

Abstract
The Escherichia coli DnaK chaperone system is a canonical heat shock protein 70 (Hsp70) chaperone system comprising Hsp70, Hsp40, and a nucleotide exchange factor. Although Hsp40 is known to facilitate the effective binding of Hsp70 to substrates, the role of Hsp40 in Hsp70-substrate interactions has not yet been fully elucidated. Using the E. coli heat shock transcription factor σ32 as a substrate in the DnaK chaperone system, we here provide new insight into the Hsp70-substrate interaction. When DnaK-σ32 complexes formed under various conditions were analyzed by gel filtration, several DnaK-σ32 complexes with different molecular masses were detected. The results indicated that multiple DnaK molecules simultaneously bind to σ32, even though it has been suggested that DnaK interacts with σ32 at a molar ratio of 1:1. Two σ32 mutants, L201D σ32 and I54A σ32, which have reduced affinities for DnaK and DnaJ (Hsp40), respectively, were used to further characterize DnaK-σ32 complex formation. Pulldown assays demonstrated that the affinity of I54A σ32 for DnaK was similar to that of wild-type σ32 in the absence of DnaJ, whereas L201D σ32 exhibited an extremely low affinity for DnaK. However, in the presence of ATP and DnaJ, the yield of DnaK eluted with L201D σ32 was much higher than that eluted with I54A σ32. These results indicate that there are multiple DnaK binding sites on σ32 and that DnaJ strongly promotes DnaK binding to any site in the presence of ATP, regardless of the intrinsic affinity of DnaK for the site.
INTRODUCTION
Hsp70 is a highly conserved protein that functions as a chaperone in the folding of newly synthesized polypeptides, the refolding of denatured proteins, the dissociation of proteins from complexes, and the degradation of abnormal proteins (1, 2, 3, 4). Hsp70 possesses ATPase activity and controls substrate association and dissociation via the following ATPase cycle. The ATP-bound form of Hsp70 has a high substrate exchange rate and low substrate affinity, whereas the ADP-bound form has a low substrate exchange rate and high substrate affinity. Hsp40 acts as a cochaperone that elevates the ATPase activity of Hsp70 through a direct interaction with Hsp70 (5); Hsp40 also functions as a chaperone by binding to unfolded polypeptides to prevent their aggregation (6). A nucleotide exchange factor promotes the dissociation of ADP bound to Hsp70, and Hsp70 subsequently returns to the ATP-bound form, reducing its substrate affinity (5, 7). Hsp70 induces conformational changes in its substrate polypeptides through this chaperone cycle, and the canonical Hsp70 chaperone system comprises Hsp70, Hsp40, and a nucleotide exchange factor. Although a model has been proposed in which Hsp40 first recognizes and binds to a substrate polypeptide and subsequently transfers it to Hsp70 (7), we recently proposed another model in which Hsp70 binds to a substrate independent of Hsp40 and the interaction between Hsp70 and Hsp40 on the substrate then leads to the stable binding of Hsp70 to the substrate (8).
The Escherichia coli heat shock transcription factor σ32 (encoded by the rpoH gene), which is required for the heat shock response, is rapidly degraded during steady-state growth at 30°C, with a half-life of 1 to 2 min (9, 10, 11). Among the five ATP-dependent proteases identified in E. coli (Lon, ClpAP, ClpXP, FtsH [HflB], and ClpYQ [HslUV]) (12), FtsH is the major protease involved in σ32 degradation (13, 14). However, other proteases are also responsible for σ32 degradation (15, 16), and an E. coli Hsp70 chaperone system comprising DnaK (Hsp70), DnaJ (Hsp40), and GrpE (nucleotide exchange factor) is required for the rapid degradation of σ32 in vivo (17, 18). It has been previously reported that σ32 forms a stable complex with DnaK and that DnaJ promotes the DnaK-σ32 interaction in the presence of ATP (19, 20, 21, 22, 23). In those reports, it was assumed that one DnaK molecule interacts with one σ32 molecule. The interaction with DnaK has been suggested to induce a conformational change in σ32 and promote its degradation by proteases (18).
Although many mutants of the DnaK chaperone system have been used to examine the Hsp70 chaperone cycle, the use of mutant substrates has been limited because the major substrates of DnaK are unfolded polypeptides that are most likely structurally heterogeneous. However, the bacteriophage P1 RepA protein that is in the native dimer form binds to both DnaK and DnaJ, and Kim et al. had located chaperone binding sites in RepA (24). As the native form of σ32 also directly interacts with both DnaK and DnaJ, σ32 may be a useful model for elucidating the function and mechanism of the Hsp70 chaperone system (8, 25, 26). A region of σ32 containing Leu201 has been reported to be a binding site for DnaK (25). We also previously isolated several in vivo-stabilizing σ32 mutants (26) and recently demonstrated that one or two substitutions at residues 47 to 55 of σ32 reduce the affinity of these mutant proteins for DnaJ (8). In that report, we suggested that two or more molecules of DnaK might bind to σ32 in the presence of ATP and DnaJ. To gain further insight into the DnaK-σ32 interaction, in the present study, we examined DnaK-σ32 complexes in greater detail. Our results indicate that σ32 contains multiple DnaK binding sites with different affinities and that DnaJ enhances DnaK binding, even at sites with low affinity for DnaK.
MATERIALS AND METHODS
Bacterial strains and plasmids.
E. coli MC4100 [F− araD Δ(argF-lac)U169 rpsL relA flbB deoC ptsF rbsR], KY1603 [MC4100 ΔrpoH30::kan zhf50::Tn10 suhX401 (λpF13-PrpoDhs-lacZ)] (27), KY1456 (MC4100 dnaJ::Tn10-42) (28), and HB101 [supE Δ(mcrC-mrr) recA ara-14 proA lacY galK rpsL xyl-5 mtl-1 leuB thi-1] were used for protein purification. MC4100 and KY1459 (MC4100 ΔdnaK52::cat) (29) were used for examining σ32 stability in vivo. The plasmid pKV1142 carries the isopropyl-β-d-thiogalactopyranoside (IPTG)-dependent trc promoter (trcp), and pKV1585 is a pKV1142 derivative that carries the weak trc promoter (26). Promoterless wild-type and mutant rpoH genes with six histidine codons located at the 3′ end were cloned downstream of the trc promoter in pKV1142 or pKV1585. Site-directed mutagenesis was used to generate plasmids carrying the rpoH gene encoding I54A σ32-His and L201D σ32-His in which six histidine residues were added to the carboxy (C) terminus. pKV1957 (pKV1142 trcp-dnaK) and pKV1961 (pKV1142 trcp-dnaJ) are pKV1142 derivatives carrying the dnaK and dnaJ genes, respectively (8). An expression vector carrying the dnaJ gene encoding H33Q DnaJ was constructed from pKV1961 using site-directed mutagenesis. The pET3a plasmid (Novagen) carries the T7 promoter for transcription by T7 RNA polymerase and was used to construct various rpoH expression plasmids for protein purification (8). In this study, novel plasmids carrying the rpoH gene encoding L201D σ32 or L201D σ32-His were constructed using site-directed mutagenesis. pKV1370 is a pACYC177 derivative that carries the kanamycin resistance gene, the trpR gene, and the dnaK-dnaJ operon under the control of the trp promoter.
Media and chemicals.
L broth (30) was used for cell growth, and ampicillin (50 μg/ml) was added when necessary. The synthetic medium used was medium E (30) supplemented with 0.5% glucose, 2 μg/ml thiamine, and all amino acids (20 μg/ml each) except methionine and tryptophan; ampicillin and kanamycin were used at a concentration of 10 μg/ml. The nonhydrolyzable ATP analogs adenosine 5′-O-(3-thiotriphosphate) (ATPγS) and adenylyl imidodiphosphate (AMP-PNP) were purchased from Biolog (Germany) and Roche Diagnostics Japan, respectively. Other chemicals were purchased from Nacalai Tesque (Japan), Wako Pure Chemicals (Japan), or Sigma-Aldrich Japan, unless otherwise specified.
