

Abstract
Rapidly after infection, live Borrelia burgdorferi, the causative agent of Lyme disease, is found within lymph nodes, causing rapid and strong tissue enlargement, a loss of demarcation between B cell follicles and T cell zones, and an unusually large accumulation of B cells. We sought to explore the mechanisms underlying these changes, as lymph tissue disruption could be detrimental for the development of robust Borrelia-specific immunity. A time course study demonstrated that the loss of the normal lymph node structure was a distinct process that preceded the strong increases in B cells at the site. The selective increases in B cell frequencies were due not to proliferation but rather to cytokine-mediated repositioning of B cells to the lymph nodes, as shown with various gene-targeted and bone marrow irradiation chimeras. These studies demonstrated that B. burgdorferi infection induced type I interferon receptor (IFNR) signaling in lymph nodes in a MyD88- and TRIF-independent manner and that type I IFNR indirect signaling was required for the excessive increases of naive B cells at those sites. It did not, however, drive the observed histopathological changes, which occurred independently also from major shifts in the lymphocyte-homing chemokines, CXCL12, CXCL13, and CCL19/21, as shown by quantitative reverse transcription-PCR (qRT-PCR), flow cytometry, and transwell migration experiments. Thus, B. burgdorferi infection drives the production of type I IFN in lymph nodes and in so doing strongly alters the cellular composition of the lymph nodes, with potential detrimental effects for the development of robust Borrelia-specific immunity.
INTRODUCTION
Following infection of its varied hosts by tick bite, Borrelia burgdorferi disseminates to numerous tissues, including lymph nodes, skin, connective tissues of muscles and bones, and the nervous tissue. This causes an array of disease manifestations, including lymphadenopathy, erythema migrans, arthritis, carditis, and neurological disease (1–3). Despite activation of B. burgdorferi-specific B and T cell immunity (4–7), the spirochetes successfully evade the immune system, in at least a subset of immunocompetent hosts, and establish a chronic nonresolving infection (8).
The lymphadenopathy induced at proximal and distal sites of infection (3) is characterized by an extremely strong increase in the lymph node cellularity, due nearly exclusively to the accumulation of B cells (7). Furthermore, the appearance of culturable B. burgdorferi and its visible presence in cortical sinuses in the lymph nodes is correlated with the disruption of the usually well-demarcated T and B cell areas and an expansion of the lymph node cortex by day 10 of infection (3, 7). Whether the B cell accumulation causes the lymph node architecture disruption or vice versa is currently unknown.
It is tempting to speculate that this loss of tissue architecture and/or the imbalance in the B cell/T cell ratios in secondary lymphoid tissues may affect the induction of appropriate adaptive immunity and thereby represent one mechanism by which B. burgdorferi can “outrun” or subvert adaptive immune responses. Indeed, the lymph nodes of B. burgdorferi-infected mice do not seem to support strong germinal centers, which are hallmarks of T cell-dependent, high-affinity B cell effector and memory responses. While germinal centers are induced within about 2 1/2 weeks of infection in the draining and nondraining lymph nodes, their frequencies and sizes are low, and the initial germinal centers disappear at about 1 month after infection. This is despite the continued presence of B. burgdorferi in these lymph nodes (references 3 and 7 and unpublished observations). Mice also do not generate appreciable numbers of long-lived bone marrow plasma cells during the first 2 months of infection (3). Understanding the signals that disrupt the structure of the lymph nodes after B. burgdorferi infection may help to identify barriers to the development of infection-induced protective B cell responses and to the induction of functional immune memory, which appears lacking even after repeat infections (9, 10).
T cell-dependent B cell responses rely on the careful orchestration of T and B cell migration within secondary lymphoid tissues, bringing antigen-specific B cells into close proximity to primed antigen-specific T cells at the edges of the T and B cell zones. This migration is regulated by the follicle-homing chemokine CXCL13 and the T cell zone chemokines CCL19/21. Upregulation of the CCL19/21 receptor CCR7 on antigen-stimulated B cells and of the CXCL13 receptor, CXCR5, on primed T cells drives their migration toward each other (11). Mice lacking one of these molecules show a block or delay in their adaptive immune responses, indicating a need for the tight regulation of these processes for optimal immune stimulation (12, 13).
B. burgdorferi is not the only pathogen whose infection causes lymph node alterations. For example, infection with Salmonella enterica serovar Typhimurium causes a loss of lymph node architecture and altered T cell/B cell ratios similar to those seen following B. burgdorferi infection. These alterations were recently shown to depend on a Toll-like receptor 4 (TLR4) signaling-dependent reduction in CCL21 and CXCL13 expression. The blockade of TLR4 signaling reversed the disruption of the tissue structure (14).
Following infection with B. burgdorferi, CXCL13 expression levels in various tissues were shown to correlate with B. burgdorferi burden (15), and in vitro stimulation of human monocytes with Borrelia garinii resulted in a TLR2-mediated induction of CXCL13 (16). Given the rapid migration of B. burgdorferi into the lymph nodes after infection (3), their presence may induce alterations in CXCL13 production or other changes in lymph node-homing chemokines that drive the tissue alteration and/or B cell accumulation. However, production of inflammatory cytokines may also affect lymph node alterations. For example, following Escherichia coli infection, mast cells were shown to produce tumor necrosis factor (TNF), causing lymph node hypertrophy (17).
This study aimed to explore the relationship between the unusually large accumulation of B cells and the alteration of the lymph node architecture after B. burgdorferi infection and the underlying mechanisms of these infection-induced changes. Our studies demonstrated that the B cell accumulation was dependent on type I interferon receptor (IFNR) signaling but independent of MyD88 and TRIF and occurred after the destruction of the lymph node architecture, which appeared to be unrelated to changes in CXCL13 or the other major known lymph node-homing chemokines.
MATERIALS AND METHODS
Borrelia burgdorferi.
