

Abstract
Conditional proteolysis is a crucial process regulating the abundance of key regulatory proteins associated with the cell cycle, differentiation pathways, or cellular response to abiotic stress in eukaryotic and prokaryotic organisms. We provide evidence that conditional proteolysis is involved in the rapid and dramatic reduction in abundance of the cyanobacterial RNA helicase, CrhR, in response to a temperature upshift from 20 to 30°C. The proteolytic activity is not a general protein degradation response, since proteolysis is only present and/or functional in cells grown at 30°C and is only transiently active at 30°C. Degradation is also autoregulatory, since the CrhR proteolytic target is required for activation of the degradation machinery. This suggests that an autoregulatory feedback loop exists in which the target of the proteolytic machinery, CrhR, is required for activation of the system. Inhibition of translation revealed that only elongation is required for induction of the temperature-regulated proteolysis, suggesting that translation of an activating factor was already initiated at 20°C. The results indicate that Synechocystis responds to a temperature shift via two independent pathways: a CrhR-independent sensing and signal transduction pathway that regulates induction of crhR expression at low temperature and a CrhR-dependent conditional proteolytic pathway at elevated temperature. The data link the potential for CrhR RNA helicase alteration of RNA secondary structure with the autoregulatory induction of conditional proteolysis in the response of Synechocystis to temperature upshift.
INTRODUCTION
The importance of conditional proteolysis in the regulation of numerous crucial cellular processes is well documented in eukaryotic systems (reviewed in reference 1). Although initially underestimated with respect to both the scope and importance in bacterial processes, the role of conditional proteolysis as a regulator of cellular physiology is becoming apparent (reviewed in reference 2–8). Proteolysis is associated with either the removal of misfolded proteins generated by heat shock or regulation of gene expression as a complement to transcriptional control (3). Although the number of prokaryotic proteins regulated by proteolysis is not extensive, they tend to be either key regulators of the cell cycle, differentiation pathways, or cellular response to abiotic stress (5). Conditional proteolysis provides directionality to physiological pathways and thus a mechanism to rapidly generate the dynamic range of proteins required for proper cellular function by maintaining protein homeostasis and balanced cell growth. Prokaryotic proteolytic regulatory pathways have been most extensively studied in Escherichia coli and the regulation of the cell cycle and developmental regulation in Caulobacter crescentus and Bacillus subtilis (3). Frequently, important aspects of the degradation system are difficult to identify, including the specific induction mechanism by which proteolysis is activated and the identity of the proteases involved (3, 4). Thus, few protein targets of specific proteases have been characterized and frequently multiple proteases are involved in the degradation (3, 4).
Conditional proteolysis provides numerous advantages, including the ability to promptly respond to changing conditions by rapidly turning a regulatory system on and subsequently off. Frequently, proteolytic targets are also subject to positive autoregulation, the two processes combining to achieve a rapid fluctuation in target levels. The resulting feedback loop allows cells to respond to changes more rapidly than achieved via a sensor-signal transduction-transcription pathway since bacterial proteins generally have long half-lives. In addition, feedback loops are frequently observed in which the proteolytic target enhances expression of a component of the proteolytic machinery (9).
Conditional proteolytic pathways are frequently associated with a rapid response to temperature stress, especially heat stress in all organisms (3, 4). In E. coli, examples include regulation of the major heat shock response regulator RpoS (sigma 38) (10, 11), the cold-induced RNA chaperone CspC (12–14), and the exoribonuclease RNase R (15, 16). Expression of these proteins exhibits common features. Basal expression occurring at a normal growth temperature is induced by heat or cold temperature stress. The response is transient, since the system shuts down upon acclimation or relief of the stress, returning to basal levels through proteolysis of the induced protein. The proteolytic target proteins are frequently molecular chaperones, associated with protein folding or RNA metabolism (6).
Proteolytic regulation of protein abundance is also observed in other bacteria, including the Gram-negative photosynthetic cyanobacteria. The genome of the cyanobacterium Synechocystis sp. strain PCC 6803 encodes 62 peptidases (17). Proteolysis in cyanobacteria has been associated with the degradation of a few specific proteins (18–24) or with stress response pathways (25–27).
Similar to other prokaryotes, cyanobacteria possess a limited number of genes whose expression is regulated by low temperature stress (28–33). However, the mechanism by which low temperature regulates expression of these genes is not known since neither a cold-specific two-component signal transduction pathway nor a low-temperature-induced sigma factor has been identified in prokaryotes (32, 33). crhR encodes a DEAD box RNA helicase whose expression in Synechocystis is regulated by conditions that alter the redox status of the electron transport chain, including temperature downshift (34–36). RNA helicases regulate gene expression via alteration of RNA secondary structure and are frequently induced by and associated with cellular response to abiotic stress (37). Low-temperature induction of CrhR involves a complex series of mechanisms operating at the posttranscriptional level, including autoregulation at a minimum of three steps (36). At 30°C, the basal levels of both crhR transcript and protein are maintained by a combination of short half-lives for both molecules. crhR transcript and protein half-lives are robustly temperature regulated, increasing dramatically in response to temperature downshift from 30 to 20°C (36). CrhR therefore belongs to the group of RNA helicases that are associated with cellular response to low temperature stress (37).
We investigated here the mechanism regulating the dramatic changes in CrhR protein abundance occurring in response to temperature shift. We utilize a combination of in vitro, in vivo, and inhibitor analyses to provide evidence that conditional proteolysis is involved in the rapid reduction in CrhR protein accumulation in response to a temperature upshift from 20 to 30°C.
MATERIALS AND METHODS
Strains and growth conditions.
The bacterial strains, plasmids, and primers used in the present study are listed in Table 1. Synechocystis sp. strain PCC 6803 was maintained on BG-11 agar supplemented with 10 mM Tricine (pH 8.0) and 0.3% sodium thiosulfate (35). Two crhR mutant strains were utilized in these studies, a partial deletion crhR mutant (crhRTR) created by insertion of a spectinomycin cassette (crhR::spec) (35) and a crhR complete deletion mutant (ΔcrhR), as described below. Corresponding antibiotics were included as required: either spectinomycin and streptomycin (50 μg/ml of each) for crhRTR or kanamycin (50 μg/ml) for the complete deletion mutant (ΔcrhR). Liquid cultures were grown at 30°C with continuous shaking (150 rpm) and bubbling with humidified air at an illumination of 50 μmol of photons m−2 s−1 (38). Cold stress was induced in mid-log-phase cells by incubation at 20°C for the indicated times.
TABLE 1.
