

Abstract
Lysostaphin represents a promising therapeutic agent for the treatment of staphylococcal infections, in particular those of methicillin-resistant Staphylococcus aureus (MRSA). However, conventional expression systems for the enzyme suffer from various limitations, and there remains a need for an efficient and cost-effective production process to facilitate clinical translation and the development of nonmedical applications. While Pichia pastoris is widely used for high-level production of recombinant proteins, there are two major barriers to the production of lysostaphin in this industrially relevant host: lack of expression from the wild-type lysostaphin gene and aberrant glycosylation of the wild-type protein sequence. The first barrier can be overcome with a synthetic gene incorporating improved codon usage and balanced A+T/G+C content, and the second barrier can be overcome by disrupting an N-linked glycosylation sequon using a broadened choice of mutations that yield aglyscosylated and fully active lysostaphin. The optimized lysostaphin variants could be produced at approximately 500 mg/liter in a small-scale bioreactor, and 50% of that material could be recovered at high purity with a simple 2-step purification. It is anticipated that this novel high-level expression system will bring down one of the major barriers to future development of biomedical, veterinary, and research applications of lysostaphin and its engineered variants.
INTRODUCTION
Lysostaphin (LST) is a glycyl-glycine zinc-dependent endopeptidase natively encoded on the pACK1 plasmid of Staphylococcus simulans (1), an environmental competitor of Staphylococcus aureus. LST is synthesized as a preproenzyme of 493 amino acids, and its pre- (36 amino acids) and pro- (211 amino acids) sequences are removed during and after secretion, respectively. Mature LST is a monomer composed of an N-terminal catalytic domain (132 amino acids), a C-terminal cell wall binding domain (102 amino acids), and a short connecting linker (13 amino acids) between the two (2). The LST enzyme selectively and efficiently degrades pentaglycine cross-links in the peptidoglycan component of S. aureus cell walls, ultimately resulting in bacterial lysis and death. Lysostaphin was discovered in the 1960s (3) and has since undergone various degrees of preclinical development and even small-scale clinical testing by different groups and organizations (4–8). Early interest in LST as a therapeutic agent waned as a result of ready access to conventional drugs, such as methicillin, but enthusiasm for LST in biomedical applications has been revived due to wide-spread antibiotic resistance and shallow antimicrobial development pipelines (9).
One barrier to LST clinical applications is the high doses required to eradicate some infections. LST appeared to show good efficacy in an unresponsive leukemia patient suffering from multidrug-resistant staphylococcal pneumonia, multiple abscesses, and cellulitis (7), but this effect required a 500-mg systemic bolus of enzyme. In another study, nasal carriers of coagulase-positive S. aureus were shown to be effectively cleared of the pathogen following intranasal LST treatment, but this effect required 2 weeks of 4-times-daily intranasal administration of a 5 mg/ml LST solution (8). Similar results at similar dosages were obtained in other human studies of nasal carriage clearance (10, 11). In a murine model of catheter-associated S. aureus biofilms, systemic LST administration was shown to clear established biofilms during a 4-day treatment regime, and a single prophylactic dose prevented subsequent biofilm formation on indwelling catheters (12). Extrapolating the effective doses to a human patient, however, would require as much as 16 or more grams of enzyme to be administered over 4 days. Thus, while LST has demonstrated consistent efficacy in animal models and even human studies, translating the effective dosages to wide-spread clinical use will require a particularly efficient production platform.
Toward this end, LST has been produced in a wide range of microbial expression hosts. One source of commercial LST is high-cell-density cultures of the native organism S. simulans, but the industrial-scale production yields from this system are withheld as proprietary information. Alternatively, expression yields from the bacterial host Escherichia coli are known to range from 10 to 20 mg/liter (13–15), and this recombinant platform is also a significant contributor to commercially sourced material. Large-scale LST production for clinical trials has been pursued with the Lactococcus lactis nisin-induced controlled expression (NICE) system (16, 17). Expression levels of 100 mg/liter were achieved in large-volume, high-cell-density fermentations, but the final purified yields were only 40 mg/liter (16). Subsequent process optimization of this system increased bioreactor expression levels to 300 mg/liter (17), but there remains considerable room for further improvement. Finally, it bears noting that LST has also been expressed in mammalian cells (18), but no effort was made toward maximizing or even quantifying yields.
Pichia pastoris has proved to be a highly successful heterologous expression host in recent years, with more than 1,000 recombinant proteins expressed as of 2009 (19). Unlike the aforementioned prokaryotic systems, P. pastoris has dedicated, high-capacity protein secretion pathways, which greatly simplify both upstream and downstream protein purification (20). Moreover, as a yeast, P. pastoris produces no endotoxins, obviating the need for arduous and inefficient endotoxin removal steps prior to biomedical applications (21). Although P. pastoris is an attractive recombinant host, the expression of bacterial proteins in these eukaryotic cells can present its own challenges. In this study, two key barriers to high-level LST expression in P. pastoris were encountered, (i) lack of detectable expression from the wild-type (WT) LST gene and (ii) aberrant glycosylation at one of two internal N-linked consensus sequons. These limitations were addressed by optimizing the gene sequence and the protein sequence, respectively.
MATERIALS AND METHODS
Regents and media.
Primers with standard desalting were ordered from IDT Technologies (Coralville, IA). Enzymes for molecular cloning were purchased from New England BioLabs (Ipswich, MA), as was Remove-iT PNGase F. Commercial LST produced in E. coli was purchased from Sigma (St. Louis, MO). All other reagents and supplies were from VWR Scientific (Philadelphia, PA), unless specifically noted.
Plasmids and strains.
P. pastoris expression vector pPIC9 and P. pastoris strain GS115 were purchased from Invitrogen (Grand Island, NY). S. aureus strain SA113 was from the American Type Culture Collection (Manassas, VA). Clinical isolates of S. aureus (strains 6445, 3425-1, and 3425-3) were the kind gift of Ambrose Cheung (Dartmouth, Hanover, NH).
Design of a synthetic lysostaphin gene (SYN lst).
An artificial gene encoding the wild-type LST enzyme was synthesized to satisfy two general objectives: (i) to substitute codons satisfying the preferred codon usage of P. pastoris for wild-type codons and (ii) to balance the A+T/G+C distribution of segments with disproportionate A+T content. In the synthetic gene, the majority of codons were replaced by the most preferred codons of P. pastoris (22). However, to disrupt long stretches of A+T bases, the second-most-frequent P. pastoris Thr and Val codons were inserted as needed. Specifically, ACC (14.5% usage) instead of ACT (22.4% usage) was occasionally used for Thr, and GTC (14.9%) instead of GTT (26.9%) was used for Val.
