

Abstract
An ultrasensitive assay utilizing high-pressure liquid chromatography and mass spectrometry detection was developed and validated for the quantification of the antiretrovirals atazanavir (ATV), darunavir (DRV), lopinavir (LPV), ritonavir (RTV), and efavirenz (EFV) in human mononuclear cell (MNC) extracts. The assay utilizes 20 μl of cellular extract that contains as few as 50,000 MNCs. The analytical range of the assay is 0.0200 to 10.0 fmol/μl for ATV, 0.0500 to 25.0 fmol/μl for DRV, LPV, and RTV, and 0.200 to 100 fmol/μl for EFV. The assay has proven to be a clinically useful tool for investigating antiretroviral drug concentrations in virologic sanctuaries where harvested cell numbers are extremely low. The assay provides a tool for investigators to explore the clinical pharmacology of strategies for prevention, treatment, and cure in pathophysiologically relevant sites.
INTRODUCTION
The human immunodeficiency virus (HIV) continues to be one of the greatest causes of mortality from an infectious disease globally. There are more than 34 million people living with HIV worldwide (1). This number continues to rise, even as the number of new infections decreases. People are living longer with HIV infection, due in part to the advent of highly active antiretroviral therapy (ART). Since 1995, antiretroviral therapy has saved 14 million life years in low- and middle-income countries (1). ART has largely rendered HIV a manageable chronic disease.
A regimen usually consisting of three drugs targeting two different points in the viral life cycle is the current standard for implementing ART (2, 3). The goal of ART is to attain durable virologic suppression (usually defined as HIV-RNA in plasma < 50 copies/ml) in individuals infected with HIV while restoring immune system functionality. With the repertoire of drugs currently available targeted at the HIV life cycle, this goal is achievable in almost every treatment-naive individual infected with HIV.
First-line treatment regimens consist of two nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs) plus a nonnucleoside reverse transcriptase inhibitor (NNRTI) or a ritonavir-boosted protease inhibitor (PI) or an integrase strand transfer inhibitor (3).
Here, we detail a novel highly sensitive and specific intracellular assay for the determination of concentrations of efavirenz (EFV), the first-line NNRTI, along with three PIs, atazanavir (ATV), darunavir (DRV), and lopinavir (LPV), and their pharmacologic boosting agent ritonavir (RTV). The assay has been utilized to investigate concentrations of these drugs in lymphoid tissue sites, including lymph nodes (LN), gut-associated lymphoid tissue (GALT), and rectum-associated lymphoid tissue (RALT), along with peripheral blood mononuclear cells (PBMC). GALT is the largest immune compartment in the body (4, 5). The GALT contains more lymphocytes than all of the secondary lymphoid organs combined (6). The GALT and RALT are established reservoirs for persistent viral replication (7). Determining drug concentrations achieved in the intracellular environment from these tissues is necessary to learn whether or not these reservoirs are a pharmacologic sanctuary and thereby play a role in the viral persistence.
MATERIALS AND METHODS
ATV sulfate, DRV, and LPV were obtained from the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH. EFV and RTV were obtained from U.S. Pharmacopeia (Rockville, MD). Efavirenz-D8 (internal standard) was a generous gift from Celerion Inc. (Lincoln, NE). ATV-13C6 (internal standard) was obtained from Bristol-Myers Squibb (New Brunswick, NJ). RTV-D8 (internal standard) was purchased from Aptochem (Montreal, Canada). Acetonitrile (liquid chromatography mass spectrometry [LC-MS] grade), methanol (Optima grade), and formic acid (99+% pure) were obtained from Fischer Scientific (Fairlawn, NJ). Type 1 water was produced in the laboratory using a Millipore MilliQ Integral 3 system (Millipore SAS, Molsheim, France). Human PBMCs were isolated from healthy donors using elutriation.