Bacterial strains, plasmids, and primers used in this study
| Strain, plasmid, or primer | Characteristics or sequence (5′–3′) | Source or reference |
|---|---|---|
| Strains | ||
| Escherichia coli | ||
| DH5α | Plasmid propagation and cloning | 54 |
| BL21(DE3)/pLysS | CrhR protein expression strain | 55 |
| BL21(pRSET-R) | His6-CrhR expression in BL21(DE3)/pLysS | 41 |
| Synechocystis sp. | ||
| PCC 6803 | Wild type | Lab strain |
| pMon-HisR | Synechocystis expressing His6-CrhR from pMon-HisR | This study |
| crhRTR | Partial deletion of crhR ORF | 35 |
| ΔcrhR | Complete deletion of crhR ORF | This study |
| Plasmids | ||
| pBSL128 | Source of kanamycin resistance cassette | 39 |
| pBS II | Cloning vector | 56 |
| pBS-ΔcrhR | pBS construct for insertional inactivation of the crhR ORF | This study |
| pRL623 | Carries three methyltransferases from cyanobacterial RM systems | 40 |
| pRSETA | Protein expression vector | Invitrogen |
| pRSET-R | Expression vector for His6-CrhR production | 41 |
| pMon 36456 | Synechocystis-E. coli shuttle plasmid | 42 |
| pMon-HisR | His6-tagged CrhR cloned in pMon 36456 | This study |
| Primers | ||
| OT1F | ACTCAGTCTAGAGTCAGTTAATCC | Forward primer, 1.5 kbp upstream of crhR |
| OT1R | ATGGAAAAGCTTAATAATTTCGG | Reverse primer, 1 bp upstream of crhR ORF |
| OT2F | AAGGTAAAGCTTGCAAACCCCGTC | Forward primer, 1.5 kbp downstream of crhR ORF |
| OT2R | GACGGACTCGAGATTGCCCATATC | Reverse primer, 1 bp downstream of crhR ORF |
| DCSR11F | TAATACGACTCACTATAGGAACCGCCGCAGACCTCAAAC | Forward primer, 818 bp into the crhR ORF |
| DC29 | CGCGGTCTAGATAATACGACTCACTATAGGGTTACTGTTGGCGATCAC | Reverse primer, 3′ end of crhR |
| LPF45 | TGTTATTGACGGTTGGTTCC | Forward primer, 51 bp upstream of crhR ORF |
| GWO74 | TTGGGAAGAATCCTTAGG | Reverse primer, 198 bp downstream of crhR ORF |
ΔcrhR mutant construction.
The 1.5-kb regions upstream and downstream of the ATG start codon (position 2887644 in the Synechocystis genome) and the TAA stop codon (position 2889122) of the crhR open reading frame (ORF) were PCR amplified from genomic DNA using the primer pairs OT1F/OT1R (52°C) and OT2F/OT2R (62°C) (Table 1) containing the indicated restriction enzyme sites. QIAquick (Qiagen, Gaithersburg, MD) purified PCR fragments were ligated with a 2.3-kb HindIII fragment carrying the kanamycin cassette isolated from pBSL128 (39) and XbaI/XhoI-cleaved pBluescript, creating pBS-ΔcrhR (see Fig. S1A in the supplemental material). pBS-ΔcrhR was confirmed by sequencing and transformed into E. coli DH5α containing the pRL623 helper plasmid (40). pBS-ΔcrhR, purified from this strain, was transformed into Synechocystis (38) and inserted into the genome by a double-crossover event as a result of homologous recombination. Complete deletion mutants were selected by screening for resistance to kanamycin (150 μg/ml) and sensitivity to cold temperature (20°C). Complete segregation was verified by PCR using two primer pairs: GWO74 and LPF45 (55°C) and DCSR11F and DC29 (60°C) (Table 1) (see Fig. S1B and C in the supplemental material) and Western analysis indicating that CrhR protein was undetectable (Fig. 1 ΔcrhR). The genomic organization in the crhR region of the Synechocystis strains is also shown in the supplemental material (Fig. S1D).
FIG 1.

CrhR protein abundance in Synechocystis. (A) The effect of temperature on CrhR accumulation in the indicated strains was tested in cultures either grown at 30°C or induced for CrhR expression by cold shock at 20°C for 2 h. (B) Effect of temperature on CrhR accumulation in strains containing pMon-HisR expressing His-CrhR from the constitutively transcribed PNIR promoter. Soluble protein (20 μg) isolated from the indicated strains and growth temperature was probed with anti-CrhR (1:5,000) and anti-E. coli Rps1 (1:5,000) antibodies and detected by Western analysis using ECL. The polypeptides detected were the 55-kDa wild-type CrhR, a truncated 27-kDa protein in the crhRTR mutant, and His-CrhR at 58 kDa. Rps1 (40 kDa) served as a loading control.
Construction of His-CrhR expressing Synechocystis strains.
An E. coli His-CrhR expression plasmid was constructed by cloning the crhR ORF into pRSET-A (Invitrogen) in frame with a polyhistidine (His6) region using BamHI and EcoRI, resulting in pRSET-R, as described by Chamot et al. (41). The His-CrhR coding sequence was isolated from pRSET-R with XbaI and EcoRI and ligated into pMon 36456 (42) using the same restriction enzyme sites. The final construct, pMon-HisR, was verified by sequencing. pMon-HisR was transformed into E. coli DH5α containing the pRL623 helper plasmid (40) and subsequently transferred into Synechocystis wild-type, ΔcrhR, and crhRTR cells by triparental mating (38; Wolfgang Hess, unpublished data). CrhR transcription in these strains will be constitutive, since activity of the PNIR promoter present in pMon 36456 is induced in response to the nitrate present in BG-11 media.
Protein manipulation.
Treated Synechocystis cells were harvested and lysed at the stated growth temperature. Soluble protein was extracted from Synechocystis cells by vortexing in the presence of glass beads as described previously (36, 38). Aliquots of the clarified soluble fraction (25 μg) were separated by SDS–10% PAGE and immunoblot detection of CrhR performed using the indicated antisera and secondary antibody (anti-rabbit IgG at 1:20,000 dilution; Sigma-Aldrich) combinations detected by enhanced chemiluminescence (ECL; Amersham). Where indicated, polyclonal antibodies against the Synechocystis CrhR and E. coli Rps1 were utilized simultaneously (1:5,000) for the parallel detection of both polypeptides. Protein loading was normalized using Bradford reagent (Bio-Rad) and the detection of ribosomal protein Rps1 (36). Representative data are shown from a minimum of two biological replicates.
CrhR degradation in vitro.
Wild-type or crhRTR cultures were grown at 30°C to mid-log phase, and CrhR protein accumulation was induced by transfer to 20°C for 2 h. One-half of the culture was transferred back to 30°C for 1 h. Cells were harvested by centrifugation at the growth temperature, either 20 or 30°C, and proteins were isolated. Aliquots from both growth temperatures were mixed and incubated at 30°C for the indicated times. Alternatively, protein lysates from a single growth temperature were incubated at 30°C as a control. Aliquots (20 μg of total protein) of the mixtures were removed at the indicated times and immediately quenched by the addition of one-third volume of 3× SDS-PAGE loading dye (125 mM Tris [pH 6.8], 4% [wt/vol] SDS, 20% [vol/vol] glycerol, 10% [vol/vol] β-mercaptoethanol, and 0.02% bromophenol blue). The samples were separated by SDS–10% PAGE, and CrhR was detected by Western blotting. His-CrhR, affinity purified from E. coli BL21(DE3)/pLysS as described previously (41), was included when indicated. The amount of purified His-CrhR exogenously added to each experiment was empirically determined by Western analysis to approximate the level of native CrhR present in each experiment.
CrhR abundance in vivo.