Cloning of lst genes.
The WT lst gene was amplified from S. simulans with primers WT-F and WT-R (Table 1). The SYN lst gene was synthesized by Shanghai Xuguan Biotechnology Development Company (Shanghai, China) and amplified with primers SYN-F and SYN-R (Table 1). The chimeric and aglycosylated lst genes were constructed by splice overlap extension PCR using the primers listed in Table 1. Briefly, paired 5′ and 3′ fragments of the WT or SYN lst genes were first amplified using one external primer and one internal primer each, and the resulting fragments were then spliced together in an overlap reaction using external primers. For example, two fragments for the N125Q point mutant (with a change of N to Q at position 125) were generated by amplifying SYN lst with (i) SYN-F and N125Q-R and (ii) SYN-R and N125Q-F. The resulting PCR products were then mixed at an equimolar ratio, and the mixture was used as the template in a subsequent splicing overlap reaction using primers SYN-F and SYN-R. All PCRs were performed using Phusion high-fidelity DNA polymerase. The LST-encoding genes were digested with XhoI and EcoRI, ligated into similarly digested pPIC9, and transformed by electroporation into E. coli DH5α [F− ϕ80lacZΔM15 Δ(lacZYA-argF) U169 recA1 endA1 hsdR17 (rK− mK+) phoA supE44 λ− thi-1 gyrA96 relA1]. This fuses LST in frame with the alpha mating factor (αMF) secretion signal from Saccharomyces cerevisiae. Clones bearing the expression vector with the insert were selected by PCR using external primers (either WT-F with WT-R or SYN-F with SYN-R) and confirmed by DNA sequencing.
TABLE 1.
Primers used in this study
| Primer | Sequence (5′ to 3′) |
|---|---|
| WT-F | ATCGCTCGAGAAAAGAGCTGCAACACAT |
| WT-R | CGATGAATTCTTACTTTATAGTTCCCCA |
| Chi1-F | CAAAGAATGGTTAATTCATTTTCCCAATCCACCGCTCAAGAC |
| Chi1-R | GTCTTGAGCGGTGGATTGGGAAAATGAATTAACCATTCTTTG |
| Chi2-F | CAAAGAATGGTCAACTCCTTCTCAAATTCAACTGCCCAAGAT |
| Chi2-R | ATCTTGGGCAGTTGAATTTGAGAAGGAGTTGACCATTCTTTG |
| Chi3-F | GAAGCTGGTTGGAGTAATTACGGAGGTGGTAACCAAATCGGTTTGATC |
| Chi3-R | GATCAAACCGATTTGGTTACCACCTCCGTAATTACTCCAACCAGCTTC |
| Chi4-F | GAGGCTGGTTGGTCCAACTACGGTGGAGGTAATCAAATAGGTCTTATT |
| Chi4-R | AATAAGACCTATTTGATTACCTCCACCGTAGTTGGACCAACCAGCCTC |
| Chi5-F | AAATATAATGTTAAAGTAGGAGATTACGTCAAGGCTGGTCAAATCATC |
| Chi5-R | GATGATTTGACCAGCCTTGACGTAATCTCCTACTTTAACATTATATTT |
| Chi6-F | AAGTACAACGTCAAGGTCGGTGACTATGTCAAAGCTGGTCAAATAATC |
| Chi6-R | GATTATTTGACCAGCTTTGACATAGTCACCGACCTTGACGTTGTACTT |
| Chi7-F | GGTCTTATTGAAAATGATGGAGTGCACAGACAATGGTACATGCACTTG |
| Chi7-R | CAAGTGCATGTACCATTGTCTGTGCACTCCATCATTTTCAATAAGACC |
| Chi8-F | GGTTTGATCGAGAACGACGGTGTCCATAGACAATGGTATATGCATCTA |
| Chi8-R | TAGATGCATATACCATTGTCTATGGACACCGTCGTTCTCGATCAAACC |
| Syn-F | ATCGCTCGAGAAAAGAGCTGCTACCCAC |
| Syn-R | CGATGAATTCTTACTTGATGGTACCCCA |
| N125Q-F | AACTCCTTCTCCCAATCCACCGCTCAA |
| N125Q-R | TTGAGCGGTGGATTGGGAGAAGGAGTT |
| N125D-F | AACTCCTTCTCCGACTCCACCGCTCAA |
| N125D-R | TTGAGCGGTGGAGTCGGAGAAGGAGTT |
| N125S-F | AACTCCTTCTCCTCCTCCACCGCTCAA |
| N125S-R | TTGAGCGGTGGAGGAGGAGAAGGAGTT |
| S126P-F | AACTCCTTCTCCAACCCAACCGCTCAA |
| S126P-R | TTGAGCGGTTGGGTTGGAGAAGGAGTT |
| T127A-F | TTCTCCAACTCCGCTGCTCAAGACCCA |
| T127A-R | TGGGTCTTGAGCAGCGGAGTTGGAGAA |
| Alfa-F | ATGAGATTTCCTTCAATTTTTACTG |
| 5Syn-R | CACCGTCGTTCTCGATCAAACCGA |
| FullSyn-R | CTTGATGGTACCCCACAAGACACC |
| 5WT-R | CTCCATCATTTTCAATAAGACCTA |
| FullWT-R | CTTTATAGTTCCCCAAAGAACACC |
| AOX1-F | ATTGCCGGAAGATTGGCAAACTTG |
| AOX1-R | AAAACGATTTGCTTTCTAGCACGG |
| N125NNK-F | AACTCCTTCTCCNNKTCCACCGCTCAA |
| N125NNK-R | TTGAGCGGTGGAMNNGGAGAAGGAGTT |
P. pastoris expression.
The pPIC9 expression vectors harboring sequenced lst genes were digested with SacI prior to electroporation into P. pastoris strain GS115. P. pastoris transformants were initially grown on solid MD medium (1.34% yeast nitrogen base [YNB], 0.00004% biotin, 2% dextrose, 1% agar), then cultured in BMGY medium (1.0% yeast extract, 2.0% peptone, 1.34% YNB, 0.00004% biotin, 1.0% glycerol, 100 mM phosphate buffer, pH 6.0), and finally induced in BMMY medium (1.0% yeast extract, 2.0% peptone, 1.34% YNB, 0.00004% biotin, 0.5% methanol, 100 mM phosphate buffer, pH 6.0). LST-expressing clones were identified by SDS-PAGE analysis of culture supernatants.
RT-PCR.
