

Abstract
Lignans, a class of dimeric phenylpropanoid derivative found in plants, such as whole grains and sesame and flax seeds, have anticancer activity and can act as phytoestrogens. The lignans secoisolariciresinol and matairesinol can be converted in the mammalian proximal colon into enterolactone and enterodiol, respectively, which reduce the risk of breast and colon cancer. To establish an efficient bioconversion system to generate matairesinol from pinoresinol, the genes encoding pinoresinol-lariciresinol reductase (PLR) and secoisolariciresinol dehydrogenase (SDH) were cloned from Podophyllum pleianthum Hance, an endangered herb in Taiwan, and the recombinant proteins, rPLR and rSDH, were expressed in Escherichia coli and purified. The two genes, termed plr-PpH and sdh-PpH, were also linked to form two bifunctional fusion genes, plr-sdh and sdh-plr, which were also expressed in E. coli and purified. Bioconversion in vitro at 22°C for 60 min showed that the conversion efficiency of fusion protein PLR-SDH was higher than that of the mixture of rPLR and rSDH. The percent conversion of (+)-pinoresinol to matairesinol was 49.8% using PLR-SDH and only 17.7% using a mixture of rPLR and rSDH. However, conversion of (+)-pinoresinol by fusion protein SDH-PLR stopped at the intermediate product, secoisolariciresinol. In vivo, (+)-pinoresinol was completely converted to matairesinol by living recombinant E. coli expressing PLR-SDH without addition of cofactors.
INTRODUCTION
Lignans, a large group of secondary metabolites found in plants, have a broad range of medicinal functions. They have been identified in more than 60 families of vascular plants (1) and are biosynthesized by the dimerization of two molecules of phenylpropanoids. Different types of phenylpropanoid subunits, different coupling orientations, and complicated modifications result in a wide diversity of lignan structures. Lignans are also a major class of phytoestrogens with estrogen-like structures (2) and antioxidant activity (3). Orally administered lignans have positive effects on breast cancer, osteoporosis, and colon cancer (4).
The phytoestrogens pinoresinol, secoisolariciresinol, and matairesinol have been found in whole grains, vegetables, and fruits (5). Secoisolariciresinol and matairesinol are converted into the enterolignans enterodiol and enterolactone by microflora in the proximal colon in mammals, and these two enterolignans are absorbed in the colon and enter the body (6). The presence of enterolignans in serum can reduce the risk of breast, prostate, and colon cancer (7, 8). Enterolignans have weak estrogen-like activity and can compete with estrogen for binding to estrogen receptors and inhibit the enzymes involved in estrogen anabolism (9). High serum enterolignan levels can reduce the risks of acute coronary syndrome (10). The concentrations of enterolignans in serum and urine are indicators of plant lignan intake (11).
Podophyllotoxin, a type of aryltetralin lignan, is the precursor of the anticancer drugs teniposide and etoposide (12–14). Pinoresinol-lariciresinol reductase (PLR), an NADPH-dependent enzyme, is the key enzyme in podophyllotoxin biosynthesis (Fig. 1) (15). The phenylpropanoid dimer (+)-pinoresinol is sequentially converted into (+)-lariciresinol and (−)-secoisolariciresinol by PLR, and then (−)-secoisolariciresinol is converted into (−)-matairesinol by the NAD+-dependent secoisolariciresinol dehydrogenase SDH (16). This, in turn, is converted into podophyllotoxin by several unidentified enzymes.
FIG 1.
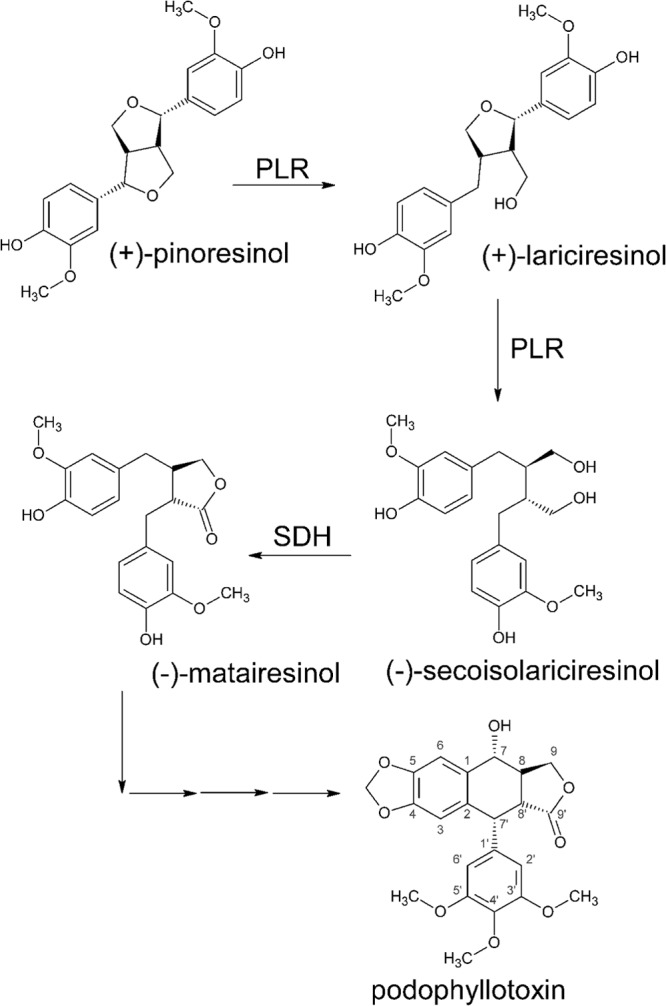
Biosynthesis pathway of podophyllotoxin.
