NS3-4A protease activity in pooled GT6a replicon cells. Cells were plated at a concentration of 5 × 104 cells per well in 96-well plates, and NS3-4A protease activity was determine at 24 h post-cell seeding. RFUs, relative fluorescence units. (C) Expression of NS5A protein in pooled GT6a replicon cells. Pooled GT6a-Neo 1C or Lunet cells were stained with anti-NS5A antibody (red) and Hoechst 33342 dye (blue; indicates nuclei). Parental 1C cells were stained as a negative control.
To confirm that the selected colonies harbored replicating GT6a replicons, colonies were pooled and assessed for intracellular NS3-4A expression by measuring NS3-4A protease activity (Fig. 2B) and for NS5A expression by immunofluorescence staining (Fig. 2C). Both the pooled GT6a-neo 1C and GT6a-neo Lunet colonies had readily detectable NS3-4A protease activity, producing signal-to-background ratios >100-fold and signals within 2-fold of those produced by a highly adapted GT1b replicon (28). Similarly, NS5A expression was also readily detectable when the pooled cells were stained with an anti-NS5A antibody (Fig. 2C). Together, these results suggest that self-replicating GT6a subgenomic replicons were successfully established in these stable colonies.
Identification of adaptive mutations for GT6a subgenomic replicons.
To determine whether adaptive mutations were acquired by the replicons in the selected colonies, 11 Lunet and 12 1C stable colonies were isolated and expanded for genotypic analysis. A diverse set of mutations were identified in the viral nonstructural genes (the full list of emerging mutations is provided in Table S2 in the supplemental material). Seventeen of 23 clones had single mutations in the NS3 gene, including one with an additional mutation in the NS5A gene. Five clones had a mutation in the NS4A gene, and one had a single mutation in the NS5B gene. Furthermore, all replicon clones retained the NS5A amino acid substitution S232I, suggesting that this substitution may be essential for GT6a replicon replication.
Interestingly, the single mutations E30V and K272R in NS3 and K34R in NS4A appeared independently at least twice. To determine if these three mutations enhanced replicon replication, they were introduced individually into a GT6a renilla luciferase gene-neo construct (GT6a-RlucNeo) (Fig. 1B) encoding the S232I amino acid substitution. In vitro-transcribed RNA from each mutant was electroporated into permissive Huh-7 Lunet cells and subjected to G418 selection until colonies were visible (Fig. 3). Incorporation of renilla luciferase into the GT6a-Neo replicon resulted in a significant reduction of stable colony formation (35 colonies for the GT6a-Neo replicon versus 1 colony for the GT6a-RlucNeo replicon). Importantly, all three of the putative adaptive mutations significantly enhanced replication of GT6a replicons, as evidenced by the formation of a large number of stable colonies relative to the number with the parental GT6a-RlucNeo replicon (Fig. 3). These data indicate that the E30V and K272R mutations in NS3 and the K34R mutation in NS4A function as adaptive mutations for GT6a replicon replication.
FIG 3.

NS3 and NS4A substitutions enhance replication of GT6a replicons. In vitro-transcribed GT6a-RlucNeo replicon RNAs, from either the parental replicon or replicons engineered to encode the indicated NS3 substitution (E30V or K272R) or NS4A substitution (K34R), were transfected into Lunet cells and subjected to selection by G418 (0.5 mg/ml). G418-resistant colonies harboring GT6a replicons emerged after approximately 3 weeks and were visualized by staining with crystal violet. All replicons encode the NS5A amino acid substitution S232I.
Establishment of luciferase-encoding GT6a replicon transient-transfection replication systems.
Transient-transfection replication systems facilitate rapid and direct analysis of replication efficiencies and kinetics of HCV RNAs, independent of clonal cell differences. A GT6a replicon transient-replication construct was generated by replacing the RlucNeo gene in the parental GT6a-RlucNeo replicon with an Rluc reporter gene that is much shorter and under translational control of the poliovirus IRES (Pi-IRES) instead of the HCV IRES (GT6a-PiRluc) (Fig. 1C) (35). The three adaptive mutations identified in NS3 (E30V and K272R) and NS4A (K34R) were introduced individually into the GT6a-PiRluc replicon encoding the S232I amino acid substitution in NS5A. The impact of each mutation on replication efficiency and kinetics was determined, and the results are shown in Fig. 4. In vitro-transcribed GT6a-PiRluc parental replicon RNA and replicon RNAs with the indicated mutations, along with the highly adapted GT1b-PiRluc replicon, were electroporated into both Huh-7 Lunet and 1C cells. HCV replication was quantified through measurement of luciferase activity in cell lysates over 7 days. All mutations in either NS3 or NS4A significantly enhanced GT6a replicon replication, as evidenced by the higher luciferase activities (>30-fold) of the mutants than those of the parental GT6a-PiRluc construct and the replication-defective NS5B polymerase GND mutant. Particularly in 1C cells, although the GT6a-PiRluc replicons with adaptive mutations had slower replication kinetics than the GT1b-PiRluc replicon, their replication capacities eventually all reached a level comparable to that of GT1b-PiRluc by day 5 after transfection. These results indicate that all three adaptive mutations, in combination with the S232I mutation in NS5A, could support transient-transfection replication studies of GT6a replicons in both Huh-7 Lunet and 1C cells.