A 5′ fragment or full-length LST mRNA was detected by reverse transcription (RT)-PCR using forward primer WT-F or SYN-F and reverse primer 5WT-R, 5SYN-R, FullWT-R, or FullSYN-R (Table 1). As an internal positive control, a fragment of alcohol oxidase 1 (AOX1) mRNA was detected with primers AOX1-F and AOX1-R.
Bioreactor culture.
For pilot-scale production of LST, P. pastoris was cultured in a 2-liter bioreactor as described previously (23). Briefly, a 3-stage fed-batch culture process was employed. First, P. pastoris was cultured at 30°C in 1.5 liters of low-salt BSM medium (per liter, 20 ml 85% phosphoric acid, 0.2 g calcium sulfate, 5 g potassium sulfate, 4 g magnesium sulfate-7H2O, 1 g potassium hydroxide, 30 g glycerol). After the initial glycerol had been exhausted, as identified by a dissolved oxygen spike, the culture density was increased to an optical density at 600 nm (OD600) of approximately 100 using a constant glycerol feed rate of 15 ml/h for 6 h. Finally, the culture temperature was decreased to 20°C and the expression of lysostaphin was induced by a bolus injection of methanol to a final concentration of 0.5%. A controlled methanol feed was initiated upon full induction of the AOX1 promoter, indicated by a steady decrease in dissolved oxygen concentration with the feeding of methanol. A controlled dissolved oxygen (DO-stat) strategy was then employed to maintain 20% dissolved oxygen by adjusting the methanol feed rate (24).
Lysostaphin purification.
LST was precipitated from culture supernatants by 25% polyethylene glycol (PEG) precipitation at room temperature, resuspended in 20 mM NaHPO4 buffer, pH 7.5, and bound to a HiPrep SP Sepharose fast flow 16/10 prepacked column that had been equilibrated with the same buffer. Following a 10-column-volume wash with the same buffer, LST was eluted with a 0-to-250 mM NaCl gradient, occurring over 10 column volumes. The purity and concentration of eluted LST was determined by SDS-PAGE gel densitometry, using a standard curve generated with commercially sourced LST.
MIC assay.
The MIC of LST was determined by the microplate method (25). Using 96-well polystyrene plates, 100-μl aliquots of P. pastoris culture supernatant were serially diluted in tryptic soy broth (TSB), while 100-μl aliquots of purified LST were serially diluted in phosphate-buffered saline (PBS; 2.7 mM KCl, 1.5 mM KH2PO4, 8.9 mM Na2HPO4, 136.9 mM NaCl, pH 7.4) supplemented with 0.1% bovine serum albumin. Each well was then inoculated with 100 μl of ∼106 CFU/ml S. aureus strain SA113 in TSB or Mueller-Hinton broth (BD) supplemented with 2% NaCl, yielding a total volume of 200 μl per well. Microplates were then incubated at 37°C for 24 h. The inhibitory activity in culture supernatants was assessed as the MIC50, the treatment dilution yielding 50% inhibition of growth. The inhibitory activity of purified LST was determined by the MIC0, the enzyme concentration yielding complete inhibition of growth. Both MIC0 and MIC50 were quantified by measuring light scattering at 650 nm in a microplate reader. All assays were performed in duplicate.
Turbidity assay.
S. aureus strain SA113 was cultured overnight in TSB, pelleted by centrifugation, and washed once with PBS. The cells were then resuspended in PBS, and this stock bacterial suspension was aliquoted into replicate wells of a 96-well flat-bottomed polystyrene plate (Nunc 269620) (100 μl, A650 = 0.8 in the plate). Lysis reactions were initiated by adding purified LST to a final concentration of 5 μg/ml using a 12-channel Pipetman. The kinetics of bacterial lysis were followed by measuring the light scattering at 650 nm every minute for a total of 30 min. The activity of the various LST constructs was defined as the time to reach half the starting optical density (TOD50) of the initial bacterial suspension.
Thermostability assay.
The relative thermostability of LST constructs was determined by differential scanning fluorimetry on an Applied Biosystems ABI 7500 fast real-time PCR system, essentially as described previously (26). Proteins and SYPRO orange were diluted in PBS, and fluorescence was quantified at 1-degree increments from 25 to 94°C.
PNGase F treatment.
One microliter of 10× G7 reaction buffer and 1 μl of Remove-iT PNGase F were added to 8 μl of culture supernatant. The reaction mixture was incubated for 1 h at 37°C, and the reaction products were analyzed by SDS-PAGE.
Saturation mutagenesis of LST residue 125.
LST amino acid 125 was subjected to saturation mutagenesis by splice overlap extension PCR using the SYN lst gene template and degenerate internal primers (NNK codon, where N = A, G, C, or T and K = G or T) (Table 1). The 32-member gene library was cloned and transformed into P. pastoris as described above. Transformants were spread on YPM agar medium (1% yeast extract, 2% peptone, 1% methanol, 1% agar) and incubated at 30°C for 2 days. An overnight culture of S. aureus strain SA113 was diluted in PBS to an OD600 of 1.0, and this bacterial suspension was mixed in a 1:10 ratio with molten top agar (0.5 yeast extract, 1% peptone, 1% NaCl, 0.75% agar). The indicating top agar was poured onto the YPM yeast plates, and the plates were incubated at 37°C for 10 h. Yeast clones expressing active enzymes were identified by their characteristic halo or zone of clearance. Halo-forming colonies were picked through the top agar, their cognate lst genes were amplified with primers SYN-F and SYN-R (Table 1), and the PCR products were sequenced.
Bioinformatics analysis.
Sequence alignment was performed using ClustalW (www.ebi.ac.uk/Tools/msa/clustalw2/). Codon analysis was performed using CodonW (codonw.sourceforge.net). The frequency of optimum codons (Fop) is defined by the formula (number of optimal codons)/(total number of codons). Its values range from 0 (when a gene contains no optimal codons) to 1 (when a gene is composed entirely of optimal codons).
Gene sequence deposition.
The optimized SYN lst gene sequence, encoding the wild-type LST enzyme, has been deposited in GenBank with the accession number KF724949.
RESULTS
Gene sequence optimization enables high-level expression of LST in P. pastoris.
In an effort to obtain high expression yields from yeast, the WT lst gene was amplified from S. simulans, cloned into P. pastoris expression vector pPIC9, and transformed into P. pastoris strain GS115. This construct was devoid of the native LST prepro sequence and was instead fused to the αMF prepro sequence from S. cerevisiae. Following induction of the GS115 host, however, no LST enzyme could be detected by SDS-PAGE analysis of shake flask culture supernatants (Fig. 1A, WT lane).
FIG 1.