Wild-type or crhRTR cultures were grown at 30°C to mid-log phase, and the culture transferred to 20°C for 24 h and then returned to 30°C for a further 24 h. Aliquots were taken at the indicated times, cells were collected by centrifugation at the growth temperature, and pellets were stored at −86°C. When indicated, quantification of the CrhR protein levels was performed using ImageJ analysis (43), with E. coli Rps1 abundance as a loading control, as previously described (36).
In vivo degradation in the presence of translation inhibitors.
Synechocystis wild-type cells were grown to mid-log phase at 30°C and transferred to 20°C for 2 h to induce CrhR protein accumulation in the absence of translation inhibition. The induced culture was divided into four independent cultures, to which chloramphenicol (250 μg/ml), kanamycin (200 μg/ml), or nothing (control) was added. Incubation was continued at 20°C for a further 1 h to ensure that the antibiotics were inhibiting translation. The cultures were then transferred back to 30°C for 4 h. Aliquots for protein isolation were harvested at the indicated times before and after either cold induction or antibiotic treatment, and the cell pellets were stored at −86°C.
In vitro degradation in the presence of protease inhibitor cocktail.
Wild-type and crhRTR cultures were grown to mid-log phase at 30°C and transferred to 20°C for 2 h to induce CrhR protein accumulation. One-half of the culture was transferred back to 30°C for 1 h to induce the CrhR degradation machinery. Soluble protein was extracted from each culture in the absence of protein inhibitor cocktail, and one-half of the 30°C extract was subsequently treated with bacterial protease inhibitor cocktail (Sigma-Aldrich) at 30°C for 30 min, as recommended by the manufacturer. Soluble protein isolated from cells grown at 20°C was mixed with an equal amount of protein from cells grown at 30°C that were either not treated (−) or treated with the protease inhibitor cocktail (+). Aliquots corresponding to 25 μg of protein were sampled at the indicated times, and CrhR was detected by Western analysis.
RESULTS
Initially, the temperature regulation of CrhR expression was examined in the three Synechocystis strains used in the present study—i.e., the wild-type, crhRTR, and ΔcrhR strains—and in the three strains transformed with pMon-HisR, expressing His6-CrhR from the constitutively transcribed PNIR promoter (Fig. 1). The partial deletion crhR mutant (crhRTR) was created by insertion of a spectinomycin cassette into the unique PmlI site (i.e., 2888319), 38 amino acids downstream of motif III, encoding the SAT box, as described previously (35). Expression of the remaining 673 bp of the crhR ORF is predicted to produce a polypeptide of 25,295 Da. This polypeptide is biochemical inactive in vitro, being unable to catalyze either RNA unwinding or annealing (D. Chamot and G. W. Owttrim, unpublished data), activities previously shown to be catalyzed by the native CrhR protein (41). As we have shown previously (36), CrhR levels in wild-type cells increase ∼10-fold in response to a temperature downshift, whereas the abundance of the CrhRTR polypeptide obtained from the truncated mutant, crhRTR, remains relatively constant at this maximal level irrespective of temperature (Fig. 1, WT and crhRTR). CrhR protein was not detected in the complete crhR deletion strain, ΔcrhR, verifying complete inactivation of the crhR gene (Fig. 1, ΔcrhR). In contrast, extrachromosomal expression of His-CrhR from the PNIR promoter, constitutively active in nitrate-containing BG-11, resulted in a divergent expression pattern (Fig. 1). Maximal His-CrhR accumulation, equivalent to the level observed in wild-type cells at 20°C, was only observed in the ΔcrhR (pMon-HisR) strain grown for prolonged periods at both 30 and 20°C (Fig. 1 ΔcrhR+His-CrhR). His-CrhR expression was reduced in both wild-type (pMon-HisR) and crhRTR (pMon-HisR) cells compared to that in ΔcrhR (pMon-HisR) cells. In crhRTR (pMon-HisR) cells, His-CrhR only accumulated at 20°C and, in conjunction, the level of the truncated CrhRTR polypeptide was also reduced at 30°C. The observations suggest that CrhR RNA helicase activity is required for the differential temperature regulation of CrhR accumulation, reducing CrhR levels at the elevated temperature. Regulation of CrhR degradation in response to temperature upshift was further investigated using in vivo and in vitro assays.
To initiate determination of the mechanism generating the differential expression of CrhR, we first determined CrhR protein accumulation in response to temperature shift over extended periods in wild-type and crhRTR cells. Consistent with our previous observations (36), wild-type CrhR protein accumulated from a basal level at 30°C to a maximum level within 3 h in response to a temperature downshift to 20°C (Fig. 2A). CrhR abundance remained enhanced and constant during prolonged incubation at 20°C, up to 24 h. A subsequent temperature upshift, returning the culture to 30°C, resulted in a rapid decrease in CrhR to basal levels within 3 h. As expected, CrhRTR levels were elevated above wild-type basal levels at 30°C and increased less significantly in response to the initial temperature downshift (Fig. 2B). In contrast to wild-type cells, CrhRTR levels did not decrease in response to the subsequent temperature upshift to 30°C, with levels decreasing slightly after prolonged incubation at 30°C for 24 h. CrhRTR abundance eventually decreases (compare Fig. 2B, 0 h versus 24 h 30°C); however, the response takes an extended time compared to the rate observed in wild-type cells. The kinetics of CrhR decrease in wild-type cells in response to the temperature upshift to 30°C was further investigated over a short time course. CrhR abundance decreases rapidly in response to the temperature upshift (Fig. 2C). Quantification of CrhR levels, shown in Fig. 2C, indicates that CrhR degrades in a linear fashion and that degradation initiates within 10 min upon temperature upshift, with 50% of the protein present at 20°C being lost within 75 min (Fig. 2D). The levels of Rps1, used as a loading control, remain relatively constant during these experiments, suggesting that the observed decrease in CrhR levels is not a global phenomenon. The results imply that proteolysis is a major contributor to the basal levels of CrhR protein observed at 30°C and that the rapid decrease in CrhR abundance is a CrhR-dependent phenomenon.
FIG 2.

Time course of temperature-dependent CrhR accumulation. (A) Extended time course of CrhR abundance in wild-type (WT) and (B) crhRTR cells grown at 30°C to mid-log phase (zero time at 20°C), transferred to 20°C for 24 h, and then transferred back to 30°C for 24 h. (C) Short time course. Wild-type cells were grown at 30°C to mid-log phase (zero time at 20°C), and the culture was transferred to 20°C. Aliquots were harvested for protein extraction at the indicated times. CrhR abundance in a wild-type culture cold shocked at 20°C for 180 min is included as a control. Wild-type CrhR protein (55 kDa) or CrhR-TR protein (27 kDa) and the loading control Rps1 protein (40 kDa) were identified by Western analysis as described in Fig. 1. (D) Quantification of the CrhR levels utilizing Rps1 levels as loading control was performed using ImageJ. The relative abundance compared to that observed at 30°C is shown.