A new aliquot of a low-passage, clonal strain of Borrelia burgdorferi sensu stricto (cN40) was grown in modified Barbour-Stoenner-Kelley II medium (18) at 33°C, and inocula were enumerated with a Petroff-Hauser bacterial counting chamber (Baxter Scientific, McGaw Park, IL) before infection of mice.
Mice and infections.
Eight- to 12-week-old C57BL/6J (B6), B6.CB17-Prkdcscid/SzJ (SCID), and B6.129P2(C)-Cd19tm1(cre)Cgn/J (CD19.Cre) female or male mice were purchased from the Jackson Laboratory. Breeders of mice lacking MyD88 (MyD88−/−) on a B6 background were kindly provided by Richard Flavell (Yale University, CT), B6 mice lacking the type I IFN receptor chain 1 (IFNAR1−/−) were provided by Murali-Krishna Kaja (Emory University, GA), IFNAR1-floxed B6 mice bred to CD19.Cre mice were provided by Ulrich Kalinke (Medical School Hannover, Germany), and MyD88−/− × TRIF−/− B6 mice were provided by Greg Barton (University of California, Berkeley). B cell-specific IFNAR1 knockout mice (CD19.IFNAR−/−) were maintained by crossing CD19.Cre heterozygotes with IFNAR1-floxed mice. The Cre− littermates were used as controls.
Irradiation bone marrow chimeras were generated by reconstituting lethally irradiated B6 or IFNAR1−/− mice (n = 4/group; 800 rads of full-body irradiation) with total bone marrow (2 × 107 cells/mouse) from either B6 or IFNAR−/− donor mice. Irradiated chimeras were given Bactrim (sulfamethoxazole-trimethoprim) at 1.2 mg/ml sulfamethoxazole and 0.25 mg/ml trimethoprim in the drinking water (ad lib) for 6 weeks. Antibiotic treatment was stopped 1 week prior to infection with tissue-adapted spirochetes. All mice were kept under conventional housing conditions in microisolator cages. Overexposure to carbon dioxide was used to euthanize mice. All studies were conducted according to protocols approved by the University of California, Davis, Animal Use and Care Committee.
Experimental mice were infected with host-adapted B. burgdorferi cN40 as previously described (3). This was to target a particular draining lymph node, which is difficult to do with tick infections but avoids the use of culture-grown B. burgdorferi, as its antigenic profile differs significantly from that of tick-transmitted Borrelia. Briefly, SCID mice were infected subcutaneously (s.c.) with 104 culture grown B. burgdorferi organisms. After a minimum of 14 days, SCID mice were euthanized, and their ears were cleaned with 70% ethanol (EtOH) followed by Nolvasan (Pfizer) and then removed. Small pieces of infected ear tissue were inserted subcutaneously in the right hind legs of recipient mice. For sham “infection,” ear tissue from uninfected SCID mice was cleaned and inserted as described above. For immunization, mice received 5 × 104 sonicated B. burgdorferi cN40 organisms in 100 μl at a 1:1 ratio with complete Freund's adjuvant s.c. into the right leg through a small incision. The right inguinal lymph nodes were the draining lymph nodes evaluated for both immunization and infection.
Flow cytometry.
Single-cell suspensions from inguinal lymph nodes were stained as previously described (3). To determine the frequency of innate cells in the lymph node, whole inguinal lymph nodes were digested in Dulbecco modified Eagle medium (DMEM) plus 10% NCS plus 50 U/ml DNase I (Worthington Biochemical) plus 250 U/ml collagenase type I (Worthington Biochemical) in C tubes (Miltenyi-Biotec), followed by dissociation on the GentleMACS (Miltenyi-Biotec). In brief, Fc receptors were blocked with 10 μg/ml anti-CD16/32, clone 2.4G2 (in-house generated), followed by staining with the following antibody conjugations, used at predetermined optimal concentrations: CXCR5-biotin, CXCR4-biotin, and CD45RA-Alexa 647 (all from BD Biosciences); CD3-PacificBlue, CD8-PacificBlue, CD19-PacBlue, CD19-Cy5-phycoerythrin (PE), CD3-Alexa 750, and CD69-PE (all from eBioscience); CD4-PacificBlue, CD19-fluorescein isothiocyanate (FITC), CD8a-allophycocyanin (APC), CD8-Cy5.5-PE, CD4-FITC, CD4-PE, and CD11c-PE, (all in-house generated); and CD19-Cy5.5-APC and streptavidin-Qdot605 (all from Invitrogen). Chemokine receptor staining was performed at 37°C for 30 min. All other steps were performed on ice for 20 min. Dead cells were identified by staining with live/dead violet stain (Invitrogen by Life Technologies) on ice for 30 min.
To quantify in vivo cell proliferation, incorporation of bromodeoxyuridine (BrdU) into the DNA of dividing cells was measured by injecting BrdU at 1 mg/100 μl in phosphate-buffered saline (PBS) intraperitoneally (i.p.) every other day, beginning on the day of infection. Cell surface staining was done as described above. Cells were then fixed with Cytofix/Cytoperm and Perm/Wash (BD Biosciences), followed by freezing in 90% fetal bovine serum (FBS)–10% dimethyl sulfoxide (DMSO) overnight at −80°C. BrdU staining was performed following the manufacturer's instructions (BD Pharmingen FITC BrdU Flow kit [BD Biosciences]). Data acquisition was performed on a 13-color FACSAria instrument (BD Biosciences) (19). Data were analyzed using FlowJo software (a kind gift from Adam Treister).
Ex vivo transwell migration studies.