FIG 4.
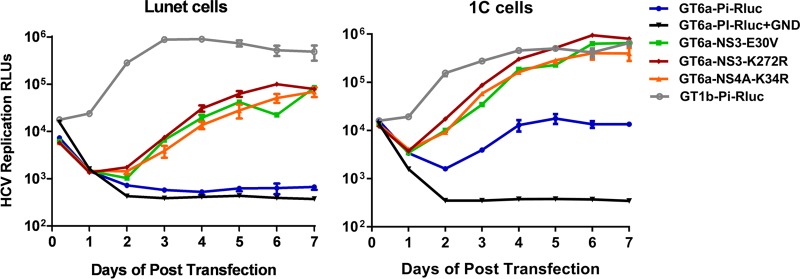
NS3 and NS4A substitutions confer high-level GT6a HCV replication. NS3 (E30V and K272R) and NS4A (K34R) substitutions were introduced into the GT6a-PiRluc replicon, harboring the S232I substitution in NS5A. In vitro-transcribed replicon RNAs were transfected into either Lunet or 1C cells and plated in 96-well plates at a density of 5 × 103 cells/well. Renilla luciferase activity (relative light units [RLUs]) was measured as a marker of HCV replication at the indicated time points. A replication-defective NS5B polymerase GND mutant was included as a negative control. The background of the assay was ∼100 RLUs. All values represent the means for two independent assays, with standard errors.
Interestingly, the replication levels of all GT6a-PiRluc replicons were approximately 10-fold lower in Huh-7 Lunet cells than in 1C cells (Fig. 4). This is in contrast to the case for the GT1b-PiRluc replicon, which replicates at similar levels in these two Huh-7-derived cell lines, suggesting that 1C cells are likely more permissive than Lunet cells for GT6a replicon replication. To determine whether the adaptive functions of the selected mutations were dependent on the NS5A amino acid substitution S232I, we reverted the S232I mutation back to the wild type (S232) in GT6a-PiRluc replicons encoding the NS3 K272R or NS4A K34R adaptive mutation. Figure 5 shows that in the absence of the NS5A S232I mutation, while the wild-type sequence GT6a replicon had a very weak replication activity (∼5-fold above that of the GND negative control), neither the NS3 K272R mutation nor the NS4A K34R mutation alone enhanced the basal replication of the GT6 replicon. In contrast, the S232I mutation alone could boost replicon replication ∼10-fold. Together, these data suggest that the NS5A S232I mutation is critical and that the K272R mutation in NS3 and the K34R mutation in NS4A are synergistic with the S232I mutation for GT6a replicon replication.
FIG 5.
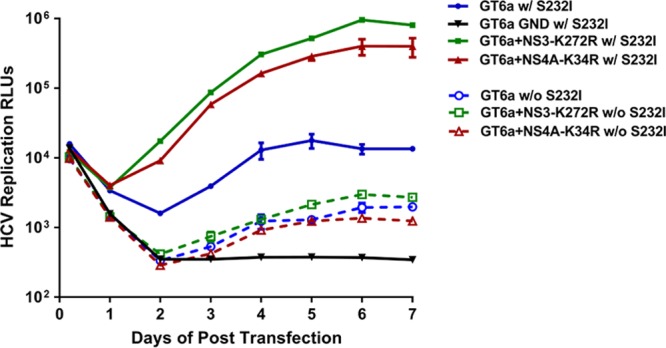
The NS5A amino acid substitution S232I is required for efficient GT6a HCV replication. The NS3 K272R and NS4A K34R substitutions were introduced individually into the GT6a-PiRluc replicon, with or without the S232I substitution in NS5A. In vitro-transcribed replicon RNAs were transfected into 1C cells and plated in 96-well plates at a density of 5 × 103 cells/well. Renilla luciferase activity (RLUs) was measured as a marker of HCV replication at the indicated time points. A replication-defective NS5B polymerase GND mutant was included as a negative control. The background of the assay was ∼100 RLUs. All values represent the means for two independent assays, with standard errors.