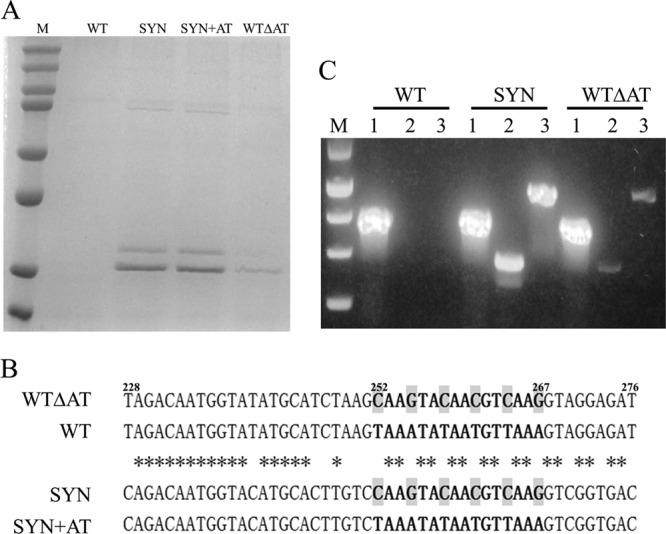
Gene sequence optimization enables high-level expression of LST in P. pastoris. (A) Comparison of LST expression levels from different recombinant genes. Lanes: M, protein marker (top to bottom, 250, 150, 100, 75, 50, 37, 25, and 20 kDa); WT, WT lst gene; SYN, SYN lst gene; SYN+AT, the A+T-rich region (nt 252 to 267) from the WT lst gene was introduced into the SYN lst gene; WTΔAT, the A+T-rich region (nt 252 to 267) of the WT lst gene was replaced by the corresponding sequence from the SYN lst gene. (B) Sequence alignment of a region from WT lst (nt 228 to 276) that caused the most significant decrease in expression when transplanted into the SYN lst gene. Asterisks indicate nucleotides that are identical among all four gene sequences. Shaded nucleotides are the six critical bases whose mutations were sufficient to activate LST expression. (C) Full-length LST mRNA and a shorter 5′ fragment were detected by RT-PCR. A fragment of AOX1 mRNA was used as an internal amplification and loading control. Lanes: M, DNA marker (top to bottom, 1,500, 1,000, 750, 500, and 250 bp); 1, fragment of AOX1; 2, 5′ fragment of LST mRNA; 3, full-length LST mRNA.
Initial speculation tied the lack of any detectable expression to differential codon bias in the bacterial gene versus the yeast host. Specifically, the frequency of optimal P. pastoris codons (Fop) within the WT lst gene is as low as 0.388. Moreover, a closer inspection of the WT lst gene revealed an extraordinarily high A+T content (62.6% overall). To address both putative issues simultaneously, an artificial gene (SYN lst) having an increased Fop of 0.978 and a decreased A+T content of 47.3% was synthesized. In contrast to WT lst, P. pastoris expression from the SYN lst gene yielded substantial quantities of enzyme in the culture supernatant (∼80 mg/liter in shake flask culture) (Fig. 1A, SYN lane).
Fine mapping of problematic gene subsequences.
To identify specific regions of the WT lst gene that were responsible for poor expression, a panel of chimeric WT-SYN genes was constructed (Fig. 2A). While the complete lack of expression from WT lst could not be traced to any single region of the gene, it was found that detrimental sequences were not evenly distributed (Fig. 2B). Importantly, the most critical determinant of poor expression yields was confined to a 49-bp segment from nucleotide (nt) 228 to 276 (Chi8).
FIG 2.
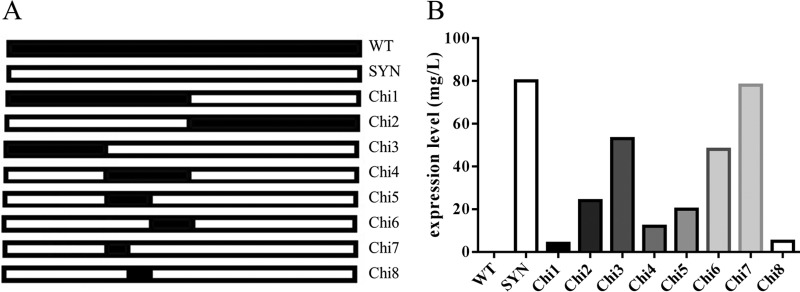
Mapping of dominant detrimental sequences in the WT lst gene. (A) Schematic representation of the chimeric genes examined (WT lst is in black and SYN lst is in white). (B) Levels of LST expression from the panel of chimeric genes, as measured by quantitative densitometry of SDS-PAGE protein bands.
A detailed comparison of this WT lst sub-sequence with that of the SYN lst gene revealed that the key differences were those within a narrow A+T-rich region (from nt 252 to 267) (Fig. 1B). To confirm the regulatory role of this 21-bp A+T-rich region, the corresponding sequence in the optimized SYN lst gene was replaced by the counterpart sequence from WT lst, yielding construct SYN+AT (Fig. 1B). Additionally, the same A+T-rich segment of the WT lst gene was replaced with the corresponding SYN lst sequence, thereby yielding construct WTΔAT, the inverse of gene SYN+AT. Analysis of culture supernatants by SDS-PAGE revealed that replacement of the A+T-rich region in WTΔAT resulted in enhanced LST expression, albeit at lower levels than from SYN lst itself (Fig. 1A). In contrast, introduction of the A+T-rich region into the SYN+AT construct did not reduce expression compared to that of SYN lst.
Putative mechanism for compromised WT lst expression.
It was speculated that the A+T-rich region was influencing expression by acting as a premature transcription terminator. To probe this proposed mechanism, RT-PCR was used to detect mRNAs corresponding to the full-length WT lst and SYN lst genes or shorter 5′ fragments thereof. Consistent with the hypothesis, expression levels were found to correlate well with the relative abundance of mRNA. In particular, no LST-specific mRNA was detected for the WT lst gene, a high mRNA level was detected for the SYN lst gene, and a low mRNA level was detected for the WTΔAT construct (Fig. 1C).
Aberrant glycosylation of the WT LST protein.
To evaluate the potential of P. pastoris for pilot-scale production of LST, P. pastoris encoding SYN lst was cultured in a 2-liter bioreactor. The expression levels from bioreactor cultivations were ∼500 mg/liter, and approximately 50% of this material could be recovered in high purity by PEG precipitation and cation exchange chromatography. However, LST from both shake flask and bioreactor culture migrated as doublets in SDS-PAGE (Fig. 3A, left). The two species coeluted from cation exchange chromatography columns as a single peak (Fig. 3B), and reducing the mass of enzyme loaded on the column did not increase chromatographic resolution (data not shown). The higher-molecular-mass material was thought to result from aberrant N-linked glycosylation, and indeed, the upper band disappeared from SDS-PAGE after PNGase F treatment (Fig. 3A, right). Consistent with prior observations that glycosylated LST from mammalian cells is inactive (27), fast protein liquid chromatography fractions containing higher proportions of glycosylated LST (Fig. 3B, lanes 1 to 3) were less active in turbidometric assays (data not shown). Thus, there was a need to generate aglycosylated variants to take full advantage of the P. pastoris expression host.
