

Abstract
Quorum sensing of Sinorhizobium meliloti relies on N-acyl-homoserine lactones (AHLs) as autoinducers. AHL production increases at high population density, and this depends on the AHL synthase SinI and two transcriptional regulators, SinR and ExpR. Our study demonstrates that ectopic expression of the gene rne, coding for RNase E, an endoribonuclease that is probably essential for growth, prevents the accumulation of AHLs at detectable levels. The ectopic rne expression led to a higher level of rne mRNA and a lower level of sinI mRNA independently of the presence of ExpR, the AHL receptor, and AHLs. In line with this, IPTG (isopropyl-β-d-thiogalactopyranoside)-induced overexpression of rne resulted in a shorter half-life of sinI mRNA and a strong reduction of AHL accumulation. Moreover, using translational sinI-egfp fusions, we found that sinI expression is specifically decreased upon induced overexpression of rne, independently of the presence of the global posttranscriptional regulator Hfq. The 28-nucleotide 5′ untranslated region (UTR) of sinI mRNA was sufficient for this effect. Random amplification of 5′ cDNA ends (5′-RACE) analyses revealed a potential RNase E cleavage site at position +24 between the Shine-Dalgarno site and the translation start site. We postulate therefore that RNase E-dependent degradation of sinI mRNA from the 5′ end is one of the steps mediating a high turnover of sinI mRNA, which allows the Sin quorum-sensing system to respond rapidly to changes in transcriptional control of AHL production.
INTRODUCTION
Quorum sensing (QS) is a communication system enabling bacteria to coordinate gene expression relative to population density (1). Important cellular functions, such as biofilm formation and production of virulence factors, depend on QS (2, 3). In Gram-negative bacteria, the autoinducers are frequently of the acyl-homoserine lactone (AHL) class, and the paradigm for studying AHL-based QS is the LuxRI system of Vibrio fischeri (1, 4). Typically, transcriptional regulators belonging to the LuxR-type family recognize AHLs, and the resulting protein/AHL complex alters expression of multiple target genes, including that of the AHL synthase gene. This perception of appropriate AHL concentrations happens when AHLs are initially produced at a low basal rate. With increasing population density, the AHL concentration reaches a critical level, whereupon the LuxR/AHL complex dramatically stimulates the expression of the gene coding for the LuxI AHL synthase. This is the basis of a positive feedback which generates a burst in AHL production. The increased number of LuxR/AHL complexes then coordinates changes in global gene expression in the bacterial population.
Throughout the phylum Proteobacteria, many factors have been found to control AHL production and accumulation at the levels of transcription, translation, and protein activity. Some examples of such factors are the transcriptional repressors of luxI in Vibrio and Pseudomonas (1, 5). In Agrobacterium tumefaciens, an anti-activator protein binds to the LuxR-type transcriptional activator and increases its proteolysis (6, 7). QS can also be quenched by enzymes such as lactonases, which degrade the AHL. Two different lactonases (encoded by attM and aiiB) were found in A. tumefaciens (8). Small regulatory RNAs (sRNAs) have also been found to regulate QS (9, 10). Typically, sRNAs interact with mRNAs with the help of the RNA chaperone Hfq and influence the translation rate and/or half-life of the mRNA targets. Usually both the sRNA and the mRNA are degraded in an RNase E-dependent manner (11–13). However, the direct role of RNases in QS had not been explored so far.
In this study, we were interested in the role of RNase E in QS in Sinorhizobium meliloti, a soil alphaproteobacterium performing nitrogen fixation in symbiosis with leguminous plants. Features important for the interaction between S. meliloti and its host plant, such as motility, the ability to form a biofilm, and production of exopolysaccharides are regulated by QS (14–16). S. meliloti produces at least five different AHLs with long carbon chains (containing 12, 14, 16, and 18 C atoms) via a single LuxI-type synthase, SinI (17), although only those with 14 to 16 carbons can complement the disruption of sinI (18, 19). Transcription of sinI is controlled by the LuxR-type transcriptional regulators SinR and ExpR (Fig. 1A) (20–22). With increasing population density, the concentration of AHLs reaches a threshold value of 1 nM, leading to the activation of ExpR, which induces strong expression of sinI. This positive feedback rapidly generates an elevated production rate of AHLs (23). A second feedback mechanism is activated at higher AHL concentrations (∼40 nM), which appear to cap production of AHLs to the μM range (23, 24). Both feedback mechanisms are sensitive to specific AHL levels and depend upon the AHL receptor ExpR, which acts as a transcriptional activator of sinI expression (positive feedback) and repressor of sinR expression (negative feedback) (Fig. 1A). The ExpR-DNA binding sites enabling this transcriptional control have been identified, along with another 30 binding sites throughout the S. meliloti genome (24). To date, the regulation of QS in S. meliloti has been studied mainly at the level of transcription, and little is known about factors acting posttranscriptionally. Recently, it was found that sinI mRNA levels are higher in an hfq mutant of S. meliloti (25), strongly suggesting the involvement of an sRNA and possibly of RNase E in the Hfq-dependent regulation of this gene (11, 12). To address this question, we decided to study the impact of RNase E on AHL accumulation.
FIG 1.
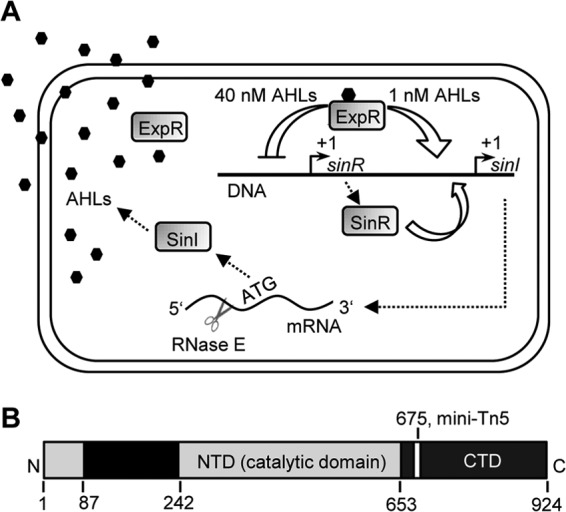
Quorum sensing and RNase E in S. meliloti. (A) Schematic representation of the Sin quorum-sensing system in S. meliloti. The role of the transcription factors SinR and ExpR on the expression of the autoinducer (AHL) synthase SinI was elucidated previously (20–24). SinR and SinI are expressed from the same locus on the chromosome. SinR is necessary for the efficient expression of the sinI gene and is independent of AHLs. ExpR senses the AHLs (octagons). At AHL concentrations of approximately 1 nM, ExpR activates the expression of SinI, leading to a strong increase in the AHL concentration (positive feedback loop). At 40 nM AHLs, ExpR negatively influences the expression of sinR, leading to low sinI expression (negative feedback loop) (23). The results of this work show that RNase E (scissors) specifically targets the 5′ UTR of sinI mRNA. (B) RNase E domains (NTD, N-terminal domain; CTD, C-terminal domain) and mini-Tn5 insertion position in S. meliloti 2011. The rne gene of S. meliloti encodes a protein comprising 924 amino acid residues. Gray bars represent regions with homology to the catalytically active, N-terminal half of RNase E of E. coli (26). Black bars represent regions without homology to RNase E of E. coli. These regions are most probably involved in the formation of the S. meliloti degradosome (41). The mini-Tn5 insertion in strain 2011rne::Tn5 is in codon 675 of rne (44).
RNase E is an endoribonuclease with major importance for the decay of mRNA in bacteria (most recently reviewed in reference 26). In E. coli, its catalytic activity is located in the N-terminal domain. The C-terminal, unstructured domain serves as a scaffold for the assembly of a multiprotein complex, the degradosome, which contains the 3′-5′ exoribonuclease polynucleotide phosphorylase (PNPase), an RNA helicase, enolase, and other minor proteins (27). Hfq also interacts with the C-terminal domain of RNase E and thereby participates in the sRNA-based regulation of gene expression in Escherichia coli (12). In Rhizobium leguminosarum, Hfq is also associated with RNase E and the degradosome, where it is necessary for the RNase E-dependent activation of the translation of NifA, the major transcriptional regulator of nitrogen fixation (28).
Different bacteria contain various compositions of RNA-degrading multiprotein complexes. However, some common characteristics, such as the association of exo- and endoribonucleases with RNA helicases and a specific subcellular localization, seem to be important for bacterial RNA metabolism (27, 29, 30). RNase E and the degradosome are bound to the cytoplasmic membrane in E. coli (31). The degradosome of Bacillus subtilis, which lacks RNase E, is also bound to the membrane. This degradosome contains PNPase, an RNA helicase, enolase, and other proteins and is organized by the endoribonuclease RNase Y (32–34). RNase E-containing degradosomes were also isolated from three alphaproteobacteria, Rhodobacter capsulatus, Caulobacter crescentus, and R. leguminosarum (28, 35, 36). In addition to RNase E, the degradosome of R. capsulatus contains two RNA helicases, the transcriptional terminator factor Rho, and substoichiometric amounts of PNPase. The degradosome of C. crescentus contains PNPase, an RNA helicase, and aconitase, while an RNA helicase and Hfq were found in the degradosome of R. leguminosarum together with other proteins. RNase E and the degradosome of S. meliloti have not yet been studied.
The N-terminal domain of RNase E is highly conserved and essential for growth of E. coli under most conditions, while mutants lacking the C-terminal domain are viable (37–40). In Streptomyces coelicolor, however, RNase E is nonessential and structurally shuffled: the catalytic domain is located in the central part of the polypeptide, while regions at the termini are involved in the interaction with PNPase (41). Bioinformatic analyses revealed an insertion of a putative degradosome-scaffold region into the putative catalytic N-terminal domain of RNase E in S. meliloti (41) (Fig. 1B). The availability of a S. meliloti Rm2011 rne mutant with a mini-Tn5 transposon insertion (44) (Fig. 1B) prompted us to analyze the role of RNase E in QS. In this study, we show that RNase E affects the production of AHLs in S. meliloti and provide evidence that the 5′ untranslated region (UTR) of sinI mRNA is a specific target of RNase E independent of Hfq.
MATERIALS AND METHODS
Strains and cultivation methods.