Podophyllum pleianthum, a herb traditionally used in Taiwan for treatment of snake bite and wound healing, contains podophyllotoxin. Due to its slow growth and overcollection, it is now critically endangered (17). In this study, the genes plr and sdh were cloned from P. pleianthum Hance (plr-PpH and sdh-PpH, respectively), fused, and heterologously expressed in E. coli to establish a high-efficiency system for the conversion of (+)-pinoresinol to matairesinol.
MATERIALS AND METHODS
Chemicals.
The solvents and chemicals used were reagent or high-performance liquid chromatography (HPLC) grade. (+)-Pinoresinol was purchased from Arbonova (Turku, Finland); (+)-lariciresinol, secoisolariciresinol, matairesinol, NADPH, NAD, dithiothreitol (DTT), and imidazole were from Sigma-Aldrich; and acetonitrile was from Merck. TRIzol reagent and the rapid amplification of cDNA ends (RACE) system were from Invitrogen; the OneStep reverse transcription-PCR (RT-PCR) kit, pQE-30 Xa, and nickel-nitrilotriacetic acid (Ni-NTA) agarose were from Qiagen; and the restriction enzymes were from New England BioLabs.
Cloning of plr-PpH and sdh-PpH.
Aseptic P. pleianthum Hance was grown in B5 medium according to a previous report (17), and the leaves were collected, frozen in liquid nitrogen, and stored at −80°C. Total RNA was extracted using TRIzol reagent. The primer sequences are listed in Table S1 in the supplemental material. Based on a multiple-sequence alignment of PLR-Fi1 (GenBank accession number U81158) (15), PLR-Tp1 (AF242503) (18), PLR-Tp2 (AF242504) (18), PLR-La1 (AJ849358) (19), and PLR-Lu1 (AJ849359) (19), the degenerate primers PLRFOR1, PLRFOR2, and PLRREV2 were designed to amplify the conserved part of plr. RT-PCR was performed using a OneStep RT-PCR kit with PLRFOR1 and oligo(dT), and the PCR product was further amplified using PLRFOR2 and PLRREV2. The resulting amplicon was sequenced, and RACE PCR was performed to amplify the cDNA from the conserved sequence to the 5′ or 3′ terminal. For 5′ RACE, the primers PLR-GSP1-5′, PLR-GSP2-5′, and PLR-GSP3-5′ were designed, and the 5′ terminal was synthesized using a 5′ RACE system for the RACE kit (Invitrogen), while PLR-GSP1-3′ was used for 3′ RACE PCR. The coding region of plr-PpH was amplified using PLRF and PLRR.
The gene sdh-PpH was amplified using a OneStep RT-PCR kit with forward primer SDHF and reverse primer SDHR, which were designed based on GenBank accession number AF352735 (16).
plr cDNA was amplified using primers sacPLRF and pstPLRR and introduced into the expression vector pQE-30 Xa at the SacI and PstI restriction sites to generate pQE-PLR. sdh-PpH cDNA was amplified using primers kpnSDHF and pstSDHR and introduced into the pQE-30 Xa vector at the KpnI and PstI restriction sites to generate pQE-SDH. pQE-PLR and pQE-SDH were transformed into E. coli M15 for heterologous expression.
Generation of the plr-sdh and sdh-plr constructs.
The proteins PLR-PpH and SDH-PpH were linked using a (GGGGS)4 protein linker. Splicing by overlap extension (SOEing) PCR was performed to connect the cDNAs for plr-PpH, sdh-PpH, and the linker. Five primers, P-S1, P-S2, P-S3, P-S4, and Link, were designed to amplify plr-(linker)-sdh. P-S1 and P-S2 were used to amplify the plr gene, and P-S3 and P-S4 were used to amplify the sdh gene. The product of P-S3 and P-S4 was further amplified using Link and P-S4 to obtain linker-sdh. The PCR products containing plr and linker-sdh were mixed together and another 15 cycles of PCR performed without adding primers. The product was finally amplified with P-S1 and P-S4 to generate plr-(linker)-sdh.
sdh-(linker)-plr was also amplified using SOEing PCR. Five primers, S-P1, S-P2, S-P3, S-P4, and Link, were designed, and S-P1 and S-P2 were used to amplify sdh, while S-P3 and S-P4 were used to amplify plr, which was further amplified using S-P4 and Link to generate linker-plr. Sdh and linker-plr were mixed, and another 15-cycle PCR was performed without primer addition and the product amplified using S-P1 and S-P4 to generate sdh-(linker)-plr.
plr-sdh and sdh-plr were introduced into the pQE-30 Xa vector at the SacI and PstI restriction sites to generate pQE-PLR-SDH and pQE-SDH-PLR (see Fig. S1 in the supplemental material), and E. coli M15 was transformed with the vectors for heterologous protein expression.
Heterologous expression and protein purification.