FIG 3.
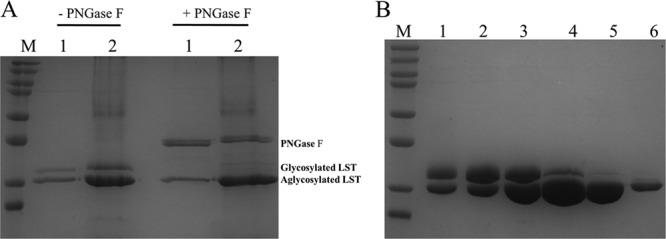
LST secreted from P. pastoris is aberrantly glycosylated. (A) LST from P. pastoris culture supernatants migrates as a doublet (left), and treatment with PNGase F eliminates the higher-molecular-mass upper band (right). Lanes: M, protein marker (top to bottom, 250, 150, 100, 75, 50, 37, 25, and 20 kDa); 1, 10 μl of supernatant of flask culture; 2, 10 μl of supernatant of bioreactor culture. (B) Glycosylated and unglycosylated LST cannot be separated during cation exchange chromatography. Lanes: M, protein marker (top to bottom, 250, 150, 100, 75, 50, 37, 25, and 20 kDa); 1 to 6, different fractions collected from a single peak during chromatographic purification.
Construction and preliminary analysis of aglycosylated LST variants.
LST contains two consensus N-linked glycosylation sequons, one at position 125 (N125-S126-T127) and the other at position 232 (N232-K233-T234). When the latter was disrupted with an N232Q point mutation, the variant protein continued to migrate as a doublet in SDS-PAGE (data not shown). This result indicated that N125 was the site of aberrant glycosylation in P. pastoris. Additional indirect evidence for this conclusion is also provided by the recent demonstration that position N125 is the site of glycosylation in mammalian cells (28). The N125 glycosylation sequon was disrupted via a typical strategy wherein a conservative N→Q, N→D, or N→S point mutation was introduced at position 125. Each of these mutations successfully abolished P. pastoris glycosylation of LST, and the expression yields of the three variants were comparable to that of the wild-type protein (Fig. 4A). Surprisingly, however, MIC50 analysis of culture supernatants showed the aglycoslyated N125Q, N125S, and N125D variants to exhibit approximately 10-, 20- and 40-fold-lower activity levels, respectively, than native LST produced in P. pastoris (Fig. 4B).
FIG 4.
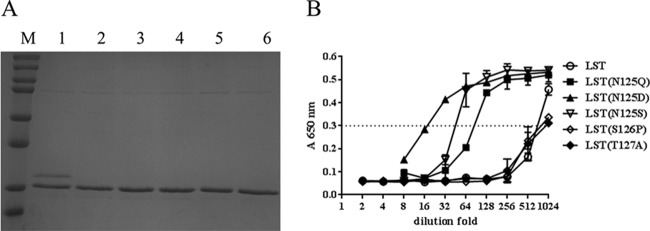
Expression of aglycosylated LST from P. pastoris. (A) Expression of aglycosylated LST variants. Lanes: M, protein marker (top to bottom, 250, 150, 100, 75, 50, 37, 25, and 20 kDa); 1, LST; 2, LST(N125Q); 3, LST(N125D); 4, LST(N125S); 5, LST(S126P); 6, LST(T127A). All variants exhibited similar expression levels. (B) Activity assays of aglycosylated LST variants. MIC assays with S. aureus SA113 were performed on 2-fold serial dilutions of culture supernatants. The OD650 readings for each enzyme dilution series are shown, and a dashed line indicates the reading corresponding to the MIC50. Error bars represent standard deviations.
To comprehensively assess the functional plasticity of LST residue 125, a small gene library was constructed by saturation mutagenesis of the corresponding codon. Screening of approximately 300 transformants (representing 10-fold coverage of the library) on indicating medium yielded only 4 active colonies able to generate halos (Fig. 5A). SDS-PAGE analysis showed that the LST secreted from these 4 colonies migrated as doublets (Fig. 5B), indicating that each mutant encoded the native asparagine residue at position 125. Further evidence of the strict requirement for an asparagine at residue 125 was found by aligning the LST amino acid sequence with those of two functional homologs (Ale-1 and LytM) (Fig. 5C).
FIG 5.
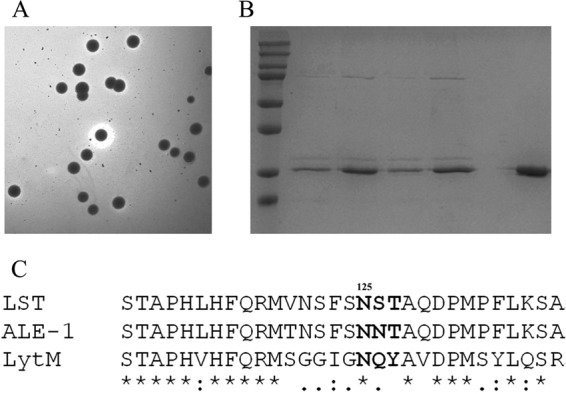
Asparagine at position 125 is essential for the full activity of LST. (A) An LST saturation mutagenesis library at position 125 was screened by plate halo assay, and all functional clones were picked for further analysis. (B) SDS-PAGE analysis of LST secreted from the halo-forming colonies. Lanes: M, protein marker (top to bottom, 250, 150, 100, 75, 50, 37, 25, and 20 kDa); 1st to 4th, LST secreted from the halo-forming colonies; 5th, commercial LST purchased from Sigma. (C) Sequence alignment of LST with two related staphylocidal lysins (Ale-1 and LytM) shows that N125 is conserved in all three enzymes. Asterisks indicate positions having one identical residue, colons indicate positions occupied by residues having strongly similar properties, periods indicate positions occupied by residues having weakly similar properties, and gaps indicate positions occupied by dissimilar residues.
Given the unexpected and unfavorable outcome from the mutation at position 125, two alternative aglycosylated variants, one at position 126 (S126P) and one at position 127 (T127A), were constructed, expressed, and analyzed. Similar to the results of mutations at N125, the LST(S126P) and LST(T127A) variants were uniformly aglycosylated and were expressed at levels comparable to the wild-type enzyme (Fig. 4A). Importantly, both of the new variants possessed bactericidal activity equivalent to that of wild-type LST (Fig. 4B).