Protein purification.
All the protein purification steps were performed at 4°C.
Wild-type and mutant σ32 and wild-type and mutant σ32-His were purified from KY1603 cells harboring pET3a derivatives as described previously (8). It has been already shown that both I54A σ32 and L201D σ32 bind to RNA polymerase in vitro (8, 25) and that I54A σ32 functions as a transcription initiation factor in vivo (26).
DnaK was purified from HB101 cells harboring pKV1957 as described previously (8), except that ammonium sulfate treatment and chromatography with a heparin column were omitted. Additionally, buffer A (50 mM Tris-HCl [pH 7.5], 1 mM EDTA, 1 mM dithiothreitol, and 10% [vol/vol] glycerol) was used for all steps.
DnaJ was purified from MC4100 cells harboring pKV1961. The cells were grown to late log phase in L broth containing 50 μg/ml ampicillin at 30°C, and DnaJ synthesis was induced with 1 mM IPTG. The cells were harvested after 1 h, resuspended in buffer J1 (50 mM Tris-HCl [pH 7.5], 1 mM EDTA, 10 mM dithiothreitol [DTT], 10% sucrose, 1 mg/ml lysozyme, and 0.6% [wt/vol] Brij 58) (31), maintained on ice for 45 min, and disrupted by sonication. The resulting lysate was centrifuged at 75,000 × g for 90 min. The pellet was resuspended in buffer J3 (50 mM Tris-HCl [pH 7.5], 2 mM β-mercaptoethanol, 10% sucrose, 100 mM KCl, and 0.5% [vol/vol] Triton X-100) and gently rotated for 60 min. After centrifugation at 75,000 × g for 90 min, the supernatant was loaded onto a HiTrap Q-Sepharose column (GE Healthcare) equilibrated with buffer J3. The flowthrough fraction was loaded onto an Ni2+-nitrilotriacetic acid (NTA) agarose (Qiagen) column equilibrated with buffer J3 because the authentic DnaJ protein weakly binds to Ni2+-NTA agarose. After washing with buffer J4 (50 mM Tris-HCl [pH 7.5], 2 mM β-mercaptoethanol, 10% sucrose, and 500 mM NaCl), DnaJ was eluted with buffer J4 containing 50 mM imidazole. DnaJ was concentrated and stored at −70°C. H33Q DnaJ was purified from KY1456 cells harboring the pKV1961 derivative in the same manner.
All the purified proteins were >90% pure, as estimated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by staining with Coomassie brilliant blue (CBB). The purified proteins were quantified using Bradford protein assays (Bio-Rad). Because the functional form of DnaJ is dimeric, the indicated concentrations of DnaJ are those of the dimer form.
Pulldown assay.
To determine interactions among σ32-His, DnaK, and DnaJ, pulldown assays were performed, as described previously (8). In brief, σ32-His and DnaK were incubated in buffer H [20 mM Tris-HCl (pH 7.5), 200 mM KCl, 10% glycerol, and 5 mM Mg(CH3COO)2] with or without DnaJ in the presence of 1 mM nucleotide at 30°C for 30 min. The protein complexes were precipitated with Ni2+-NTA agarose (Qiagen), and the proteins were subjected to SDS-PAGE and detected by CBB staining. Gel images were captured using an LAS3000 image analyzer (GE Healthcare), and the protein bands were quantitated with Multi Gauge software (GE Healthcare).
Gel filtration.
Gel filtration analyses were performed as described previously (8). In brief, σ32 and DnaK were incubated in buffer G [20 mM Tris-HCl (pH 7.5), 200 mM KCl, 1 mM DTT, 0.1 mM EDTA, 10% (vol/vol) glycerol, and 5 mM Mg(CH3COO)2] with or without DnaJ in the presence of 1 mM nucleotide at 30°C for 30 min. After addition of molecular mass standards (β-amylase, 200 kDa; apo-transferrin, 81 kDa; and carbonic anhydrase, 29 kDa), the mixture was loaded on a Superdex 200 gel filtration column (GE Healthcare). Aliquots (15 μl each) from eluted fractions (500 μl each) were subjected to SDS-PAGE. Proteins were detected by staining with Oriole fluorescent gel stain reagent (Bio-Rad). Gel images were captured using an LAS3000 image analyzer, and the protein bands were quantitated with Multi Gauge software. The fraction numbers of the eluted proteins were as follows: β-amylase (200 kDa), fraction 26; apo-transferrin (81 kDa), fraction 29; DnaK (69 kDa), fraction 28; σ32 (33 kDa), fractions 32 and 33; and carbonic anhydrase (29 kDa), fractions 33 and 34.
σ32 stability in vivo.
Cells were grown to mid-log phase in L broth or synthetic medium at 30°C. To inhibit protein synthesis, chloramphenicol or spectinomycin was added to the cultures to a final concentration of 100 μg/ml or 1 mg/ml, respectively. The samples were collected at intervals and treated with the same volume of 10% triacetic acid. Whole-cell extracts were subjected to SDS-PAGE, and σ32, DnaK, GroEL, and DnaJ were detected by immunoblotting with their respective antisera. Images were captured using an LAS3000 image analyzer, and the respective bands were quantitated with Multi Gauge software.
Other methods.
Nucleic acid manipulation (32), site-directed mutagenesis (33), SDS-PAGE (30), and immunoblotting (15) were performed as described previously.
RESULTS
DnaK forms multiple DnaK-σ32 complexes with different molecular masses.
Although it has been assumed that DnaK interacts with σ32 at a molar ratio of 1:1 (20, 21, 22, 23), the results of our previous study suggested that multiple DnaK molecules simultaneously bind to σ32 (8). Thus, the present study was conceived to understand the DnaK-substrate interaction in more detail. After establishing that there are multiple DnaK binding sites on σ32, we investigated the differences in the binding properties of these sites. Pulldown assays were performed with σ32-His to establish the conditions for examining the interaction between DnaK and σ32; the DnaJ concentration varied up to 1.2 μM, whereas the DnaK and σ32 concentrations remained constant at 3.2 and 0.4 μM, respectively (Fig. 1). In the presence of ATP, the amount of DnaK that coeluted with σ32-His markedly increased by more than 10-fold as the DnaJ concentration increased (Fig. 1A and C). In contrast, the yield of eluted DnaK increased by only 2-fold in the presence of ADP (Fig. 1B and C), suggesting that ATP-bound DnaK binds to σ32 in a manner that is different from that for ADP-bound DnaK. The yield of σ32-His decreased as the DnaJ concentration increased in the presence of ATP (Fig. 1A). Because the phenomenon was not observed in the case of the mixture containing ADP (Fig. 1B), it is probable that more frequent interactions between ATP-bound DnaK and σ32-His at higher concentrations of DnaJ sterically inhibit the approach of Ni2+-NTA agarose to σ32-His. When a mixture containing 0.4 μM σ32-His, 3.2 μM DnaK, 1.2 μM DnaJ, and 1 mM ATP was incubated at 30°C for 30 min and centrifuged at 20,400 × g for 10 min, σ32-His was hardly detected in the pellet fraction (data not shown). This indicates that the reduced yield of σ32-His did not result from denaturation or aggregation of σ32-His under the experimental conditions used.
FIG 1.