Analysis was performed using a Nexera ultra-high-performance LC (UPLC) (Shimadzu, Kyoto, Japan) system consisting of two LC-30AD pumps, a SIL-30AC autosampler, a CTO-30A column oven, and a CBM-20A system controller coupled to an API 6500 (Applied Biosystem Sciex, Foster City, CA) triple-quadrupole mass spectrometer. The mass spectrometer was operated in multiple reaction monitoring (MRM) mode, and ionization was performed via electrospray in both positive and negative ion modes. Data acquisition was performed using Analyst 1.6 software (Applied Biosystem Sciex, Foster City, CA). Cells used in the assay were counted on a Countess cell counter (Invitrogen, Life Technologies Corp., Grand Island, NY).
Human PBMCs were collected, counted, and then lysed with 70% methanol to create a lysed cellular blank matrix at an extracted cell density of 2 million cells per ml.
ATV, DRV, EFV, LPV, and RTV reference powders were weighed and diluted with the appropriate amount of methanol to create 2.00 mg/ml primary stock solutions. Primary stock solutions were subsequently diluted in methanol to create an 8-point combined spiking standard curve ranging from 0.0200 to 10.0 fmol/μl for ATV, 0.0500 to 25.0 fmol/μl for DRV, LPV, and RTV, and 0.200 to 100 fmol/μl for EFV.
Quality control (QC) samples were prepared by spiking stock solutions, made from unique weighings in a fashion similar to that used for the standard curve primary stocks, into a 2 million/ml lysed PBMC matrix at concentrations of 0.0600, 0.500, and 8.00 fmol/μl for ATV, 0.150, 1.25, and 20.0 fmol/μl for DRV, LPV, and RTV, and 0.600, 5.00, and 80.0 fmol/μl for EFV. QC samples were split into polypropylene vials and stored at −80°C until used. A separate lower limit of quantitation (LLOQ) sample was prepared to test sensitivity at 0.0200 fmol/μl for ATV, 0.0500 fmol/μl for DRV, LPV, and RTV, and 0.200 fmol/μl for EFV.
ATV-13C6 (ATV-IS), EFV-D8 (EFV-IS), and RTV-D8 (RTV-IS) powders were weighed and diluted with the appropriate amount of methanol to create 1.00 mg/ml primary stock solutions. Primary stocks were subsequently diluted in acetonitrile to make working internal standard concentrations of 1.50, 2.50, and 13.5 pmol/μl for ATV-IS, EFV-IS, and RTV-IS, respectively. DRV and LPV utilized ATV-IS and RTV-IS, respectively, as isotopic internal standards were not available for DRV and LPV at the time of assay development.
Samples that were stored for analysis were removed from the freezer and thawed at room temperature. Samples were then mixed using a vortex device and centrifuged at 13,000 relative centrifugal force (RCF) for 5 min. Microcentrifuge tubes (1.5 ml) were used for precipitating all samples. Standard tubes were spiked with 20 μl of the appropriate spiking standard solution level and 20 μl of blank lysed cellular matrix. Blank tubes received 20 μl of blank lysed cellular matrix and 20 μl of methanol. QC and clinical samples were divided into aliquots at a volume of 20 μl and spiked with 20 μl of methanol. A 20-μl volume of working internal standard was added to each tube except for the blanks, which received 20 μl of acetonitrile.
All samples were precipitated with 50 μl 0.1% formic acid in acetonitrile, capped, mixed using a vortex device, and incubated at 5°C for 1 h to facilitate precipitation. After incubation, all samples were centrifuged at 13,000 RCF for 5 min. A 90-μl volume of supernatant was transferred to a new microcentrifuge tube containing 70 μl of high-performance LC (HPLC)-grade water and mixed using a vortex device, and the resulting 160-μl volume was transferred to HPLC vials (Waters, Milford, MA) for LC-MS injection.
Chromatographic separation was achieved using an ACE C18 column (Advanced Chromatography Technologies Ltd., Aberdeen, Scotland) (3 by 100 mm). The mobile phase consisted of acetonitrile:water:formic acid (60:40:0.1 [vol/vol/vol]), the flow rate was 500 μl/min, the analytical column was maintained at 40°C, and the injection volume was 40 μl. The total run time was 5 min per sample. MS conditions are shown in Table 1.
TABLE 1.