Investigation of the mechanism reducing CrhR protein levels at 30°C in the three genetic backgrounds was analyzed in vitro, as depicted in Fig. 3A. Mixing cell lysates from wild-type cells grown at 20°C, a source of detectable CrhR, and 30°C, a potential source of active degradation machinery, resulted in a decrease in native CrhR levels over time (Fig. 3B). In contrast, a similar decrease was not observed when wild-type extracts obtained from cells grown at 20°C were incubated in the absence of 30°C extract (Fig. 3C), suggesting that the active degradation machinery is present only in protein lysates isolated from wild-type cells grown at 30°C.
FIG 3.

In vitro CrhR degradation using wild-type or crhRTR extracts. (A) Schematic of an in vitro mixing experiment in which cell lysate from Synechocystis cells grown at 30°C serves as a potential source of active degradation machinery, and lysate from 20°C serves as a source of CrhR protein detectable by Western analysis. (B) Wild-type extracts. A 1:1 mixture of soluble protein, extracted from wild-type (WT) cells grown at 30 and 20°C, was incubated at 30°C, and aliquots were taken at the indicated times. (C) The in vitro degradation experiment described in panel B was repeated using only soluble protein isolated from wild-type cells grown at 20°C. (D) crhR mutant extracts. A 1:1 mixture of soluble protein, extracted from the partial crhR mutant (crhRTR) grown at 30 and 20°C, was incubated at 30°C, and aliquots were taken at the indicated times. (E) The in vitro degradation experiment described in panel D was repeated using only soluble protein isolated from crhRTR cells grown at 20°C. Wild-type CrhR (55-kDa) or crhRTR (27-kDa) protein was identified by Western analysis.
The results presented in Fig. 1 and 2 indicate that the CrhRTR protein is not degraded in response to a temperature upshift from 20 to 30°C in crhRTR cells. In vitro mixing experiments confirmed this result using protein extracts from crhRTR cells. In contrast to the wild-type results, the 27-kDa CrhRTR protein was not degraded over the course of the experiment in the presence (Fig. 3D) or absence (Fig. 3E) of extract from cells grown at 30°C.
The lack of CrhRTR degradation could result from either the absence of a degradation signal in the truncated CrhRTR protein or the lack of functional degradation machinery in the crhRTR mutant. These possibilities were investigated by repeating the in vitro mixing experiments utilizing protein extracts isolated from wild-type and crhRTR cells. The level of CrhRTR decreased progressively in the presence of extract obtained from wild-type cells grown at 30°C (Fig. 4A). In contrast, wild-type CrhR was not degraded by extracts obtained from crhRTR cells grown at 30°C (Fig. 4B). These results suggest that a degradation signal is not missing in the CrhRTR peptide and that functional degradation machinery is not present in the absence of CrhR activity. Thus, it appears that functional CrhR degradation machinery is only observed in wild-type cells grown at 30°C and is absent from crhRTR cells grown at either 30 or 20°C.
FIG 4.

In vitro CrhR degradation using mixtures of wild-type and crhRTR extracts. The effect of mixing protein extracts from wild-type and crhRTR cells grown at 20 and 30°C was tested. (A) A mixture of soluble protein, extracted from wild-type (WT) cells grown at 30°C (source of active degradation machinery) and crhRTR grown at 20°C (source of the potential 27-kDa CrhRTR protein substrate), was incubated at 30°C, and CrhRTR (27 kDa) levels were detected by Western analysis in aliquots taken at the indicated times. (B) The reverse of the experiment described in panel A was performed using cell lysates from crhRTR grown at 30°C (source of the potential 27-kDa CrhRTR protein substrate) and wild-type cells grown at 20°C (source of detectable native CrhR). CrhR (55 kDa) levels were detected by Western analysis in aliquots taken at the indicated times.
These results were confirmed and extended by determining if exogenously added His-CrhR, affinity purified from E. coli, is degraded in vitro in the presence of protein extracts obtained from wild-type and crhRTR cells grown at both temperatures (see Fig. S2 in the supplemental material). Exogenously added His-CrhR only decreased in the presence of protein lysate obtained from wild-type cells grown at 30°C (see Fig. S2A in the supplemental material). Exogenously added His-CrhR, and also wild-type CrhR, was not degraded in the presence of either wild-type protein lysate obtained from wild-type cells grown at 20°C (see Fig. S2B in the supplemental material) or protein lysate obtained from crhRTR cells grown at either 30 or 20°C (see Fig. S2C and D in the supplemental material, respectively).
It was of interest to extend these observations to the in vivo situation. His-CrhR was therefore expressed from the nitrate-inducible promoter (Pnir) in the three Synechocystis strains transformed with pMon-HisR. In these strains, Pnir transcriptional activity, and thus His-CrhR expression, should be constitutive in response to the nitrate present in the BG-11 growth media used for the experiments. In wild-type cells expressing His-CrhR, native CrhR increased in response to a temperature downshift to 20°C and then rapidly decreased upon transfer back to 30°C, remaining at basal levels for the duration of incubation at 30°C (Fig. 5, WT [pMon-HisR]), as also shown above for wild-type cells (Fig. 2, WT). His-CrhR levels exhibit a significantly different pattern in wild-type cells. As expected from the constitutive transcriptional activity of the PNIR promoter, a maximum level of His-CrhR accumulation was observed after prolonged growth at 30°C (zero time at 20°C). This level was not altered by a subsequent temperature downshift to 20°C, similar to that observed in ΔcrhR (pMon-HisR) cells in Fig. 1, indicating that the PNIR promoter is not responsive to temperature fluctuation. Transfer of WT (pMon-HisR) to 30°C resulted in a rapid decrease in native CrhR abundance, as expected; however, unexpectedly, His-CrhR abundance also decreased with kinetics similar to that observed for the native CrhR. In distinct contrast to native CrhR levels, the constitutively transcribed His-CrhR then progressively increased in abundance starting ∼2.5 h after transfer to 30°C, quickly reaching the maximum level (Fig. 5, WT [pMon-HisR]). These results are indicative of the CrhR degradation pathway being transiently active for only 2 to 3 h after temperature upshift.
FIG 5.
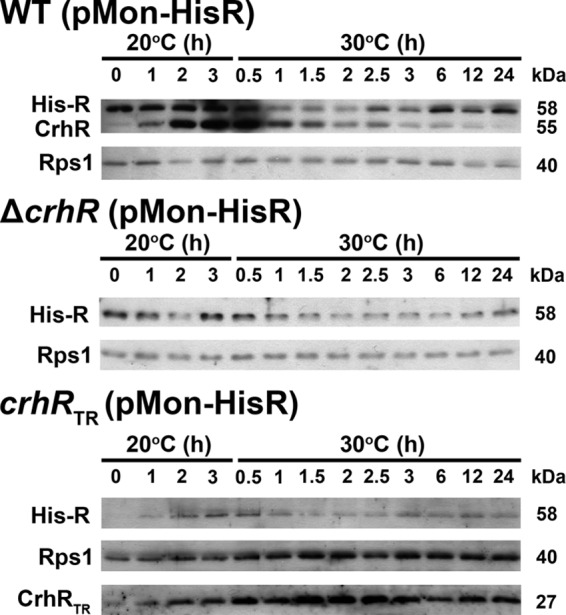
CrhR abundance in Synechocystis strains expressing His-CrhR in vivo. The abundance and effect of His-CrhR, expressed from the constitutive promoter PNIR, were examined. Cells corresponding to the wild type (WT [pMon-HisR]), the complete crhR deletion mutant (ΔcrhR [pMon-HisR]), and the partial crhRTR mutant (crhRTR [pMon-HisR]) transformed with pMon-HisR were grown at 30°C to mid-log phase (zero time at 20°C). Cultures were then transferred to 20°C for 3 h to induce maximal native CrhR protein (55 kDa) accumulation and then transferred back to 30°C for 24 h. Aliquots were taken at the indicated times for isolation of soluble protein. His-tagged CrhR (58 kDa), wild-type CrhR (55 kDa), truncated CrhRTR (27 kDa), and Rps1 (40 kDa) were identified by Western analysis.