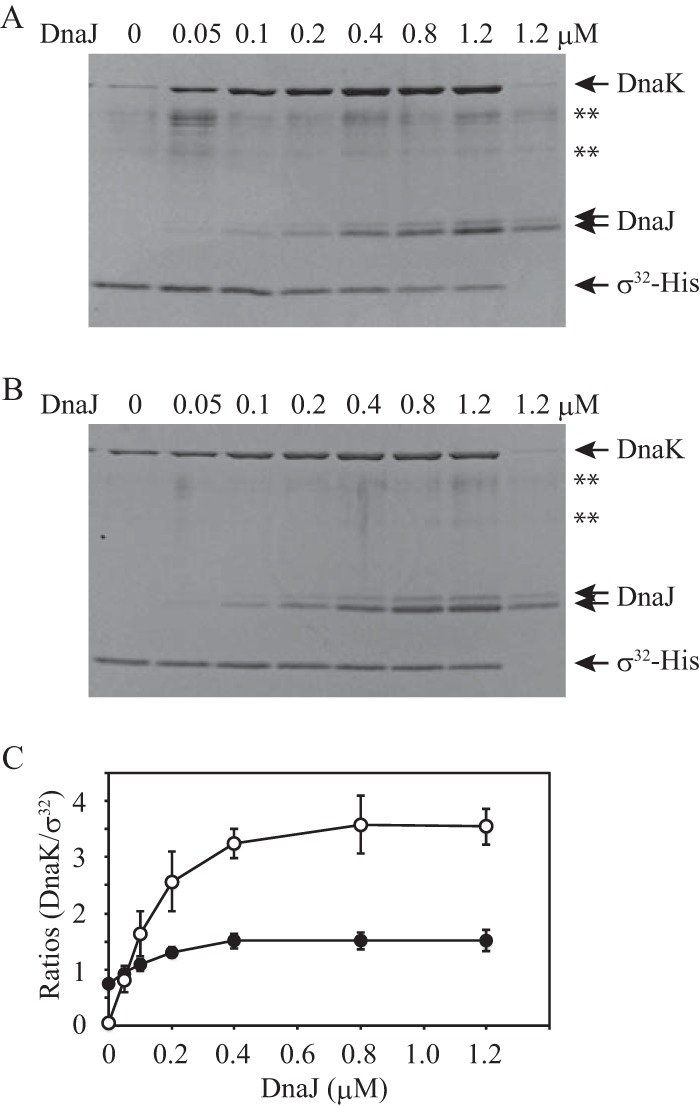
Pulldown assays for complexes including σ32-His, DnaK, and DnaJ. (A and B) σ32-His (0.4 μM) was incubated with DnaK (3.2 μM) and various concentrations of DnaJ (0 to 1.2 μM) at 30°C for 30 min in the presence of 1 mM ATP (A) or 1 mM ADP (B). No σ32-His was added to the control mixture (the rightmost lane in each panel). DnaJ is observed as two bands due to the modification during electrophoresis. **, nonspecific bands which often appeared even in lanes in which no protein was loaded. (C) DnaK and σ32-His bands were quantified. The band intensity for DnaK in the control lane was subtracted from the band intensity for DnaK in each lane, and the result was divided by the band intensity for σ32-His in each lane. The ratios were plotted against the DnaJ concentration. Note that values on the y axis do not indicate molar ratios. The mean values from three experiments are shown with standard errors (SE) (error bars). Open circles, ATP; closed circles, ADP.
Rodriguez et al. previously reported that the region of σ32 that includes Leu201 is directly involved in the interaction with DnaK (25). To further characterize the DnaK-σ32 complexes, we performed gel filtration assays with mixtures containing DnaK and σ32 (wild-type σ32 or L201D σ32) with or without DnaJ in the presence of ADP (Fig. 2). In the absence of DnaJ, the amount of L201D σ32 detected in fractions 32 and 33 (in which free σ32 is eluted) was larger than that of wild-type σ32. This result is consistent with a previous report indicating that the affinity of L201D σ32 for DnaK is lower than that of wild-type σ32 (25). In the presence of DnaJ, the difference between the wild-type and mutant was also observed, even though complexes with higher molecular masses were detected for wild-type σ32. Notably, σ32 in complexes was primarily eluted in fraction 26, regardless of whether DnaJ was absent or present. According to the standard curve constructed based on the elution profiles of the standard proteins and the core RNA polymerase (389 kDa; fraction 22), the deduced molecular mass of the complexes eluted in fraction 26 was 169 kDa, which is larger than the expected molecular mass of the DnaK-σ32 complex (102 kDa). However, free DnaK (69 kDa) was eluted in fraction 28, which corresponds to a deduced molecular mass of 107 kDa, and the only complexed σ32 peak was present in fractions 25 to 28. Therefore, we conclude that the complexes eluted in fraction 26 comprised one DnaK molecule and one σ32 molecule. A similar phenomenon, in which free DnaK was eluted in higher-molecular-weight fractions than in theory, was also observed in a previous study (23).
FIG 2.

Gel filtration analyses of σ32 interacting with DnaK and DnaJ in the presence of ADP. (A) Wild-type or L201D σ32 (0.4 μM) was incubated with DnaK (3.2 μM), DnaJ (0 or 0.4 μM), and ADP (1 mM) at 30°C for 30 min, and the mixture was analyzed by gel filtration. Aliquots from eluted fractions were analyzed by SDS-PAGE, followed by fluorescent staining. Only portions including σ32 bands are shown. A total of 23 fractions (fractions 18 to 40) in each analysis were divided into two groups (fractions 18 to 36 and fractions 30 to 40) and electrophoresed separately. The proteins in fractions 30 to 36 were loaded on both gels and used to correct difference in the band intensity between the two gels. (B) The amount of σ32 is presented as the percentage of the total σ32 recovered. Circles, wild-type σ32; squares, L201D σ32.
Next, to dissect the DnaK-σ32 interaction in the presence of ATP, complexes formed in the presence of 0, 0.05, and 0.4 μM DnaJ were analyzed by gel filtration (Fig. 3). In the absence of DnaJ, σ32 in complexes was detected in fractions 26 and 27. These results are similar to those obtained in the presence of ADP (Fig. 2), though the amount of σ32 in complexes was less than that detected in the presence of ADP. The amount of wild-type σ32 in complexes was slightly larger than the amount of L201D σ32 in complexes. At 0.05 μM DnaJ, increased amounts of both wild-type and L201D σ32 were detected in fraction 26. Because the concentration of DnaJ was lower than that of σ32, this result indicates that DnaJ catalytically promotes the formation of DnaK-σ32 complexes. Intriguingly, a considerable amount of wild-type σ32 was eluted in fractions 24 and 25, suggesting that another DnaK molecule bound to σ32. Compared to that in the presence of 0.05 μM DnaJ, the amount of free σ32 was much lower for both wild-type and mutant in the presence of 0.4 μM DnaJ, and σ32 was eluted in earlier fractions (fractions 22 to 25). This tendency was clearly observed for wild-type σ32, and the molecular mass of the complexes containing wild-type σ32 was larger than that of the complexes containing L201D σ32. These results strongly suggest that there are multiple DnaK binding sites on σ32 and that one site is located in the region containing Leu201.
FIG 3.

Gel filtration analyses of σ32 interacting with DnaK and DnaJ in the presence of ATP. (A) Wild-type or L201D σ32 (0.4 μM) was incubated with DnaK (3.2 μM), DnaJ (0, 0.05, and 0.4 μM), and ATP (1 mM) at 30°C for 30 min, and the mixture was analyzed by gel filtration as for Fig. 2. (B) The amount of σ32 is presented as the percentage of the total σ32 recovered. Circles, wild-type σ32; squares, L201D σ32.
To measure the stoichiometry of σ32, DnaK, and DnaJ in the complexes, the proteins in fractions 20 to 24 from the gel filtration assay containing wild-type σ32 were subjected to electrophoresis along with serial dilutions of the purified proteins (Fig. 4). Because both DnaK and DnaJ tend to self-aggregate and be eluted in high-molecular-weight fractions, fraction 20 was used as a control, and the amounts of DnaK and DnaJ in fraction 20 were then subtracted from those in fractions 22 to 24 to calculate the molar ratios (Table 1). The results showed that two to three DnaK molecules were bound to σ32, whereas less than half of one DnaJ molecule was detected per molecule of σ32.
FIG 4.