The recombinant E. coli M15 strains were grown overnight at 37°C with shaking at 130 rpm in LB medium containing 100 ppm of ampicillin and 50 ppm of kanamycin (LBAK medium), and then the bacteria were inoculated into 100 ml of fresh LBAK medium in a Hinton flask and the cultures grown under the same conditions to an optical density at 600 nm (OD600) of 0.6, when 0.01 mM isopropyl-β-d-thiogalactopyranoside (IPTG) was added to induce heterologous protein expression. After 9 h of induction at 25°C with shaking at 130 rpm, the bacteria were harvested by centrifugation at 4,000 × g for 30 min at room temperature. The pellets from 100 ml of bacterial culture were resuspended in 10 ml of lysis buffer (50 mM NaHPO4, 300 mM NaCl, 10 mM imidazole, pH 8.0), and the suspensions were sonicated in an ice bath to break the cells. Cell debris then was removed by centrifugation at 14,000 × g for 30 min at 4°C. The target proteins in the supernatants were purified using Ni-NTA agarose according to the manufacturer's instructions and stored at −20°C after addition of 50% (wt/vol) glycerol. SDS-PAGE (12.5% polyacrylamide) followed by Western blotting with monoclonal anti-polyhistidine antibody (Sigma) and goat anti-mouse alkaline phosphatase (AP)-conjugated antibody was used to determine the molecular weight of the recombinant proteins.
Enzyme assay.
The enzyme activity assay for recombinant PLR (rPLR) was modified from that in a previous report (20). The assay mixture (250 μl) containing 55 μM, 110 μM, 167 μM, 223 μM, and 279 μM (+)-pinoresinol or (+)-lariciresinol, 2.5 mM NADPH, 10 μg of rPLR, and 0.1 M potassium phosphate buffer, pH 7.1, was incubated at 30°C for 20 min. The kinetics of rPLR were determined by the disappearance of the substrate, (+)-pinoresinol or (+)-lariciresinol. The enzyme activity assay for recombinant SDH (rSDH) was modified from that in a previous report (16). The assay mixture (250 μl) containing 0.4 μg of rSDH; 15 μM, 45 μM, 75 μM, 105 μM, 135 μM, and 165 μM (−)-secoisolariciresinol; 4 mM NAD; and 20 mM Tris buffer containing 5 mM DTT, pH 8.8, was incubated at 20°C for 4 min. The kinetics of rSDH were determined by the formation of the product, matairesinol. Kinetic parameters were determined from Lineweaver-Burk plots for both rPLR and rSDH. The assay conditions for the complete transformation of (+)-pinoresinol to matairesinol were determined using rPLR and rSDH. Assay mixtures containing 0.19 mM (+)-pinoresinol, 1 mM NADPH, 0.6 mM NAD, and 5 mM DTT in 20 mM Tris buffer at different pHs (7.0, 8.0, and 8.8) were tested at a temperature of 22°C, 26°C, or 30°C for 1 h. The conditions chosen for assay of the fusion proteins were 10 μg of PLR-SDH or SDH-PLR, 0.19 mM (+)-pinoresinol, 1 mM NADPH, 0.6 mM NAD, and 20 mM Tris buffer containing 5 mM DTT, pH 8.0, at 22°C for 1 h. To monitor the time course of conversion in vivo in bacteria, the assay mixture containing 2 × 109 or 1 × 1010 CFU of bacteria, 50 μM (+)-pinoresinol, and 20 mM Tris buffer, pH 8.0, in LB medium was shaken vigorously at 22°C for 0.5, 1, 2, or 3 h.
HPLC analysis.
The assay mixtures described above were extracted three times with 300 μl of ethyl acetate, air dried, and redissolved in 200 μl of methanol for HPLC analysis using an Ascentis C18 HPLC column (particle size, 5 μm; column dimensions, 25 cm by 4.6 mm) in an HPLC system equipped with an SCL-10A system controller, SPD-M10A photodiode array detector, SIL-10AD autosampler, and LC-10AT pumps (all from Shimadzu, Kyoto, Japan). A binary buffer system (solvent A water containing 0.01% phosphoric acid, solvent B containing acetonitrile) was used to create the mobile-phase gradient. The analytes were separated using 25% solvent B for the first 25 min and then linear gradients of 25 to 43% solvent B for 18 min, 43 to 55% for 3 min, 55 to 70% for 8 min, 70 to 25% for 2 min, and 25% for 4 min at a flow rate of 1 ml/min (20) with detection at 280 nm.
Nucleotide sequence accession numbers.
Newly determined sequence data were deposited in GenBank under accession numbers KJ000044 and KJ000045.
RESULTS
Cloning and sequence analysis.
A segment of plr in P. pleianthum Hance was amplified by RT-PCR based on the conserved part of the plr sequences in Forsythia intermedia (U81158), Thuja plicata (AF242503 and AF242503), Linum album (AJ849358), and Linum usitatissimum (AJ849359), resulting in a 320-bp cDNA fragment, the deduced protein sequence of which showed high similarity to those of all of the PLRs described above and that determined later for Linum perenne (20). A 933-bp cDNA containing the full-length coding sequence, plr-Pp (GenBank accession number KJ000045), was cloned by 5′- and 3′-RACE PCR. The putative protein contained 311 amino acids. The deduced amino acid sequence for PLR-PpH showed 75.2% identity and 85% similarity with the PLR of Forsythia intermedi (see Fig. S2 in the supplemental material). The SIM alignment tool was used to examine the sequence for various motifs. A putative NADPH binding domain, GxxGxxG, was found in the PLR-PpH sequence, as was the case in the PLR of Forsythia intermedi.