In this work we used the laboratory strain S. meliloti Rm2011 (referred to here as 2011), which is closely related to the first sequenced S. meliloti strain Rm1021 (42, 43). Its isogenic RNase E mutant 4.07.G10 originates from a mini-Tn5 library (44). The transposon is inserted downstream of the putative catalytic domain, in the 675th codon of the gene rne (SMc01336) (41, 42) (Fig. 1B). Since strain 2011 is a wild type in respect to rne but is an ExpR-deficient mutant with an insertion element in the expR gene (21, 23), it is referred to as parental strain 2011 in this study. The mini-Tn5 RNase E mutant is referred to as 2011rne::Tn5. To mimic the mini-Tn5 insertion, we used the pK18mob2 suicide vector carrying a 902-bp internal fragment from position 1123 to 2024 of the 2,775-nucleotide (nt) rne gene. Following homologous recombination between the plasmid and the S. meliloti chromosome, the rne gene was disrupted by the insertion of pK18mob2 at nucleotide position 2025 (675th codon). This mutant was named 2011rne675.
S. meliloti was cultivated on tryptone-yeast (TY) plates or in liquid TY cultures (45) with appropriate antibiotics (streptomycin, 250 μg μl−1; neomycin, 120 μg μl−1; gentamicin, 20 μg μl−1; and tetracycline, 20 μg μl−1). Routinely, 50 ml S. meliloti culture was grown semiaerobically in 100-ml Erlenmeyer flasks at 140 rpm and 30°C. For the experiments whose results are shown in Fig. 5 to 7, 100-μl cultures in a 96-well microtiter plate (Greiner) were grown at 30°C and 200 rpm in modified MOPS (morpholinepropanesulfonic acid)-buffered minimal medium containing 48 mM MOPS (adjusted to pH 7.2 with KOH), 55 mM mannitol, 21 mM sodium glutamate, 1 mM MgSO4, 250 mM CaCl2, 37 mM FeCl3, 48 mM H3BO3, 10 mM MnSO4, 1.0 mM ZnSO4, 0.6 mM NaMoO4, 0.3 mM CoCl2, 4.1 mM biotin, and 0.1 mM K2HPO4. Escherichia coli was grown in LB broth. E. coli JM109 and E. coli DH5α were used for standard cloning methods (46). Plasmids were transferred from E. coli S17-1 to S. meliloti by diparental conjugation (47). Bacterial strains and their relevant characteristics are listed in Table 1.
FIG 5.

Induced overexpression of either rne or truncated rne leads to low SinI expression. (A) Schematic of the translational egfp fusions in the plasmids pLK64, pLK60, and pLK61 (not to scale). Included are the plasmids containing the sinI promoter, the 5′ UTR, and the sinI coding regions of the indicated lengths followed by the egfp ORF. The transcriptional start site (+1 TSS) and the start codon (ATG) are marked. For further descriptions, see Table 1 and the text. (B) Fluorescence from the parental strain 2011 carrying pLK01 (promoterless egfp, background fluorescence), pLK64, pLK60, or pLK61 was measured in the presence and absence of IPTG-induced overexpression of rne (pWBrne) or truncated rne (pWBrne675). Included as a control is the empty vector pSRK-Gm lacking rne. All three sinI-egfp fusions (pLK64, pLK60, and pLK61) produced less fluorescence in response to overexpression of either rne or truncated rne. (C) As for panel B, except that strain 2011rne675 was used. Once again, all three sinI-egfp fusions produced less fluorescence in response to either rne or truncated rne overexpression. Error bars indicate variations from 4 cultures.
FIG 7.

The 5′ UTR of sinI is sufficient for the decrease in sinI expression when rne is overexpressed. (A) Schematic of the translational egfp fusions in the plasmids pLK65, pLK002, pLKrec01, and pLKrec02. Indicated are the plasmids along with the egfp fusions containing sinI-, sinR-, or cspA3-specific promoters, 5′ UTRs, and/or coding regions. For additional descriptions, see Fig. 5. (B) Measurement of fluorescence in strain Sm2B3001 (sinI+ expR+) from the indicated plasmids with and without IPTG-induced overexpression of rne from pBSrne. A comparison between the two synthetic constructs, pLKrec01 and pLKrec02 (see Fig. 4 for an explanatory scheme), shows that rne overexpression does not greatly affect cspA3′-egfp expression from the sinI promoter (pLKrec02) but the sinI′-egfp expression from the cspA3 promoter (pLKrec01) does show a clear decrease upon rne overexpression. Error bars indicate variations from 4 cultures.
TABLE 1.
Strains and plamids used in this study
| Strain or plasmid | Description | Reference or source |
|---|---|---|
| Strains | ||
| E. coli JM109 | endA1 recA1 gyrA96 thi hsdR17 (rK− mK+) relA1 supE44 Δ(lac-proAB) | 75 |
| E. coli DH5α | F− endA1 supE44 thi-1 recA1 gyrA96 relA1 deoR Δ(lacZYA-argF)U169 | 76 |
| E. coli S17-1 | E. coli 294; Thi RP4-2-Tc::Mu-Km::Tn7 integrated into the chromosome | 47 |
| E. coli MT102(pJBA89) | pUC18Not-luxR-PluxI -RBSII-gfp(ASV)-T0-T1; expresses EGFP upon addition of AHLs; Apr | 56 |
| S. meliloti 2011 | Contains insertion sequence within expR gene; Nxr Smr | 43 |
| S. meliloti 2011rne::Tn5 | 2011 derivative, RNase E mutant 4.07.G10 with mini-Tn5 inserted in the 675th codon of rne (SMc01336); Smr Nmr | 44 |
| S. meliloti 2011rne675 | 2011 derivative; RNase E mutant with a suicide vector pK18mobII inserted in the 675th codon of rne; Smr Kmr | This study |
| S. meliloti Sm2B3001 | 2011 derivative with restored expR gene on the chromosome | 77 |
| S. meliloti Sm2B4001 | sinI mutant of Sm2B3001 | 23 |
| S. meliloti Sm2011dhfqGmLR | 2011 derivative, Δhfq mutant, Gmr | 54 |
| A. tumefaciens NTL4(pZLR4) | Expresses beta-galactosidase upon addition of AHLs; Gmr | 58 |
| Plasmids | ||
| pK18mob2 | Suicide vector; mob lacZ Kmr | 78 |
| pK1123-2024 | pK18mobII carrying an internal fragment of rne, nt 1123–2024; Kmr | Stefan Meyer |
| pK79-926 | pK18mobII carrying an internal fragment of rne, nt 79–926; Kmr | Stefan Meyer |
| pPHU231 | pRK290 with a 388-bp HaeII insert containing pUC18 polylinker; Tcr | 79 |
| pLK01 | pPHU231 with a promoterless egfp; Tcr | 50 |
| pLK60 | pLK64 derivative without sinI codons; Tcr | This study |
| pLK61 | pPHU231 containing sinIp-sinI′-egfp translational fusion; allows expression of full-length SinI fused to EGFP; Tcr | This study |
| pLK64 | pPHU231 containing sinIp-sinI-egfp translational fusion, allows the expression of a SinI′-EGFP containing the first 9 amino acid residues of SinI; Tcr | 50 |
| pLK65 | pPHU231 containing sinRp-sinR′-egfp translational fusion; Tcr | 23 |
| pLK002 | pPHU231 containing cspA3p-cspA3′-egfp translational fusion; Tcr | This study |
| pLKrec01 | pLK64 derivative containing cspA3 promoter instead of the sinI promoter; Tcr | This study |
| pLKrec02 | pLK002 derivative containing sinI promoter instead of the cspA3 promoter; Tcr | This study |
| pSRK-Km and -Gm | Broad-host-range expression vectors with tightly regulated, IPTG-inducible lac promoter; Kmr or Gmr | 49 |
| pWBrne | pSRK-Gm containing rne; Gmr | This study |
| pWBrne675 | pSRK-Gm containing rne (codons 1–675); Gmr | This study |
| pBSrne | pSRK-Km containing rne; Kmr | This study |
| pRK415 | Tcr broad-host-range expression vector; the lac promoter is constitutive in S. meliloti | 48 |
| pRKrne | pRK415 containing rne with a C-terminal streptavidin tag-coding sequence; Tcr | This study |
| pDrive | PCR cloning kit | Qiagen |
Plasmid construction.
The plasmids used in this work are listed in Table 1, and the primers used for cloning are listed in Table S1 in the supplemental material. For disruption of the C-terminal region of rne, see the strain descriptions above. For disruption of the N-terminal region of rne, an 848-bp region of rne from positions 79 to 926 was cloned into the suicide vector pK18mob2. No mutants were obtained following the homologous recombination procedure with this construction.
For complementation, the rne gene (excluding UTRs) was cloned between the HindIII and KpnI restriction sites of the broad-host-range vector pRK415 (48). The resulting plasmid, pRKrne, allows the expression of RNase E-streptavidin from a lac promoter, which is constitutively active in S. meliloti. Furthermore, the pSRK plasmids (49) were used for construction of pBSrne (Kmr) and pWBrne (Gmr), which allow induced overexpression of rne in S. meliloti.
Construction of plasmids pLK64 and pLK65 with sinI′-egfp and sinR′-egfp translational fusions were previously described (23, 50). The plasmid pLK64 contains the promoter region of sinI, the 5′ UTR, and the first 9 codons of sinI fused to egfp. Two derivatives of pLK64 were also constructed in which the sinI codons were omitted (pLK60) and in which all sinI codons were included in the fusion to egfp (pLK61). Similarly, pLK002 was constructed, which contains the promoter and the 5′ UTR of cspA3 in a translational fusion to egfp (24). Synthetic derivatives of the pPHU231-based plasmids pLK64 and pLK002 were constructed by swapping the 5′ UTR of the sinI mRNA with the 5′ UTR of the cspA3 mRNA in each of the translational fusions (pLKrec01 and pLKrec02).
Isolation and analysis of nucleic acids.