Patterns of His-CrhR abundance similar to those observed in wild-type cells were also detected in ΔcrhR (pMon-HisR; Fig. 5, crhR [pMon-HisR]) and crhRTR (pMon-HisR; Fig. 5, crhRTR [pMon-HisR]) cells. The return to maximal levels at 30°C was, however, delayed in comparison with native CrhR, possibly a result of the reduced levels of His-CrhR accumulation compared to native CrhR at 20°C (36). This delay was also observed for the return of CrhRTR to basal levels in the crhRTR (pMon-HisR) strain (Fig. 5, crhRTR [pMon-HisR]). This indicates that His-CrhR is partially complementing the CrhR degradation defect in both crhR mutant strains.
As an initial investigation into the degradation system itself, we sought to determine whether de novo protein synthesis was required for the appearance of CrhR degradation activity in response to a temperature upshift. Antibiotics known to affect different steps in translation were utilized: kanamycin (Km) binding to the 30S ribosomal subunit inhibits translation initiation, while chloramphenicol (Cm) binding to the 50S subunit blocks translation elongation. In the absence of the translation inhibitors, the expected initial increase in CrhR abundance was observed in response to a temperature downshift from 30 to 20°C (Fig. 6, 20°C at 0, 1, and 2 h). Subsequent to the temperature upshift to 30°C, inclusion of the antibiotics produced divergent effects. Although CrhR levels decreased in kanamycin-treated cells (Fig. 6, +Km), similar to untreated cells (Fig. 6, −Km and −Cm), CrhR remained elevated in cells treated with chloramphenicol (Fig. 6, +Cm). The results suggest that translation elongation but not initiation is required for CrhR degradation in response to a temperature upshift.
FIG 6.

Effect of translation inhibitors on in vivo degradation. A single wild-type Synechocystis culture was grown at 30°C to mid-log phase. The induced culture was transferred to 20°C for 2 h to induce CrhR accumulation. The induced culture was divided into four equal aliquots, and either chloramphenicol (+Cm, 250 μg/ml), kanamycin (+Km, 200 μg/ml), or nothing (−Cm and −Km, the two no-antibiotic controls) was added to the individual cultures. Incubation was continued for a further 1 h at 20°C to allow the antibiotic inhibition of translation to take effect. Aliquots were harvested for protein extraction after 0 h (no low temperature induction), 1 and 2 h in the absence of inhibitors, and after 3 h, i.e., after 1 h in the presence of the inhibitors, of cold induction at 20°C. All four cultures were subsequently transferred to 30°C, and aliquots were harvested at 0.5, 1, 2, 3, and 4 h for protein extraction. Proteins were extracted from the aliquots taken at the indicated times, and CrhR (55 kDa) was identified by Western analysis.
The effect of a bacterial protease cocktail on the in vitro degradation of CrhR was determined to initiate identification of the protease associated with the proteolytic pathway. Wild-type extracts from cells grown at 30°C were incubated in the presence or absence of a bacterial protease inhibitor cocktail and subsequently mixed with extracts obtained from cells grown at 20°C. As shown in Fig. 7, CrhR abundance in the mixtures decreased in the absence of the inhibitor cocktail (Fig. 7, −), while a similar decrease was not observed in the presence of the cocktail (Fig. 7, +). This indicates that a component of the cocktail inhibits the CrhR degradation machinery.
FIG 7.

Effect of bacterial protease inhibitors on in vitro degradation. Wild-type cells were grown at 30°C to mid-log phase and transferred to 20°C for 2 h to induce CrhR protein accumulation. Subsequently, one-half of the culture transferred back to 30°C for 1 h to induce the CrhR degradation machinery. Soluble protein was extracted from each culture, and one-half of the 30°C extract was treated with bacterial protease inhibitor cocktail (Sigma-Aldrich) at 30°C for 30 min, as recommended by the manufacturer. Protein aliquots (25 μg) obtained from cells grown at 30°C, with (+) or without (−) inhibitor cocktail, were mixed with equivalent protein aliquots from the 20°C extracts. The mixtures were incubated at 30°C for 4 h, and aliquots were removed at the indicated times for CrhR (55 kDa) detection by Western analysis.
DISCUSSION
We provide evidence for a conditional proteolytic mechanism that regulates the posttranslational accumulation of the RNA helicase, CrhR, in the cyanobacterium Synechocystis sp. strain PCC 6803. The proteolytic mechanism is intimately linked to the differential temperature-dependent expression of CrhR (36). We demonstrate that the elevated abundance of CrhR observed at 20°C is rapidly reduced to basal levels in response to temperature upshift from 20 to 30°C by degradation machinery that is only active at 30°C. The proteolytic activity is also transiently active, being functional for only 2 to 3 h after the temperature upshift, and is inactivated with similar kinetics to CrhR degradation, implying that a component of the proteolytic machinery is a target for degradation. Functional CrhR RNA helicase activity is also required for its own degradation, as CrhR abundance remains elevated, even at 30°C in crhR mutants. Rapid deactivation of the proteolytic machinery is crucial, since it provides the ability to rapidly induce CrhR accumulation in response to a subsequent downshift in temperature. The process involves conditional proteolysis since it does not occur under all conditions and responds to a specific environmental cue, temperature. In addition, CrhR degradation does not appear to be a general response to temperature upshift, since Rps1 levels were not affected, and we would predict that it does not involve the removal of misfolded proteins caused by temperature stress. Maximal heat shock protein expression occurs at 42.5°C in Synechocystis (44), which is significantly above the temperature at which CrhR degradation is observed. We cannot rule out, however, the potential that thermal denaturation of CrhR at 30°C is involved in the process, similar to the temperature-induced misfolding and subsequent degradation of homoserine transsuccinylase HTS (MetA) at temperatures above 25°C in E. coli (45). These observations indicate that an autoregulatory feedback loop exists between the CrhR RNA helicase and both the induction and inactivation of the proteolytic mechanism. Feedback loops between global regulators and components of the proteolytic machinery are frequently observed in prokaryotes, for example, RpoS (sigma 32) regulation in E. coli (5).
To date, only a limited number of examples of conditional proteolysis have been reported in prokaryotic systems and even fewer in response to temperature change. Similar to the decrease in CrhR abundance reported here, MetA (45) and CspC (13) are degraded in response to a temperature upshift, resulting in the regulation of cell growth and induction of heat shock gene expression, respectively. Conversely, degradation maintains RpoH (sigma 32) at basal levels at normal growth temperatures, in the absence of heat shock (46).