A 834-bp cDNA containing the full-length sdh-PpH coding sequence (GenBank accession number KJ000044) was also identified based on the sdh sequence from Forsythia intermedia (AF352735). The putative protein contained 278 amino acids. The deduced amino acid sequence of SDH-PpH showed 98.2% identity and 99% similarity with that in Podophyllum peltatum and contained a conserved NAD binding domain, GxGGxG (see Fig. S3 in the supplemental material).
Functional expression in E. coli.
The gene plr-PpH was heterologously expressed in E. coli M15 with an N-terminal 6-histidine tag. The recombinant PLR containing a 4-kDa 6-His tag and Xa factor had an apparent molecular mass of 39 kDa on a polyvinylidene difluoride (PVDF) membrane (Fig. 2A). Different concentrations of (+)-pinoresinol or (+)-lariciresinol were added as the substrate for an enzyme kinetic assay using 10 μg of rPLR and 2.5 mM NADPH in 250 μl of assay mixture at 30°C and pH 7.1, and the Km and Vmax were found to be 19.8 μM and 7.34 μmol h−1 mg1 of protein for (+)-pinoresinol and 37.3 μM and 3.12 μmol h−1 mg1 of protein for (+)-lariciresinol, respectively.
FIG 2.

Expression and functional analysis of rPLR, rSDH, and the fusion proteins. (A) Western blot detection of the recombinant proteins with anti-His antibody. Lane 1 is rPLR (∼39 kDa), lane 2 is rSDH (∼34 kDa), lane 3 is PLR-SDH fusion protein (∼65 kDa), and lane 4 is SDH-PLR fusion protein (∼65 kDa). NC is the protein extract from wild-type E. coli M15 as the negative control. (B) Bioconversion efficiency under different conditions using rPLR and rSDH (1-h reaction). The concentration of (+)-pinoresinol is indicated by white bars, that of lariciresinol by light gray bars, that of secoisolariciresinol by dark gray bars, and that of matairesinol by black bars. The assay mixture without protein was used as the negative control. (C and D) HPLC traces at 280 nm of lignan compounds extracted from the 1-h bioconversion assay mixture using the two different fusion proteins. The substrate (+)-pinoresinol was converted to the final product, matairesinol, using fusion protein PLR-SDH (C) but not using SDH-PLR (D). (E) Percent bioconversion of matairesinol using a mixture of rPLR and rSDH or PLR-SDH fusion protein. The results using PLR-SDH are shown by dark gray bars and those using the rPLR and rSDH mixture by light gray bars. The error bars represent standard deviations.
SDH-PpH was also heterologously expressed in E. coli M15 with an N-terminal 6-His tag. The recombinant protein containing the His tag and Xa-factor had an apparent molecular mass of about 34 kDa on PVDF membrane (Fig. 2A). Different concentrations of (−)-secoisolariciresinol were converted into matairesinol using 0.4 μg of rSDH and 4 mM NAD in a 250-μl assay mixture at 20°C, pH 8.8, and the Km and Vmax were found to be 231 μM and 13.3 μmol min−1 mg1 of protein, respectively. The UV absorption spectra for the products were shown to confirm the identities (see Fig. S4 in the supplemental material).
Functional expression of the PLR-SDH fusion protein.
Two fusion protein constructs, plr-sdh and sdh-plr, were designed. The proteins were linked via a (GGGGS)4 protein linker to maintain flexibility. The molecular mass of the fusion proteins PLR-SDH and SDH-PLR, including the N-terminal His tag and Xa-factor, was about 65 kDa on SDS gels (Fig. 2A).
To determine the best reaction conditions for rPLR and rSDH, different combinations of buffers (20 mM Tris buffer at pH 7.0, 8.0, or 8.8) and temperatures (20°C, 26°C, and 30°C) were tested using 30 μg of each of the recombinant proteins, 0.19 mM (+)-pinoresinol, 1 mM NADPH, and 0.6 mM NAD. As shown in Fig. 2B, the results show that a higher conversion was obtained at a lower reaction temperature and that the efficiencies of formation of matairesinol and conversion of (+)-pinoresinol were both highest in Tris buffer, pH 8.0, at 22°C; these conditions were chosen for enzyme assay of the fusion proteins.
As shown in Fig. 2C and D, (+)-pinoresinol was successfully converted into matairesinol by PLR-SDH but not by SDH-PLR. SDH activity was lost in SDH-PLR, with the result that secoisolariciresinol, the product of PLR, accumulated in the reaction mixture. Time course conversion tests using PLR-SDH or the mixture of rPLR and rSDH are shown in Fig. 2E. At 60 min, the percent conversion of (+)-pinoresinol to matairesinol using PLR-SDH was 49.8%, higher than that of 17.7% using the rPLR and rSDH mixture.
In vivo bioconversion by PLR-SDH.