To assess the chemokine responsiveness of lymph node cells, right inguinal lymph nodes were removed at various time points after infection and processed into single-cell suspensions. Lymph nodes from noninfected mice served as controls. Cells were resuspended in RPMI 1640–0.5% bovine serum albumin (BSA) and loaded into the upper well of a transwell plate (5-μm polycarbonate membrane [Costar; Corning]) at 1 × 107 cells/ml in quadruplicate. CXCL13 and CXCL12 were added at 2.5 μg/ml and 1 μg/ml, respectively, into the lower chambers. The transwell plates were placed in an incubator at 37°C with 5% CO2 for 2 h. Cells were collected from the bottom chamber and stained with anti-CD4, -CD8, and -CD19 and propidium iodide for live/dead discrimination. Prior to staining, 5 μl of 15-μm polystyrene beads (Polysciences) was added to each tube. The ratio of lymphocytes to bead count was used for calculation of relative cell numbers in each well. Data acquisition was performed on a 4-color FACSCalibur (BD Biosciences) instrument. The percentage of transmigrated lymphocytes was calculated as a ratio: (tested sample lymphocyte/bead × 100)/(ratio lymphocyte/bead input).
Quantitative reverse transcription-PCR (qRT-PCR).
To assess gene expression for selected cytokines, the right inguinal lymph nodes from day 4 B. burgdorferi-infected, noninfected, or B. burgdorferi-immunized mice (one lymph node from individual mice; n = 4/group) were collected and immediately processed. Total RNA was extracted with the RNeasy Minikit (Qiagen) according to the manufacturer's instructions using Miltenyi-Biotec M tubes. cDNA was prepared by using random hexamers (Promega) with SuperScript II (Invitrogen). Amplification was performed with Clontech polymerase and the commercial primer probes CXCL13, CCL2, interleukin-2 (IL-2), IL-4, SphK1, TNF, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Applied Biosystems), using a Prism 7700 instrument (Applied Biosystems). Data were normalized for expression relative to the expression of GAPDH.
To determine gene expression for lymph node-homing chemokines, inguinal lymph nodes were collected before and at 10 days (the peak of lymph node cellularity) and 22 days (when germinal centers were present) after infection (n = 4/group) and kept frozen at −80°C until being processed. Total RNA was extracted as described above, and a low-density array was performed by real-time RT-PCR as previously described (7). In brief, first-strand cDNA was synthesized from total RNA using the QuantiTect reverse transcription kit (Qiagen) according to the manufacturer's instructions. All samples were analyzed for the presence of 18S rRNA in order to determine the efficiency of the nucleic acid extraction and amplification. The synthesized cDNA template from each sample was added to 2× Universal PCR Master Mix (Applied Biosystems) in 100-μl reaction mixtures. The mixture was added to each line of a microfluidic low-density array card (Applied Biosystems) and amplified using an ABI Prism 7900HT sequence detection system (Applied Biosystems). Primers and probes were designed using Primer Express software (Applied Biosystems) (CCL19 [forward, GGGTGCTAATGATGCGGAAG; reverse, GGTGAACACAACAGCAGGCA]) or supplied by Applied Biosystems (CCL21, CXCL13, CXCL12, and GAPDH). As a reference gene to normalize transcriptional activity of mouse cytokines, the GAPDH gene was used.
To determine gene expression of type I IFN and inducible genes, inguinal lymph nodes were collected from day 0 uninfected mice, at days 10 and 15 after B. burgdorferi infection with host-adapted spirochetes, and at days 10 and 15 after sham infection with tissue from uninfected SCID mice. Whole lymph nodes were collected and processed immediately. Total RNA was extracted with the RNeasy Minikit (Qiagen) according to manufacturer's instructions after homogenization of tissue. cDNA was prepared by using random hexamers (Promega) with SuperScript II (Invitrogen). Amplification was performed with Clontech polymerase and the commercial primer probes IFIT1, IFIT2, IFN-α4, and IFN-β (Applied Biosystems), using a Prism 7700 instrument (Applied Biosystems). Data were normalized for expression relative to the expression of GAPDH.
Enzyme-linked immunosorbent spot (ELISPOT) assay.
To probe for B. burgdorferi-specific antibody-producing cells, 96-well plates (Multiscreen HA filtration; Millipore) were coated overnight with a cocktail of four immunodominant recombinant B. burgdorferi cN40-specific proteins, decorin binding protein A (DbpA), outer surface protein C (OspC), arthritis-related protein (Arp), and Borrelia membrane protein A (BmpA), as previously described (3). A total of 5 × 105 cells in medium (RPMI 1640, 292 μg/ml l-glutamine, 100 μg/ml of penicillin and streptomycin, 10% heat-inactivated fetal calf serum [FCS], and 0.03 M 2-mercaptoethanol [2-ME]) were placed in the starting well and 2-fold serially diluted after the plates were blocked with PBS–4% BSA for 1 h. Cells were incubated at 37°C overnight and then lysed with water. Antibody binding was revealed by treatment with biotin-conjugated anti-IgM or IgG (Southern Biotech) in PBS–2% BSA for 2 h and streptavidin-horseradish peroxidase (SA-HRP) (Vector Laboratories) in PBS–2% BSA for 1 h, with 3-amino-9-ethylcarbazole (Sigma-Aldrich) as the substrate. Plates were dried, and spots were counted in all wells with cell dilutions resulting in visible and countable spots using an iSpot Fluorospot reader system (AID). Mean numbers ± standard deviations (SD) were calculated from cell counts of all wells with countable spots.
Histology.
Lymph nodes were fixed in neutral buffered formalin, embedded in paraffin, sectioned at 4-μm thickness, and then stained with hematoxylin and eosin (H&E). Images were taken at the indicated magnification with a Zeiss AxioShop/AxioCam using AxioVision 4.8 software. Other lymph nodes were frozen on dry ice in Tissue-Tek OCT (Sakura) and stored at −80°C until sectioned. Five-micrometer sections were cut on a Leica cryostat onto Superfrost/Plus slides (Fisher Scientific), immediately placed into ice-cold acetone for 2 min, and dried at room temperature for 1 to 2 h before staining with hematoxylin and eosin.
Statistical analysis.