Detailed analysis of highly active aglycosylated LST variants.
The two most highly active aglycosylated variants, LST(S126P) and LST(T127A), were expressed in 2-liter bioreactors and purified to homogeneity following the same production process as for wild-type LST (Fig. 6A). The expression levels and final purified yields of both aglycosylated variants were similar to those of wild-type LST. The activities and thermostabilities of aglycosylated LST variants were compared with those of commercially sourced LST as a reference. In kinetic turbidometric assays, the TOD50 values of LST(S126P), LST(T127A), and commercial LST were 5, 7, and 17 min, respectively (Fig. 6B). In antibacterial assays against a small panel of clinical isolates (strains 6445, 3425-1, and 3425-3) and a laboratory strain (SA113), both variants exhibited MIC values equivalent to those of the commercial enzyme (Fig. 6C). With respect to structural stability, LST(S126P) showed a single apparent melting temperature (Tm) of 57°C and LST(T127A) a single Tm of 59°C (Fig. 6D). This is in contrast to commercially sourced wild-type LST, which exhibited two distinct transitions, one at 47°C and another at 60°C.
FIG 6.

Detailed analysis of aglycosylated LST variants. (A) SDS-PAGE analysis of enzyme expression and purification. Lanes: M, marker; 1, 10 μl of supernatant from shake flask culture; 2, 10 μl of supernatant from bioreactor culture; 3, 10 μg of purified protein. (B) Kinetic analysis of S. aureus lysis in microplate turbidometric assays. The horizontal line provides a visual guide to assess TOD50. (C) Antibacterial efficacy as measured by MIC assay. An S. aureus laboratory strain (SA113) and three clinical isolates were assessed for growth inhibition by serial dilutions of purified enzymes. The OD650 readings after 24 h of growth show that, while the different strains exhibited different MIC0 values (P = 0.0008, two-way analysis of variance), wild-type LST and the engineered variants did not exhibit significantly different MIC0s for any given strain (P = 0.8281), and neither was there a significant interaction between the different enzyme treatments and different strains (P = 0.6529). (D) Melting temperature analysis by differential scanning fluorimetry. The engineered aglycoslyated variants exhibit a single transition, whereas commercial wild-type LST exhibits two distinct transitions, one at a substantially lower temperature. Error bars represent standard deviations from three experiments.
DISCUSSION
Because of its lack of endotoxin and high inherent capacity for recombinant protein expression and secretion, P. pastoris is becoming increasingly attractive as a cost-effective production platform for therapeutic proteins. In some cases, however, biotherapeutic protein expression levels have been found to be low to undetectable (29). Protein secretion from P. pastoris is a multiple-step process involving transcription, translation, protein folding, and translocation through the secretory pathway. Any one of these steps may be rate limiting for a given protein target (30, 31). Thus, while P. pastoris has numerous advantages for biotherapeutic protein production, obtaining high levels of expression with recalcitrant targets can be a difficult problem.
Gene sequence optimization has been a widely effective measure to increase heterologous protein expression levels in P. pastoris (32–36), but the mechanism behind this strategy has seldom been studied. The results presented herein are consistent with prior insights that premature transcription termination may result from short segments of severe nucleotide bias within some native genes (37–39). Thus, targeted disruption of these regions may represent an efficient and cost-effective strategy for gene optimization compared to the whole-gene synthesis strategy. In the current study, the comparison of expression levels from the WT, WTΔAT, SYN, and SYN+AT constructs indicates that the core 21-bp A+T-rich sequence is necessary but not sufficient for a putative premature transcription termination signal. Notably, replacement of a mere six nucleotides was sufficient to activate expression from the otherwise incompetent WT lst gene.
Aberrant glycosylation is another frequently encountered barrier to high levels of production of functional proteins from P. pastoris. As shown here, this is indeed the case for the antibacterial enzyme LST. In most instances, destruction of the glycosylation sequon is accomplished by replacing the N-linked asparagine residue with glutamine, aspartic acid, serine, or alanine (40–43). In the current study of LST glycosylation, all mutations at the N-linked N125 residue led to significantly decreased enzyme activity, whereas mutations at two adjacent residues (S126 and T127) yielded fully functional aglycosylated variants. The severely compromised activity of the N125 mutants was especially surprising given the fact that residue 125 is thought to be located at the C-terminal end of the catalytic domain, and prior studies of key LST catalytic residues did not identify N125 (2). Molecular modeling of the LST catalytic domain suggested that substitutions at position 125 caused minimal structural perturbation and affected the peptide backbone even less than mutations at positions 126 and 127 (data not shown). Therefore, an understanding of the strict requirement for an asparagine at position 125 awaits more-specific and in-depth mechanistic and structural analyses. Regardless, the current study demonstrates an unexpected positional constraint for mutations able to disrupt an N-linked glycosylation sequon while maintaining protein function.
It has been shown that transgenic animals expressing LST have enhanced resistance to staphylococcal infections (44). However, transgenic expression of the native LST enzyme is suboptimal, as aberrant glycosylation was found to abolish its activity (27). To address this issue, aglycosylated LST variants with decreased activity (such as N125Q) have been used in transgenic animal production (27, 45). Notably, only animals expressing the highest LST(N125Q) levels were completely resistant to staphylococcal infection. We speculate that the optimized aglycosylated LST variants described here would greatly enhance the utility of LST in transgenic animals, and more generally, we anticipate that other functionally sensitive proteins will benefit from a broadened choice of mutations able to disrupt undesirable N-linked glycosylation sequons.
In conclusion, an efficient high-level-production system has been constructed for LST in the yeast host P. pastoris. By a combination of gene sequence, protein sequence, and bioprocess optimization, expression levels of 500 mg/liter and final purified yields of 250 mg/liter have been obtained in laboratory-scale bioreactors. These high yields and the ease of purification for the secreted, endotoxin-free enzyme should facilitate future development of LST applications, particularly those relating to treating bacterial infections in mammals.
ACKNOWLEDGMENTS
This work was supported by the National Institute of Allergy and Infectious Diseases, NIH, through grant number 1R21AI098122 (awarded to K.E.G. and C.B.-K.). H.Z. was supported in part by funds from the Dartmouth Cystic Fibrosis Research Development Program.
We thank the reviewers for their insightful comments and assistance improving the manuscript.