Antiviral activities of known HCV inhibitors against GT6a.
To evaluate GT6a replicons for antiviral inhibitor screening, we assessed the responses of GT6a-PiRluc replicons in parallel with the well-established GT1b-PiRluc replicon against five different classes of HCV inhibitors (Table 1). 1C cells were transiently transfected with GT6a-PiRluc-NS3-K272R or GT1b-PiRluc HCV RNA and seeded into 384-well plates for antiviral assays in a high-throughput assay format. Compounds were added to the cells and incubated for 72 h, after which luciferase activity was measured to determine replication levels. Fifty percent effective concentrations (EC50s) were generated successfully for all inhibitors and are shown in Table 1. Similar results were also generated with the GT6a-PiRluc-NS4A-K34R replicon (EC50 values varied <2-fold from those generated with GT6a-PiRluc-NS3-K272R [data not shown]). CsA, which targets host cyclophilin A, and the nucleoside NS5B inhibitors sofosbuvir and 2-CMeA had similar activities against GT6a and GT1b replicons. The nonnucleoside inhibitors targeting thumb site I (MK-3281) and palm site IV (HCV-796) of NS5B also had similar potencies against GT6a and GT1b replicons. In contrast, the thumb site II inhibitors filibuvir and GS-9669 and the palm site III/IV inhibitor tegobuvir were approximately 30-, 40-, and 200-fold less active, respectively, against GT6a replicons than against GT1b replicons. The NS5A inhibitor daclatasvir had slightly less antiviral activity (∼7-fold) against GT6a replicons than against GT1b replicons; ledipasvir was active against GT6a replicons, but approximately 300-fold less than the case for GT1b replicons. The lower antiviral activity of ledipasvir against GT6a replicons was not specific to 1C cells, because similar EC50 values were obtained with Lunet cells, and ledipasvir had equivalent activities against GT1b replicons in 1C and Lunet cells (data not shown). The NS3 protease inhibitors currently approved for treatment of GT1 HCV, boceprevir and telaprevir, each had similar activities against GT6a and GT1b replicons. MK-5172, a potentially pan-genotypic protease inhibitor, maintained potency against GT6a replicons, within a 4-fold range of its potency against GT1b replicons. Other protease inhibitors, including BILN-2061 and GS-9451, were less active against GT6a replicons than against GT1b replicons (17- and 8-fold, respectively). Overall, these results confirm that the GT6a replicon system is a valuable tool for evaluating a wide range of HCV inhibitors against GT6a replicons.
TABLE 1.
Antiviral activities of HCV inhibitors against GT6a repliconsa
| Drug class | Compound | EC50 (nM) |
GT6a/GT1b EC50 ratio | |
|---|---|---|---|---|
| GT6a replicon | GT1b replicon | |||
| NS3 protease inhibitors | Telaprevir | 208 ± 41 | 428 ± 22 | 0.5 |
| Boceprevir | 185 ± 42 | 181 ± 42 | 1.0 | |
| GS-9451 | 67 ± 15 | 8.3 ± 1.6 | 8.1 | |
| MK-5172 | 3.0 ± 0.4 | 0.74 ± 0.07 | 4.1 | |
| BILN-2061 | 17 ± 1.6 | 0.99 ± 0.1 | 17 | |
| NS5A inhibitors | Ledipasvir | 1.1 ± 0.08 | 0.004 ± 0.003 | 275 |
| Daclatasvir | 0.11 ± 0.05 | 0.016 ± 0.010 | 6.9 | |
| Nucleoside NS5B inhibitors | Sofosbuvir | 22 ± 6.5 | 98 ± 38 | 0.2 |
| 2-CMeA | 22 ± 8 | 86 ± 0.1 | 0.3 | |
| Nonnucleoside NS5B inhibitors | GS-9669 | 736 ± 18 | 3.5 ± 0.3 | 210 |
| MK-3281 | 1,311 ± 635 | 534 ± 47 | 2.5 | |
| Tegobuvir | 110 ± 21 | 2.6 ± 0.2 | 42 | |
| Filibuvir | 3,536 ± 93 | 112 ± 13 | 32 | |
| HCV-796 | 21 ± 0.5 | 20 ± 5.5 | 1.1 | |
| Host-targeting drugs | Cyclosporine | 65 ± 4.4 | 81 ± 1.4 | 0.8 |
Replicons GT6a-PiRluc-NS3-K272R (with S232I mutation in NS5A) and GT1b-PiRluc were transiently transfected into 1C cells, and antiviral assays were performed in a high-throughput 384-well format. All EC50s represent averages for at least two independent experiments, with standard deviations.