Statistical analysis was performed using the Student t test or one-way analysis of variance (ANOVA) with the Dunnett posttest with help from Prism 5 software (GraphPad Software). A P value of 0.05 was considered statistically significant.
RESULTS
Rapid loss of lymph node architecture following B. burgdorferi infection.
Strong regional lymphadenopathy (i.e., lymph node enlargement) is a common early manifestation of infection with B. burgdorferi in both humans (20) and mice (3). As outlined above, by day 10 after infection these enlarged lymph nodes had lost B cell follicles and T cell zones and amassed B cells with frequencies reaching over 70% in some cases, while T cell numbers were unchanged (7). We aimed to determine whether the massive accumulation of B cells drives the disruption of the tissue architecture or vice versa by determining the temporal relationship between these two phenomena.
Histological evaluations of lymph nodes draining the site of infection with host-adapted B. burgdorferi demonstrated that B cell follicles first began to deteriorate on day 5 of infection. Nondraining lymph nodes showed a similar pattern, but the changes were somewhat delayed, consistent with the delayed infection of the other lymph nodes (3). By day 7, follicles were no longer discernible (Fig. 1A). In contrast to lymph nodes from day 10 B. burgdorferi-infected mice, those from the immunized mice showed typical lymph node architecture and the development of germinal centers within B cell follicles (Fig. 1B), suggesting a connection between the presence of live spirochetes in the lymph nodes and their deterioration.
FIG 1.

Alteration of tissue architecture and B cell accumulation in the lymph node following infection with host-adapted B. burgdorferi. (A) H&E staining of lymph nodes on days 3 to 8 postinfection. Black arrows indicate B cell follicles (original magnification, ×10), representative of n = 2/group. (B) H&E staining of lymph nodes from mice taken at day 10 immunization (left) or infection (right) with B. burgdorferi. White arrows indicate germinal centers with B cell follicles, and black arrows indicate primary B cell follicles without germinal centers (original magnification, ×10), representative of n = 2/group. (C) Shown are representative 5% contour plots and outliers generated by flow cytometry on draining inguinal lymph nodes collected at the indicated times after infection and stained to determine the frequency of CD3+ CD4+ T cells, CD19+ B cells, CD4− CD19− cells (live, single cells and CD19− CD3− CD8−). Numbers represent mean ± SD of the gated cells (n = 4). Data are representative of two independent experiments.
B cell frequency increases in these lymph nodes occurred only after the morphological alterations had taken place. A previous extensive time course study showed that B cell frequencies spiked sharply on day 8 (7). Consistent with those previous data, we show here that on day 4 of infection, the last day the lymph nodes showed strong T and B cell zone demarcations (Fig. 1A), CD19+ B cell frequency changes were modest compared to the changes seen on day 10 (Fig. 1C). The subsequent increases in B cell frequencies were accompanied by a corresponding drop in CD4 T cell frequencies. Absolute numbers of lymph node T cells, however, remained steady over time, while B cell numbers increased strongly (7). Thus, the data demonstrate that the B. burgdorferi infection-induced destruction of the lymph node architecture precedes the accumulation of CD19+ B cells at that site.
Loss of architecture and B cell accumulation are not attributable to CXCL13.
The lymph node is organized in various zones through localized expression of a small number of chemokines. The B cell follicles are established by production of the chemokine CXCL13 and its corresponding receptor, CXCR5, on B cells and T follicular helper cells. T cell zones are established by expression of CCL19/21 and its corresponding receptor, CCR7, on T cells as well as activated B cells. Plasma cells are drawn to medullary cord areas of the lymph node via expression of CXCR4, which binds to the chemokine CXCL12. Interestingly, previous evidence indicated a correlation between B. burgdorferi tissue burden and CXCL13 production (15, 21, 22). Therefore, we investigated to what extent changes in CXCL13 or the other major known lymph node-homing chemokines might underlie these infection-induced lymph node changes. We first assessed mRNA levels by qRT-PCR at the peak of the B cell accumulation (day 10) and at a later time point (day 22) after B. burgdorferi infection, when lymph nodes showed some germinal center responses (7). Our studies found no changes in CCL21, CXCL12, or CXCL13 mRNA levels at either time point compared to those in lymph nodes from noninfected mice, while CCL19 expression was modestly increased (Fig. 2). This increase in the T cell-homing chemokine was surprising, given the loss of the T cell zones and the low number of T cells in the lymph node at these time points, but it cannot explain the observed strong increases in B cell accumulation, as only a very small fraction of B cells expressed CCR7 in the lymph nodes on day 6 after infection (see Fig. S2 in the supplemental material). Overall these data indicated that the loss of lymph node architecture and increase in B cell accumulation were not correlated with strong alterations in expression of major lymph node-homing chemokines, including CXCL13 or its receptors.
FIG 2.
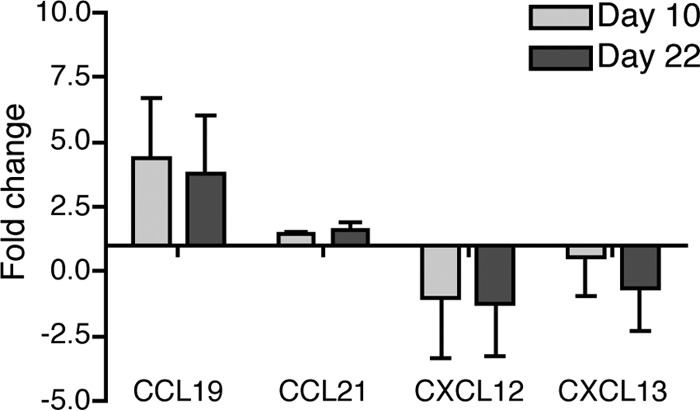
Lymph node-homing chemokine expression following infection with host-adapted B. burgdorferi. Shown are mean relative expression levels + SD of the indicated chemokines measured by qRT-PCR in lymph nodes of mice infected for 10 or 22 days compared to lymph nodes from uninfected mice (n = 4/group). Data are normalized for RNA input using the GAPDH housekeeping gene.