MRM monitoring mass spectrometer conditions
| Analyte | Mode | Q1 mass (m/z) | Q3 mass (m/z) | Dwell time (ms) | DP (V) | CE (V) | CXP (V) | EP (V) | IS (V) | Temp (°C) | GS1 | GS2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ATV | + | 705.4 | 168.2 | 100 | 120.0 | 101.0 | 13.0 | 10.0 | 5,500 | 450 | 80 | 70 |
| ATV-IS | + | 711.5 | 168.1 | 100 | 120.0 | 101.0 | 13.0 | 10.0 | 5,500 | 450 | 80 | 70 |
| DRV | + | 548.3 | 392.3 | 100 | 100.0 | 18.5 | 16.0 | 10.0 | 5,500 | 450 | 80 | 70 |
| EFV | − | 313.9 | 244.0 | 300 | −30.0 | −22.0 | −16.0 | −10.0 | −4,500 | 650 | 50 | 90 |
| EFV-IS | − | 322.0 | 251.1 | 300 | −30.0 | −22.0 | −16.0 | −10.0 | −4,500 | 650 | 50 | 90 |
| LPV | + | 629.4 | 447.3 | 100 | 84.0 | 19.5 | 12.0 | 10.0 | 5,500 | 450 | 80 | 70 |
| RTV | + | 721.4 | 268.2 | 100 | 95.0 | 37.0 | 14.0 | 10.0 | 5,500 | 450 | 80 | 70 |
| RTV-IS | + | 729.4 | 276.3 | 100 | 95.0 | 37.0 | 14.0 | 10.0 | 5,500 | 450 | 80 | 70 |
Method validation was performed according to FDA guidelines for industry (8). Assessments performed in the validation included standard curve performance, intra- and interday precision and accuracy (QCs), stability, dilution, matrix effect, and selectivity.
Intra- and interday precision and accuracy were assessed over 3 days with 6 replicates of samples at 0.0200 fmol/μl ATV, 0.0500 fmol/μl DRV, LPV, and RTV, and 0.200 fmol/μl EFV (LLOQ), as well as at 0.0600 fmol/μl ATV, 0.150 fmol/μl DRV, LPV, and RTV, and 0.600 fmol/μl EFV (low QC), 0.500 fmol/μl ATV, 1.25 fmol/μl DRV, LPV, and RTV, and 5.00 fmol/μl EFV (middle QC), and 8.00 fmol/μl ATV, 20.0 fmol/μl DRV, LPV, and RTV, and 80.0 fmol/μl EFV (high QC). The accuracy of the QCs was determined by the percent deviation (%Dev) from the expected concentrations of the samples. Precision was determined by the coefficient of variation percentage (%CV) of the analyzed QCs.
Stability assessments were conducted at n = 3 for each level and condition. Acceptable percent difference from controls kept at −80°C was ±15%. Freeze-thaw (F/T) stability was assessed by cycling QC samples at the low and high concentrations out of the freezer for at least 3 h and refreezing the samples for at least 12 h 3 times. Short-term (ST) stability was tested by removing a set of low and high QC samples and placing them on the bench for 24 h at room temperature prior to extraction against controls. Postpreparative stability (PPS) quality control samples were prepared by extracting low and high samples and placing them into the autosampler cooled to 15°C and were analyzed for accuracy after 5 days. Controls were prepared fresh and compared with the previously extracted samples. Primary stock solution stability assessments were performed by comparing stock solutions stored at −20°C with freshly prepared stock solutions.
Dilution integrity was established in the following manner: samples were prepared with concentrations above the analytical range at 30.0 fmol/μl for ATV, 75.0 fmol/μl for DRV, LPV, and RTV, and 300 fmol/μl for EFV and diluted in lysed cellular matrix. Selectivity was assessed by comparing five different lots of lysed cellular PBMC matrix with and without spiked analyte.
Matrix effect (ME), recovery (RE), and process efficiency (PE) were determined at the QC concentrations (low, medium, and high) for each analyte in five different lots of lysed PBMC matrix. Peak area counts from extracted (set 3) preparations were compared with neat samples prepared in the dilution and protein crash solvents (set 1) and postextraction spiked (set 2) blank preparations. ME was calculated as follows: set 2/set 1 × 100. RE was calculated as follows: set 3/set 2 × 100. PE was calculated as follows: set 3/set 1 × 100. We employed a method for LC-MS matrix effect determination adapted from work by Matuszewski et al. (9) where unweighted linear regression slopes were generated for each of the 5 lots of matrix and compared (%CV).