A further conversion assay was performed using living recombinant E. coli. (+)-Pinoresinol was successfully converted to the final product, matairesinol, by the living bacteria expressing PLR-SDH in LB medium, pH 8.0, with or without addition of cofactors NADPH and NAD at 22°C (Fig. 3). This result suggested that the enzyme reactions could take place in the living cells without external addition of cofactors. Cultures containing 2 × 109 or 1 × 1010 CFU of bacteria were used to synthesize (−)-matairesinol in the absence of added cofactors. As shown in Fig. 4, the substrate (+)-pinoresinol was almost completely (2 × 109 CFU) or completely (1 × 1010 CFU) converted into (−)-matairesinol by living cells without the addition of any cofactors in 2 h.
FIG 3.
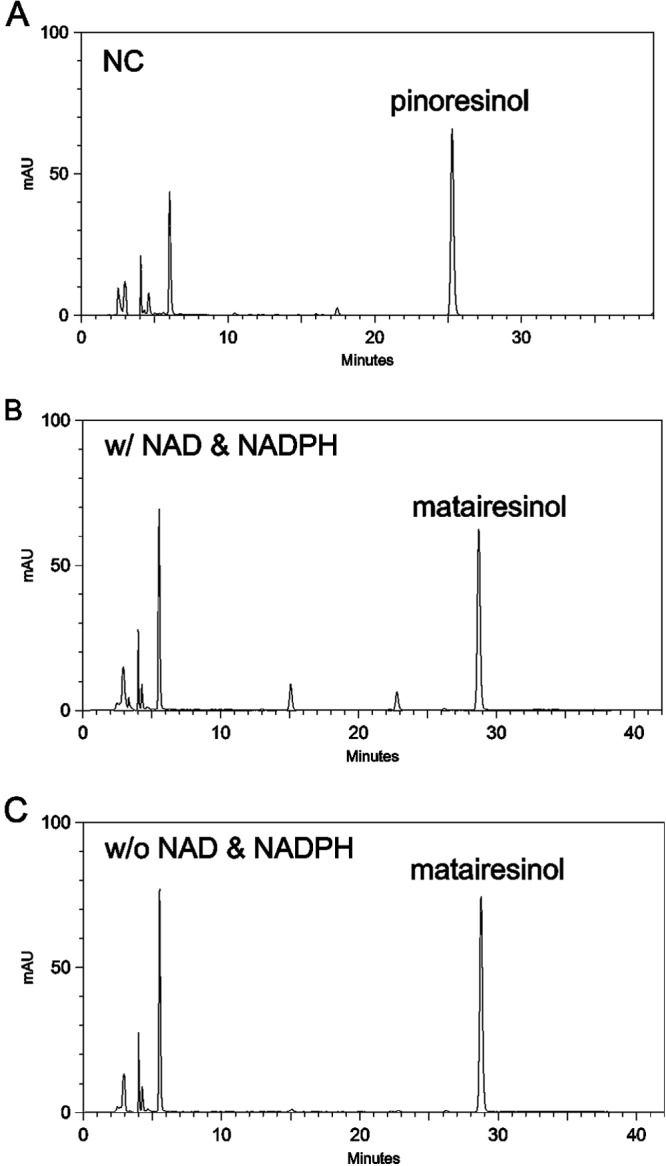
HPLC traces at 280 nm of lignan compounds extracted from a conversion assay using living recombinant E. coli. The substrate (+)-pinoresinol was coincubated for 6 h with wild-type E. coli (A) or with E. coli expressing PLR-SDH in the presence (B) or absence (C) of cofactors. w/, with; w/o, without.
FIG 4.
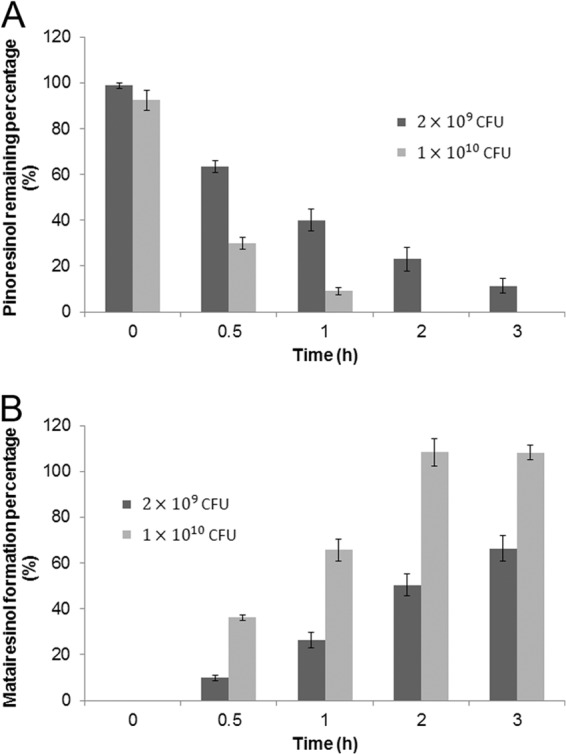
Time course of in vivo bioconversion of (+)-pinoresinol to matairesinol by E. coli M15 transformed with pQE-PLR-SDH. (A) Percentage of substrate (+)-pinoresinol remaining. (B) Production of the final product, matairesinol. The enzyme assay was performed at two different concentrations of bacteria, as indicated by the dark gray and light gray bars. The error bars represent standard deviations.