Total DNA was isolated by the method of Masterson et al. (51). To isolate RNA for RT-PCR analysis or 5′-RACE, 1 ml of S. meliloti cultures grown to an optical density at 600 nm (OD600) of 1.3 was added to 1 ml of RNAprotect bacterial reagent (Qiagen). Cells were harvested by centrifugation (6,000 × g for 10 min at 4°C) and resuspended in the lysis buffer provided with the RNeasy minikit (Qiagen). After the addition of acid-washed glass beads (Sigma), cells were disrupted in a Tissuelyser (Retsch) for 50 s. Glass beads were removed by centrifugation. RNA was isolated with the RNeasy minikit (Qiagen), treated with RNase-free DNase (Invitrogen), and resuspended in water.
The primers employed for analyzing relative mRNA amounts of genes using real-time quantitative reverse transcription-PCR (qRT-PCR) are listed in Table S2 in the supplemental material. For normalization of mRNA levels, the rpoB gene, which encodes the beta subunit of RNA polymerase of S. meliloti, was used. Conditions for qRT-PCR were as previously described (52). We used a one-step RT-PCR kit (Qiagen) and added 4 ng μl−1 of total RNA into the reaction mixture. SYBR green I (Sigma) was diluted at 1:100,000 in the master mix to detect double-stranded DNA. Relative expression of a gene in the mutant strain was calculated relative to expression in the parental strain and relative to rpoB (53). Similarly relative mRNA levels were calculated before and after addition of IPTG (isopropyl-β-d-thiogalactopyranoside) to cultures. PCR efficiencies of primer pairs were determined using serial dilutions of RNA (see Table S2 in the supplemental material). At least two biologically independent experiments were performed, each with two technical replicates.
mRNA half-lives were determined as previously described (54), with the following modifications. A S. meliloti culture was grown to an OD600 of 0.5 and split into two flasks, and to one of the flasks 1 mM IPTG was added to induce ectopic expression of RNase E. No IPTG was added to the flask with the control culture. Transcription was stopped 60 min later by the addition of rifampin (500-μg ml−1 final concentration; stock concentration, 30 mg ml−1 in methanol). Cells were harvested at time points of 0, 3, and 6 min by adding 1 ml of the culture to 1 ml of RNAprotect bacterial reagent (Qiagen). RNA was isolated with an RNeasy minikit (Qiagen) as described above and treated with Turbo DNA-free (Ambion). mRNA levels were determined by qRT-PCR as described above, using 16S rRNA as the reference (55). Half-lives were calculated from linear-log graphs of time after rifampin addition against relative mRNA amounts.
AHL and eGFP detection.
AHLs were extracted 10 min from 1 ml bacterial culture supernatant with 0.3 ml chloroform. Extracts were evaporated, and the remaining pellet was resuspended in 30 μl of acetone. The detection of AHLs was done with two different systems. First, E. coli MT102 (pJBA89) expressing enhanced green fluorescent protein (eGFP) upon addition of AHLs and detecting a range of AHLs from C6-HSL to oxo-C14-HSL was used (56). Reporter bacteria were grown on LB medium with specific antibiotics. Ten μl of acetone extract was dropped on the bacterial lawn. Fluorescence was observed 4 h after incubation using filter with an excitation wavelength of 480/40 nm and an emission wavelength of 510 nm. The second method for detection of AHLs is based on A. tumefaciens NTL4(pZLR4) expressing beta-galactosidase from an AHL/TraR-dependent promoter (57, 58). A. tumefaciens grown with 40 μg ml−1 gentamicin was mixed with MGM agar containing 11 g Na2HPO4, 3 g KH2PO4, 0.5 g NaCl, 1 g glutamate, 10 g mannitol, 1 μg biotin, 0.25 mM CaCl2, and 1 mM MgSO4 per liter without gentamicin. X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) was added to a final concentration of 80 μg ml−1. Two μl of the AHL extracts were spotted onto the agar, and the plate was incubated at 32°C overnight. A blue color indicated the detection of AHLs.
Rapid amplification of 5′ cDNA ends.
For the determination of 5′ ends of RNA by rapid amplification of 5′ cDNA ends (5′-RACE), cells were grown in TY medium to an OD600 of 1.0. Ectopic expression of RNase E was induced by addition of 1 mM IPTG. Cells were harvested 20 min, 40 min, and 60 min after induction. No IPTG was added to the control cultures. 5′-RACE was performed as described previously (59) with primers described by McIntosh et al. (50).
RESULTS
The C-terminal region of RNase E is nonessential.
Loss of the rne gene in E. coli is lethal under most conditions (37, 40). However, insertion mutations in the C-terminal coding region of rne, which codes for the nonessential macromolecular-interaction domain, are growth permissive (39). The availability of a S. meliloti 2011rne::Tn5 mutant, in which a mini-Tn5 transposon is inserted in the C-terminal region (675th codon) of the rne gene (44) (Fig. 1B), suggests an arrangement similar to that in E. coli. In this study, we created another RNase E mutant which carries the suicide vector pK18mobII, also inserted in the 675th codon of rne. This mutant strain, 2011rne675, was viable, like the mini-Tn5 mutant, confirming that the C-terminal domain of S. meliloti RNase E is nonessential. However, attempts to insert pK18mobII into the N-terminal coding region of rne (309th codon) failed to produce any colonies. Therefore, we used the 2011rne::Tn5 and 2011rne675 mutants to study the effect of RNase E on AHL production. In addition, plasmids bearing rne with a constitutive (pRKrne) and an IPTG-inducible lac promoter (pWBrne and pBSrne) were used to study the effect of ectopic expression of rne on AHL production.
Overexpression of rne affects AHL accumulation.
To address the question of whether RNase E regulates quorum sensing in S. meliloti, AHLs harvested from the 2011rne::Tn5 mutant and the mutant containing pRKrne were compared to AHLs from the 2011 parent strain using an AHL sensor system with a GFP reporter in E. coli (Fig. 2B). AHLs were extracted from supernatants of cultures at OD600 of 1.3 (Fig. 2A) and added to the E. coli reporter strain, and fluorescence was measured. Similar fluorescence levels were observed for the AHL extracts from the transposon mutant and the 2011 parent strain, while constitutive ectopic expression of the rne gene from pRKrne resulted in a dramatic reduction of fluorescence (Fig. 2B). We postulated that this extremely low fluorescence reflects a strongly reduced AHL production in strain 2011rne::Tn5 (pRKrne) due to overproduction of RNase E.
FIG 2.
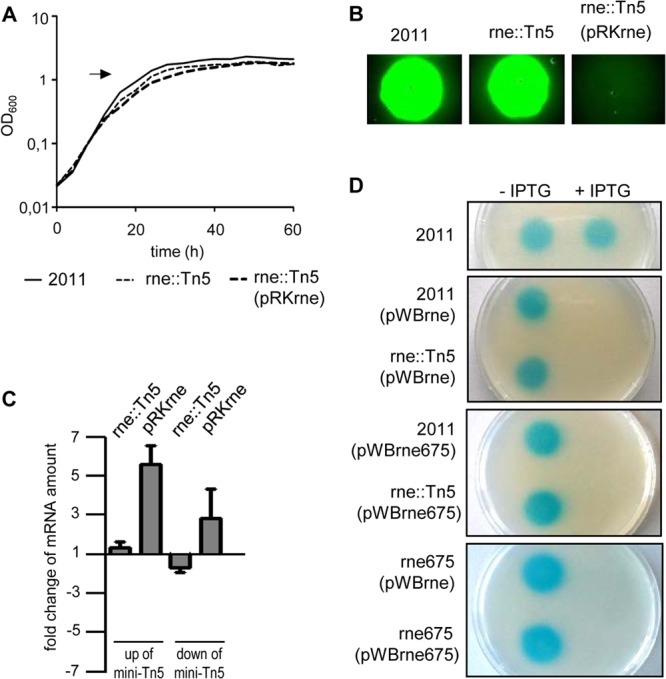
Overexpression of rne dramatically decreases AHL accumulation. (A) Growth curves of the expR-deficient parent strain S. meliloti 2011, the RNase E mutant 2011rne::Tn5, and the RNase E mutant containing pRKrne. AHLs were extracted from supernatants of cultures grown to an OD600 of 1.3 (arrow). (B) The extracted AHLs were detected with a GFP reporter system in E. coli MT102 (pJBA89). Shown is the fluorescence of the reporter strain grown with AHLs extracted from the indicated S. meliloti cultures. The spots were on the same plate. (C) Real time RT-PCR analysis of the rne gene, encoding RNase E. Levels of rne mRNA in the mutant 2011rne::Tn5 and in the mutant containing pRKrne were compared to the levels in the parental strain, 2011. Two primer pairs targeting rne gene at locations upstream and downstream of the mini-Tn5 insertion in the RNase E mutant were used. Results are from three independent experiments with two technical replicates. Error bars indicate the standard deviations (SDs). (D) Detection of AHLs in cultures of strain 2011 and its isogenic mutants 2011rne::Tn5 and 2011rne675 grown without (−) or with (+) 1 mM IPTG. The strains contain pWBrne or pWBrne675 as indicated, with rne or truncated rne under the control of an inducible lac promoter. Detection was performed with the A. tumefaciens reporter strain NTL4(pZLR4).
To test whether ectopic expression of the S. meliloti rne gene results in an elevated accumulation of rne mRNA, qRT-PCR analysis of rne was performed. Two primer pairs annealing to different rne regions (downstream and upstream of the mini-Tn5 insertion in strain 2011rne::Tn5) were used to analyze total RNA isolated from cultures at an OD600 of 1.3. Consistent with the location of the primer annealing sites with respect to the mini-Tn5 insertion (Fig. 1B), the level of the C-terminal coding region of the mRNA from downstream of the mini-Tn5 insertion was lower in the 2011rne::Tn5 mutant than in the parental 2011 strain (Fig. 2C). However, the N-terminal coding region of rne mRNA upstream of the mini-Tn5 insertion was not changed in the mutant compared to the parental strain 2011. The level of rne mRNA increased in the mutant carrying the plasmid pRKrne compared to the parental strain 2011 (Fig. 2C). We conclude that ectopic expression of rne does indeed lead to increased rne mRNA accumulation and that the mini-Tn5 insertion does not greatly alter the abundance of mRNA from the N-terminal region of rne.