The in vivo experiments presented here also revealed that the increase in CrhR protein abundance in response to a temperature downshift from 30 to 20°C is a CrhR-independent phenomenon that does not require functional CrhR RNA helicase activity. This is indicated by the observation that increased CrhR abundance is observed in both wild-type and crhRTR mutant cells. In contrast, the decrease in CrhR abundance in response to a temperature upshift from 20 to 30°C observed in both the wild type and also His-CrhR-expressing strains is CrhR dependent, since the decrease is not observed in crhRTR mutant cells. These results imply that the cellular response to the temperature shift is occurring via two independent pathways in Synechocystis: a CrhR-independent pathway associated with the initial cold sensing and molecular response leading to the induction of crhR expression and a CrhR-dependent pathway required for inducing activity of the CrhR proteolytic machinery at high temperature.
Why would a cyanobacterium require multiple levels of control over the abundance of a RNA helicase, including proteolysis, in response to temperature shift? The system described here is reminiscent of, but not identical to, the regulation of and by cspC during heat shock in E. coli (13). The two systems have common elements in that they both involve RNA-binding proteins whose expression is attenuated in response to temperature upshift by proteolysis. In the case of CspC, there is also a corresponding decrease in the stability of mRNAs that interact with CspC, thus abrogating the heat shock response (13). This does not appear to be the case for the crhR transcript, since crhR accumulation is only transiently enhanced for 2 to 3 h after a downshift to 20°C (36). Furthermore, transient crhR transcript accumulation is not observed in the crhRTR mutant, indicating that crhR transcript is not destabilized in the absence of CrhR (36). Although CrhR does not appear to protect the crhR transcript from degradation at low temperature, it could potentially protect other transcripts.
Cyanobacterial genomes do not encode E. coli-like Csp's and thus CrhR may perform a role complementary to CspC. Thus, CrhR may function as an RNA chaperone, protecting bound mRNAs from degradation and/or translation elongation at low temperatures. It is possible that the double-stranded RNA (dsRNA) unwinding and RNA annealing activities catalyzed by CrhR in vitro (41) facilitate these functions in vivo, removing RNA secondary structure and/or bound proteins that block translation elongation at 30°C, allowing rapid translation of the interacting mRNAs. RNA helicases dissociate from their substrates after unwinding (47), potentially releasing CrhR protein and its RNA targets, making them available for degradation and effectively suppressing the response. Although the RNA targets for CrhR are not known, it is possible that the mechanism targets heat shock genes and/or photosynthetic genes, since the major physiological effect associated with crhR mutation is a reduction in photosynthetic fixation of carbon dioxide (35). Enhanced expression of CrhR-bound mRNAs at 20°C could potentially contribute to the severely compromised cell growth and survival observed in the absence of functional CrhR (35).
This scenario implies a novel role for a RNA helicase in response to elevated temperature that is contrary to the classical unwinding of thermodynamically stabilized dsRNAs at low temperatures (48). Indirect support for aspects of this hypothesis can be inferred from the function of RNA helicases in other organisms. RNA helicases are associated with enhanced transcript stability in both prokaryotic (49, 50) and eukaryotic (51) systems, and inactivation of a DEAD box RNA helicase related to CrhR, the deaD gene, impairs heat shock gene expression in E. coli (52). At the protein level, proteolysis appears to be associated with RNA helicase activity since stabilization of RIG-1, a DExD/H box RNA helicase, is crucial for its physiological function (53).
Induction of CrhR proteolysis occurs within 10 min of temperature upshift, with CrhR performing a crucial role in the induction of active proteolytic machinery. Insight into how the rapid activation is achieved was obtained from determination of the requirement for de novo protein synthesis in the induction process. Unexpectedly, inhibition of translation initiation did not affect induction of the degradation machinery, whereas inhibition of elongation abolished CrhR proteolysis. This indicates that de novo protein synthesis of an unknown factor is required for activation of the proteolytic machinery; however, translation of the crucial factor had already initiated at 20°C.
Examples of proteases associated with a cyanobacterial response to abiotic stress have been reported. Mutational analysis has indicated that a site-2 protease (S2P) is associated with acid acclimation (27), that three Deg/HtrA-related proteases are involved in high temperature and light stress in different subcellular locations (26), and that FtsH2 functions in osmoregulation by degrading GgpS (20). It would be of interest to determine whether one of these proteases is involved in CrhR regulation. With respect to the signal that elicits degradation, CrhR does not appear to have a degradation signal in the C-terminal region, since CrhRTR is degraded by protein extracts isolated from wild-type cells grown at 30°C. Although this is potentially unexpected due to the fact that AAA+ proteases recognize a degradation tag, a degradation sequence has also not been identified in RpoH (sigma 32) (6). Potentially similar to CrhR, MetA degradation requires an unidentified signal at the N terminus that does not involve the N-end rule and is not catalyzed by the tested proteases Lon, HslVU, and ClpP (45).
We have provided here the first evidence for a posttranslational mechanism that stringently regulates RNA helicase expression via conditional proteolysis. The mechanism links the potential for a CrhR-catalyzed alteration of the RNA secondary structure with the autoregulatory induction of conditional proteolysis in the response of Synechocystis to temperature upshift. Identification of the degradation machinery and the mechanism by which temperature and CrhR are associated with its regulation will provide further unique insights, facilitating the manipulation of temperature regulation of gene expression in cyanobacteria for biotechnological applications.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by grant number 171319 from the Natural Sciences and Engineering Research Council of Canada to G.W.O.
We thank Wolfgang Hess for providing the Synechocystis triparental mating protocol, Albert R. R. Rosana for assistance with ImageJ analysis, and Monsanto (St. Louis, MO) for providing pMon 36456. E. coli Rps1 antibody was generously provided by P. Baumann (University of California, Davis).
Footnotes
Published ahead of print 7 February 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01362-13.