DISCUSSION
The enzyme PLR has been identified in Forsythia intermedia (15), Thuja plicata (18), Linum perenne (20), L. album, and L. usitatissimum (19). We cloned plr cDNA from P. pleianthum Hance. This may be the first report of a PLR identified in the genus Podophyllum. Podophyllum is the major plant source of podophyllotoxin. The plr-PpH sequence showed high nucleotide sequence similarity with other plr sequences. Besides PLRs, other lignan reductases, such as phenylcoumaran benzylic ether reductases (21) and isoflavone reductases (22, 23), also show high sequence similarity with PLRs. All of the lignan reductases contain a conserved NADPH-binding domain, GxxGxxG, in a region close to the N terminus, and this NADPH-binding domain was also present in the deduced amino sequence of PLR-PpH. Although pinoresinol stereospecificity was initially reported to be determined by Leu164, Phe272, and Gly268 (24), these residues were soon found not to be sufficient (19). Thus, the existence of Gly268 and another aromatic amino acid, Tyr272, instead of Phe in the PLR-PpH sequence is not sufficient to explain the (+)-pinoresinol substrate stereospecificity and suggests that other residues are also involved. The Km value of rPLR for pinoresinol was 37.3 μM, which was close to that of PLR-Fi1 and PLR-Fi2 (27 and 23 μM, respectively) (15). However, the Km for lariciresinol was 37.3 μM, which was lower than that of PLR-Fi1 and PLR-Fi2 (121 and 123 μM, respectively) (15). It seems the affinity of rPLR to lariciresinol was slightly higher than those of the PLRs in F. intermedia.
The enzyme SDH has been identified in F. intermedia and P. peltatum (16), and its cDNA (sdh-PpH) was cloned from P. pleianthum Hance in the present study. The deduced amino acid sequence showed high similarity with other SDH sequences, and the conserved NAD-binding domain GxGGxG and the amino acid residues Ser153, Tyr167, and Lys171, predicted to be involved in the SDH catalytic center (25), were found to be present. The Km value of rSDH was 231 μM, which was higher than that of SDH-Pp (160 μM) (16). This suggests that the substrate affinity of rSDH was lower than that of SDH in P. peltatum. The recombinant protein was able to convert (−)-secoisolariciresinol into matairesinol. Since no PLR and SDH active on the opposite stereoisomers were found, our results suggest that (+)-pinoresinol, (+)-lariciresinol, (−)-secoisolariciresinol, and (−)-matairesinol are involved in the podophyllotoxin biosynthesis pathway in P. pleianthum Hance.
Fusion proteins were designed to simplify the bioconversion process from (+)-pinoresinol to matairesinol. The enzyme reaction conditions used for the fusion proteins were pH 8.0 (between the values used for rPLR and rSDH individually) and a relatively low reaction temperature of 22°C. This low temperature is not surprising, since P. pleianthum Hance grows in medium- to high-altitude mountainous areas. PLR activity was seen with both the PLR-SDH and SDH-PLR fusion proteins, whereas SDH activity was seen only with PLR-SDH. This suggests that the C-terminal region of SDH is important for its enzyme function. A previous report of the crystal structure of SDH also suggested that substrate-enzyme binding requires the involvement of a C-terminal flexible arm (25). Thus, a decreased flexibility in the C terminus of SDH might be the reason for its disrupted function in SDH-PLR, and this may be overcome if a longer and more flexible protein linker was used.
The percent conversion of (+)-pinoresinol to matairesinol catalyzed by PLR-SDH was higher than that using a mixture of rPLR and rSDH. The distance from PLR to SDH was restricted in the fusion protein by the flexible protein linker (GGGGS)4. This may make it easier for (−)-secoisolariciresinol, the product of PLR and the substrate for SDH, to move from PLR to SDH, suggesting that a metabolic channel, in a broad sense, was formed in the fusion protein to increase conversion efficiency, as suggested previously (26–31). The same phenomenon of an increase in conversion efficiency in fusion proteins has been reported for isoflavone synthase-chalcone isomerase (32) and trehalose-6-phosphate synthetase-trehalose-6-phosphate phosphatase (33) fusion proteins. We also found that the substrate (+)-pinoresinol was converted into matairesinol in medium containing living E. coli transformants expressing PLR-SDH without addition of cofactors, such as NAD and NADPH, suggesting that the recombinant enzymes utilize the cofactors produced by the living cells. This might result from the substrate entering the living cell or broken cells releasing enzyme and cofactors into the culture medium. To distinguish between these two processes, further experiments are needed. In conclusion, we have established an E. coli bioconversion system for the efficient conversion of plant lignan (+)-pinoresinol to matairesinol without the addition of cofactors to save time and cost.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by grant 96-2313-B-002-070-MY3 from the National Science Council, Executive Yuan, Taiwan, Republic of China.
Footnotes
Published ahead of print 21 February 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03397-13.