Working spiking standards were initially prepared and routinely used throughout the validation process (i.e., frozen, thawed, and maintained at room temperature). A new set of working spiking standards were prepared 56 days later from a fresh set of primary stock solutions and compared with the initial working spiking standard preparation at the lowest concentration.
RESULTS
A weighted linear regression curve (1/concentration2) was determined to best represent the concentration/detector response relationship for ATV, DRV, EFV, LPV, and RTV in PBMCs. The limit of reliable quantitation was set at the concentration of the lowest nonzero standard. Back-calculated calibration curve standard concentrations (see Table S1 in the supplemental material) and standard curve parameters supported the LLOQ. The interbatch and intrabatch (Table 2) precision and accuracy results for LLOQ samples met predetermined acceptance and method validation criteria. Results for all matrix effect and recovery tests are shown in Table S3. Long-term stability of QC samples was initially established at 56 days and will continue to be updated. ATV, DRV, EFV, LPV, and RTV were stable (within 10%) in methanol at −20°C for at least 176, 261, 261, 261, and 261 days, respectively (see Table S2). Samples assessing dilution integrity quantitated within ±15% of the nominal concentration meeting acceptance criteria. QC preparation with cell densities ranging from 10,000 to 20 million mononuclear cells/ml did not alter analytical results (data not shown).
TABLE 2.
Intra- and interbatch precision and accuracy
| Antiretroviral | Parameter | Theoretical (fmol/μl) | Intrabatch precision and accuracy |
Interbatch precision and accuracy |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
2 |
3 |
Mean (fmol/μl) | %CV | %Dev | n | ||||||||||||
| Mean (fmol/μl) | %CV | %Dev | n | Mean (fmol/μl) | %CV | %Dev | n | Mean (fmol/μl) | %CV | %Dev | n | |||||||
| ATV | LLOQ | 0.02 | 0.0207 | 3.4 | 3.3 | 6 | 0.0189 | 17.3 | −5.4 | 6 | 0.0223 | 7.3 | 11.5 | 6 | 0.0206 | 11.7 | 3 | 18 |
| Low | 0.06 | 0.0614 | 1.7 | 2.4 | 6 | 0.058 | 9 | −3.3 | 6 | 0.065 | 3.1 | 8.4 | 6 | 0.0615 | 7 | 2.5 | 18 | |
| Middle | 0.5 | 0.494 | 2.5 | −1.2 | 6 | 0.496 | 3.8 | −0.7 | 6 | 0.496 | 4 | −0.8 | 6 | 0.496 | 3.3 | −0.9 | 18 | |
| High | 8 | 7.97 | 1.7 | −0.4 | 6 | 7.77 | 5.1 | −2.9 | 6 | 8.16 | 1.3 | 2 | 6 | 7.97 | 3.6 | −0.4 | 18 | |