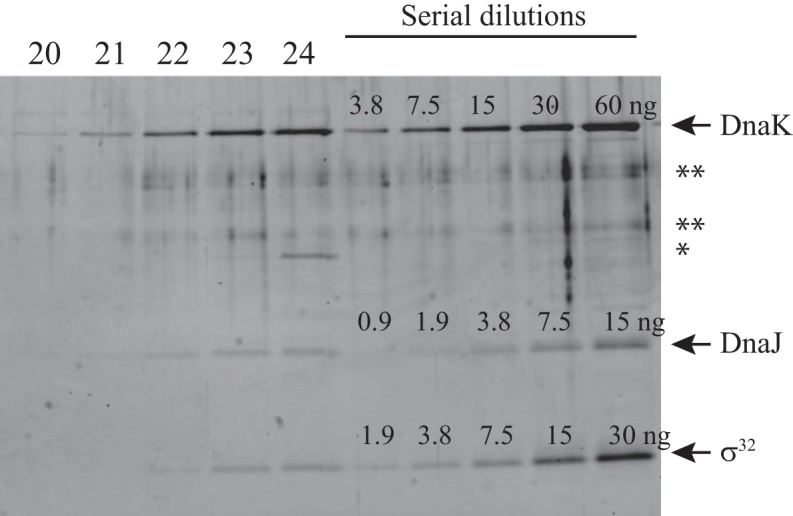
Measurement of stoichiometry. The proteins in fractions 20 to 24 of the gel filtration analysis (conditions, 3.2 μM DnaK, 0.4 μM DnaJ, 0.4 μM wild-type σ32, and 1 mM ATP) were electrophoresed with serial dilutions of the purified proteins and detected by staining with Oriole fluorescent reagent. The gel image was captured with an LAS3000 image analyzer. The amounts of the purified proteins are shown above each protein band. *, β-amylase added as a molecular mass standard; **, nonspecific bands which often appeared even in lanes in which no protein was loaded.
TABLE 1.
Molar ratios in complexes
| Fraction no. | Protein | Net value (AU)a | Amt (ng)b | Molar ratioc |
|---|---|---|---|---|
| 20 | DnaK | 415,755 | 2.00 | |
| DnaJ | 47,880 | 0.20 | ||
| 22 | DnaK | 2,510,786 | 12.11 | 2.37 |
| DnaJ | 633,465 | 2.70 | 0.49 | |
| σ32 | 490,239 | 2.01 | ||
| 23 | DnaK | 4,096,529 | 19.75 | 2.43 |
| DnaJ | 781,948 | 3.33 | 0.36 | |
| σ32 | 839,782 | 3.44 | ||
| 24 | DnaK | 5,770,308 | 27.82 | 2.83 |
| DnaJ | 995,277 | 4.24 | 0.37 | |
| σ32 | 1,050,017 | 4.30 |
Data are derived from the experiment shown in Fig. 4. The intensity of the protein bands was measured in arbitrary units (AU) with Multi Gauge software.
Amounts were calculated from the standard curves drawn based on the serially diluted proteins.
The molar ratios of DnaK or DnaJ per σ32 in fractions 22 to 24 were calculated using the following formula: [(amount − amount in fraction 20)/molecular mass of DnaK (69.1 kDa) or DnaJ (82.2 kDa)]/[(σ32 amount/molecular mass of σ32 (32.5 kDa)].
L201D σ32 is promptly degraded in vivo, similar to wild-type σ32.
Wild-type σ32 is stabilized in dnaK mutants (17, 18), and σ32 mutants with a reduced affinity for DnaK are stabilized in a wild-type strain (8, 26). Based on these reports and the above results, the degradation of L201D σ32, which has a reduced affinity for DnaK, should be suppressed in comparison to that of wild-type σ32. To distinguish plasmid-encoded σ32 from chromosome-encoded wild-type σ32, plasmids encoding wild-type and mutant σ32 with six histidine residues at the C terminus were transformed into a wild-type strain of E. coli (MC4100). The level of L201D σ32-His was similar to that of wild-type σ32-His, whereas the level of I54A σ32-His was lower than that of wild-type σ32-His (Fig. 5A), because I54A σ32-His was expressed from the weak trc promoter. When the levels of major heat shock proteins (DnaK, GroEL, and DnaJ) that are upregulated by σ32 were examined by immunoblotting, they were higher in cells producing I54A σ32-His than in cells producing wild-type σ32-His, as described previously (26), whereas those in cells producing L201D σ32-His were the same as those in cells producing wild-type σ32-His (Fig. 5A). Next, the stability of σ32-His was assessed. After chloramphenicol was added to the culture medium to inhibit protein synthesis, the level of L201D σ32-His rapidly decreased at a rate that was similar to that of wild-type σ32-His, whereas I54A σ32-His degradation was suppressed as described previously (Fig. 5B) (26).
FIG 5.

In vivo stability of σ32-His. (A) MC4100 cells expressing wild-type (WT) σ32-His (from a pKV1142 derivative), L201D σ32-His (from a pKV1142 derivative), or I54A σ32-His (from a pKV1585 derivative) were grown to mid-log phase in L broth at 30°C. Sufficient amounts of σ32-His were synthesized in the absence of added IPTG. Samples were collected, whole-cell extracts were subjected to SDS-PAGE, and DnaK, GroEL, DnaJ, and σ32-His were detected by immunoblotting. MC4100 cells harboring pKV1142 were treated in a similar manner (−). DnaJ is observed as three bands due to the modification during electrophoresis. (B) The cells shown in panel A were grown to mid-log phase in L broth at 30°C. After chloramphenicol was added (100 μg/ml), samples were collected at intervals. Whole-cell extracts were analyzed by SDS-PAGE, and σ32-His bands were detected by immunoblotting. A representative result is shown (upper panel). σ32-His bands were quantified, and the mean values from three experiments are shown with SE (error bars) (lower panel). The values at time zero are defined as 1. Circles, WT σ32-His; triangles, I54A σ32-His; squares, L201D σ32-His. (C) A ΔdnaK strain (KY1459) harboring two expression vectors, pKV1370 (carrying the dnaK-dnaJ operon) and a pKV1142 derivative (carrying the WT rpoH gene with six histidine codons), was grown to mid-log phase in synthetic medium containing tryptophan at the indicated concentrations at 30°C. IPTG was not added. Samples were collected, and whole-cell extracts were analyzed by SDS-PAGE. DnaK, GroEL, DnaJ, and σ32-His were detected by immunoblotting. A wild-type strain (MC4100) that did not harbor any plasmid was treated in a similar manner. (D) KY1459 cells harboring pKV1370 and expressing WT σ32-His (from a pKV1142 derivative), L201D σ32-His (from a pKV1142 derivative), or I54A σ32-His (from a pKV1585 derivative) were grown to mid-log phase in synthetic medium containing tryptophan at 1.4 μg/ml or 40 μg/ml at 30°C in the absence of added IPTG. Samples were collected, whole-cell extracts were subjected to SDS-PAGE, and DnaK, GroEL, DnaJ, and σ32-His were detected by immunoblotting. (E) and (F) The cells shown in panel D were grown to mid-log phase in synthetic medium containing tryptophan at 1.4 μg/ml (E) or 40 μg/ml (F) at 30°C in the absence of added IPTG. Spectinomycin was added (1 mg/ml) at mid-log phase. The stability of σ32-His was analyzed as for panel B. Circles, wild-type σ32-His; triangles, I54A σ32-His; squares, L201D σ32-His. *, authentic σ32 expressed from chromosomal DNA; **, a nonspecific band.