REFERENCES
- 1.Castro MA, Gordaliza M, DelCorral JMM, SanFeliciano A. 1996. Distribution of lignanoids in the order Coniferae. Phytochemistry 41:995–1011. 10.1016/0031-9422(95)00512-9 [DOI] [Google Scholar]
- 2.Cos P, De Bruyne T, Apers S, Vanden Berghe D, Pieters L, Vlietinck AJ. 2003. Phytoestrogens: recent developments. Planta Med. 69:589–599. 10.1055/s-2003-41122 [DOI] [PubMed] [Google Scholar]
- 3.Sok DE, Cui HS, Kim MR. 2009. Isolation and bioactivities of furfuran type lignan compounds from edible plants. Recent Pat. Food Nutr. Agric. 1:87–95. 10.2174/2212798410901010087 [DOI] [PubMed] [Google Scholar]
- 4.Bleyer WA. 1997. The U.S. pediatric cancer clinical trials programmes: international implications and the way forward. Eur. J. Cancer 33:1439–1447. 10.1016/S0959-8049(97)00249-9 [DOI] [PubMed] [Google Scholar]
- 5.Adlercreutz H, Honjo H, Higashi A, Fotsis T, Hamalainen E, Hasegawa T, Okada H. 1991. Urinary excretion of lignans and isoflavonoid phytoestrogens in Japanese men and women consuming a traditional Japanese diet. Am. J. Clin. Nutr. 54:1093–1100 [DOI] [PubMed] [Google Scholar]
- 6.Borriello SP, Setchell KD, Axelson M, Lawson AM. 1985. Production and metabolism of lignans by the human faecal flora. J. Appl. Bacteriol. 58:37–43. 10.1111/j.1365-2672.1985.tb01427.x [DOI] [PubMed] [Google Scholar]
- 7.Adlercreutz H. 2002. Phyto-oestrogens and cancer. Lancet Oncol. 3:364–373. 10.1016/S1470-2045(02)00777-5 [DOI] [PubMed] [Google Scholar]
- 8.Milder IEJ, Feskens EJM, Arts ICW, Bueno de Mesquita HB, Hollman PCH, Kromhout D. 2005. Intake of the plant lignans secoisolariciresinol, matairesinol, lariciresinol, and pinoresinol in Dutch men and women. J. Nutr. 135:1202–1207 http://jn.nutrition.org/content/135/5/1202.long [DOI] [PubMed] [Google Scholar]
- 9.Kuijsten A, Arts ICW, Vree TB, Hollman PCH. 2005. Pharmacokinetics of enterolignans in healthy men and women consuming a single dose of secoisolariciresinol diglucoside. J. Nutr. 135:795–801 http://jn.nutrition.org/content/135/4/795.long [DOI] [PubMed] [Google Scholar]
- 10.Vanharanta M, Voutilainen S, Lakka TA, van der Lee M, Adlercreutz H, Salonen JT. 1999. Risk of acute coronary events according to serum concentrations of enterolactone: a prospective population-based case-control study. Lancet 354:2112–2115. 10.1016/S0140-6736(99)05031-X [DOI] [PubMed] [Google Scholar]
- 11.Lampe JW. 2003. Isoflavonoid and lignan phytoestrogens as dietary biomarkers. J. Nutr. 133(Suppl 3):956S–964S http://jn.nutrition.org/content/133/3/956S.long [DOI] [PubMed] [Google Scholar]
- 12.Hande KR. 1998. Etoposide: four decades of development of a topoisomerase II inhibitor. Eur. J. Cancer 34:1514–1521. 10.1016/S0959-8049(98)00228-7 [DOI] [PubMed] [Google Scholar]
- 13.Broomhead AJ, Dewick PM. 1990. Tumour-inhibitory aryltetralin lignans in Podophyllum versipelle, Diphylleia cymosa and Diphylleia grayi. Phytochemistry 29:3831–3837. 10.1016/0031-9422(90)85342-D [DOI] [Google Scholar]
- 14.Kupchan SM, Hemingway JC, Knox JR. 1965. Tumor inhibitors. VII. Podophyllotoxin, the active principle of Juniperus virginiana. J. Pharm. Sci. 54:659–660 [DOI] [PubMed] [Google Scholar]
- 15.Dinkova-Kostova AT, Gang DR, Davin LB, Bedgar DL, Chu A, Lewis NG. 1996. (+)-Pinoresinol/(+)-lariciresinol reductase from Forsythia intermedia. Protein purification, cDNA cloning, heterologous expression and comparison to isoflavone reductase. J. Biol. Chem. 271:29473–29482 [DOI] [PubMed] [Google Scholar]
- 16.Xia ZQ, Costa MA, Pelissier HC, Davin LB, Lewis NG. 2001. Secoisolariciresinol dehydrogenase purification, cloning, and functional expression. Implications for human health protection. J. Biol. Chem. 276:12614–12623. 10.1074/jbc.M008622200 [DOI] [PubMed] [Google Scholar]
- 17.Chen CY, Lee KT, Liu TH, Liu WH. 2004. Callus induction of Podophyllum pleianthum Hance and the detection of podophyllotoxin. Taiwan Nong Ye Hua Xue Yu Shi Pin Ke Xue 42:412–420 http://www.airitilibrary.com/Publication/alDetailedMesh?DocID=16052471-200412-42-6-412-420-a [Google Scholar]