Footnotes
Published ahead of print 21 February 2014
REFERENCES
- 1.Gargis AS, Tate AH, Heath LS, Heath HE, Leblanc PA, Sloan GL. 2010. Complete nucleotide sequences of plasmids pACK1 and pACK3 from Staphylococcus simulans biovar Staphylolyticus. Plasmid 64:104–109. 10.1016/j.plasmid.2010.05.002 [DOI] [PubMed] [Google Scholar]
- 2.Bardelang P, Vankemmelbeke M, Zhang Y, Jarvis H, Antoniadou E, Rochette S, Thomas NR, Penfold CN, James R. 2009. Design of a polypeptide FRET substrate that facilitates study of the antimicrobial protease lysostaphin. Biochem. J. 418:615–624. 10.1042/BJ20081765 [DOI] [PubMed] [Google Scholar]
- 3.Schindler CA, Schuhardt VT. 1964. Lysostaphin: a new bacteriolytic agent for the staphylococcus. Proc. Natl. Acad. Sci. U. S. A. 51:414–421. 10.1073/pnas.51.3.414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harrison EF, Fuquay ME, Zygmunt WA. 1975. Antigenic response to topically applied proteins. Infect. Immun. 11:309–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zygmunt WA, Tavormina PA. 1972. Lysostaphin: model for a specific enzymatic approach to infectious disease. Prog. Drug Res. 16:309–333 [DOI] [PubMed] [Google Scholar]
- 6.Walsh S, Shah A, Mond J. 2003. Improved pharmacokinetics and reduced antibody reactivity of lysostaphin conjugated to polyethylene glycol. Antimicrob. Agents Chemother. 47:554–558. 10.1128/AAC.47.2.554-558.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stark FR, Thornsvard C, Flannery EP, Artenstein MS. 1974. Systemic lysostaphin in man—apparent antimicrobial activity in a neutropenic patient. N. Engl. J. Med. 291:239–240. 10.1056/NEJM197408012910507 [DOI] [PubMed] [Google Scholar]
- 8.Harris RL, Nunnery AW, Riley HD., Jr 1967. Effect of lysostaphin on staphylococcal carriage in infants and children. Antimicrob. Agents Chemother. 7:110–112 [DOI] [PubMed] [Google Scholar]
- 9.Szweda P, Schielmann M, Kotlowski R, Gorczyca G, Zalewska M, Milewski S. 2012. Peptidoglycan hydrolases—potential weapons against Staphylococcus aureus. Appl. Microbiol. Biotechnol. 96:1157–1174. 10.1007/s00253-012-4484-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quickel KE, Jr, Selden R, Caldwell JR, Nora NF, Schaffner W. 1971. Efficacy and safety of topical lysostaphin treatment of persistent nasal carriage of Staphylococcus aureus. Appl. Microbiol. 22:446–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martin RR, White A. 1968. The reacquisition of staphylococci by treated carriers: a demonstration of bacterial interference. J. Lab. Clin. Med. 71:791–797 [PubMed] [Google Scholar]
- 12.Kokai-Kun JF, Chanturiya T, Mond JJ. 2009. Lysostaphin eradicates established Staphylococcus aureus biofilms in jugular vein catheterized mice. J. Antimicrob. Chemother. 64:94–100. 10.1093/jac/dkp145 [DOI] [PubMed] [Google Scholar]
- 13.Szweda P, Pladzyk R, Kotlowski R, Kur J. 2001. Cloning, expression, and purification of the Staphylococcus simulans lysostaphin using the intein-chitin-binding domain (CBD) system. Protein Expr. Purif. 22:467–471. 10.1006/prep.2001.1454 [DOI] [PubMed] [Google Scholar]
- 14.Szweda P, Kotłowski R, Kur J. 2005. New effective sources of the Staphylococcus simulans lysostaphin. J. Biotechnol. 117:203–213. 10.1016/j.jbiotec.2005.01.012 [DOI] [PubMed] [Google Scholar]
- 15.Sharma R, Sharma PR, Choudhary ML, Pande A, Khatri GS. 2006. Cytoplasmic expression of mature glycylglycine endopeptidase lysostaphin with an amino terminal hexa-histidine in a soluble and catalytically active form in Escherichia coli. Protein Expr. Purif. 45:206–215. 10.1016/j.pep.2005.07.025 [DOI] [PubMed] [Google Scholar]
- 16.Mierau I, Leij P, van Swam I, Blommestein B, Floris E, Mond J, Smid EJ. 2005. Industrial-scale production and purification of a heterologous protein in Lactococcus lactis using the nisin-controlled gene expression system NICE: the case of lysostaphin. Microb. Cell Fact. 4:15. 10.1186/1475-2859-4-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mierau I, Olieman K, Mond J, Smid EJ. 2005. Optimization of the Lactococcus lactis nisin-controlled gene expression system NICE for industrial applications. Microb. Cell Fact. 4:16. 10.1186/1475-2859-4-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Williamson CM, Bramley AJ, Lax AJ. 1994. Expression of the lysostaphin gene of Staphylococcus simulans in a eukaryotic system. Appl. Environ. Microbiol. 60:771–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cregg JM, Tolstorukov I, Kusari A, Sunga J, Madden K, Chappell T. 2009. Expression in the yeast Pichia pastoris. Methods Enzymol. 463:169–189. 10.1016/S0076-6879(09)63013-5 [DOI] [PubMed] [Google Scholar]
- 20.Gasser B, Prielhofer R, Marx H, Maurer M, Nocon J, Steiger M, Puxbaum V, Sauer M, Mattanovich D. 2013. Pichia pastoris: protein production host and model organism for biomedical research. Future Microbiol. 8:191–208. 10.2217/fmb.12.133 [DOI] [PubMed] [Google Scholar]
- 21.Vogl T, Hartner FS, Glieder A. 2013. New opportunities by synthetic biology for biopharmaceutical production in Pichia pastoris. Curr. Opin. Biotechnol. 24:1094–1101. 10.1016/j.copbio.2013.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao X, Huo KK, Li YY. 2000. Synonymous codon usage in Pichia pastoris. Sheng Wu Gong Cheng Xue Bao 16:308–311 [PubMed] [Google Scholar]
- 23.Zhao HL, Xue C, Wang Y, Yao XQ, Liu ZM. 2008. Increasing the cell viability and heterologous protein expression of Pichia pastoris mutant deficient in PMR1 gene by culture condition optimization. Appl. Microbiol. Biotechnol. 81:235–241. 10.1007/s00253-008-1666-0 [DOI] [PubMed] [Google Scholar]