REFERENCES
- 1.Pahl HL, Baeuerle PA. 1996. Control of gene expression by proteolysis. Curr. Opin. Cell Biol. 8:340–347. 10.1016/S0955-0674(96)80007-X [DOI] [PubMed] [Google Scholar]
- 2.Ryan KR, Shapiro L. 2003. Temporal and spatial regulation in prokaryotic cell cycle progression and development. Annu. Rev. Biochem. 72:367–394. 10.1146/annurev.biochem.72.121801.161824 [DOI] [PubMed] [Google Scholar]
- 3.Gur E, Biran D, Ron EZ. 2011. Regulated proteolysis in Gram-negative bacteria: how and when? Nat. Rev. Microbiol. 9:839–848. 10.1038/nrmicro2669 [DOI] [PubMed] [Google Scholar]
- 4.Gottesman S. 2003. Proteolysis in bacterial regulatory circuits. Annu. Rev. Cell Dev. Biol. 19:565–587. 10.1146/annurev.cellbio.19.110701.153228 [DOI] [PubMed] [Google Scholar]
- 5.Jenal U, Hengge-Aronis R. 2003. Regulation by proteolysis in bacterial cells. Curr. Opin. Microbiol. 6:163–172. 10.1016/S1369-5274(03)00029-8 [DOI] [PubMed] [Google Scholar]
- 6.Meyer AS, Baker TA. 2011. Proteolysis in the Escherichia coli heat shock response: a player at many levels. Curr. Opin. Microbiol. 14:194–199. 10.1016/j.mib.2011.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raju RM, Goldberg AL, Rubin EJ. 2012. Bacterial proteolytic complexes as therapeutic targets. Nat. Rev. Drug Discov. 11:777–789. 10.1038/nrd3846 [DOI] [PubMed] [Google Scholar]
- 8.Battesti A, Gottesman S. 2013. Roles of adaptor proteins in regulation of bacterial proteolysis. Curr. Opin. Microbiol. 16:140–147. 10.1016/j.mib.2013.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Domian IJ, Reisenauer A, Shapiro L. 1999. Feedback control of a master bacterial cell-cycle regulator. Proc. Natl. Acad. Sci. U. S. A. 96:6648–6653. 10.1073/pnas.96.12.6648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Straus D, Walter W, Gross CA. 1990. DnaK, DnaJ, and GrpE heat shock proteins negatively regulate heat shock gene expression by controlling the synthesis and stability of sigma 32. Genes Dev. 4:2202–2209. 10.1101/gad.4.12a.2202 [DOI] [PubMed] [Google Scholar]
- 11.Tomoyasu T, Gamer J, Bukau B, Kanemori M, Mori H, Rutman AJ, Oppenheim AB, Yura T, Yamanaka K, Niki H, et al. 1995. Escherichia coli FtsH is a membrane-bound, ATP-dependent protease which degrades the heat-shock transcription factor sigma 32. EMBO J. 14:2551–2560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Phadtare S, Inouye M. 2001. Role of CspC and CspE in regulation of expression of RpoS and UspA, the stress response proteins in Escherichia coli. J. Bacteriol. 183:1205–1214. 10.1128/JB.183.4.1205-1214.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shenhar Y, Rasouly A, Biran D, Ron EZ. 2009. Adaptation of Escherichi coli to elevated temperatures involves a change in stability of heat shock gene transcripts. Environ. Microbiol. 11:2989–2997. 10.1111/j.1462-2920.2009.01993.x [DOI] [PubMed] [Google Scholar]
- 14.Shenhar Y, Biran D, Ron EZ. 2012. Resistance to environmental stress requires the RNA chaperones CspC and CspE. Environ. Microbiol. Rep. 4:532–539. 10.1111/j.1758-2229.2012.00358.x [DOI] [PubMed] [Google Scholar]
- 15.Cairrão F, Cruz A, Mori H, Arraiano CM. 2003. Cold shock induction of RNase R and its role in the maturation of the quality control mediator SsrA/tmRNA. Mol. Microbiol. 50:1349–1360. 10.1046/j.1365-2958.2003.03766.x [DOI] [PubMed] [Google Scholar]
- 16.Chen C, Deutscher MP. 2010. RNase R is a highly unstable protein regulated by growth phase and stress. RNA 16:667–672. 10.1261/rna.1981010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sokolenko A, Pojidaeva E, Zinchenko V, Panichkin V, Glaser VM, Herrmann RG, Shestakov SV. 2002. The gene complement for proteolysis in the cyanobacterium Synechocystis sp. PCC 6803 and Arabidopsis thaliana chloroplasts. Curr. Genet. 41:291–310. 10.1007/s00294-002-0309-8 [DOI] [PubMed] [Google Scholar]
- 18.Gómez-Baena G, Manuel García-Fernández J, López-Lozano A, Toribio F, Diez J. 2006. Glutamine synthetase degradation is controlled by oxidative proteolysis in the marine cyanobacterium Prochlorococcus marinus strain PCC 9511. Biochim. Biophys. Acta 1760:930–940. 10.1016/j.bbagen.2006.01.016 [DOI] [PubMed] [Google Scholar]
- 19.Roberts IN, Lam XT, Miranda H, Kieselbach T, Funk C. 2012. Degradation of PsbO by the Deg protease HhoA is thioredoxin dependent. PLoS One 7:e45713. 10.1371/journal.pone.0045713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stirnberg M, Fulda S, Huckauf J, Hagemann M, Krämer R, Marin K. 2007. A membrane-bound FtsH protease is involved in osmoregulation in Synechocystis sp. PCC 6803: the compatible solute synthesizing enzyme GgpS is one of the targets for proteolysis. Mol. Microbiol. 63:86–102. 10.1111/j.1365-2958.2006.05495.x [DOI] [PubMed] [Google Scholar]
- 21.Li L, Kehoe DM. 2008. Abundance changes of the response regulator RcaC require specific aspartate and histidine residues and are necessary for normal light color responsiveness. J. Bacteriol. 190:7241–7250. 10.1128/JB.00762-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singh M, Yamamoto Y, Satoh K, Aro EM, Kanervo E. 2005. Post-illumination-related loss of photochemical efficiency of Photosystem II and degradation of the D1 protein are temperature-dependent. J. Plant Physiol. 162:1246–1253. 10.1016/j.jplph.2004.12.008 [DOI] [PubMed] [Google Scholar]
- 23.Ning D, Ye S, Liu B, Chang J. 2011. The proteolytic activation of the relNEs (ssr1114/slr0664) toxin-antitoxin system by both proteases Lons and ClpP2s/Xs of Synechocystis sp. PCC 6803. Curr. Microbiol. 63:496–502. 10.1007/s00284-011-0011-5 [DOI] [PubMed] [Google Scholar]
- 24.Lopes Pinto F, Erasmie S, Blikstad C, Lindblad P, Oliveira P. 2011. FtsZ degradation in the cyanobacterium Anabaena sp. strain PCC 7120. J. Plant Physiol. 168:1934–1942. 10.1016/j.jplph.2011.05.023 [DOI] [PubMed] [Google Scholar]
- 25.Barker M, de Vries R, Nield J, Komenda J, Nixon PJ. 2006. The deg proteases protect Synechocystis sp. PCC 6803 during heat and light stresses but are not essential for removal of damaged D1 protein during the photosystem two repair cycle. J. Biol. Chem. 281:30347–30355. 10.1074/jbc.M601064200 [DOI] [PubMed] [Google Scholar]
- 26.Miranda H, Cheregi O, Netotea S, Hvidsten TR, Moritz T, Funk C. 2013. Co-expression analysis, proteomic and metabolomic study on the impact of a Deg/HtrA protease triple mutant in Synechocystis sp. PCC 6803 exposed to temperature and high light stress. J. Proteomics 78:294–311. 10.1016/j.jprot.2012.09.036 [DOI] [PubMed] [Google Scholar]
- 27.Zhang X, Chen G, Qin C, Wang Y, Wei D. 2012. Slr0643, an S2P homologue, is essential for acid acclimation in the cyanobacterium Synechocystis sp. PCC 6803. Microbiology 158:2765–2780. 10.1099/mic.0.060632-0 [DOI] [PubMed] [Google Scholar]