DISCUSSION
GT6 HCV is most common among patients in Southeast Asia and the surrounding regions, where there is a high prevalence of HCV infection. In this study, a robust GT6a replicon was established to aid in the discovery and development of new therapies for GT6 HCV infection. A subgenomic replicon cDNA encoding a GT6a consensus sequence plus the NS5A S232I substitution was synthesized. In Huh-7-derived cells, this replicon construct consistently yielded a modest number of stable GT6a colonies and allowed identification of multiple novel adaptive mutations for GT6 replicon replication, including the E30V and K272R mutations in NS3 and the K34R mutation in NS4A. These new adaptive substitutions enabled us to establish robust luciferase-encoding GT6a replicons for reproducible quantification of HCV replication and efficient antiviral activity profiling of different classes of HCV inhibitors in a high-throughput assay format.
Multiple factors have been attributed to the difficulties in establishing HCV replicons, including significant genetic divergence between genotypes, poor basal replication levels for other genotypes, and the lack of suitable host cell lines. For example, Huh7.5, a cell line derived from Huh-7 cells, became significantly more permissive to HCV replication due to an inactivating mutation in retinoic acid-inducible gene I (RIG-I) (36). Compared to our recent GT4a replicon development, Huh-7-derived Lunet cells were more permissive to GT6a replicons, with an average of 35 colonies per transfection versus an average of 0.5 colony per transfection for GT4a replicons (Fig. 2A) (24). Nevertheless, Lunet cells were still less permissive than 1C cells for GT6a HCV replication in both colony formation assays (Fig. 2A) and transient-transfection replicon replication assays (Fig. 4). The 1C cell line was a key to our success in GT4a replicon development (24). The high permissiveness of 1C cells for GT6a replicon replication, as well as GT3a replicon replication (37), suggests that these cells very likely have some unique cellular background to support HCV replicon replication and have the potential to further contribute to the establishment of additional HCV replicons of other genotypes and subtypes.
Inclusion of the S232I substitution in NS5A (2) was essential to the success of establishing the GT6a replicons. Without the S232I substitution, no GT6a replicon colonies emerged in either 1C or Lunet cells (Fig. 2A). Furthermore, the identified GT6a adaptive substitutions required the S232I mutation to confer efficient HCV RNA replication (Fig. 5). In the absence of S232T, neither the K272R mutation in NS3 nor the K34R mutation in NS4A was sufficient to meaningfully enhance the basal replication of GT6a replicons. In contrast, the S232I substitution alone was able to boost replicon replication significantly. These results are very similar to what we observed in the establishment of genotype 3a and 4a HCV replicons (24). Together, these data indicate that the NS5A S232I substitution could confer replication competence across HCV genotypes and should be considered strongly to aid in the establishment of replicons for other HCV genotypes/subtypes that have yet to demonstrate replication in vitro.
Of the adaptive substitutions identified, the K272R mutation in NS3 helicase was the most frequently observed, appearing independently in 6 of 23 colonies analyzed. Residue K272 is located in domain 1 of the NS3 helicase protein and is highly conserved (99.9%) across all HCV genotypes listed in the European Union HCV database. Crystallography studies of the HCV NS3 RNA helicase domain complexed with a single-stranded DNA oligonucleotide revealed that K272 lies outside the central DNA binding cavity and is involved in phosphate interactions with DNA, helping to stabilize its conformation (38). Therefore, the emergence of the adaptive substitution K272R in NS3 helicase suggests that regulation of helicase activity could be involved in cell culture adaption.
The adaptive substitution K34R in NS4A was also observed frequently, appearing in 3 of 23 colonies analyzed. Residue K34 is 79% conserved, with the adapted residue R34 observed in 20% of viral sequences across all HCV genotypes listed in the European Union database. K34 is located immediately downstream of the NS4A region that forms a noncovalent complex with NS3 to stimulate protease activity (39). It has been hypothesized that residue 34 substitutions may modulate NS3-4A complex formation; however, no direct evidence exists to support this hypothesis. Interestingly, the same substitution was identified in both genotype 1a and 1b replicons (3, 21). Similarly, in our GT4a replicon development, an adaptive substitution at residue 34 of NS4A was also identified (Q34K or Q34R) and was highly efficient at enhancing replication (24). These results thus suggest that a potential protein-protein interaction involving residue 34 in NS4A could have profound effects on HCV RNA replication in cell culture across genotypes.