To confirm that rne overexpression leads to changes in AHL accumulation in S. meliloti, we used pWBrne, allowing IPTG-inducible ectopic expression of full-length rne in the parental strain 2011 and in the mutant 2011rne::Tn5. AHLs were extracted and detected with an A. tumefaciens reporter system (Fig. 2D). The experiment was also performed with pWBrne675, bearing rne codons 1 to 675 under the control of an IPTG-inducible lac promoter with similar effects: a reduction of AHLs to nondetectable levels in strains 2011 and 2011rne::Tn5 in the presence of IPTG (Fig. 2D). Similar results were obtained with the mutant strain 2011rne675 when full-length RNase E from pWBrne or the truncated RNase E from pWBrne675 was overexpressed (Fig. 2D). Altogether, these results show that overexpression of only the region encoding the N-terminal part of RNase E is sufficient for the disruption of AHL accumulation. This is consistent with the assumption that like in RNase E of E. coli, the N-terminal part of S. meliloti RNase E contains the catalytically active domain (41) (Fig. 1B).
Overexpression of rne diminishes sinI mRNA accumulation.
To determine the mechanism for the dependence of AHL accumulation on rne, real-time RT-PCR (qRT-PCR) analysis was performed on the genes encoding SinI (the AHL synthase) and SinR (the AHL-independent transcriptional activator of sinI) (24). We found that in comparison to S. meliloti 2011, the sinR mRNA levels were not changed significantly in the mutant 2011rne::Tn5 or in the pRKrne-containing mutant, which constitutively overexpresses rne (Fig. 3A). A slight decrease in the amount of sinI mRNA was detected in 2011rne::Tn5, and a strong decrease was detected in the overexpressing strain. To exclude artifacts due to stable mRNA fragments, the sinI analysis was performed with two different primer pairs with similar results (Fig. 3A). Even 20 min after induction of IPTG-induced overexpression of full-length rne in both the 2011rne::Tn5 mutant and the parental strain 2011, the levels of sinI mRNA but not of sinR mRNA were decreased (Fig. 3B and C). These data correlate with the observed decrease in AHL accumulation upon overexpression of rne (Fig. 2B and D) and fit with the hypothesis that RNase E specifically degrades sinI mRNA but not sinR mRNA. This is also consistent with the results shown in Fig. 2C, where ectopic expression of rne results in an increase in rne mRNA accumulation.
FIG 3.
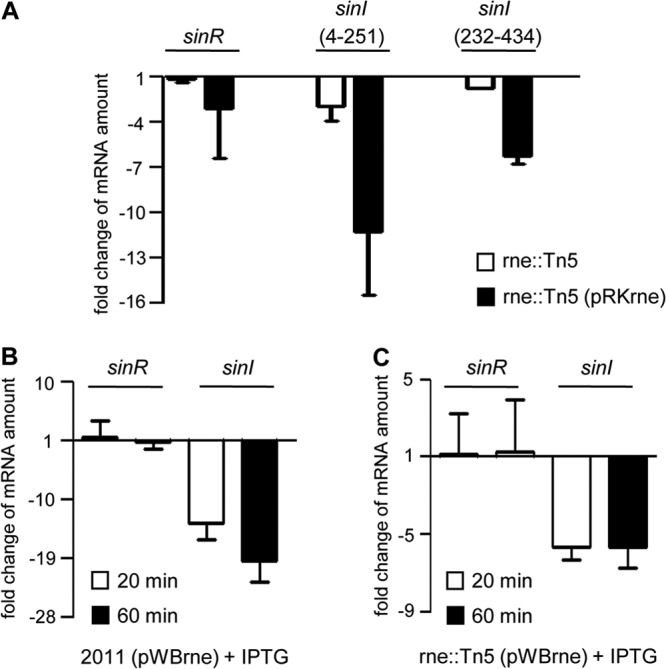
Real-time RT-PCR reveals a strong decrease in levels of sinI mRNA in strains overexpressing rne. (A) Real-time RT-PCR of sinR and sinI was performed with total mRNA isolated from the parental strain 2011, the RNase E mutant 2011rne::Tn5, and the RNase E mutant containing pRKrne. Two primer pairs amplifying nucleotides 4 to 251 and 232 to 434 of the sinI open reading frame (ORF) were used. The mRNA levels in strain 2011rne::Tn5 and strain 2011rne::Tn5 (pRKrne) were compared to the levels in the parental strain. Results are from two independent experiments with two technical replicates. An exception was the analysis of strain 2011rne::Tn5 (pRKrne), for which three biological experiments with two technical replicates were performed. Data are means and SDs. (B) Real time RT-PCR of sinR and sinI in strain 2011 (pWBrne). Samples were harvested at 20 and 60 min after addition of 1 mM IPTG to cultures grown to an OD600 of 1.3, and mRNA levels were compared to the levels before IPTG addition. (C) Real-time RT-PCR of sinR and sinI in strain 2011rne::Tn5 (pWBrne), as described for panel B.
The reduction of the steady-state amount of sinI mRNA upon overexpression of rne is most probably due to decreased sinI mRNA stability. To test whether overexpression of rne affects the half-life of sinI mRNA, mRNA stability was measured in strain 2011 (pWBrne) grown without IPTG and compared to mRNA stability in the same strain following the addition of IPTG. Relative mRNA amounts were determined 0, 3, and 6 min after the addition of rifampin, which stops RNA transcription in bacteria. We did not obtain signals for sinI mRNA in Northern blots, probably due to the small amount of this messenger (data not shown). Therefore, qRT-PCR analysis was performed to determine the relative amounts of sinI mRNA. As an internal reference, the stable 16S rRNA was used. To test the specificity of the rne effect on sinI mRNA stability, half-lives were also determined for sinR and rpoB mRNA, which was the internal reference in the qRT-PCR experiments whose results are shown in Fig. 3.
The results of the mRNA stability measurements are shown in Fig. 4. The half-life of sinI mRNA was determined with the two different primer pairs with very similar results (3.2 ± 0.4 min and 3.8 ± 0.3 min). As expected, the stability of sinI mRNA was significantly reduced upon overexpression of rne (1.9 ± 0.1 and 1.9 ± 0.2 min with each of the primer pairs, respectively) (Fig. 4A and B). In contrast, the stability of sinR and rpoB mRNAs was not affected (Fig. 4C and D). Using one of the primer pairs and cultures without IPTG, sinI mRNA stability was determined in two independent experiments at ODs of 0.5, 1.0, and 1.3. It is noteworthy that the sinI mRNA stability was comparable at all three ODs (Fig. 4A, 0 mM IPTG). Based on these data, we conclude that there is no differential regulation of sinI expression at the level of mRNA stability in strain 2011 at these three ODs and that overexpressed rne specifically decreases the stability of sinI mRNA, leading to lower steady-state amounts.
FIG 4.

Overexpression of rne specifically decreases the stability of sinI mRNA. The graphs show results of representative experiments. Unless differently stated, half-lives (t1/2) were calculated from two independent experiments, each with two technical replicates. Cells were harvested 0, 3, and 6 min after rifampin addition to 2011 (pWBrne) cultures at an OD600 of 0.5 grown with and without IPTG. Total RNA was isolated and relative mRNA levels were determined by qRT-PCR. (A) Stability of sinI mRNA was determined with the primer pair targeting nt 4 to 251 in the sinI ORF. Measurements without IPTG were performed in two independent experiments with two technical replicates at three ODs (0.5, 1.0, and 1.3) with very similar results, and the half-lives (at 0 mM IPTG) were calculated from a total of 12 measurements. (B) Stability of sinI mRNA was determined with the primer pair targeting nt 232 to 434. (C) Stability of sinR mRNA. (D) Stability of rpoB mRNA.
Overexpression of rne lowers sinI expression.
Overexpression of rne in S. meliloti leads to both low AHL levels and low sinI mRNA levels, as determined by AHL extraction and detection, and by qRT-PCR. To better understand the mechanism of rne-dependent reduction in sinI mRNA levels, we used a plasmid-based reporter system with sinI-egfp fusions. The plasmid pLK64 has been used previously to study the control of the sinI promoter (23, 24, 50). In this construct, the sinI promoter region (287 bp), the region corresponding to the 5′ UTR of sinI mRNA (28 bp), and the first 27 bp of the sinI coding sequence are fused to the ATG of egfp. In addition, two other constructs were designed: pLK61, in which the sinI promoter, the 5′ UTR, and the full-length sinI coding region were fused to egfp, and pLK60, in which only the promoter region and the 5′ UTR of sinI were fused to egfp (Fig. 5). We tested pLK61 in a sinI deletion strain, and this plasmid restored AHL production (data not shown), indicating that the SinI-EGFP fusion protein is functional. Fluorescence detected from each of the three reporter plasmids was measured in the parental strain 2011 (Fig. 5B) and in the 2011rne675 mutant (Fig. 5C) containing the empty vector control pSRK-Gm or one of the IPTG-inducible plasmids pWBrne or pWBrne675. Background fluorescence was measured using the vector pLK01, which contains a promoterless egfp.
Plasmid pLK64 produced the highest fluorescence (8,000 to 9,000 fluorescence units per unit of optical density [F/OD]), followed by pLK60 (5,000 to 6,000 F/OD). Plasmid pLK61 produced the lowest fluorescence (2,000 to 3,000 F/OD), indicating that the sinI-egfp mRNA and/or the fusion protein has lower stability. All three plasmids produced a significantly lower fluorescence upon overexpression of either rne (pWBrne) or the truncated rne (pWBrne675). This effect was similar in both the parental strain 2011 and the 2011rne675 mutant. These results are similar to those obtained via AHL extraction and qRT-PCR and are consistent with an RNase E-dependent degradation of sinI mRNA. Also notable is that this effect requires only the N-terminal part of the coding region of rne. The results also strongly suggest that as a target for RNase E, the minimal requirement is the 5′ UTR of sinI mRNA. This is remarkable in that the length of the 5′ UTR of sinI is only 28 nucleotides.
Rne influence on sinI expression is independent of expR.