- 28.Sato N. 1995. A family of cold-regulated RNA-binding protein genes in the cyanobacterium Anabaena variabilis M3. Nucleic Acids Res. 23:2161–2167. 10.1093/nar/23.12.2161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sakamoto T, Bryant DA. 1997. Temperature-regulated mRNA accumulation and stabilization for fatty acid desaturase genes in the cyanobacterium Synechococcus sp. strain PCC 7002. Mol. Microbiol. 23:1281–1292. 10.1046/j.1365-2958.1997.3071676.x [DOI] [PubMed] [Google Scholar]
- 30.Chamot D, Magee WC, Yu E, Owttrim GW. 1999. A cold shock-induced cyanobacterial RNA helicase. J. Bacteriol. 181:1728–1732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chamot D, Owttrim GW. 2000. Regulation of cold shock-induced RNA helicase gene expression in the cyanobacterium Anabaena sp. strain PCC 7120. J. Bacteriol. 182:1251–1246. 10.1128/JB.182.5.1251-1256.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suzuki I, Los DA, Kanesaki Y, Mikami K, Murata N. 2000. The pathway for perception and transduction of low-temperature signals in Synechocystis. EMBO J. 19:1327–1334. 10.1093/emboj/19.6.1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mikami K, Kanesaki Y, Suzuki I, Murata N. 2002. The histidine kinase Hik33 perceives osmotic stress and cold stress in Synechocystis sp. PCC 6803. Mol. Microbiol. 46:905–915. 10.1046/j.1365-2958.2002.03202.x [DOI] [PubMed] [Google Scholar]
- 34.Kujat SL, Owttrim GW. 2000. Redox-regulated RNA helicase expression. Plant Physiol. 124:703–714. 10.1104/pp.124.2.703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rosana AR, Ventakesh M, Chamot D, Patterson-Fortin LM, Tarassova O, Espie GS, Owttrim GW. 2012. Inactivation of a low temperature-induced RNA helicase in Synechocystis sp. PCC 6803: physiological and morphological consequences. Plant Cell Physiol. 53:646–658. 10.1093/pcp/pcs020 [DOI] [PubMed] [Google Scholar]
- 36.Rosana AR, Chamot D, Owttrim GW. 2012. Autoregulation of RNA helicase expression in response to temperature stress in Synechocystis sp. PCC 6803. PLoS One 7:e48683. 10.1371/journal.pone.0048683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Owttrim GW. 2013. RNA helicases: diverse roles in prokaryotic response to abiotic stress. RNA Biol. 10:96–110. 10.4161/rna.22638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Owttrim GW. 2012. RNA helicases in cyanobacteria: biochemical and molecular approaches. Methods Enzymol. 511:385–403. 10.1016/B978-0-12-396546-2.00018-8 [DOI] [PubMed] [Google Scholar]
- 39.Alexeyev MF, Shokolenko IN, Croughan TP. 1995. Improved antibiotic-resistance gene cassettes and omega elements for Escherichia coli vector construction and in vitro deletion/insertion mutagenesis. Gene 160:63–67. 10.1016/0378-1119(95)00108-I [DOI] [PubMed] [Google Scholar]
- 40.Elhai J, Vepritskiy A, Muro-Pastor AM, Flores E, Wolk CP. 1997. Reduction of conjugal transfer efficiency by three restriction activities of Anabaena sp. strain PCC 7120. J. Bacteriol. 179:1998–2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chamot D, Colvin KR, Kujat-Choy SL, Owttrim GW. 2005. RNA structural rearrangement via unwinding and annealing by the cyanobacterial RNA helicase, CrhR. J. Biol. Chem. 280:2036–2044. 10.1074/jbc.M409700200 [DOI] [PubMed] [Google Scholar]
- 42.Qi Q, Hao M, Ng WO, Slater SC, Baszis SR, Weiss JD, Valentin HE. 2005. Application of the Synechococcus nirA promoter to establish an inducible expression system for engineering the Synechocystis tocopherol pathway. Appl. Environ. Microbiol. 71:5678–5684. 10.1128/AEM.71.10.5678-5684.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9:671–675. 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lehel C, Wada H, Kovács E, Török Z, Gombos Z, Horváth I, Murata N, Vigh L. 1992. Heat shock protein synthesis of the cyanobacterium Synechocystis PCC 6803: purification of the GroEL-related chaperonin. Plant Mol. Biol. 18:327–336. 10.1007/BF00034959 [DOI] [PubMed] [Google Scholar]
- 45.Biran D, Gur E, Gollan L, Ron EZ. 2000. Control of methionine biosynthesis in Escherichia coli by proteolysis. Mol. Microbiol. 37:1436–1443. 10.1046/j.1365-2958.2000.02097.x [DOI] [PubMed] [Google Scholar]
- 46.Kanemori M, Nishihara K, Yanagi H, Yura T. 1997. Synergistic roles of HslVU and other ATP-dependent proteases in controlling in vivo turnover of sigma32 and abnormal proteins in Escherichia coli. J. Bacteriol. 179:7219–7225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Linder P, Jankowsky E. 2011. From unwinding to clamping: the DEAD box RNA helicase family. Nat. Rev. Mol. Cell. Biol. 12:505–516. 10.1038/nrm3154 [DOI] [PubMed] [Google Scholar]
- 48.Iost I, Bizebard T, Dreyfus M. 2013. Functions of DEAD-box proteins in bacteria: current knowledge and pending questions. Biochim. Biophys. Acta 1829:866–877. 10.1016/j.bbagrm.2013.01.012 [DOI] [PubMed] [Google Scholar]
- 49.Iost I, Dreyfus M. 1994. mRNAs can be stabilized by DEAD-box proteins. Nature 372:193–196. 10.1038/372193a0 [DOI] [PubMed] [Google Scholar]
- 50.Brandi A, Spurio R, Gualerzi CO, Pon CL. 1999. Massive presence of the Escherichia coli ‘major cold-shock protein' CspA under non-stress conditions. EMBO J. 18:1653–1659. 10.1093/emboj/18.6.1653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun M, Zhou T, Jonasch E, Jope RS. 2013. DDX3 regulates DNA damage-induced apoptosis and p53 stabilization. Biochim. Biophys. Acta 1833:1489–1497. 10.1016/j.bbamcr.2013.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jones PG, Mitta M, Kim Y, Jiang W, Inouye M. 1996. Cold shock induces a major ribosomal-associated protein that unwinds double-stranded RNA in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 93:76–80. 10.1073/pnas.93.1.76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang L, Zhao W, Zhang M, Wang P, Zhao K, Zhao X, Yang S, Gao C. 2013. USP4 positively regulates RIG-I-mediated antiviral response through deubiquitination and stabilization of RIG-I. J. Virol. 87:4507–4515. 10.1128/JVI.00031-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bethesda Research Laboratories. 1986. BRL pUC host: E. coli DH5α competent cells. Bethesda Res. Lab. Focus 8:9 [Google Scholar]
- 55.Studier FW, Moffatt BA. 1986. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. Mol. Biol. 189:113–130. 10.1016/0022-2836(86)90385-2 [DOI] [PubMed] [Google Scholar]
- 56.Alting-Mees MA, Short JM. 1989. pBluescript II: gene mapping vectors. Nucleic Acids Res. 17:9494. 10.1093/nar/17.22.9494 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.