| DRV | LLOQ | 0.05 | 0.0508 | 13.1 | 1.5 | 6 | 0.056 | 10.5 | 12 | 6 | 0.0542 | 8.9 | 8.4 | 6 | 0.0535 | 109 | 6.9 | 18 |
| Low | 0.15 | 0.154 | 5.6 | 2.8 | 6 | 0.159 | 5.7 | 6.2 | 6 | 0.153 | 4.3 | 1.9 | 6 | 0.155 | 5.3 | 3.6 | 18 | |
| Middle | 1.25 | 1.26 | 3.1 | 0.9 | 6 | 1.18 | 6.4 | −5.7 | 6 | 1.21 | 1.8 | −3 | 6 | 1.22 | 4.9 | −2.6 | 18 | |
| High | 20 | 20.2 | 3.3 | 0.9 | 6 | 18.8 | 7.8 | −5.8 | 6 | 19.9 | 2.6 | −0.7 | 6 | 19.6 | 5.6 | −1.9 | 18 | |
| EFV | LLOQ | 0.2 | 0.227 | 10.4 | 13.3 | 6 | 0.232 | 6.7 | 16.2 | 6 | 0.22 | 4.9 | 10.1 | 6 | 0.225 | 7.9 | 12.5 | 18 |
| Low | 0.6 | 0.654 | 3.5 | 9.1 | 6 | 0.647 | 1.7 | 7.9 | 6 | 0.662 | 2.1 | 10.3 | 6 | 0.654 | 2.6 | 9.1 | 18 | |
| Middle | 5 | 5.13 | 1.5 | 2.6 | 6 | 5.17 | 0.8 | 3.4 | 6 | 5.21 | 2.2 | 4.2 | 6 | 5.17 | 1.7 | 3.4 | 18 | |
| High | 80 | 81.8 | 1.1 | 2.3 | 6 | 83.1 | 1.5 | 3.9 | 6 | 85 | 3.3 | 6.2 | 6 | 83.3 | 2.6 | 4.1 | 18 | |
| LPV | LLOQ | 0.05 | 0.052 | 3.3 | 3.9 | 6 | 0.0502 | 1.9 | 0.4 | 6 | 0.051 | 3.3 | 1.9 | 6 | 0.051 | 3 | 2 | 18 |
| Low | 0.15 | 0.157 | 7.7 | 4.9 | 6 | 0.15 | 5.7 | −0.1 | 6 | 0.15 | 4.6 | 0.2 | 6 | 0.153 | 6.2 | 1.7 | 18 | |
| Middle | 1.25 | 1.28 | 8.5 | 2.6 | 6 | 1.16 | 3.5 | −7.5 | 6 | 1.15 | 2.9 | −7.7 | 6 | 1.2 | 7.5 | −4.2 | 18 | |
| High | 20 | 19.4 | 2.4 | −2.8 | 6 | 19.5 | 4.7 | −2.6 | 6 | 18.3 | 1.3 | −8.7 | 6 | 19.1 | 4.3 | −4.7 | 18 | |
| RTV | LLOQ | 0.05 | 0.0599 | 13.8 | 19.8 | 6 | 0.0571 | 3.2 | 14.1 | 6 | 0.0572 | 8.4 | 14.5 | 6 | 0.0576 | 9.7 | 15.3 | 18 |
| Low | 0.15 | 0.164 | 3.9 | 9.6 | 6 | 0.166 | 6.9 | 10.8 | 6 | 0.161 | 2.7 | 7.2 | 6 | 0.164 | 4.8 | 9.2 | 18 | |
| Middle | 1.25 | 1.28 | 4 | 2.6 | 6 | 1.27 | 2.8 | 1.9 | 6 | 1.24 | 2.4 | −0.6 | 6 | 1.27 | 3.3 | 1.3 | 18 | |
| High | 20 | 20.5 | 4.5 | 2.5 | 6 | 20.7 | 4.6 | 3.4 | 6 | 20.2 | 2.2 | 0.9 | 6 | 20.4 | 3.9 | 2.2 | 18 | |
A typical blank sample for each analyte is shown with typical LLOQ samples in Fig. 1. No endogenous interference was observed in any of the five lots screened at the retention time of any analyte at the respective transitions used. Internal standard transitions were free from interference.
FIG 1.
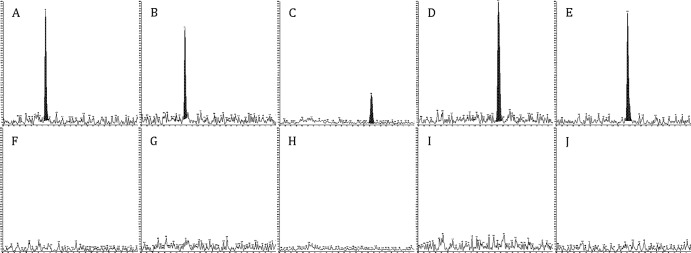
Lower limit of quantitation (LLOQ) and blank sample representative chromatography. LLOQ (A to E) and blank chromatograms (F to J) at 1,000 cps (y axis) and 5 min (x axis) are shown. A and F, ATV; B and G, DRV; C and H, EFV; D and I, LPV; E and J, RTV.