- 24.Woo SH, Park SH, Lim HK, Jung KH. 2005. Extended operation of a pressurized 75-L bioreactor for shLkn-1 production by Pichia pastoris using dissolved oxygen profile control. J. Ind. Microbiol. Biotechnol. 32:474–480. 10.1007/s10295-005-0248-8 [DOI] [PubMed] [Google Scholar]
- 25.Kusuma CM, Kokai-Kun JF. 2005. Comparison of four methods for determining lysostaphin susceptibility of various strains of Staphylococcus aureus. Antimicrob. Agents Chemother. 49:3256–3263. 10.1128/AAC.49.8.3256-3263.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niesen FH, Berglund H, Vedadi M. 2007. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2:2212–2221. 10.1038/nprot.2007.321 [DOI] [PubMed] [Google Scholar]
- 27.Kerr DE, Plaut K, Bramley AJ, Williamson CM, Lax AJ, Moore K, Wells KD, Wall RJ. 2001. Lysostaphin expression in mammary glands confers protection against staphylococcal infection in transgenic mice. Nat. Biotechnol. 19:66–70. 10.1038/83540 [DOI] [PubMed] [Google Scholar]
- 28.Huang CY, Hsu JT, Chung PH, Cheng WT, Jiang YN, Ju YT. 2013. Site-specific N-glycosylation of caprine lysostaphin restricts its bacteriolytic activity toward S. aureus. Anim. Biotechnol. 24:129–147. 10.1080/10495398.2012.760469 [DOI] [PubMed] [Google Scholar]
- 29.Sinclair G, Choy FY. 2002. Synonymous codon usage bias and the expression of human glucocerebrosidase in the methylotrophic yeast, Pichia pastoris. Protein Expr. Purif. 26:96–105. 10.1016/S1046-5928(02)00526-0 [DOI] [PubMed] [Google Scholar]
- 30.Delic M, Valli M, Graf AB, Pfeffer M, Mattanovich D, Gasser B. 2013. The secretory pathway: exploring yeast diversity. FEMS Microbiol. Rev. 37:872–914 [DOI] [PubMed] [Google Scholar]
- 31.Love KR, Politano TJ, Panagiotou V, Jiang B, Stadheim TA, Love JC. 2012. Systematic single-cell analysis of Pichia pastoris reveals secretory capacity limits productivity. PLoS One 7:e37915. 10.1371/journal.pone.0037915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakamura K, Maeda Y, Morimoto K, Katayama S, Kondo K, Nakamura S. 2013. Functional expression of amyloidogenic human stefins A and B in Pichia pastoris using codon optimization. Biotechnol. Appl. Biochem. 60:283–288. 10.1002/bab.1079 [DOI] [PubMed] [Google Scholar]
- 33.Hu H, Gao J, He J, Yu B, Zheng P, Huang Z, Mao X, Yu J, Han G, Chen D. 2013. Codon optimization significantly improves the expression level of a keratinase gene in Pichia pastoris. PLoS One 8:e58393. 10.1371/journal.pone.0058393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang JK, Liu LY, Dai JH, Li Q. 2013. de novo design and synthesis of Candida antarctica lipase B gene and α-factor leads to high-level expression in Pichia pastoris. PLoS One 8:e53939. 10.1371/journal.pone.0053939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tu Y, Wang Y, Wang G, Wu J, Liu Y, Wang S, Jiang C, Cai X. 2013. High-level expression and immunogenicity of a porcine circovirus type 2 capsid protein through codon optimization in Pichia pastoris. Appl. Microbiol. Biotechnol. 97:2867–2875. 10.1007/s00253-012-4540-z [DOI] [PubMed] [Google Scholar]
- 36.Chung BK, Lee DY. 2012. Computational codon optimization of synthetic gene for protein expression. BMC Syst. Biol. 6:134. 10.1186/1752-0509-6-134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scorer CA, Buckholz RG, Clare JJ, Romanos MA. 1993. The intracellular production and secretion of HIV-1 envelope protein in the methylotrophic yeast Pichia pastoris. Gene 136:111–119. 10.1016/0378-1119(93)90454-B [DOI] [PubMed] [Google Scholar]
- 38.Schuren FH, Wessels JG. 1998. Expression of heterologous genes in Schizophyllum commune is often hampered by the formation of truncated transcripts. Curr. Genet. 33:151–156. 10.1007/s002940050321 [DOI] [PubMed] [Google Scholar]
- 39.Sreekrishna K, Brankamp RG, Kropp KE, Blankenship DT, Tsay JT, Smith PL, Wierschke JD, Subramaniam A, Birkenberger LA. 1997. Strategies for optimal synthesis and secretion of heterologous proteins in the methylotrophic yeast Pichia pastoris. Gene 190:55–62. 10.1016/S0378-1119(96)00672-5 [DOI] [PubMed] [Google Scholar]
- 40.Zhao HL, He Q, Xue C, Sun B, Yao XQ, Liu ZM. 2009. Secretory expression of glycosylated and aglycosylated mutein of onconase from Pichia pastoris using different secretion signals and their purification and characterization. FEMS Yeast Res. 9:591–599. 10.1111/j.1567-1364.2009.00498.x [DOI] [PubMed] [Google Scholar]
- 41.Muller-Steffner H, Kuhn I, Argentini M, Schuber F. 2010. Identification of the N-glycosylation sites on recombinant bovine CD38 expressed in Pichia pastoris: their impact on enzyme stability and catalytic activity. Protein Expr. Purif. 70:151–157. 10.1016/j.pep.2009.10.003 [DOI] [PubMed] [Google Scholar]
- 42.Vinzón SE, Pirpignani ML, Nowicki C, Biscoglio de Jiménez Bonino M. 2010. Molecular cloning and expression in Pichia pastoris of a hypoallergenic antigen 5. Protein Expr. Purif. 73:23–30. 10.1016/j.pep.2010.03.029 [DOI] [PubMed] [Google Scholar]
- 43.Peraino J, Zhang H, Hermanrud CE, Li G, Sachs DH, Huang CA, Wang Z. 2012. Expression and purification of soluble porcine CTLA-4 in yeast Pichia pastoris. Protein Expr. Purif. 82:270–278. 10.1016/j.pep.2012.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Donovan DM, Kerr DE, Wall RJ. 2005. Engineering disease resistant cattle. Transgenic Res. 14:563–567. 10.1007/s11248-005-0670-8 [DOI] [PubMed] [Google Scholar]
- 45.Wall RJ, Powell AM, Paape MJ, Kerr DE, Bannerman DD, Pursel VG, Wells KD, Talbot N, Hawk HW. 2005. Genetically enhanced cows resist intramammary Staphylococcus aureus infection. Nat. Biotechnol. 23:445–451. 10.1038/nbt1078 [DOI] [PubMed] [Google Scholar]