Using robust GT6a-Pi-Rluc transient-transfection replicon systems encoding either the K272R mutation in NS3 or the K34R mutation in NS4A, we evaluated five classes of HCV inhibitors for antiviral activity against GT6a replicons in parallel with GT1b replicon in a 384-well assay format (Table 1). As expected, inhibitors targeting the NS5B active site or host factors had similar potencies against both GT1b and GT6a replicons, consistent with previous pan-genotypic studies (3, 13). Nonnucleoside NS5B polymerase, NS3 protease, and NS5A inhibitors exhibited differential activity between genotypes, depending on the compound and target binding site. Often, the fact that some compounds did not maintain potency in GT6a compared to GT1b replicon could be explained by genetic variations within the inhibitor target site between the genotypes. For example, the NS5B thumb site II inhibitor filibuvir was 32-fold less active against GT6a replicons than against GT1b replicon. Interestingly, the NS5B amino acid substitutions L419I and I482L, which are known to confer resistance to filibuvir in genotype 1b, are commonly present in GT6a NS5B sequences, and also in our GT6 replicon sequence (40). Regarding protease inhibitors, GS-9451 and BILN-2061 were less active against GT6a replicons than against GT1b replicon (8- and 17-fold, respectively). The loss of antiviral activity against GT6a replicons was likely due, at least in part, to the Q80K polymorph. The Q80K amino acid substitution mutant shows reduced susceptibilities to GS-9451 and BILN-2061 in GT1 replicons (6- and 3-fold, respectively [data not shown]) and is commonly present in GT6a NS3 sequences. Put together, these antiviral activity results prove the utility and efficiency of this GT6a replicon system for assessing activities of inhibitors across multiple classes.
Currently, a surrogate approach using chimeric replicons is used to study different GT6a genes (e.g., NS3, NS5A, and NS5B) in cell culture. These tools are limited because they often have low replication capacities and rely on the interplay of heterologous HCV sequences and proteins. The availability of this robust GT6a replicon system (in which all HCV nonstructural genes are GT6 sequences) will address the limitations of existing surrogate approaches. This GT6a replicon not only will become a valuable tool for the discovery and evaluation of HCV inhibitors targeting GT6a but also can be used in molecular virology studies and to evaluate resistant variants identified in both preclinical and clinical studies. Lastly, in conjunction with existing replicon systems, the GT6a replicon will contribute to the development of pan-genotypic HCV regimens for the treatment of HCV worldwide.
Supplementary Material
ACKNOWLEDGMENTS
All authors of this report are stockholders and employees of Gilead Sciences. This article contains information describing the in vitro activities of Gilead compounds currently in development as treatments for HCV.
M.Y. was involved in study design, data acquisition, and manuscript preparation. B.P. was involved in study design and data acquisition. K.C. was involved in analysis and interpretation of data and in manuscript preparation. R.G. and H.Y. were involved in acquisition and analysis of data. W.D. and G.C. were involved in the study concept, analysis and interpretation of data, study supervision, and manuscript preparation.
Footnotes
Published ahead of print 18 February 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01780-13.