DISCUSSION
The mechanism(s) behind persistent HIV infection is not fully understood and is the target of many research strategies for a cure. Secondary LNs and the GALT are the primary sites of HIV replication and where the latent pool of virus is maintained (10). HIV replication in these tissues causes inflammation and immune activation, collagen deposition, and T cell depletion. These immunopathologies persist after ART initiation, which raises the possibility that replication may continue in lymphoid tissues even when the plasma viral load is suppressed. Animal studies have shown low concentrations of some antiretroviral drugs in tissue compartments; lymphoid tissues have been shown to be viral reservoirs in nonhuman primate studies; and HIV RNA has been detected in human rectal samples and HIV DNA detected in GALT tissue of well-suppressed patients (11–13). In order to determine whether or not individual drugs of ART regimens are achieving therapeutic concentrations at the site of pharmacologic action, there is a need to establish validated and rigorously quality-controlled analytical assays for measuring intracellular drug concentrations in reservoir compartments.
Fletcher et al. prospectively treated 12 HIV-infected subjects with ARVs and performed multiple samplings of LN, ileum, and rectum after initiating ART to investigate the hypothesis that antiretroviral drug concentrations might be insufficient to fully suppress replication in the lymphoid tissue compartments. The complete details of this work, including sample isolation procedures, have been published elsewhere (14). In the study by Fletcher et al., 592 analyte determinations for intracellular drug concentrations in PBMCs as well as MNCs from the LN, ileum, and rectum were performed using the UPLC/MS/MS method described here. Median concentrations (fmol/106 cells) for ATV, DRV, RTV, and EFV in these subjects were as follows: for ATV in PBMC, 445, in LN, all samples were below the LLOQ, in ileum, 44, and in rectum, 446; for DRV in PBMC, 1,260, in LN, 45 (n = 1), in ileum, 1,110, and in rectum, 943; for RTV in PBMC, 256, in LN, 47, in ileum, 107, and in rectum, 515; and for EFV in PBMC, 1,464, in LN, 312, in ileum, 759, and in rectum, 535 (14). The novel finding that intracellular concentrations, particularly in the LN but also in the ileum and rectum for some drugs, were lower than those in PBMCs illustrates a role for this analytical method in investigating the intracellular pharmacology of antiretroviral drugs in reservoir compartments.
The method described here has proven to be simple and highly robust, allowing the routine analysis of large numbers of samples. This method is new, highly accurate, and specific for the intracellular measurement of the NNRTI EFV along with the commonly used PIs ATV, DRV, LPV, and RTV in MNCs. The novelty of the assay lies in the sensitivity attained with the small sample volume required for the assay.
One of the limitations to analyzing tissue samples from human subjects has been the need for large sample volumes to achieve the limits of quantitation needed to capture intracellular concentrations of antiretrovirals. The tissue harvested for our analysis was gathered via biopsies of the LN and the colon and rectum performed during colonoscopy. The biopsy technique yields small sample volumes. Additional processing required for extracting MNCs from LN, colon, and rectal tissue biopsy specimens further depletes the sample available for pharmacologic analysis. Nevertheless, we chose the MNC matrix instead of a tissue homogenate because the tissue homogenate matrix may give misleading information on intracellular drug actually available to the cell for antiretroviral activity; additionally, we can accurately count the MNC number in a sample. A tissue homogenate is comprised of extra- and intracellular fluids and particles, is easily contaminated by blood present in tissue vessels, and presents the problem that drug is not homogenously distributed to all distinct compartments of the tissue (15). Our method helps alleviate one of the traditional limitations of using isolated cells as a matrix by utilizing a sample size of as low as 50,000 mononuclear cells. From 50,000 MNCs isolated peripherally, as well as from LN, ileum, and rectum, we have attained quantifiable ARV concentrations from patients who achieved the expected plasma concentrations of the same ARVs when taking the usual doses of these drugs.