The sequenced laboratory strain Rm1021 (42) and the closely related strain 2011 have incomplete Sin QS systems due to an interruption of the expR gene by an IS element (21, 23). The lack of ExpR renders the sinI promoter insensitive to the presence of AHLs, but sinI expression and AHL production continue in a SinR-dependent manner (see Fig. 1 for the regulatory scheme). Thus, in the absence of ExpR, sinI expression and AHL production are detectable, albeit ∼3-fold weaker (23). This is because the activation of the sinI promoter by SinR is independent of AHLs (24). One advantage of using an expR mutant strain, such as 2011, is that it enables the study of the regulation of genes underlying QS in a simplified genetic background. To check whether the rne effect on sinI expression is dependent upon expR or AHLs, fluorescence from the plasmid pLK64 was compared in three genetic backgrounds: (i) in strain 2011 (expR sinI+), (ii) in its derivative Sm2B3001 (expR+ sinI+) with a restored expR gene located at its native site on the chromosome, and (iii) in strain Sm2B4001, a sinI mutant derivative of Sm2B3001 (expR+ sinI) (Fig. 6). Strains carrying pBSrne, in which rne was placed under the control of an IPTG-inducible lac promoter in plasmid pSRK-Km, were grown with and without IPTG. In all three strains, fluorescence was significantly reduced upon overexpression of rne, indicating that the rne effect on sinI expression was not dependent upon either expR or AHLs.
FIG 6.
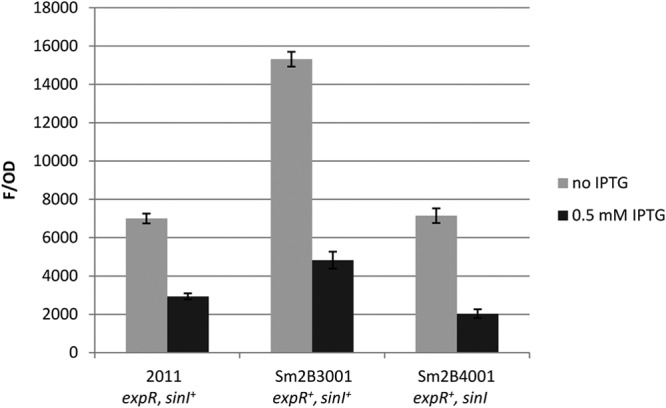
rne overexpression effect is independent of expR and AHLs. Fluorescence from the sinI-egfp fusion in pLK64 was used to determine whether the rne effect on sinI expression was dependent upon either the presence of expR or AHLs. Upon IPTG-induced overexpression of rne from plasmid pBSrne, a decrease in fluorescence was observed not only in the parental expR mutant strain 2011 but also in a derivative strain carrying a functional copy of expR (Sm2B3001) and in a second derivative strain with a functional copy of expR but without sinI (Sm2B4001, no AHLs). Error bars indicate variations from 4 cultures.
The 5′ UTR of sinI mRNA is a specific target of RNase E.
The influence of RNase E on sinI is related to the region encompassing the promoter, the 5′ UTR, and the first nine codons of sinI (Fig. 5). To better understand the underlying mechanisms, additional fusions with the reporter egfp gene were used (Fig. 7A), and fluorescence was measured after cultivation of the strains with and without overexpression of rne (Fig. 7B). Overexpression of rne did not reduce fluorescence from the vector pLK65, which contains the promoter and 5′ UTR of sinR fused to the translation start of egfp. Therefore, rne does not appear to control the expression of sinR. As an additional control, we used the plasmid pLK002, which contains a fusion of the cspA3 promoter and 5′ UTR to egfp (Fig. 7A). The gene cspA3 encodes a cold shock protein, and its promoter is QS independent (24). The expression of the cspA3′-egfp fusion was also almost unaffected by rne overexpression (Fig. 7B). Altogether, these experiments are in agreement with the conclusion that sinI mRNA is a specific target of RNase E.
To confirm that RNase E specifically targets the 5′ UTR of sinI mRNA, we used a synthetic approach and fused the cspA3 promoter to the 5′ UTR of sinI followed by egfp (pLKrec01). Additionally, the promoter of sinI was fused to the 5′ UTR of cspA3 followed by egfp (pLKrec02) (for a fusion scheme, see Fig. 7A). Fluorescence in strain Sm2B3001 (sinI+ expR+) bearing these plasmids was measured with and without IPTG-induced expression of rne. Upon rne overexpression, fluorescence from pLKrec02 essentially did not decrease, while fluorescence from pLKrec01 did decrease (Fig. 7B). These results, together with data in Fig. 5 demonstrating the rne-dependent decrease of fluorescence from pLK60 lacking sinI codons, show that the 5′ UTR of sinI mRNA is sufficient for downregulation of sinI expression upon overexpression of rne. This confirms that the 5′ UTR of sinI is a specific target of RNase E.
RNase E is expressed as a streptavidin-tagged fusion protein from pRKrne. However, we were not able to isolate the protein for an in vitro determination of the putative cleavage site in the 5′ UTR of sinI. Therefore, we decided to use an in vivo approach. The 5′ ends of sinI mRNA were detected via 5′-RACE analysis in strains 2011 and 2011rne::Tn5 and following overexpression of rne from pWBrne. The results are summarized in Fig. 8 and also in Table S3 in the supplemental material. A total of 24 5′ ends were detected in RNA extracted from strain 2011 without pWBrne and without IPTG. Nine of these were mapped to the previously determined transcriptional start site (TSS; +1) of sinI (50), six to position +24 in the 5′ UTR of the transcript (between the Shine-Dalgarno sequence and the start codon), and nine within the coding region of sinI, mostly at position +359. The 5′ ends downstream of the TSS probably correspond to degradation intermediates. Similar results were obtained from the control strain 2011, which was grown with IPTG but lacked pWBrne. Ten of the 23 mapped 5′ ends from strain 2011 with pWBrne but without IPTG corresponded to the TSS of sinI, four were found at position +24, and the rest mapped to different internal positions in the sinI mRNA (Fig. 8; also, see Table S3 in the supplemental material). However, upon IPTG-induced overexpression of rne (2011 with pWBrne and IPTG), the number of 5′ ends detected at position +1 dropped to zero, six were at position +24, and the remainder were at positions within the coding region of sinI.
FIG 8.

RNase E is necessary for occurrence of processed 5′ ends in the first half of the sinI mRNA and probably cleaves at position +24 in the 5′ UTR of the transcript. (A) Mapping of 5′ ends by RACE. Results are from at least two biologically independent 5′-RACE experiments with strains 2011, 2011 (pWBrne), and the RNase E mutant 2011rne::Tn5 (pWBrne). 5′ ends were mapped for cultures with (+) and without (−) IPTG. The total number of the sequenced clones (experimentally determined 5′ ends), the number and the positions (indicated above the mRNA sequence; +1 is the TSS) of 5′ ends in the 5′ UTR of sinI mRNA, and the number of 5′ ends at variable positions in the coding region of the transcript are shown. The Shine-Dalgarno sequence is in bold; the start codon is in italics. For detailed information, see Table S1 in the supplemental material. (B) Proposed secondary structure (Mfold [74]) of the 5′ UTR of sinI mRNA with the RNase E cleavage site at position +24.
The comparison between strains 2011 and 2011(pWBrne) without IPTG suggests that even in the absence of IPTG, RNase E is slightly expressed from pWBrne. This leaky expression without IPTG does not block the AHL production (Fig. 2D) but possibly leads to the occurrence of multiple internal 5′ ends at various positions in the sinI transcript (Fig. 8; also, see Table S3 in the supplemental material). A stronger scattering of internal 5′ ends was observed in the corresponding IPTG-induced culture of strain 2011 (pWBrne), supporting the view that overexpression of rne leads to their occurrence. These results are consistent with strong degradation of sinI mRNA upon overexpression of rne. When analyzed temporally (see Table S3), the data show that the proportion of 5′ ends downstream of +1 increased with increasing exposure to IPTG-induced rne expression. Thus, increased expression of rne leads to an increased degradation of sinI mRNA in the 5′-3′ direction.
For the 2011rne::Tn5 mutant carrying pWBrne, a total of 24 5′ ends corresponding to the TSS (+1) of sinI were detected in the absence of IPTG (Fig. 8). This indicates that our method for detecting 5′ ends does not include premature stops during cDNA synthesis from mRNA and supports the conclusion that the internal 5′ ends represent RNase E-mediated degradation intermediates of sinI mRNA. In IPTG-induced 2011rne::Tn5 (pWBrne) cultures, internal 5′ ends were detected in addition to the TSS. Six out of 11 internal 5′ ends mapped to position +24 in the 5′ UTR of sinI mRNA (from a total of 26 analyzed clones) (Fig. 8).
In summary, results presented in Fig. 8 show that overexpression of rne leads to a faster degradation of sinI mRNA, and that position +24 in the 5′ UTR of sinI mRNA is a potential RNase E cleavage site. Furthermore, full-length RNase E is necessary for occurrence of processed 5′ ends in the region analyzed by 5′-RACE.
RNase E acts on the 5′ UTR of sinI independently of Hfq.
An RNase E cleavage in the 5′ UTR of bacterial mRNAs is often mediated by trans-encoded sRNAs, and the sRNA-mRNA interaction is usually Hfq dependent (12). Hfq-dependent RNase E cleavage in the 5′ UTR of nifA mRNA was also found in R. leguminosarum (28). Thus, a similar mechanism may operate at the 5′ UTR of sinI. To check for the involvement of Hfq, we used a 2011Δhfq mutant (54). We detected larger AHL amounts in cultures of this mutant than in the parental strain, 2011 (data not shown), in agreement with the previously reported higher sinI mRNA and AHL levels in the absence of a functional hfq gene in S. meliloti (25). The plasmids pLK64 (containing the sinI′-egfp translational fusion) and pWBrne (containing rne under the control of an inducible lac promoter) were introduced into the 2011Δhfq mutant. The strain was grown with and without IPTG in two independent experiments, and fluorescence was measured. In the presence of IPTG, fluorescence was reduced 2.4-fold. This is comparable to the reduction of fluorescence in the presence of IPTG in the parental strain 2011 containing the same plasmids (Fig. 5B, data for pLK64). We conclude that overexpression of rne negatively influences sinI expression in an Hfq-independent manner.