To examine the dependence of L201D σ32-His degradation on DnaK, we used a ΔdnaK mutant strain (KY1459) in which the dnaK gene was partially replaced with a chloramphenicol resistance gene (cat) (34). The replacement of the dnaK gene results in a barely detectable level of DnaJ expression from the downstream gene in the dnaK-dnaJ operon due to a polar effect (35). Expression vectors carrying the rpoH genes with six histidine codons could not be transformed into the ΔdnaK strain, potentially reflecting the toxic effect of stabilizing both σ32 and σ32-His, because high activity of σ32 is deleterious to cell growth (e.g., the gene encoding I54A σ32, which is a hyperactive σ32, cannot be cloned under control of the intact trc promoter). Therefore, these plasmids were cotransformed with an expression vector carrying the dnaK-dnaJ operon (pKV1370); because the operon is under the control of the trp promoter, expression is inhibited after the addition of tryptophan to the medium. When tryptophan was added at a final concentration of 40 μg/ml, the DnaK level in the mutant strain was reduced by 30 to 50% after three generations compared to that in the wild-type strain, although the DnaJ level was not reduced less than that in the wild-type strain (Fig. 5C and D). In contrast, the levels of plasmid-encoded σ32-His and chromosome-encoded authentic σ32 increased due to stabilization, which in turn resulted in an increased level of GroEL (Fig. 5C and D). We next examined the levels of σ32-His after the addition of spectinomycin to inhibit protein synthesis. A control experiment was performed with 1.4 μg/ml tryptophan to readily detect unstable wild-type σ32-His. I54A σ32-His was very stable in the presence of 1.4 μg/ml tryptophan, whereas both wild-type and L201D σ32-His were unstable (Fig. 5E). Although the DnaK level at 1.4 μg/ml tryptophan was slightly higher than that in the wild-type strain (Fig. 5C and D), the half-life of wild-type σ32-His was longer in KY1459 than in the wild-type strain (Fig. 5B and E). Because lower concentrations of tryptophan (1 and 1.2 μg/ml) resulted in lower levels of wild-type σ32-His in KY1459 (Fig. 5C), rapid degradation of wild-type σ32-His seems to require larger amounts of DnaK and DnaJ in KY1459 than in the wild-type strain. An increased level of wild-type σ32-His expressed from the plasmid (a pKV1142 derivative) may affect its stability more markedly in KY1459 than in the wild-type strain. Similar results were obtained for L201D σ32-His (Fig. 5A, B, D, and E and data not shown). At 40 μg/ml tryptophan, both wild-type and L201D σ32-His were strongly stabilized, similar to I54A σ32-His (Fig. 5F). This result indicates that the in vivo degradation of L201D σ32-His was dependent on the DnaK chaperone system, even though the affinity of L201D σ32 for DnaK was reduced compared to that of wild-type σ32. These findings suggest that σ32 stability can be regulated by DnaK binding to any one of multiple sites.
DnaJ promotes DnaK binding to multiple sites on σ32 in the presence of ATP.
Using the I54A σ32-His and L201D σ32-His mutants, pulldown assays were performed to examine the effects of DnaJ on the DnaK-σ32 interaction under various conditions (Fig. 6). Regardless of the nucleotide used, the amounts of DnaK that coeluted with wild-type σ32-His and I54A σ32-His were comparable in the absence of DnaJ, whereas much less DnaK was coeluted with L201D σ32-His than with wild-type σ32-His. This difference between wild-type σ32 and L201D σ32 was more clear than that indicated by the gel filtration analyses (Fig. 2 and 3), strongly suggesting that the region containing Leu201 is the primary target site of DnaK in the absence of DnaJ. In the presence of DnaJ and ADP, the amounts of DnaK that coeluted with wild-type σ32-His and L201D σ32-His gradually increased with increasing DnaJ concentration, whereas the amount of DnaK that coeluted with I54A σ32-His was not altered. Interestingly, in the presence of DnaJ and ATP, more DnaK was coeluted with L201D σ32-His than with I54A σ32-His, though the yield of DnaK was less than that obtained with wild-type σ32-His. Because more DnaJ was coeluted with wild-type σ32-His and L201D σ32-His than with I54A σ32-His, these results indicate that DnaJ binding to σ32 strongly enhances DnaK binding to sites that intrinsically exhibit a low affinity for DnaK, in addition to DnaK binding to the site containing Leu201.
FIG 6.
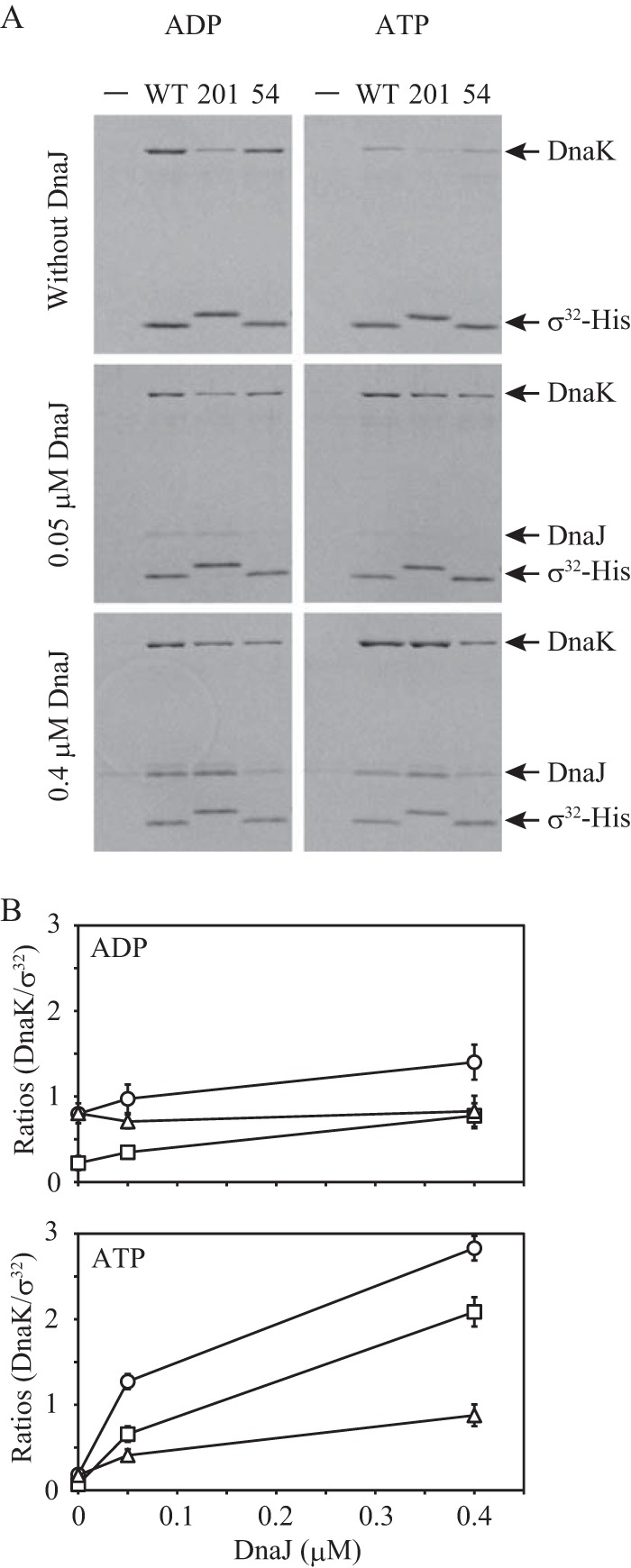
Pulldown assays for complexes of σ32-His, DnaK, and DnaJ. (A) σ32-His (0.4 μM) was incubated with DnaK (3.2 μM) and DnaJ (0, 0.05, and 0.4 μM) at 30°C for 30 min in the presence of 1 mM ADP (left panels) or 1 mM ATP (right panels). No σ32-His was added to the control mixture (−). Representative results are shown. (B) The results were analyzed as for Fig. 1C. Note that values on the y axis do not indicate molar ratios. The mean values from three experiments are shown with SE (error bars). Circles, wild-type σ32-His; triangles, I54A σ32-His; squares, L201D σ32-His.
DnaK binding to each site on σ32 requires the hydrolysis of DnaK-bound ATP through interactions with DnaJ.
The results described above demonstrate that multiple DnaK molecules simultaneously bind to σ32 in the presence of ATP and DnaJ. Because the binding efficiency of DnaK to multiple sites on σ32 was low in the presence of ADP (Fig. 2 and 6), it is possible that some ATP-bound DnaK molecules bind to σ32 without hydrolyzing ATP. Therefore, it is important to examine whether the binding of multiple DnaK molecules to σ32 requires the hydrolysis of ATP bound to each DnaK.