Given those results, we sought to evaluate more broadly cytokines and chemokines and their receptors using a qRT-PCR-based array screen of 84 related genes. We compared expression in lymph nodes of nonmanipulated mice with that in lymph nodes of infected mice just prior to the structural changes (i.e., day 4 of B. burgdorferi infection) and in “typically” activated lymph nodes on day 4 after s.c. immunization with sonicated B. burgdorferi in complete Freund's adjuvant. As shown, these lymph nodes exhibited normal lymph node histology and manifested germinal centers by day 10 (Fig. 1B). The array indicated a lack of significant differences between lymph nodes from infected and immunized mice for most lymph node-homing chemokines (see Table S1 in the supplemental material). Those that showed differences in the array screen were analyzed further by qRT-PCR (Fig. 3).
FIG 3.
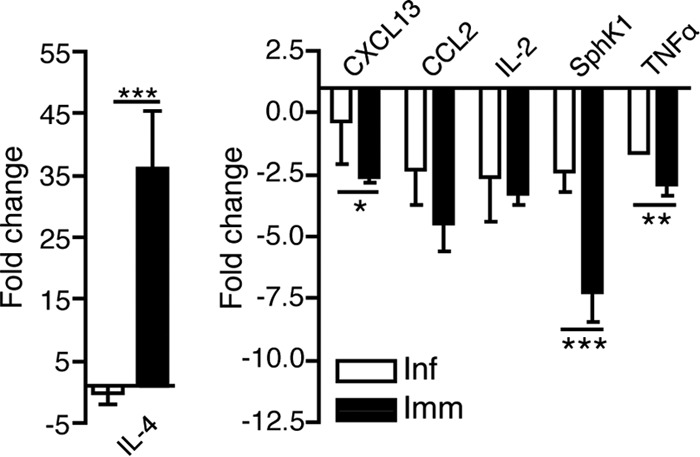
Gene expression changes in lymph nodes after infection and immunization with B. burgdorferi. Shown are mean relative expression levels + SD of cytokines measured by qRT-PCR of lymph nodes of mice infected or immunized for 4 days compared to lymph nodes from uninfected mice (n = 4/group). Data are normalized for RNA input using the GAPDH housekeeping gene. Statistical analysis used one-way ANOVA with the Dunnett posttest: *, P < 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
The strongest observed difference was an induction of IL-4 in B. burgdorferi-immunized but not in infected mice (Fig. 3, left panel). This difference is unlikely to be responsible for the lymph node architecture changes in infection. Other notable changes all marked reductions in gene expression with activation (Fig. 3, right panel). Three genes were confirmed by qRT-PCR to be differentially expressed, those for CXCL13, SphK1, and TNF-α. In each case the immunized mice showed stronger reductions in expression of these genes (Fig. 3). The reduction in CXCL13 was unexpected given the large number of B cells in the lymph node and the findings by others that CXCL13 expression levels in various tissues correlated with B. burgdorferi burden (15). SphK1 is a kinase necessary for sphingosine-1-phosphate (S1P) production and was shown previously to be induced by stimulation with bacterial lipoproteins (23).
Mast cell-produced tumor necrosis factor (TNF) was shown previously to cause lymph node hypertrophy following infection with Escherichia coli (17). However, TNF expression was reduced after B. burgdorferi infection, albeit to a smaller degree than after immunization. Additionally, mice deficient in TNF-α showed no difference in B cell expansion or lymph node architecture on day 10 of infection compared to controls (see Fig. S1 in the supplemental material). Together, the gene expression analysis showed that genes previously associated with alterations of lymph node architecture are unlikely to be involved in the B. burgdorferi infection-induced destruction of the lymph node.
Lymph node B cells from B. burgdorferi-infected mice respond to CXCL12 and CXCL13 with migration in vitro.
Given that the lymph node-homing chemokine expression appeared similar to those seen after immunizations that resulted in robust immune responses, we next sought to determine if infection with B. burgdorferi altered the ability of lymph node cells to respond to chemokine signaling with migration, as this could represent another mechanism to explain the loss of T and B cell zone demarcations. Protein expression analysis showed that the number of CD19+ B cells expressing CXCR4 significantly increased throughout the first 10 days of infection (Fig. 4A). This was due to the appearance of three distinct populations of CXCR4-expressing B cells: CD138+ and CD138− BLIMP-1+ plasma cells/plasmablasts (Fig. 4C and data not shown) and small numbers of germinal center B cells (Fig. 4D). Chemotaxis experiments using in vitro transwell assays demonstrated that a higher fraction of B cells from infected lymph nodes at day 10 of infection than of control B cells migrated toward CXCL12 (Fig. 4E), consistent with the observed increased numbers of CXCR4+ B cells in the population. The reduced migration of B cells from day 4 infected lymph nodes toward the CXCR4 ligand CXCL12 (Fig. 4E) may indicate that these B cells were refractory to ex vivo stimulation via CXCL12.
FIG 4.