- 18.Fujita M, Gang DR, Davin LB, Lewis NG. 1999. Recombinant pinoresinol-lariciresinol reductases from western red cedar (Thuja plicata) catalyze opposite enantiospecific conversions. J. Biol. Chem. 274:618–627. 10.1074/jbc.274.2.618 [DOI] [PubMed] [Google Scholar]
- 19.von Heimendahl CB, Schäfer KM, Eklund P, Sjöholm R, Schmidt TJ, Fuss E. 2005. Pinoresinol-lariciresinol reductases with different stereospecificity from Linum album and Linum usitatissimum. Phytochemistry 66:1254–1263. 10.1016/j.phytochem.2005.04.026 [DOI] [PubMed] [Google Scholar]
- 20.Hemmati S, Schmidt TJ, Fuss E. 2007. (+)-Pinoresinol/(−)-lariciresinol reductase from Linum perenne Himmelszelt involved in the biosynthesis of justicidin B. FEBS Lett. 581:603–610. 10.1016/j.febslet.2007.01.018 [DOI] [PubMed] [Google Scholar]
- 21.Gang DR, Kasahara H, Xia ZQ, Vander Mijnsbrugge K, Bauw G, Boerjan W, Van Montagu M, Davin LB, Lewis NG. 1999. Evolution of plant defense mechanisms. Relationships of phenylcoumaran benzylic ether reductases to pinoresinol-lariciresinol and isoflavone reductases. J. Biol. Chem. 274:7516–7527 [DOI] [PubMed] [Google Scholar]
- 22.Paiva NL, Edwards R, Sun YJ, Hrazdina G, Dixon RA. 1991. Stress responses in alfalfa (Medicago sativa L.) 11. Molecular cloning and expression of alfalfa isoflavone reductase, a key enzyme of isoflavonoid phytoalexin biosynthesis. Plant Mol. Biol. 17:653–667 [DOI] [PubMed] [Google Scholar]
- 23.Paiva NL, Sun Y, Dixon RA, Van Etten HD, Hrazdina G. 1994. Molecular cloning of isoflavone reductase from pea (Pisum sativum L.): evidence for a 3R-isoflavanone intermediate in (+)-pisatin biosynthesis. Arch. Biochem. Biophys. 312:501–510. 10.1006/abbi.1994.1338 [DOI] [PubMed] [Google Scholar]
- 24.Min T, Kasahara H, Bedgar DL, Youn B, Lawrence PK, Gang DR, Halls SC, Park H, Hilsenbeck JL, Davin LB, Lewis NG, Kang C. 2003. Crystal structures of pinoresinol-lariciresinol and phenylcoumaran benzylic ether reductases and their relationship to isoflavone reductases. J. Biol. Chem. 278:50714–50723. 10.1074/jbc.M308493200 [DOI] [PubMed] [Google Scholar]
- 25.Youn B, Moinuddin SG, Davin LB, Lewis NG, Kang C. 2005. Crystal structures of apo-form and binary/ternary complexes of Podophyllum secoisolariciresinol dehydrogenase, an enzyme involved in formation of health-protecting and plant defense lignans. J. Biol. Chem. 280:12917–12926. 10.1074/jbc.M413266200 [DOI] [PubMed] [Google Scholar]
- 26.Spivey HO, Ovádi J. 1999. Substrate channeling. Methods 19:306–321. 10.1006/meth.1999.0858 [DOI] [PubMed] [Google Scholar]
- 27.Pettersson H, Olsson P, Bulow L, Pettersson G. 2000. Kinetics of the coupled reaction catalysed by a fusion protein of yeast mitochondrial malate dehydrogenase and citrate synthase. Eur. J. Biochem. 267:5041–5046. 10.1046/j.1432-1327.2000.01558.x [DOI] [PubMed] [Google Scholar]
- 28.Pettersson H, Pettersson G. 1999. Mechanism of metabolite transfer in coupled two-enzyme reactions involving aldolase. Eur. J. Biochem. 262:371–376. 10.1046/j.1432-1327.1999.00386.x [DOI] [PubMed] [Google Scholar]
- 29.Pettersson H, Pettersson G. 2001. Kinetics of the coupled reaction catalysed by a fusion protein of beta-galactosidase and galactose dehydrogenase. Biochim. Biophys. Acta 1549:155–160. 10.1016/S0167-4838(01)00252-7 [DOI] [PubMed] [Google Scholar]
- 30.Spivey HO, Merz JM. 1989. Metabolic compartmentation. Bioessays 10:127–130. 10.1002/bies.950100409 [DOI] [PubMed] [Google Scholar]
- 31.Westerhoff HV, Welch GR. 1992. Enzyme organization and the direction of metabolic flow: physicochemical considerations. Curr. Top. Cell. Regul. 33:361–390 [DOI] [PubMed] [Google Scholar]
- 32.Tian L, Dixon RA. 2006. Engineering isoflavone metabolism with an artificial bifunctional enzyme. Planta 224:496–507. 10.1007/s00425-006-0233-0 [DOI] [PubMed] [Google Scholar]
- 33.Seo HS, Koo YJ, Lim JY, Song JT, Kim CH, Kim JK, Lee JS, Choi YD. 2000. Characterization of a bifunctional enzyme fusion of trehalose-6-phosphate synthetase and trehalose-6-phosphate phosphatase of Escherichia coli. Appl. Environ. Microbiol. 66:2484–2490. 10.1128/AEM.66.6.2484-2490.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.