We chose to validate the method using peripherally collected MNCs as a surrogate matrix for MNCs collected from other anatomical compartments (LN, ileum, and rectum). Harvesting sufficient MNCs from specific locations in the body such as LN, ileum, and rectum to complete a full method of cross-validation is impractical from both clinical and ethical perspectives. The FDA guidance on bioanalytical method validation specifically allows for “physiologically appropriate proxy matrices” to be used for tissues of limited availability (8). The method achieves validated LLOQs of 0.0200 fmol/μl (14.1 pg/ml) for ATV, 0.0500 fmol/μl (27.4, 31.4, and 36 pg/ml) for DRV, LPV, and RTV, and 0.200 fmol/μl (63.1 pg/ml) for EFV. To our knowledge, these are the lowest validated LC-MS concentrations of these antiretrovirals in intracellular compartments published to date. D'Avolio et al. previously published details of an LC-MS assay that included ATV, DRV, LPV, RTV, and EFV, with an LLOQ of 0.1 ng/ml for each analyte (16).
The method has many potential areas of use, from investigating ART pharmacokinetics-pharmacodynamics in viral reservoirs of HIV to determining long-term patient medication compliance, as intracellular half-lives of many antiretrovirals are longer than plasma half-lives (17–21). The ability to use a small sample aliquot of 20 μl and cell numbers as low as 50,000 cells allows studies in human subjects that might not otherwise be possible because of the limited size and number of biopsy specimens in, for example, lymphatic tissues which can be obtained. This tool, therefore, should open new avenues of pharmacologic research, in particular, to investigate strategies for a functional cure of HIV infection.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by P01 AI074340 (to C.V.F.) from the National Institutes of Allergy and Infectious Diseases.
Footnotes
Published ahead of print 10 March 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02551-13.
REFERENCES
- 1.UNAIDS. 2012. Global report 2012: UNAIDS report on the global AIDS epidemic. http://www.unaids.org/en/resources/publications/2012/name,76121,en.asp
- 2.World Health Organization. 2013. Consolidated guidelines on general HIV care and the use of antiretroviral drugs for treating and preventing HIV infection: recommendations for a public health approach. World Health Organization, Geneva, Switzerland [Google Scholar]
- 3.U.S. Department of Health and Human Services. 2013. Panel on antiretroviral guidelines for adults and adolescents. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. http://aidsinfo.nih.gov/contentfiles/lvguidelines/AdultandAdolescentGL.pdf
- 4.Mehandru S, Poles MA, Tenner-Racz K, Horowitz A, Hurley A, Hogan C, Boden D, Racz P, Markowitz M. 2004. Primary HIV-1 infection is associated with preferential depletion of CD4+ T lymphocytes from effector sites in the gastrointestinal tract. J. Exp. Med. 200:761–770. 10.1084/jem.20041196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mehandru S, Tenner-Racz K, Racz P, Markowitz M. 2005. The gastrointestinal tract is critical to the pathogenesis of acute HIV-1 infection. J. Allergy Clin. Immunol. 116:419–422. 10.1016/j.jaci.2005.05.040 [DOI] [PubMed] [Google Scholar]
- 6.Nagler-Anderson C. 2001. Man the barrier! strategic defences in the intestinal mucosa. Nat. Rev. Immunol. 1:59–67. 10.1038/35095573 [DOI] [PubMed] [Google Scholar]
- 7.Chun T-W, Nickle DC, Justement JS, Meyers JH, Roby G, Hallahan CW, Kottilil S, Moir S, Mican JM, Mullins JI. 2008. Persistence of HIV in gut-associated lymphoid tissue despite long-term antiretroviral therapy. J. Infect. Dis. 197:714–720. 10.1086/527324 [DOI] [PubMed] [Google Scholar]
- 8.U.S. Department of Health and Human Services Food and Drug Administration. 2001. Guidance for industry, bioanalytical methods validation. http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf
- 9.Matuszewski B, Constanzer M, Chavez-Eng C. 2003. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal. Chem. 75:3019–3030. 10.1021/ac020361s [DOI] [PubMed] [Google Scholar]