B. burgdorferi infection does not impair chemokine-directed B cell migration. (A to D) Representative 5% contour plots with outliers, indicating mean frequencies of cell populations stained as indicated following gating for live, single-lymphocyte-size cells from lymph nodes of noninfected and day 10 B. burgdorferi-infected mice (n = 4). Small graphs are for fluorescence-minus-one (FMO) control stains. (A and B) Line graphs indicate mean frequencies + SD (n = 4/time point) of CXCR4+ cells at the indicated time points before and after infection (A) and means + SD (n = 4/time point) of median fluorescence intensities (MFIs) for CXCR5+ staining on CD19+ B cells at the indicated times after infection with B. burgdorferi (B). (C and D) Mean frequencies ± SD of CXCR4+ CD138− non-germinal center B cells and CXCR4+ CD138+ plasma cells as percentages of CD19+ B cells (C) and of germinal center B cells, identified as CD24hi CD38lo CD19+ CD45R+ (D, left), and CXCR4 and CXCR5 expression by germinal center B cells (n = 6) (D, right). (E and F) Mean frequencies + SD from quadruplicate cultures of migrated CD19+ cells compared to total input cells following ex vivo transwell migration experiments with chemokine ligand CXCL12 (E) or CXCL13 (F, left) and frequencies + SD of migrated B cells from day 10-infected, day 10-immunized, and control lymph nodes (F, right). Statistical analysis used one-way ANOVA with the Dunnett posttest: *, P < 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
As expected, nearly all B cells expressed CXCR5, and therefore we measured the median fluorescence intensity (MFI) of CXCR5 staining on lymph node B cells over the course of B. burgdorferi infection. A significant short-term drop in the MFI of CXCR5, the receptor for CXCL13, was noted on day 4 of infection. By day 6, expression levels had rebounded, and they remained steady thereafter (Fig. 4B). This reduction on day 4 correlated with the strong increases in the expression of its ligand, CXCL13, on that day (Fig. 3) and thus is likely due to CXCR5 receptor internalization. With regard to CXCR5 signaling, B cells from infected mice and those from noninfected controls showed similar migration toward CXCL13 at days 4 to 8 postinfection, but the B cells from infected mice showed significant increased migration on day 10 (Fig. 4F). However, the increases on day 10 were also observed with B cells from day 10 immunized mice (Fig. 4F).
Overall, these data suggest that B cells from lymph nodes of B. burgdorferi-infected mice were able to migrate in response to chemokine receptor signaling through CXCR4 and -5. The appearance of small numbers of CCR7+ B cells (see Fig. S2 in the supplemental material) further suggested that chemokine receptor expression or functionality was not affected by the infection and thus that these signaling pathways are unlikely to contribute to the observed lymph node changes.
B cell accumulation in lymph nodes after B. burgdorferi infection is dependent on type I IFN signaling.
Innate signaling via TLR4 was shown previously to be associated with histological changes in lymph nodes of Salmonella-infected mice (14), suggesting that innate signals might also be responsible for the changes seen following B. burgdorferi infection. However, mice deficient in the TLR adapter molecule MyD88 or in both MyD88 and TRIF showed high B cell frequencies (Fig. 5A and B) similar to those of wild-type mice on day 10 postinfection, and MyD88−/− mice showed the same level of destruction of the lymph node structure as wild-type mice after Borrelia infection (Fig. 5C). Thus, TLR signaling did not appear to play a role in the observed alterations.
FIG 5.

B cell accumulation is type I IFN dependent but MyD88 and TRIF independent. (A) Mean frequencies + SD of CD19+ B cells, CD4+ T cells, and CD8+ T cells as assessed by flow cytometry in wild-type (WT) and MyD88−/− mice (n = 8/group) at day 10 postinfection. n.s., not significant. (B) Mean frequencies and SD of CD19+ B cells, CD4+ T cells, and CD8+ T cells as assessed by flow cytometry in WT and MyD88−/− × TRIF−/− mice (n = 4/group) on day 15 postinfection. (C) H&E staining of representative WT, IFNAR−/−, and MyD88−/− mice (n = 2/group) at day 10 postinfection (magnification, ×10). (D) Mean relative expression levels (+ SEM) compared to GAPDH of type I IFN-induced genes (IFIT1 and IFIT2), IFN-α4, and IFN-β measured by qRT-PCR of lymph nodes from day 0 (uninfected, black bars), day 10 and 15 B. burgdorferi-infected (white bars), and sham-infected (gray bars) WT mice (n = 4/group). (E) Frequencies + SD of plasmacytoid DC in WT mice (n = 8/group) at day 10 after infection or immunization with B. burgdorferi lysate in adjuvant. (F) Mean frequencies + SD (n = 4 to 8/group) of CD19+ B cells in draining lymph nodes of WT (dashed line) and IFNAR−/− (solid line) mice at the indicated times after infection.
The innate effector cytokine family, type I IFN, was reportedly increased in the blood of humans and at the site of erythema migrans following B. burgdorferi infection (24). It also seems to play a role in B. burgdorferi infection-induced arthritis development in mice (25). Infection with B. burgdorferi also induces TLR2-dependent and -independent IFN-β production by bone marrow-derived macrophages (26). Indeed, much of its production after B. burgdorferi infection appears to be induced independently of MyD88 and TRIF signaling (27). Together the data indicated that type I IFN is strongly induced by B. burgdorferi infection but might be induced independently of TLR signaling. Indeed, qRT-PCR analysis on lymph nodes of B. burgdorferi-infected mice as well as sham-infected mice showed a strong induction of two type I IFN-induced genes, IFIT1 and IFIT2, as well as modestly increased gene expression for IFN-α4 and IFN-β on day 10 of infection compared to controls (Fig. 5D). The increases in IFN expression were not associated with an increase in the frequencies of plasmacytoid dendritic cells (pDC) (CD3−, CD19−, CD45RA+ CD11c− [28]), which are known strong producers of IFN-α (Fig. 5E).
To determine if type I IFN played a role in the strong B cell accumulation seen after B. burgdorferi infection, we compared B cell numbers in lymph nodes from control mice and IFNAR−/− mice, which lack all responsiveness to type I IFN. While the infection caused increases in cell numbers and B cell frequencies in wild-type and IFNAR−/− mice (Fig. 5F), the rate of B cell accumulation in the IFNAR−/− mice was significantly lower than that in the infected controls (Fig. 5F), causing a reduction in the total numbers of lymph node cells in IFNAR−/− mice compared to their wild-type counterparts (data not shown). However, the lack of IFNAR signaling did not affect the lymph node structure, as IFNAR−/− mice showed the same level of tissue destruction as the wild-type mice on day 10 of infection (Fig. 5C). Thus, we conclude that MyD88- and TRIF-independent type I IFN signaling was required for the overshooting B cell accumulation that follows the destruction of the lymph node architecture after B. burgdorferi infection.