REFERENCES
- 1.Negro F, Alberti A. 2011. The global health burden of hepatitis C virus infection. Liver Int. 31(Suppl 2):1–3. 10.1111/j.1478-3231.2011.02537.x [DOI] [PubMed] [Google Scholar]
- 2.Lavanchy D. 2011. Evolving epidemiology of hepatitis C virus. Clin. Microbiol. Infect. 17:107–115. 10.1111/j.1469-0691.2010.03432.x [DOI] [PubMed] [Google Scholar]
- 3.Ghany MG, Strader DB, Thomas DL, Seeff LB. 2009. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology 49:1335–1374. 10.1002/hep.22759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yee HS, Chang MF, Pocha C, Lim J, Ross D, Morgan TR, Monto A. 2012. Update on the management and treatment of hepatitis C virus infection: recommendations from the Department of Veterans Affairs Hepatitis C Resource Center Program and the National Hepatitis C Program Office. Am. J. Gastroenterol. 107:669–689. 10.1038/ajg.2012.48 [DOI] [PubMed] [Google Scholar]
- 5.Kuiken C, Simmonds P. 2009. Nomenclature and numbering of the hepatitis C virus. Methods Mol. Biol. 510:33–53. 10.1007/978-1-59745-394-3_4 [DOI] [PubMed] [Google Scholar]
- 6.Cornberg M, Razavi HA, Alberti A, Bernasconi E, Buti M, Cooper C, Dalgard O, Dillion JF, Flisiak R, Forns X, Frankova S, Goldis A, Goulis I, Halota W, Hunyady B, Lagging M, Largen A, Makara M, Manolakopoulos S, Marcellin P, Marinho RT, Pol S, Poynard T, Puoti M, Sagalova O, Sibbel S, Simon K, Wallace C, Young K, Yurdaydin C, Zuckerman E, Negro F, Zeuzem S. 2011. A systematic review of hepatitis C virus epidemiology in Europe, Canada and Israel. Liver Int. 31(Suppl 2):30–60. 10.1111/j.1478-3231.2011.02539.x [DOI] [PubMed] [Google Scholar]
- 7.Germer JJ, Mandrekar JN, Bendel JL, Mitchell PS, Yao JD. 2011. Hepatitis C virus genotypes in clinical specimens tested at a national reference testing laboratory in the United States. J. Clin. Microbiol. 49:3040–3043. 10.1128/JCM.00457-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sievert W, Altraif I, Razavi HA, Abdo A, Ahmed EA, Alomair A, Amarapurkar D, Chen CH, Dou X, El Khayat H, Elshazly M, Esmat G, Guan R, Han KH, Koike K, Largen A, McCaughan G, Mogawer S, Monis A, Nawaz A, Piratvisuth T, Sanai FM, Sharara AI, Sibbel S, Sood A, Suh DJ, Wallace C, Young K, Negro F. 2011. A systematic review of hepatitis C virus epidemiology in Asia, Australia and Egypt. Liver Int. 31(Suppl 2):61–80. 10.1111/j.1478-3231.2011.02540.x [DOI] [PubMed] [Google Scholar]
- 9.Nguyen NH, Vutien P, Trinh HN, Garcia RT, Nguyen LH, Nguyen HA, Nguyen KK, Nguyen MH. 2010. Risk factors, genotype 6 prevalence, and clinical characteristics of chronic hepatitis C in Southeast Asian Americans. Hepatol. Int. 4:523–529. 10.1007/s12072-010-9181-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chao DT, Abe K, Nguyen MH. 2011. Systematic review: epidemiology of hepatitis C genotype 6 and its management. Aliment. Pharmacol. Ther. 34:286–296. 10.1111/j.1365-2036.2011.04714.x [DOI] [PubMed] [Google Scholar]
- 11.Fung J, Lai CL, Hung I, Young J, Cheng C, Wong D, Yuen MF. 2008. Chronic hepatitis C virus genotype 6 infection: response to pegylated interferon and ribavirin. J. Infect. Dis. 198:808–812. 10.1086/591252 [DOI] [PubMed] [Google Scholar]
- 12.Gane E, Stedman C, Hyland R, Sorensen R, Symonds W, Hindes R, Berrey M. 2012. Once daily GS-7977 plus ribavirin in HCV genotypes 1–3: the ELECTRON Trial, poster 1113. 47th Annu. Meet. Eur. Assoc. Study Liver [Google Scholar]
- 13.Hassanein T, Nelson DR, Lawitz E, Dieterich D, Jacobson I, Baker C. 2011. PSI-7977 with PEG/RBV elicits rapid declines in HCV RNA in patients with HCV GT-4 and GT-6, abstr 50 HEPDart. [Google Scholar]