First, we tested whether direct interactions between individual DnaK molecules and DnaJ are required for DnaK binding to multiple sites on σ32. DnaJ contains a specific domain known as the J domain that is conserved among Hsp40 family proteins and includes the HPD motif, His33-Pro34-Asp35. DnaJ interacts with DnaK through this HPD motif and promotes the ATPase activity of DnaK, and amino acid substitutions in this motif result in weak binding of DnaK to substrates, such as σ32 (22, 36). We constructed a dnaJ expression vector in which the histidine codon (CAC) at position 33 was replaced with a glutamine codon (CAG). When the DnaJ-σ32 interaction was examined by pulldown assays, the amount of H33Q DnaJ that coeluted with σ32-His was similar to that of wild-type DnaJ (Fig. 7A), indicating that the H33Q mutation does not appreciably affect the DnaJ-σ32 interaction. However, when the DnaK-σ32 interaction was examined in the presence of H33Q DnaJ and ATP, the amount of DnaK that coeluted with wild-type σ32-His was markedly reduced and did not increase, even in the presence of a high concentration of H33Q DnaJ (Fig. 7B); this amount was much smaller than that detected in the presence of ADP (Fig. 6). These results indicate that the binding of multiple DnaK molecules to multiple sites on σ32 requires the direct interaction of each DnaK molecule with DnaJ.
FIG 7.
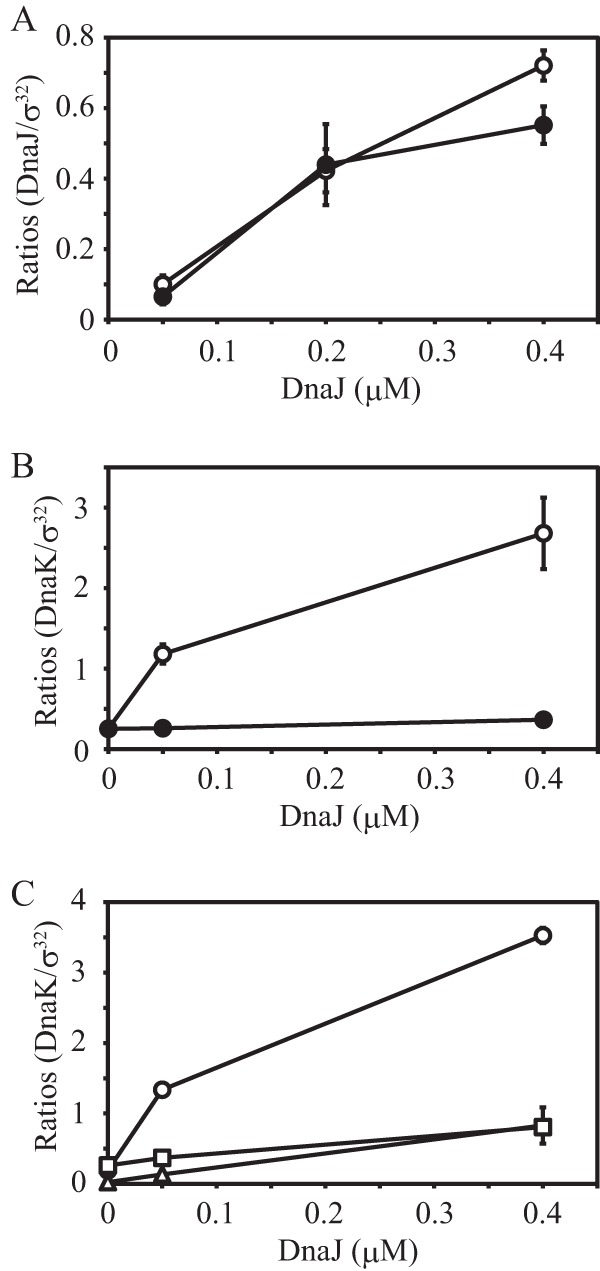
Pulldown assays in the presence of the H33Q DnaJ mutant or nonhydrolyzable ATP analogs. (A) Wild-type σ32-His (0.4 μM) was incubated with wild-type or H33Q DnaJ (0.05, 0.2 and 0.4 μM) at 30°C for 30 min. The pulldown assays were performed, and the proteins were subjected to SDS-PAGE. DnaJ and σ32 bands were quantified, and the band intensity for DnaJ was divided by the band intensity of σ32. The ratios are plotted against the DnaJ concentration. Note that values on the y axis do not indicate molar ratios. The mean values from three experiments are shown with SE (error bars). Open circles, wild-type DnaJ; closed circles, H33Q DnaJ. (B) Wild-type σ32-His (0.4 μM) was incubated with DnaK (3.2 μM) and wild-type or H33Q DnaJ (0, 0.05, and 0.4 μM) at 30°C for 30 min in the presence of 1 mM ATP. The pulldown assays were performed, and the results were analyzed as for Fig. 1C. The mean values from three experiments are shown with SE (error bars). Open circles, wild-type DnaJ; closed circles, H33Q DnaJ. (C) Wild-type σ32-His (0.4 μM) was incubated with DnaK (3.2 μM) and DnaJ (0, 0.05, and 0.4 μM) at 30°C for 30 min in the presence of 1 mM nucleotide. Pulldown assays were performed, and the results were analyzed as for Fig. 1C. The mean values from three experiments are shown with SE (error bars). Circles, ATP; triangles, ATPγS; squares, AMP-PNP.
We next examined whether the binding of multiple DnaK molecules to σ32 requires the hydrolysis of DnaK-bound ATP by performing pulldown assays with two nonhydrolyzable ATP analogs, ATPγS and AMP-PNP (Fig. 7C). In the presence of either ATP analog, the amount of DnaK that coeluted with wild-type σ32-His was low, even at 0.4 μM DnaJ. This result indicates that the binding of multiple DnaK molecules to multiple sites on σ32 requires the hydrolysis of the ATP bound to each DnaK molecule.
DISCUSSION
σ32 is rapidly degraded in cells, with a half-life of 1 to 2 min (9), and the DnaK chaperone system has been implicated in this rapid degradation (17, 18). It has been suggested that DnaK binds to σ32 at a molar ratio of 1:1 and induces a conformational change (18, 20, 21, 22, 23). In the present study, we dissected the DnaK-σ32 interaction using L201D σ32 and I54A σ32 mutants and observed that DnaK forms various complexes with σ32 in the presence of ATP and DnaJ. Based on these results, particularly the data from our gel filtration analyses (Fig. 2 to 4), we suggest that three regions on σ32 are responsible for DnaK binding, with one being the site including Leu201. Although the other two sites remain to be delineated, they appear to be important for the regulation of the intracellular σ32 level by the DnaK chaperone system (Fig. 5).
ADP-bound DnaK binds to the site containing Leu201, independent of DnaJ. Thus, the role of DnaJ in the DnaK-σ32 interaction at this site is to accelerate the ATPase activity of DnaK, and the direct binding of DnaJ to σ32 is dispensable; i.e., DnaK binding to σ32 requires only the cochaperone activity of DnaJ and not its chaperone activity.
DnaK can stably bind to the other sites only in the presence of DnaJ, and there are two possible mechanisms to explain DnaK binding to these sites. In the first mechanism, these sites are occluded inside the σ32 molecule, and the binding of DnaJ triggers their exposure to the outside environment. If hidden DnaK binding sites on σ32 are exposed upon DnaJ binding, then multiple ADP-bound DnaK molecules would be expected to bind to multiple sites on σ32 in the presence of DnaJ. However, this was not observed: DnaJ binding was not sufficient to promote the stable binding of multiple ADP-bound DnaK molecules to σ32 (Fig. 2 and 6). In the second mechanism, DnaJ promotes the binding of ATP-bound DnaK to low-affinity sites on σ32 through the formation of a ternary complex (DnaK-DnaJ-σ32) in which a weak DnaK-σ32 interaction is stabilized through both DnaJ-σ32 and DnaK-DnaJ interactions. The interaction of DnaK with DnaJ is ATP dependent (36), and the structure of ATP-bound DnaK markedly differs from that of the ADP-bound form (37, 38). These observations support the second mechanism. Because the association rate constant of ATP-bound DnaK for a substrate is higher than that of nucleotide-free DnaK (39), which has properties that are similar to those of the ADP-bound form, the faster binding of ATP-bound DnaK might also contribute to efficient DnaK-DnaJ interactions on σ32. Based on the second mechanism, we propose a model in which the multiple DnaK binding sites on σ32 differ in their affinity for DnaK and DnaK binding to sites with low affinity is markedly enhanced by simultaneous DnaJ binding (Fig. 8A). Furthermore, the present study suggests that experiments with ADP-bound or nucleotide-free DnaK are not sufficient to determine the substrate specificity of DnaK because ATP-bound DnaK stably binds to sites with an intrinsic low affinity for DnaK in the presence of DnaJ. To clarify the mechanism of the DnaK-σ32 interaction in more detail, all the DnaK binding sites on σ32 should be identified.