Type I IFN-mediated accumulation of B cells occurs independently of direct B cell signaling.
Previous studies have shown that B. burgdorferi causes mitogenic stimulation of naive B cells in vitro (29, 30), and many Ki67+ cells; i.e., cycling cells are present in the draining lymph nodes of B. burgdorferi-infected mice at day 10 of infection (3). To determine whether B cell proliferation may be differentially affected by innate signaling, we compared the rates of B cell proliferation in wild-type and MyD88−/− mice with that in IFNAR−/− mice by fluorescence-activated cell sorter (FACS) analysis of lymph node cells from mice infected with B. burgdorferi for 10 days and repeatedly injected with BrdU. The rate of B cell proliferation, as measured by the frequencies of BrdU+ cells, was similar in all strains of mice and indeed was not any higher than that of the T cells in the lymph nodes (Fig. 6A), which do not significantly increase (7). Therefore, the differential accumulation of B cells in wild-type and IFNAR−/− mice cannot be explained by different rates of B cell proliferation.
FIG 6.

Type I IFNR signaling affects B cell accumulation extrinsically. (A) Top, representative contour plots of BrdU+ CD19+ B cells from wild-type (WT), MyD88−/−, and IFNAR−/− mice, indicating the percentage of recently divided cells at day 10 of infection. FMO, fluorescence-minus-one control stain. The bar chart indicates mean frequencies of BrdU+ cells of the indicated cell populations in WT and MyD88−/− mice. *, P < 0.05; **, P ≤ 0.01; n.s., not significant. (B) Mean frequency + SD of CD69+ B cells in WT mice (n = 4 to 8/time point) before and at the indicated times after infection as measured by flow cytometry. (C) Mean numbers + SD (n = 8/group) of the indicated cell populations in draining lymph nodes of B cell-specific IFNAR−/− (CD19.IFNAR−/−) mice and control littermates at day 10 postinfection. (D) Mean frequencies + SD of IgM (left) and IgG (right) antibody-secreting cells calculated per B cells in draining lymph nodes of WT and IFNAR−/− mice (n = 4/group) on day 10 of infection as assessed by ELISPOT with a pool of B. burgdorferi recombinant proteins (DbpA, Arp, OspC, and BmpA) and flow cytometry.
Type I IFN signaling can regulate lymph node cell numbers following immunization by inducing CD69 expression on lymphocytes, which in turn blocks S1P1-mediated egress from lymph nodes, leading to strong cell retention (31). However, when we studied the rate of CD69 expression on B cells within B. burgdorferi-infected lymph nodes, we found only small frequencies of CD69-expressing B cells over a 15-day time course (Fig. 6B), even on day 10, the height of IFIT1 and IFIT2 expression (Fig. 5D), suggesting that type I IFN does not regulate B cell retention via CD69 and S1P1. The indirect effect of type I IFN on B cell accumulation/retention was confirmed in studies in which we infected mice that lack the IFNAR only on B cells (CD19. IFNAR−/− mice [32]). These mice showed no reduction in lymph node B cell frequencies after Borrelia infection compared to wild-type controls (Fig. 6C).
Finally, while type I IFN signaling drives the B cell accumulation in the lymph node, it does not affect the Borrelia-specific antibody-secreting cell numbers, as these remained unaffected at their peak on day 10 (Fig. 6D). This was consistent with our previous data showing that the accumulating lymph nodes B cells were mainly naive follicular cells (7), which do not participate in the immune response. What long-term consequences type I IFN signaling may have for the quality or magnitude of the B cell response to B. burgdorferi remains to be studied further. We conclude that B. burgdorferi infection-induced histopathological changes are induced prior to and by mechanisms other than the type I IFN-mediated signaling events, which force an exacerbated naive B cell accumulation in affected lymph nodes by an unknown B cell extrinsic signaling pathway.
DISCUSSION
We demonstrate that infection with B. burgdorferi results in the loss of the lymph node's normal tissue architecture within the first week after infection, which is then followed a few days later by the IFNR signaling-dependent accumulation of unusually large numbers of naive B cells at that site. We propose that the induction of apparently aberrant innate signals in lymph nodes represents an evasion strategy of B. burgdorferi designed to disrupt the development of protective immune responses. This may explain the rapid loss of germinal centers in lymph nodes of B. burgdorferi-infected mice (7) and/or the lack of adequate long-term immune protection observed following infection (9, 33).
Consistent with our previous study (7), we show here that the increase in lymph node cellularity after infection with B. burgdorferi is due nearly exclusively to the accumulation of naive B cells. We further provide evidence that the B cell accumulation in the draining lymph nodes is not due to their excessive proliferation (Fig. 5E) or to the overexpression of the B cell-homing chemokine CXCL13 (Fig. 2 and 3). Instead, it is mediated by type I IFN signaling (Fig. 5). While IFNAR−/− mice had reduced frequencies of B cells in their lymph nodes, the frequencies of Borrelia-specific antibody-secreting cells were similar to those in wild-type mice (Fig. 5G). This indicates that the IFN-mediated increase in B cell numbers in wild-type mice does not benefit the induction of specific B cell immunity but may contribute further to the destruction of the normal lymph node tissue organization and/or the interaction of antigen-specific T and B cells and ultimately the development of robust germinal center responses and highly protective humoral immunity.