- 14.Dunford L, Carr MJ, Dean J, Waters A, Nguyen LT, Ta Thi TH, Thi LA, Do HD, Thi TT, Nguyen HT, Diem Do TT, Luu QP, Connell J, Coughlan S, Hall WW, Nguyen Thi LA. 2012. Hepatitis C virus in Vietnam: high prevalence of infection in dialysis and multi-transfused patients involving diverse and novel virus variants. PLoS One 7:e41266. 10.1371/journal.pone.0041266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manns MP, McHutchison JG, Gordon SC, Rustgi VK, Shiffman M, Reindollar R, Goodman ZD, Koury K, Ling M, Albrecht JK. 2001. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet 358:958–965. 10.1016/S0140-6736(01)06102-5 [DOI] [PubMed] [Google Scholar]
- 16.Liverton N. 2010. MK-5172, the 1st HCV protease inhibitor with potent activity against resistance mutations in vitro, abstr 39 45th Annu. Meet. Eur. Assoc. Study Liver [Google Scholar]
- 17.Scheel TK, Gottwein JM, Mikkelsen LS, Jensen TB, Bukh J. 2011. Recombinant HCV variants with NS5A from genotypes 1–7 have different sensitivities to an NS5A inhibitor but not interferon-alpha. Gastroenterology 140:1032–1042. 10.1053/j.gastro.2010.11.036 [DOI] [PubMed] [Google Scholar]
- 18.Bartenschlager R. 2005. The hepatitis C virus replicon system: from basic research to clinical application. J. Hepatol. 43:210–216. 10.1016/j.jhep.2005.05.013 [DOI] [PubMed] [Google Scholar]
- 19.Blight KJ, McKeating JA, Marcotrigiano J, Rice CM. 2003. Efficient replication of hepatitis C virus genotype 1a RNAs in cell culture. J. Virol. 77:3181–3190. 10.1128/JVI.77.5.3181-3190.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kato T, Date T, Miyamoto M, Furusaka A, Tokushige K, Mizokami M, Wakita T. 2003. Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology 125:1808–1817. 10.1053/j.gastro.2003.09.023 [DOI] [PubMed] [Google Scholar]
- 21.Lohmann V, Hoffmann S, Herian U, Penin F, Bartenschlager R. 2003. Viral and cellular determinants of hepatitis C virus RNA replication in cell culture. J. Virol. 77:3007–3019. 10.1128/JVI.77.5.3007-3019.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110–113. 10.1126/science.285.5424.110 [DOI] [PubMed] [Google Scholar]
- 23.Blight KJ, Kolykhalov AA, Rice CM. 2000. Efficient initiation of HCV RNA replication in cell culture. Science 290:1972–1974. 10.1126/science.290.5498.1972 [DOI] [PubMed] [Google Scholar]
- 24.Peng B, Yu M, Xu S, Lee YJ, Tian Y, Yang H, Chan K, Mo H, McHutchison J, Delaney W, IV, Cheng G. 2013. Development of robust hepatitis C virus genotype 4 subgenomic replicons. Gastroenterology 144:59.e6–61.e6. 10.1053/j.gastro.2012.09.033 [DOI] [PubMed] [Google Scholar]
- 25.Saeed M, Gondeau C, Hmwe S, Yokokawa H, Date T, Suzuki T, Kato T, Maurel P, Wakita T. 2013. Replication of hepatitis C virus genotype 3a in cultured cells. Gastroenterology 144:56.e7–58.e7. 10.1053/j.gastro.2012.09.017 [DOI] [PubMed] [Google Scholar]
- 26.Saeed M, Scheel TK, Gottwein JM, Marukian S, Dustin LB, Bukh J, Rice CM. 2012. Efficient replication of genotype 3a and 4a hepatitis C virus replicons in human hepatoma cells. Antimicrob. Agents Chemother. 56:5365–5373. 10.1128/AAC.01256-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Friebe P, Boudet J, Simorre JP, Bartenschlager R. 2005. Kissing-loop interaction in the 3′ end of the hepatitis C virus genome essential for RNA replication. J. Virol. 79:380–392. 10.1128/JVI.79.1.380-392.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robinson M, Yang H, Sun SC, Peng B, Tian Y, Pagratis N, Greenstein AE, Delaney WE., IV 2010. Novel hepatitis C virus reporter replicon cell lines enable efficient antiviral screening against genotype 1a. Antimicrob. Agents Chemother. 54:3099–3106. 10.1128/AAC.00289-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheng G, Chan K, Yang H, Corsa A, Pokrovskii M, Paulson M, Bahador G, Zhong W, Delaney W., IV 2011. Selection of clinically relevant protease inhibitor-resistant viruses using the genotype 2a hepatitis C virus infection system. Antimicrob. Agents Chemother. 55:2197–2205. 10.1128/AAC.01382-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Link JO, Taylor JG, Xu L, Mitchell M, Guo H, Liu H, Kato D, Kirschberg T, Sun J, Squires N, Parrish J, Keller T, Yang ZY, Yang C, Matles M, Wang Y, Wang K, Cheng G, Tian Y, Mogalian E, Mondou E, Cornpropst M, Perry J, Desai MC. 11 December 2013. Discovery of ledipasvir (GS-5885): a potent, once-daily oral NS5A inhibitor for the treatment of hepatitis C virus infection. J. Med. Chem. 10.1021/jm401499g [DOI] [PubMed] [Google Scholar]