FIG 8.
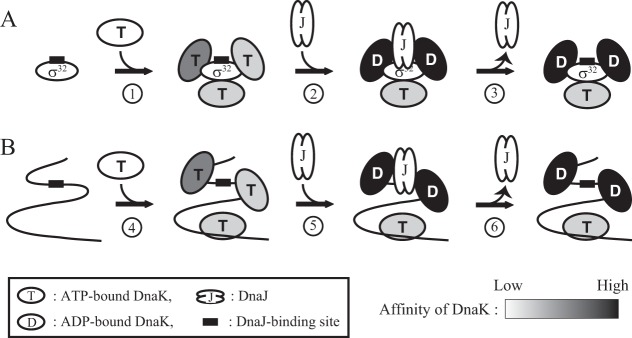
Model of the DnaK binding reaction. The numbers in the circles below the arrows indicate each reaction. The shades of black in DnaK indicate the intensity of the affinity for a binding site. (A) Model of DnaK binding to σ32. Multiple DnaK molecules rapidly associate with and dissociate from σ32 independent of DnaJ (reaction 1). When DnaK is the ATP-bound form, the affinity of the binding sites for DnaK varies among the sites. The simultaneous binding of DnaK and DnaJ on the same σ32 molecule stabilizes the DnaK-σ32 interaction and promotes the ATPase activity of DnaK (reaction 2), and ADP-bound DnaK tightly binds σ32. Subsequently, DnaJ dissociates from the complex (reaction 3). (B) Model of DnaK binding to an unfolded polypeptide. Multiple ATP-bound DnaK molecules rapidly associate with and dissociate from an unfolded polypeptide (such as newly synthesized polypeptides) independent of DnaJ (reaction 4). When a DnaJ binding site is exposed on the exterior of the polypeptide, DnaK can interact with DnaJ on the same polypeptide, and ATP bound to DnaK is hydrolyzed (reaction 5). The resulting ADP-bound DnaK stably binds to the polypeptide, and DnaJ dissociates from the complex (reaction 6).
The model shown in Fig. 8A can be expanded to general substrates of DnaK or to unfolded proteins (Fig. 8B). Because the amount of DnaK in the cell greatly exceeds that of DnaJ (2), it is expected that ATP-bound DnaK cycles between associating with and dissociating from various sites on a polypeptide without ATP hydrolysis. When the DnaJ binding site on the polypeptide is exposed and DnaJ binds to this site, DnaK molecules in the vicinity of DnaJ stably bind to the polypeptide through the DnaK-DnaJ interaction, which supports the weak binding of DnaK to a low-affinity site. As long as the DnaJ binding site is not hidden inside the polypeptide, DnaK repeatedly binds to the substrate. Because DnaK is a key factor in the DnaK chaperone system, it must bind to a wide spectrum of amino acid sequences on various polypeptides. Thus, for DnaK to effectively function as a chaperone, DnaK binding sites must be relatively nonspecific. The stable binding of DnaK to a polypeptide depends on the binding of DnaJ to a specific site(s) on the same polypeptide.
DnaJ belongs to the Hsp40 family (also known as the J protein family). J protein family members are classified into several types based on the presence or absence of a substrate (also termed client) binding domain (3), and J proteins lacking client binding activity stimulate the ATPase activity of Hsp70 without binding to its substrate. The conclusion of the present study that the formation of a ternary complex (DnaK-DnaJ-σ32 [or substrate]) promotes DnaK binding to a polypeptide with weak affinity for DnaK can also be applied to the functions of J proteins lacking a client binding domain, because in those cases, J proteins might not be free in cells but might bind to a molecule with which substrates of Hsp70 interact to form a quaternary complex, such as the J protein-ribosome-substrate (newly synthesized polypeptide)-Hsp70 complex (40). The formation of such a complex could ensure the Hsp70-Hsp40 interaction. Six J protein family members have been identified in E. coli, and 41 members have been identified in humans; in contrast, only three and 11 Hsp70 family members have been identified in E. coli and humans, respectively. Thus, organisms might have evolved Hsp40 family members with various substrate specificities to meet the demand for Hsp70 activities.
ACKNOWLEDGMENT
We thank Takumi Nishiuchi for help with the DNA sequence analysis.
Footnotes
Published ahead of print 14 February 2014
REFERENCES
- 1.Mayer MP, Bukau B. 2005. Hsp70 chaperones. Cellular functions and molecular mechanism. Cell. Mol. Life Sci. 62:670–684. 10.1007/s00018-004-4464-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Genevaux P, Georgopoulos C, Kelley WL. 2007. The Hsp70 chaperone machines of Escherichia coli: a paradigm for the repartition of chaperone functions. Mol. Microbiol. 66:840–857. 10.1111/j.1365-2958.2007.05961.x [DOI] [PubMed] [Google Scholar]
- 3.Kampinga HH, Craig EA. 2010. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 11:579–592. 10.1038/nrm2941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hartl FU, Bracher A, Hayer-Hartl M. 2011. Molecular chaperones in protein folding and proteostasis. Nature 475:324–332. 10.1038/nature10317 [DOI] [PubMed] [Google Scholar]
- 5.Liberek K, Marszalek J, Ang D, Georgopoulos C, Zylicz M. 1991. Escherichia coli DnaJ and GrpE heat shock proteins jointly stimulate ATPase activity of DnaK. Proc. Natl. Acad. Sci. U. S. A. 88:2874–2878. 10.1073/pnas.88.7.2874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Langer T, Lu C, Echols H, Flanagan J, Hayer MK, Hartl FU. 1992. Successive action of DnaK, DnaJ and GroEL along the pathway of chaperone-mediated protein folding. Nature 356:683–689. 10.1038/356683a0 [DOI] [PubMed] [Google Scholar]
- 7.Szabo A, Langer T, Schröder H, Flanagan J, Bukau B, Hartl FU. 1994. The ATP hydrolysis-dependent reaction cycle of the Escherichia coli Hsp70 system—DnaK, DnaJ and GrpE. Proc. Natl. Acad. Sci. U. S. A. 91:10345–10349. 10.1073/pnas.91.22.10345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Suzuki H, Ikeda A, Tsuchimoto S, Adachi K, Noguchi A, Fukumori Y, Kanemori M. 2012. Synergistic binding of DnaJ and DnaK chaperones to heat shock transcription factor σ32 ensures its characteristic high metabolic instability. Implications for heat shock protein 70 (Hsp70)-Hsp40 mode of function. J. Biol. Chem. 287:19275–19283. 10.1074/jbc.M111.331470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Straus DB, Walter WA, Gross CA. 1987. The heat shock response of E. coli is regulated by changes in the concentration of σ32. Nature 329:348–351. 10.1038/329348a0 [DOI] [PubMed] [Google Scholar]
- 10.Yura T, Kanemori M, Morita MT. 2000. The heat shock response: regulation and function, p 3–18 In Storz G, Hengge-Aronis R. (ed), Bacterial stress responses. ASM Press, Washington, DC [Google Scholar]
- 11.Guisbert E, Yura T, Rhodius VA, Gross CA. 2008. Convergence of molecular, modeling, and systems approaches for an understanding of the Escherichia coli heat shock response. Microbiol. Mol. Biol. Rev. 72:545–554. 10.1128/MMBR.00007-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gottesman S. 2003. Proteolysis in bacterial regulatory circuits. Annu. Rev. Cell Dev. Biol. 19:565–587. 10.1146/annurev.cellbio.19.110701.153228 [DOI] [PubMed] [Google Scholar]