DISCUSSION
RNase E is an endoribonuclease with major importance for the decay of mRNA in bacteria (26). RNase E is essential for growth of E. coli and M. smegmatis under standard laboratory conditions (27, 40, 60). The role of RNase E in S. meliloti has not been analyzed so far, although it is likely to be essential as well, since our attempts to insert pK18mob2 in the rne region encoding the N-terminal domain of RNase E were not successful, while insertions into the region encoding the C-terminal domain were. Mutants 2011rne::Tn5 and 2011rne674 are not strongly impaired in their growth (Fig. 2A, growth of 2011rne::Tn5). This can be explained by the assumption that these mutants express a truncated, catalytically active RNase E lacking (a part of) the domain responsible for the interaction with other components of the degradosome (26, 38). Both insertions are at codon 675 of rne, downstream of the region encoding the putative catalytic RNase E domain (Fig. 1B). This view is supported by the qRT-PCR analyses of rne regions upstream and downstream of the mini-Tn5 insertion and by the fact that the mutant strains 2011rne::Tn5 and 2011rne674 are both viable and capable of AHL production (Fig. 2).
Studies on the RNase E of E. coli have revealed RNase E as a potent autoregulator (reviewed in reference 26). When RNase E activity exceeds the demands for RNA processing and turnover, the rne mRNA becomes a target for degradation. The 5′ UTR of rne mRNA and a functional C-terminal domain of RNase E are important for this autoregulation (26, 61). The ectopic expression of the S. meliloti rne coding region led to elevated rne mRNA levels (Fig. 2C) and increased degradation of sinI mRNA (Fig. 4). Ectopically expressed rne may escape a potential autoregulation due to the lack of native nontranscribed regions with regulatory functions.
Previous studies revealed the involvement of sRNAs in the control of translation and mRNA levels of transcriptional regulators of QS in Vibrio and Pseudomonas species. These sRNAs influence the expression of luxR-like transcriptional regulators or other QS-dependent genes, but not directly the autoinducer synthase (62, 63). Although it can be assumed that endoribonucleases such as RNase E and RNase III contribute to the adjustment of mRNA levels by QS sRNAs, so far this was not demonstrated experimentally. Indeed, there is little experimental evidence for a role for ribonucleases in the control of bacterial QS systems. An exception is the work by Luo and Farrand (64) showing that an RNase D homolog is important for the expression of TraR, a LuxR-type transcriptional factor in A. tumefaciens.
Overexpression of rne results in enhanced degradation of sinI mRNA. Our data show that this negative effect is (at least partly) due to a specific, Hfq-independent cleavage of RNase E in the 5′ UTR of sinI mRNA. The Hfq-independent status of this cleavage does not exclude the involvement of an sRNA, since a trans-encoded, Hfq-independent sRNA was shown to regulate the expression of photosynthesis genes in the alphaproteobacterium Rhodobacter sphaeroides (65).
Although overexpression of rne specifically destabilizes sinI mRNA, no differences in the stability of sinI mRNA at different points of the growth curve were detected when rne was not overexpressed. This shows that RNase E cleavage in the 5′ UTR is an important factor in the turnover of sinI mRNA but is not modulated under the tested conditions. The importance of RNase E for the turnover of sinI mRNA is demonstrated by the lack of processed 5′ ends in the 5′ half of this mRNA in the 2011rne::Tn5 mutant (Fig. 8; also, see Table S3 in the supplemental material). Despite the reduction in degradation events in the 5′ half of sinI mRNA in this mutant, the total level of sinI mRNA was not increased in comparison to the wild type (Fig. 3A). This is suggestive of an unknown, alternative degradation pathway(s) which also contributes to the degradation of sinI mRNA in the mutant.
Generally, mRNA degradation in bacteria is triggered by dephosphorylation of the primary transcript or by an internal endonucleolytic cleavage (66). Since many bacterial RNases, including RNase E, RNase G (which shows homology to the N-terminal part of RNase E and exhibits similar substrate specificity), RNase J, and RNase Y, prefer monophosphorylated substrates, dephosphorylation by a pyrophosphatase or an endonucleolytic cleavage strongly destabilizes the target transcripts, which are then degraded in a concerted action by endo- and exoribonucleases (reviewed in reference 66). While the exoribonucleolytic degradation proceeds only in the 3′-5′ direction in E. coli, 5′-3′ degradation by RNase J takes place in B. subtilis (67). S. meliloti harbors RNase E but not RNase G (42). In addition, it harbors RNase J, which is responsible for the 5′-end maturation of rRNA (68), and the 3′-5′ exoribonucleases RNase R and PNPase (42). The 5′ ends which we detected by 5′-RACE resulted from either endonucleolytic cleavages or exoribonucleolytic decay in a 5′-3′ direction. Based on our 5′-RACE data, we suggest that sinI mRNA decay includes endonucleolytic cleavages at positions +24 and +359, the two internal positions at which 5′ ends were found in independent experiments. It is not clear whether the scattered 5′ ends, represented by the detection of single (nonduplicated) events at multiple positions within the sinI mRNA (see Table S3 in the supplemental material), result directly from increased RNase E activity in the cell or from increased accessibility of mRNA for exoribonucleolytic 5′-3′ degradation.
One question that arose in the course of this study was whether the overexpression of rne somehow regulates gene expression that leads to AHL degradation independently of its effect on sinI expression. However, when we added synthetic AHLs to cultures overexpressing rne and lacking the sinI gene (and therefore incapable of producing endogenous AHLs), we saw no difference in the amount of AHLs recovered compared to cultures not overexpressing rne (our unpublished data). This is consistent with our conclusions that RNase E affects AHL accumulation through targeting the mRNA of sinI. However, we cannot rule out other mechanisms by which RNase E affects AHL accumulation, since multiple pathways of mRNA degradation and interdependence of RNases are known for other bacteria (69–71).
In this study, we have shown that RNase E specifically targets the 5′ UTR of sinI mRNA at position +24. This seems to be an efficient cleavage site for regulation of sinI expression, since it is located immediately after the Shine-Dalgarno sequence, preventing translation of the mRNA and destabilizing the transcript. This fits very well with previous observations. For example, a mathematical model of the S. meliloti Sin system has been described which correlates predicted and observed behavior of the Sin system using the activity of the sinI promoter as the output and the relative abundance of ExpR, SinR, and AHLs as various inputs (72). In that study, one basic assumption necessary for a workable model of the Sin system is that the gene products of both sinR and sinI should be rapidly degraded, allowing a finely tuned transcriptional control of AHL production that is sensitive to AHL levels. Consistent with this, the half-lives of both sinR and sinI mRNAs are in the range of typical mean chemical half-lives of RNA measured in bacteria (between 2.4 min in Prochlorococcus and 6.8 min in E. coli) (73). With RNase E, we have identified and reported the first factor which is specifically involved in the turnover of sinI mRNA.
In summary, our data strongly suggest that RNase E is essential in S. meliloti. It can be assumed that in this species, as in other bacteria, RNase E influences many cellular processes. We show that RNase E is one of the factors involved in the degradation of the AHL synthase transcript sinI and that the 5′ UTR of sinI is a specific target of RNase E. These findings open the door to understanding the posttranscriptional mechanisms influencing the expression of QS-related genes in S. meliloti.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Deutsche Forschungsgemeinschaft (DFG), grant Ev42/4-1 (associated with SPP 1258), to E.E.-H. K.B. is a member of IRTG 1384 “Enzymes and multienzyme complexes acting on nucleic acids” supported by DFG. Work of A.S., E.S., and K.H.K. was supported by Bundesanstalt für Landwirtschaft und Ernährung (BLE) grant Nr. 2811NA033. M.M. and A.B. acknowledge support from the LOEWE program of the State of Hessen, Germany (in the framework of the Center for Synthetic Microbiology, SYNMIKRO, Marburg) and from the DFG (SPP 1258, grant Be2121/5-2). P.C. is supported by the Development and Promotion of Science and Technology talents project (DPST), the Royal Thai Government.
Footnotes
Published ahead of print 31 January 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01471-13.