- 31.Shih IH, Vliegen I, Peng B, Yang H, Hebner C, Paeshuyse J, Purstinger G, Fenaux M, Tian Y, Mabery E, Qi X, Bahador G, Paulson M, Lehman LS, Bondy S, Tse W, Reiser H, Lee WA, Schmitz U, Neyts J, Zhong W. 2011. Mechanistic characterization of GS-9190 (tegobuvir), a novel nonnucleoside inhibitor of hepatitis C virus NS5B polymerase. Antimicrob. Agents Chemother. 55:4196–4203. 10.1128/AAC.00307-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sheng XC, Appleby T, Butler T, Cai R, Chen X, Cho A, Clarke MO, Cottell J, Delaney WE, IV, Doerffler E, Link J, Ji M, Pakdaman R, Pyun HJ, Wu Q, Xu J, Kim CU. 2012. Discovery of GS-9451: an acid inhibitor of the hepatitis C virus NS3/4A protease. Bioorg. Med. Chem. Lett. 22:2629–2634. 10.1016/j.bmcl.2012.01.017 [DOI] [PubMed] [Google Scholar]
- 33.Fenaux M, Eng Leavitt SSA, Lee YJ, Mabery EM, Tian Y, Byun D, Canales E, Clarke MO, Doerffler E, Lazerwith SE, Lew W, Liu Q, Mertzman M, Morganelli P, Xu L, Ye H, Zhang J, Matles M, Murray BP, Mwangi J, Hashash A, Krawczyk SH, Bidgood AM, Appleby TC, Watkins WJ. 2013. Preclinical characterization of GS-9669, a thumb site II inhibitor of the hepatitis C virus NS5B polymerase. Antimicrob. Agents Chemother. 57:804–810. 10.1128/AAC.02052-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sofia MJ, Bao D, Chang W, Du J, Nagarathnam D, Rachakonda S, Reddy PG, Ross BS, Wang P, Zhang HR, Bansal S, Espiritu C, Keilman M, Lam AM, Steuer HM, Niu C, Otto MJ, Furman PA. 2010. Discovery of a beta-d-2′-deoxy-2′-alpha-fluoro-2′-beta-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J. Med. Chem. 53:7202–7218. 10.1021/jm100863x [DOI] [PubMed] [Google Scholar]
- 35.Friebe P, Lohmann V, Krieger N, Bartenschlager R. 2001. Sequences in the 5′ nontranslated region of hepatitis C virus required for RNA replication. J. Virol. 75:12047–12057. 10.1128/JVI.75.24.12047-12057.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sumpter R, Jr, Loo YM, Foy E, Li K, Yoneyama M, Fujita T, Lemon SM, Gale M., Jr 2005. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J. Virol. 79:2689–2699. 10.1128/JVI.79.5.2689-2699.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu M, Corsa AC, Xu S, Peng B, Gong R, Lee YJ, Chan K, Mo H, Delaney W, IV, Cheng G. 2013. In vitro efficacy of approved and experimental antivirals against novel genotype 3 hepatitis C virus subgenomic replicons. Antiviral Res. 100:439–445. 10.1016/j.antiviral.2013.08.018 [DOI] [PubMed] [Google Scholar]
- 38.Kim JL, Morgenstern KA, Griffith JP, Dwyer MD, Thomson JA, Murcko MA, Lin C, Caron PR. 1998. Hepatitis C virus NS3 RNA helicase domain with a bound oligonucleotide: the crystal structure provides insights into the mode of unwinding. Structure 6:89–100. 10.1016/S0969-2126(98)00010-0 [DOI] [PubMed] [Google Scholar]
- 39.Yao N, Reichert P, Taremi SS, Prosise WW, Weber PC. 1999. Molecular views of viral polyprotein processing revealed by the crystal structure of the hepatitis C virus bifunctional protease-helicase. Structure 7:1353–1363. 10.1016/S0969-2126(00)80025-8 [DOI] [PubMed] [Google Scholar]
- 40.Herlihy KJ, Graham JP, Kumpf R, Patick AK, Duggal R, Shi ST. 2008. Development of intergenotypic chimeric replicons to determine the broad-spectrum antiviral activities of hepatitis C virus polymerase inhibitors. Antimicrob. Agents Chemother. 52:3523–3531. 10.1128/AAC.00533-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.