- 13.Herman C, Thévenet D, D'Ari R, Bouloc P. 1995. Degradation of σ32, the heat shock regulator in Escherichia coli, is governed by HflB. Proc. Natl. Acad. Sci. U. S. A. 92:3516–3520. 10.1073/pnas.92.8.3516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tomoyasu T, Gamer J, Bukau B, Kanemori M, Mori H, Rutman AJ, Oppenheim AB, Yura T, Yamanaka K, Niki H, Hiraga S, Ogura T. 1995. Escherichia coli FtsH is a membrane-bound, ATP-dependent protease which degrades the heat-shock transcription factor σ32. EMBO J. 14:2551–2560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kanemori M, Nishihara K, Yanagi H, Yura T. 1997. Synergistic roles of HslVU and other ATP-dependent proteases in controlling in vivo turnover of σ32 and abnormal proteins in Escherichia coli. J. Bacteriol. 179:7219–7225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kanemori M, Yanagi H, Yura T. 1999. Marked instability of the σ32 heat shock transcription factor at high temperature. Implications for heat shock regulation. J. Biol. Chem. 274:22002–22007 [DOI] [PubMed] [Google Scholar]
- 17.Tilly K, Spence J, Georgopoulos C. 1989. Modulation of stability of the Escherichia coli heat shock regulatory factor σ32. J. Bacteriol. 171:1585–1589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Straus D, Walter W, Gross CA. 1990. DnaK, DnaJ, and GrpE heat shock proteins negatively regulate heat shock gene expression by controlling the synthesis and stability of σ32. Genes Dev. 4:2202–2209. 10.1101/gad.4.12a.2202 [DOI] [PubMed] [Google Scholar]
- 19.Gamer J, Bujard H, Bukau B. 1992. Physical interaction between heat shock proteins DnaK, DnaJ, and GrpE and the bacterial heat shock transcription factor σ32. Cell 69:833–842. 10.1016/0092-8674(92)90294-M [DOI] [PubMed] [Google Scholar]
- 20.Liberek K, Galitski TP, Zylicz M, Georgopoulos C. 1992. The DnaK chaperone modulates the heat shock response of Escherichia coli by binding to the σ32 transcription factor. Proc. Natl. Acad. Sci. U. S. A. 89:3516–3520. 10.1073/pnas.89.8.3516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liberek K, Georgopoulos C. 1993. Autoregulation of the Escherichia coli heat shock response by the DnaK and DnaJ heat shock proteins. Proc. Natl. Acad. Sci. U. S. A. 90:11019–11023. 10.1073/pnas.90.23.11019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liberek K, Wall D, Georgopoulos C. 1995. The DnaJ chaperone catalytically activates the DnaK chaperone to preferentially bind the σ32 heat shock transcriptional regulator. Proc. Natl. Acad. Sci. U. S. A. 92:6224–6228. 10.1073/pnas.92.14.6224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gamer J, Multhaup G, Tomoyasu T, McCarty JS, Rüdiger S, Schönfeld H-J, Schirra C, Bujard H, Bukau B. 1996. A cycle of binding and release of the DnaK, DnaJ and GrpE chaperones regulates activity of the Escherichia coli heat shock transcription factor σ32. EMBO. J. 15:607–617 [PMC free article] [PubMed] [Google Scholar]
- 24.Kim S-Y, Sharma S, Hoskins JR, Wickner S. 2002. Interaction of the DnaK and DnaJ chaperone system with a native substrate, P1 RepA. J. Biol. Chem. 277:44778–44783. 10.1074/jbc.M206176200 [DOI] [PubMed] [Google Scholar]
- 25.Rodriguez F, Arsène-Ploetze F, Rist W, Rüdiger S, Schneider-Mergener J, Mayer MP, Bukau B. 2008. Molecular basis for regulation of the heat shock transcription factor σ32 by the DnaK and DnaJ chaperones. Mol. Cell 32:347–358. 10.1016/j.molcel.2008.09.016 [DOI] [PubMed] [Google Scholar]
- 26.Horikoshi M, Yura T, Tsuchimoto S, Fukumori Y, Kanemori M. 2004. Conserved region 2.1 of Escherichia coli heat shock transcription factor σ32 is required for modulating both metabolic stability and transcriptional activity. J. Bacteriol. 186:7474–7480. 10.1128/JB.186.22.7474-7480.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kusukawa N, Yura T. 1988. Heat shock protein GroE of Escherichia coli: key protective roles against thermal stress. Genes Dev. 2:874–882. 10.1101/gad.2.7.874 [DOI] [PubMed] [Google Scholar]
- 28.Ishiai M, Wada C, Kawasaki Y, Yura T. 1992. Mini-F plasmid mutants able to replicate in Escherichia coli deficient in the DnaJ heat shock protein. J. Bacteriol. 174:5597–5603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagai H, Yuzawa H, Kanemori M, Yura T. 1994. A distinct segment of the σ32 polypeptide is involved in DnaK-mediated negative control of the heat shock response in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 91:10280–10284. 10.1073/pnas.91.22.10280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tobe T, Ito K, Yura T. 1984. Isolation and physical mapping of temperature-sensitive mutants defective in heat-shock induction of proteins in Escherichia coli. Mol. Gen. Genet. 195:10–16. 10.1007/BF00332716 [DOI] [PubMed] [Google Scholar]
- 31.Cajo GC, Horne BE, Kelley WL, Schwager F, Georgopoulos C, Genevaux P. 2006. The role of the DIF motif of the DnaJ (Hsp40) co-chaperone in the regulation of the DnaK (Hsp70) chaperone cycle. J. Biol. Chem. 281:12436–12444. 10.1074/jbc.M511192200 [DOI] [PubMed] [Google Scholar]
- 32.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 33.Sawano A, Miyawaki A. 2000. Directed evolution of green fluorescent protein by a new versatile PCR strategy for site-directed and semi-random mutagenesis. Nucleic Acids Res. 28:e78. 10.1093/nar/28.16.e78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paek K-H, Walker GC. 1987. Escherichia coli dnaK null mutants are inviable at high temperature. J. Bacteriol. 169:283–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bukau B, Walker GC. 1989. Cellular defects caused by deletion of the Escherichia coli dnaK gene indicate roles for heat shock protein in normal metabolism. J. Bacteriol. 171:2337–2346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wall D, Zylicz M, Georgopoulos C. 1995. The conserved G/F motif of the DnaJ chaperone is necessary for the activation of the substrate binding properties of the DnaK chaperone. J. Biol. Chem. 270:2139–2144. 10.1074/jbc.270.5.2139 [DOI] [PubMed] [Google Scholar]
- 37.Zhuravleva A, Clerico EM, Gierasch M. 2012. An interdomain energetic tug-of-war creates the allosterically active state in Hsp70 molecular chaperones. Cell 151:1296–1307. 10.1016/j.cell.2012.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kityk R, Kopp J, Sinning I, Mayer MP. 2012. Structure and dynamics of the ATP-bound open conformation of Hsp70 chaperones. Mol. Cell 48:863–874. 10.1016/j.molcel.2012.09.023 [DOI] [PubMed] [Google Scholar]
- 39.Schmid D, Baici A, Gehring H, Christen P. 1994. Kinetics of molecular chaperone action. Science 263:971–973. 10.1126/science.8310296 [DOI] [PubMed] [Google Scholar]
- 40.Willmund F, del Alamo M, Pechmann S, Chen T, Albanèse V, Dammer EB, Peng J, Frydman J. 2013. The cotranslational function of ribosome-associated Hsp70 in eukaryotic protein homeostasis. Cell 152:196–209. 10.1016/j.cell.2012.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]