- 10.Pantaleo G, Graziosi C, Butini L, Pizzo PA, Schnittman SM, Kotler DP, Fauci AS. 1991. Lymphoid organs function as major reservoirs for human immunodeficiency virus. Proc. Natl. Acad. Sci. U. S. A. 88:9838–9842. 10.1073/pnas.88.21.9838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Di Mascio M, Srinivasula S, Bhattacharjee A, Cheng L, Martiniova L, Herscovitch P, Lertora J, Kiesewetter D. 2009. Antiretroviral tissue kinetics: in vivo imaging using positron emission tomography. Antimicrob. Agents Chemother. 53:4086–4095. 10.1128/AAC.00419-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horiike M, Iwami S, Kodama M, Sato A, Watanabe Y, Yasui M, Ishida Y, Kobayashi T, Miura T, Igarashi T. 2012. Lymph nodes harbor viral reservoirs that cause rebound of plasma viremia in SIV-infected macaques upon cessation of combined antiretroviral therapy. Virology 423:107–118. 10.1016/j.virol.2011.11.024 [DOI] [PubMed] [Google Scholar]
- 13.North TW, Higgins J, Deere JD, Hayes TL, Villalobos A, Adamson L, Shacklett BL, Schinazi RF, Luciw PA. 2010. Viral sanctuaries during highly active antiretroviral therapy in a nonhuman primate model for AIDS. J. Virol. 84:2913–2922. 10.1128/JVI.02356-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fletcher CV, Staskus K, Wietgrefe SW, Rothenberger M, Reilly C, Chipman JG, Beilman GJ, Khoruts A, Thorkelson A, Schmidt TE, Anderson J, Perkey K, Stevenson M, Perelson AS, Douek DC, Haase AT, Schacker TW. 27 January 2014. Persistent HIV-1 replication is associated with lower antiretroviral drug concentrations in lymphatic tissues. Proc. Natl. Acad. Sci. U. S. A. 10.1073/pnas.1318249111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mouton JW, Theuretzbacher U, Craig WA, Tulkens PM, Derendorf H, Cars O. 2008. Tissue concentrations: do we ever learn? J. Antimicrob. Chemother. 61:235–237. 10.1093/jac/dkm476 [DOI] [PubMed] [Google Scholar]
- 16.D'Avolio A, Simiele M, Siccardi M, Baietto L, Sciandra M, Oddone V, Stefani FR, Agati S, Cusato J, Bonora S. 2011. A HPLC-MS method for the simultaneous quantification of fourteen antiretroviral agents in peripheral blood mononuclear cell of HIV infected patients optimized using medium corpuscular volume evaluation. J. Pharm. Biomed. Anal. 54:779–788. 10.1016/j.jpba.2010.10.011 [DOI] [PubMed] [Google Scholar]
- 17.Baheti G, Kiser JJ, Havens PL, Fletcher CV. 2011. Plasma and intracellular population pharmacokinetic analysis of tenofovir in HIV-1-infected patients. Antimicrob. Agents Chemother. 55:5294–5299. 10.1128/AAC.05317-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ford J, Boffito M, Wildfire A, Hill A, Back D, Khoo S, Nelson M, Moyle G, Gazzard B, Pozniak A. 2004. Intracellular and plasma pharmacokinetics of saquinavir-ritonavir, administered at 1,600/100 milligrams once daily in human immunodeficiency virus-infected patients. Antimicrob. Agents Chemother. 48:2388–2393. 10.1128/AAC.48.7.2388-2393.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moyle G, Boffito M, Fletcher C, Higgs C, Hay PE, Song IH, Lou Y, Yuen GJ, Min SS, Guerini EM. 2009. Steady-state pharmacokinetics of abacavir in plasma and intracellular carbovir triphosphate following administration of abacavir at 600 milligrams once daily and 300 milligrams twice daily in human immunodeficiency virus-infected subjects. Antimicrob. Agents Chemother. 53:1532–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nascimbeni M, Lamotte C, Peytavin G, Farinotti R, Clavel F. 1999. Kinetics of antiviral activity and intracellular pharmacokinetics of human immunodeficiency virus type 1 protease inhibitors in tissue culture. Antimicrob. Agents Chemother. 43:2629–2634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Piliero P, Shachoy-Clark A, Para M, Preston S, Lou I, Drusano G, Stein D, Yuen G. 2003. A study examining the pharmacokinetics of abacavir and the intracellular carbovir triphosphate (GSK protocol CNA10905). Abstr. 43rd Intersci. Conf. Antimicrob. Agents Chemother., p 14–17 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.