REFERENCES
- 1.Fuqua WC, Winans SC, Greenberg EP. 1994. Quorum sensing in bacteria: the LuxR-LuxI family of cell-density-responsive transcriptional regulators. J. Bacteriol. 176:269–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davies DG, Parsek MR, Pearson JP, Iglewski BH, Costerton JW, Greenberg EP. 1998. The involvement of cell-to-cell signals in the development of a bacterial biofilm. Science 280:295–298. 10.1126/science.280.5361.295 [DOI] [PubMed] [Google Scholar]
- 3.Zhu J, Miller MB, Vance RE, Dziejman M, Bassler BL, Mekalanos JJ. 2002. Quorum-sensing regulators control virulence gene expression in Vibrio cholerae. Proc. Natl. Acad. Sci. U. S. A. 99:3129–3134. 10.1073/pnas.052694299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ng WL, Bassler BL. 2009. Bacterial quorum-sensing network architectures. Annu. Rev. Genet. 43:197–222. 10.1146/annurev-genet-102108-134304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Venturi V, Rampioni G, Pongor S, Leoni L. 2011. The virtue of temperance: built-in negative regulators of quorum sensing in Pseudomonas. Mol. Microbiol. 82:1060–1070. 10.1111/j.1365-2958.2011.07890.x [DOI] [PubMed] [Google Scholar]
- 6.Costa ED, Chai Y, Winans SC. 2012. The quorum-sensing protein TraR of Agrobacterium tumefaciens is susceptible to intrinsic and TraM-mediated proteolytic instability. Mol. Microbiol. 84:807–815. 10.1111/j.1365-2958.2012.08037.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hwang I, Smyth AJ, Luo ZQ, Farrand SK. 1999. Modulating quorum sensing by antiactivation: TraM interacts with TraR to inhibit activation of Ti plasmid conjugal transfer genes. Mol. Microbiol. 34:282–294. 10.1046/j.1365-2958.1999.01595.x [DOI] [PubMed] [Google Scholar]
- 8.Haudecoeur E, Tannières M, Cirou A, Raffoux A, Dessaux Y, Faure D. 2009. Different regulation and roles of lactonases AiiB and AttM in Agrobacterium tumefaciens C58. Mol. Plant Microbe Interact. 22:529–537. 10.1094/MPMI-22-5-0529 [DOI] [PubMed] [Google Scholar]
- 9.Lenz DH, Mok KC, Lilley BN, Kulkarni RV, Wingreen NS, Bassler BL. 2004. The small RNA chaperone Hfq and multiple small RNAs control quorum sensing in Vibrio harveyi and Vibrio cholerae. Cell 118:69–82. 10.1016/j.cell.2004.06.009 [DOI] [PubMed] [Google Scholar]
- 10.Bejerano-Sagie M, Xavier KB. 2007. The role of small RNAs in quorum sensing. Curr. Opin. Microbiol. 10:189–198. 10.1016/j.mib.2007.03.009 [DOI] [PubMed] [Google Scholar]
- 11.Massé E, Escorcia FE, Gottesman S. 2003. Coupled degradation of a small regulatory RNA and its mRNA targets in Escherichia coli. Genes Dev. 17:2374–2383. 10.1101/gad.1127103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morita T, Maki K, Aiba H. 2005. RNase E-based ribonucleoprotein complexes: mechanical basis of mRNA destabilization mediated by bacterial noncoding RNAs. Genes Dev. 19:2176–2186. 10.1101/gad.1330405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Storz G, Vogel J, Wassarman KM. 2011. Regulation by small RNAs in bacteria: expanding frontiers. Mol. Cell 43:880–891. 10.1016/j.molcel.2011.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.González JE, Marketon MM. 2003. Quorum sensing in nitrogen-fixing rhizobia. Microbiol. Mol. Biol. Rev. 67:574–592. 10.1128/MMBR.67.4.574-592.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marketon MM, Glenn SA, Eberhard A, González JE. 2003. Quorum sensing controls exopolysaccharide production in Sinorhizobium meliloti. J. Bacteriol. 185:325–331. 10.1128/JB.185.1.325-331.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoang HH, Gurich N, González JE. 2008. Regulation of motility by the ExpR/Sin quorum-sensing system in Sinorhizobium meliloti. J. Bacteriol. 190:861–871. 10.1128/JB.01310-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Teplitski M, Eberhard A, Gronquist MR, Gao M, Robinson JB, Bauer WD. 2003. Chemical identification of N-acyl homoserine lactone quorum-sensing signals produced by Sinorhizobium meliloti strains in defined medium. Arch. Microbiol. 180:494–497. 10.1007/s00203-003-0612-x [DOI] [PubMed] [Google Scholar]
- 18.Gao M, Coggin A, Yagnik K, Teplitski M. 2012. Role of specific quorum-sensing signals in the regulation of exopolysaccharide II production within Sinorhizobium meliloti spreading colonies. PLoS One 7:e42611. 10.1371/journal.pone.0042611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Llamas I, Keshavan N, González JE. 2004. Use of Sinorhizobium meliloti as an indicator for specific detection of long-chain N-acyl homoserine lactones. Appl. Environ. Microbiol. 70:3715–3723. 10.1128/AEM.70.6.3715-3723.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marketon MM, Gronquist MR, Eberhard A, González JE. 2002. Characterization of the Sinorhizobium meliloti sinR/sinI locus and the production of novel N-acyl homoserine lactones. J. Bacteriol. 184:5686–5695. 10.1128/JB.184.20.5686-5695.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pellock BJ, Teplitski M, Boinay RP, Bauer WD, Walker GC. 2002. A LuxR homolog controls production of symbiotically active extracellular polysaccharide II by Sinorhizobium meliloti. J. Bacteriol. 184:5067–5076. 10.1128/JB.184.18.5067-5076.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao M, Chen H, Eberhard A, Gronquist MR, Robinson JB, Rolfe BG, Bauer WD. 2005. sinI- and expR-dependent quorum sensing in Sinorhizobium meliloti. J. Bacteriol. 187:7931–7944. 10.1128/JB.187.23.7931-7944.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McIntosh M, Meyer S, Becker A. 2009. Novel Sinorhizobium meliloti quorum sensing positive and negative regulatory feedback mechanisms respond to phosphate availability. Mol. Microbiol. 74:1238–1256. 10.1111/j.1365-2958.2009.06930.x [DOI] [PubMed] [Google Scholar]
- 24.Charoenpanich P, Meyer S, Becker A, McIntosh M. 2013. Temporal expression program of quorum sensing-based transcription regulation in Sinorhizobium meliloti. J. Bacteriol. 195:3224–3236. 10.1128/JB.00234-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao M, Barnett MJ, Long SR, Teplitski M. 2010. Role of the Sinorhizobium meliloti global regulator Hfq in gene regulation and symbiosis. Mol. Plant Microbe Interact. 23:355–365. 10.1094/MPMI-23-4-0355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mackie GA. 2013. RNase E: at the interface of bacterial RNA processing and decay. Nat. Rev. Microbiol. 11:45–57. 10.1038/nrmicro2930 [DOI] [PubMed] [Google Scholar]
- 27.Carpousis AJ, Luisi BF, McDowall KJ. 2009. Endonucleolytic initiation of mRNA decay in Escherichia coli. Prog. Mol. Biol. Transl. Sci. 85:91–135. 10.1016/S0079-6603(08)00803-9 [DOI] [PubMed] [Google Scholar]
- 28.Zhang Y, Hong G. 2009. Post-transcriptional regulation of NifA expression by Hfq and RNase E complex in Rhizobium leguminosarum bv. viciae. Acta Biochim. Biophys. Sin. (Shanghai) 41:719–730. 10.1093/abbs/gmp060 [DOI] [PubMed] [Google Scholar]
- 29.Evguenieva-Hackenberg E, Klug G. 2009. RNA degradation in Archaea and Gram-negative bacteria different from Escherichia coli. Prog. Mol. Biol. Transl. Sci. 85:275–317. 10.1016/S0079-6603(08)00807-6 [DOI] [PubMed] [Google Scholar]
- 30.Evguenieva-Hackenberg E, Roppelt V, Lassek C, Klug G. 2011. Subcellular localization of RNA degrading proteins and protein complexes in prokaryotes. RNA Biol. 8:49–54. 10.4161/rna.8.1.14066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khemici V, Poljak L, Luisi BF, Carpousis AJ. 2008. The RNase E of Escherichia coli is a membrane-binding protein. Mol. Microbiol. 70:799–813. 10.1111/j.1365-2958.2008.06454.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Commichau FM, Rothe FM, Herzberg C, Wagner E, Hellwig D, Lehnik-Habrink M, Hammer E, Völker U, Stülke J. 2009. Novel activities of glycolytic enzymes in Bacillus subtilis: interactions with essential proteins involved in mRNA processing. Mol. Cell. Proteomics 8:1350–1360. 10.1074/mcp.M800546-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shahbabian K, Jamalli A, Zig L, Putzer H. 2009. RNase Y, a novel endoribonuclease, initiates riboswitch turnover in Bacillus subtilis. EMBO J. 28:3523–3533. 10.1038/emboj.2009.283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lehnik-Habrink M, Newman J, Rothe FM, Solovyova AS, Rodrigues C, Herzberg C, Commichau FM, Lewis RJ, Stülke J. 2011. RNase Y in Bacillus subtilis: a natively disordered protein that is the functional equivalent of RNase E from Escherichia coli. J. Bacteriol. 193:5431–5441. 10.1128/JB.05500-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jäger S, Fuhrmann Heck O, Hebermehl C, Schiltz M, Rauhut E, Klug R, G. 2001. An mRNA degrading complex in Rhodobacter capsulatus. Nucleic Acids Res. 29:4581–4588. 10.1093/nar/29.22.4581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hardwick SW, Chan VS, Broadhurst RW, Luisi BF. 2011. An RNA degradosome assembly in Caulobacter crescentus. Nucleic Acids Res. 39:1449–1459. 10.1093/nar/gkq928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goldblum K, Apririon D. 1981. Inactivation of the ribonucleic acid-processing enzyme ribonuclease E blocks cell division. J. Bacteriol. 146:128–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaberdin VR, Miczak A, Jakobsen JS, Lin-Chao S, McDowall KJ, von Gabain A. 1998. The endoribonucleolytic N-terminal half of Escherichia coli RNase E is evolutionarily conserved in Synechocystis sp. and other bacteria but not the C-terminal half, which is sufficient for degradosome assembly. Proc. Natl. Acad. Sci. U. S. A. 95:11637–11642. 10.1073/pnas.95.20.11637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kido M, Yamanaka K, Mitani T, Niki H, Ogura T, Hiraga S. 1996. RNase E polypeptides lacking a carboxyl-terminal half suppress a mukB mutation in Escherichia coli. J. Bacteriol. 178:3917–3925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tamura M, Moore CJ, Cohen SN. 2013. Nutrient dependence of RNase E essentiality in Escherichia coli. J. Bacteriol. 195:1133–1141. 10.1128/JB.01558-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee K, Cohen SN. 2003. A Streptomyces coelicolor functional orthologue of Escherichia coli RNase E shows shuffling of catalytic and PNPase-binding domains. Mol. Microbiol. 48:349–360. 10.1046/j.1365-2958.2003.03435.x [DOI] [PubMed] [Google Scholar]
- 42.Galibert F, Finan TM, Long SR, Puhler A, Abola P, Ampe F, Barloy-Hubler F, Barnett MJ, Becker A, Boistard P, Bothe G, Boutry M, Bowser L, Buhrmeseter J, Cadieu E, Capela D, Chain P, Cowie A, Davis RW, Dreano S, Federspiel NA, Fisher RF, Gloux S, Godrie T, Goffeau A, Golding B, Gouzy J, Gurjal M, Hernandez-Lucas I, Hong A, Huizar L, Hyman RW, Jones T, Kahn D, Kahn ML, Kalman S, Keating DH, Kiss E, Komp C, Lelaure V, Masuy D, Palm C, Peck MC, Pohl TM, Portetelle D, Purnelle B, Ramsperger U, Surzycki R, Thebault P, Vandenbol M, Vorholter FJ, Weidner S, Wells DH, Wong K, Yeh KC, Batut J. 2001. The composite genome of the legume symbiont Sinorhizobium meliloti. Science 293:668–672. 10.1126/science.1060966 [DOI] [PubMed] [Google Scholar]
- 43.Casse F, Boucher C, Julliot JS, Michel M, Denarie J. 1979. Identification and characterization of large plasmids in Rhizobium meliloti using agarose-gel electrophoresis. J. Gen. Microbiol. 113:229–242. 10.1099/00221287-113-2-229 [DOI] [Google Scholar]
- 44.Pobigaylo N, Wetter D, Szymczak S, Schiller U, Kurtz S, Meyer F, Nattkemper TW, Becker A. 2006. Construction of a large signature-tagged mini-Tn5 transposon library and its application to mutagenesis of Sinorhizobium meliloti. Appl. Environ. Microbiol. 72:4329–4337. 10.1128/AEM.03072-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beringer JE. 1974. R factor transfer in Rhizobium leguminosarum. J. Gen. Microbiol. 84:188–198. 10.1099/00221287-84-1-188 [DOI] [PubMed] [Google Scholar]
- 46.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 47.Simon R, Priefer U, Pühler A. 1982. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram-negative bacteria. Biotechnology 1:784–791 [Google Scholar]
- 48.Keen NT, Tamaki S, Kobayashi D, Trollinger D. 1988. Improved broad-host-range plasmids for DNA cloning in gram-negative bacteria. Gene 70:191–197. 10.1016/0378-1119(88)90117-5 [DOI] [PubMed] [Google Scholar]
- 49.Khan SR, Gaines J, Roop RM, II, Farrand SK. 2008. Broad-host-range expression vectors with tightly regulated promoters and their use to examine the influence of TraR and TraM expression on Ti plasmid quorum sensing. Appl. Environ. Microbiol. 74:5053–5062. 10.1128/AEM.01098-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McIntosh M, Krol E, Becker A. 2008. Competitive and cooperative effects in quorum-sensing-regulated galactoglucan biosynthesis in Sinorhizobium meliloti. J. Bacteriol. 190:5308–5317. 10.1128/JB.00063-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Masterson RV, Prakash RK, Atherly AG. 1985. Conservation of symbiotic nitrogen fixation gene sequences in Rhizobium japonicum and Bradyrhizobium japonicum. J. Bacteriol. 163:21–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Glaeser J, Klug G. 2005. Photo-oxidative stress in Rhodobacter sphaeroides: protective role of carotenoids and expression of selected genes. Microbiology 151:1927–1938. 10.1099/mic.0.27789-0 [DOI] [PubMed] [Google Scholar]
- 53.Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45. 10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Voss B, Hölscher M, Baumgarth B, Kalbfleisch A, Kaya C, Hess WR, Becker A, Evguenieva-Hackenberg E. 2009. Expression of small RNAs in Rhizobiales and protection of a small RNA and its degradation products by Hfq in Sinorhizobium meliloti. Biochem. Biophys. Res. Commun. 390:331–336. 10.1016/j.bbrc.2009.09.125 [DOI] [PubMed] [Google Scholar]
- 55.Nogales J, Domínguez-Ferreras A, Amaya-Gómez CV, van Dillewijn P, Cuéllar V, Sanjuán J, Olivares J, Soto MJ. 2010. Transcriptome profiling of a Sinorhizobium meliloti fadD mutant reveals the role of rhizobactin 1021 biosynthesis and regulation genes in the control of swarming. BMC Genomics 11:157. 10.1186/1471-2164-11-157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Andersen JB, Heydorn A, Hentzer M, Eberl L, Geisenberger O, Christensen BB, Molin S, Givskov M. 2001. gfp-based N-acyl homoserine-lactone sensor systems for detection of bacterial communication. Appl. Environ. Microbiol. 67:575–585. 10.1128/AEM.67.2.575-585.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shaw PD, Ping G, Daly SL, Cha C, Cronan JE, Jr, Rinehart KL, Farrand SK. 1997. Detecting and characterizing N-acyl-homoserine lactone signal molecules by thin-layer chromatography. Proc. Natl. Acad. Sci. U. S. A. 94:6036–6041. 10.1073/pnas.94.12.6036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cha C, Gao P, Chen YC, Shaw PD, Farrand SK. 1998. Production of acyl-homoserine lactone quorum-sensing signals by gram-negative plant-associated bacteria. Mol. Plant Microbe Interact. 11:1119–1129. 10.1094/MPMI.1998.11.11.1119 [DOI] [PubMed] [Google Scholar]
- 59.Nuss AM, Glaeser J, Klug G. 2009. RpoHII activates oxidative-stress defense systems and is controlled by RpoE in the singlet oxygen-dependent response in Rhodobacter sphaeroides. J. Bacteriol. 191:220–230. 10.1128/JB.00925-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Taverniti V, Forti F, Ghisotti D, Putzer H. 2011. Mycobacterium smegmatis RNase J is a 5′-3′ exo-/endoribonuclease and both RNase J and RNase E are involved in ribosomal RNA maturation. Mol. Microbiol. 82:1260–1276. 10.1111/j.1365-2958.2011.07888.x [DOI] [PubMed] [Google Scholar]
- 61.Schuck A, Diwa A, Belasco JG. 2009. RNase E autoregulates its synthesis in Escherichia coli by binding directly to a stem-loop in the rne 5′-untranslated region. Mol. Microbiol. 72:470–478. 10.1111/j.1365-2958.2009.06662.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bardill JP, Zhao X, Hammer BK. 2011. The Vibrio cholerae quorum sensing response is mediated by Hfq-dependent sRNA/mRNA base pairing interactions. Mol. Microbiol. 80:1381–1394. 10.1111/j.1365-2958.2011.07655.x [DOI] [PubMed] [Google Scholar]
- 63.Romeo T, Vakulskas CA, Babitzke P. 2013. Post-transcriptional regulation on a global scale: form and function of Csr/Rsm systems. Environ. Microbiol. 15:313–324. 10.1111/j.1462-2920.2012.02794.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Luo ZQ, Farrand SK. 2001. The Agrobacterium tumefaciens rnd homolog is required for TraR-mediated quorum-dependent activation of Ti plasmid tra gene expression. J. Bacteriol. 183:3919–3930. 10.1128/JB.183.13.3919-3930.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mank NN, Berghoff BA, Hermanns YN, Klug G. 2012. Regulation of bacterial photosynthesis genes by the small noncoding RNA PcrZ. Proc. Natl. Acad. Sci. U. S. A. 109:16306–16311. 10.1073/pnas.1207067109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Belasco JG. 2010. All things must pass: contrasts and commonalities in eukaryotic and bacterial mRNA decay. Nat. Rev. Mol. Cell Biol. 11:467–478. 10.1038/nrm2917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mathy N, Bénard L, Pellegrini O, Daou R, Wen T, Condon C. 2007. 5′-to-3′ exoribonuclease activity in bacteria: role of RNase J1 in rRNA maturation and 5′ stability of mRNA. Cell 129:681–692. 10.1016/j.cell.2007.02.051 [DOI] [PubMed] [Google Scholar]
- 68.Madhugiri R, Evguenieva-Hackenberg E. 2009. RNase J. is involved in the 5′ end maturation of 16S rRNA and 23S rRNA in Sinorhizobium meliloti. FEBS Lett. 583:2339–2342. 10.1016/j.febslet.2009.06.026 [DOI] [PubMed] [Google Scholar]
- 69.Arraiano CM, Cruz AA, Kushner SR. 1997. Analysis of the in vivo decay of the Escherichia coli dicistronic pyrF-orfF transcript: evidence for multiple degradation pathways. J. Mol. Biol. 268:261–272. 10.1006/jmbi.1997.0962 [DOI] [PubMed] [Google Scholar]
- 70.Carzaniga T, Briani F, Zangrossi S, Merlino G, Marchi P, Dehò G. 2009. Autogenous regulation of Escherichia coli polynucleotide phosphorylase expression revisited. J. Bacteriol. 191:1738–1748. 10.1128/JB.01524-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rische T, Klug G. 2012. The ordered processing of intervening sequences in 23S rRNA of Rhodobacter sphaeroides requires RNase J. RNA Biol. 9:343–350. 10.4161/rna.19433 [DOI] [PubMed] [Google Scholar]
- 72.McIntosh M, Czuppon P, Best K, Becker A, Pfaffelhuber P. 2013. Modeling quorum sensing in Sinorhizobium meliloti. Int. J. Biomath. Biostat. 2:59–74 [Google Scholar]
- 73.Evguenieva-Hackenberg E, Klug G. 2011. New aspects of RNA processing in prokaryotes. Curr. Opin. Microbiol. 14:587–592. 10.1016/j.mib.2011.07.025 [DOI] [PubMed] [Google Scholar]
- 74.Mathews DH, Sabina J, Zuker JM, Turner DH. 1999. Expanded sequence dependence of thermodynamic parameters provides robust prediction of RNA secondary structure. J. Mol. Biol. 288:911–940. 10.1006/jmbi.1999.2700 [DOI] [PubMed] [Google Scholar]
- 75.Yanisch-Perron C, Vieira J, Messing J. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33:103–119. 10.1016/0378-1119(85)90120-9 [DOI] [PubMed] [Google Scholar]
- 76.Grant SG, Jessee J, Bloom FR, Hanahan D. 1990. Differential plasmid rescue from transgenic mouse DNAs into Escherichia coli methylation-restriction mutants. Proc. Natl. Acad. Sci. U. S. A. 87:4645–4649. 10.1073/pnas.87.12.4645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bahlawane C, McIntosh M, Krol E, Becker A. 2008. Sinorhizobium meliloti regulator MucR couples exopolysaccharide synthesis and motility. Mol. Plant Microbe Interact. 21:1498–1509. 10.1094/MPMI-21-11-1498 [DOI] [PubMed] [Google Scholar]
- 78.Tauch A, Zheng Z, Pühler A, Kalinowski J. 1998. Corynebacterium striatum chloramphenicol resistance transposon Tn5564: genetic organization and transposition in Corynebacterium glutamicum. Plasmid 40:126–139. 10.1006/plas.1998.1362 [DOI] [PubMed] [Google Scholar]
- 79.Hübner P, Willison JC, Vignais PM, Bickle TA. 1991. Expression of regulatory nif genes in Rhodobacter capsulatus. J. Bacteriol. 173:2993–2999 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.