The molecular mechanism underlying the here-discovered IFNR signaling effects on selective B cell accumulation remains to be explored. IFN signaling has been linked previously to the regulation of lymphocyte egress from secondary lymphoid tissues. Naive lymphocytes exit the lymph node via S1P receptors, mainly S1P1, driven by a primarily endothelial cell-generated S1P gradient (34, 35). Type I IFN induces the upregulation of CD69 on B cells (31), which in turn can block the S1P-mediated egress from the lymph nodes by forming a complex with S1P1 (31). However, only small frequencies (about 5%) of B cells expressed CD69 after B. burgdorferi infection (Fig. 6C), and deletion of IFNAR1 only on B cells did not reduce the infection-induced B cell accumulation (Fig. 6D), while deletion on all cells strongly reduced overall B cell frequencies (Fig. 5B). Thus, IFNR signaling acts on cells other than B cells to affect B cell accumulation. Preliminary data with bone marrow chimeras between wild-type and IFNAR−/− mice suggest that other bone marrow-derived cells may be responsible (Fig. 6E).
The delicate balance of S1P and its receptor S1P1 on B cells remains to be explored further, as S1P1 is sensitive to downregulation upon binding to its ligand (36). Previous reports have shown that type I IFN-responsive endothelial cells produce S1P in the lymph node (35, 37). SphK1, a kinase necessary for S1P production and known to be upregulated after stimulation with bacterial lipoproteins (23), may increase S1P production in IFN-responding endothelial cells. Indeed, SphK1 expression was reduced to a lesser extent in infected mice than in immunized mice (Fig. 3). Relative increases in S1P may cause internalization of the S1P1 receptor on the B cells, blocking egress from the lymph node.
Type I IFN is produced in response to infections with both extracellular and intracellular pathogens, including infections with B. burgdorferi (24, 25, 38, 39). Type I IFN is found in skin at the site of the erythema migrans as well as in sera of humans infected with B. burgdorferi (24). Blockade of type I IFN reduced Lyme arthritis in mouse studies (25). Induction of type I IFN production is one downstream effect of TLR stimulation. Previous studies have indicated roles for MyD88 and TRIF in the type I IFN response of bone marrow-derived macrophages following B. burgdorferi stimulation (40). Our data show, however, that the B. burgdorferi infection-induced B cell accumulation is dependent on type I IFNR signaling but independent of the major TLR adapter proteins MyD88 and TRIF (Fig. 5). This is consistent with previous studies showing that type I IFN-responsive gene induction following stimulation of bone marrow-derived macrophages with B. burgdorferi was MyD88, TRIF, and Nod2 independent but IRF3 dependent (27), although a role for Nod2 was suggested by others (41). Furthermore, studies with human cells have suggested a role for various TLRs in B. burgdorferi-induced IFN production (42, 43). Thus, it appears likely that multiple pathways lead to type I IFN production following B. burgdorferi infection, depending on the major cell type involved. As we show here, this can occur independently of TLR engagement.
Infection with numerous pathogens causes lymph node architecture alteration. As we show here, type I IFN or the adapter molecules MyD88 and TRIF play no role in lymph node remodeling following B. burgdorferi infection. This is in contrast to infection with Salmonella, for which the mechanisms have been linked to TLR4-induced downregulation of CXCL13 and CCL21 (14). Thus, B. burgdorferi must use a different mechanism to cause very similar lymph node changes. We also failed to find any evidence for suppression of chemokines, beyond those observed after immunization (Fig. 2 and 3), indicating that changes in architecture and an increase in B cell numbers are unrelated to CXCL13, CCL21, CXCR4, CXCR5, or CCR7 expression or the migratory capacity of B cells toward these chemokines (Fig. 4; see Fig. S2 in the supplemental material). Furthermore, we found no evidence for an involvement of TNF-α (Fig. 3; see Fig. S1 in the supplemental material), which had been linked previously to lymph node hypertrophy during infection with Escherichia coli (17). Together the data reveal that different pathogens exploit distinct innate response pathways with the common purpose to disrupt the normal lymph node architecture.
The disruption of the lymph node architecture might significantly impact the effectiveness of the lymph node response. In the case of Lyme disease, we previously reported that they lack strong and sustained germinal centers. Others demonstrated the lack of strong functional immunity following infection with B. burgdorferi, which permits reinfections even with the same strain of B. burgdorferi (9, 33). By revealing the two-step process that leads to the disruption of the lymph node architecture and the subsequent IFN-driven accumulation of B cells, we identify distinct mechanisms that B. burgdorferi employs to interact with and potentially to manipulate the host response to its advantage. A better understanding of these processes might ultimately allow interventions that can boost the host's own immune system to enable bacterial clearance.
Supplementary Material
ACKNOWLEDGMENTS
We thank Abigail Spinner for help in operating the BD FACSAria and Adam Treistar (TreeStar Inc.) for FlowJo software.
This work was supported by an NIH/NIAID grant (AI073911) to N.B. and S.W.B. and by a T32 training grant (AI060555) to C.J.H.
Footnotes
Published ahead of print 22 January 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00969-13.
REFERENCES
- 1.Steere AC. 1989. Lyme disease. N. Engl. J. Med. 321:586–596. 10.1056/NEJM198908313210906 [DOI] [PubMed] [Google Scholar]
- 2.Steere AC. 2001. Lyme disease. N. Engl. J. Med. 345:115–125. 10.1056/NEJM200107123450207 [DOI] [PubMed] [Google Scholar]
- 3.Tunev SS, Hastey CJ, Hodzic E, Feng S, Barthold SW, Baumgarth N. 2011. Lymphoadenopathy during lyme borreliosis is caused by spirochete migration-induced specific B cell activation. PLoS Pathog. 7:e1002066. 10.1371/journal.ppat.1002066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kang I, Barthold SW, Persing DH, Bockenstedt LK. 1997. T-helper-cell cytokines in the early evolution of murine Lyme arthritis. Infect. Immun. 65:3107–3111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barthold SW, Bockenstedt LK. 1993. Passive immunizing activity of sera from mice infected with Borrelia burgdorferi. Infect. Immun. 61:4696–4702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ganapamo F, Dennis VA, Philipp MT. 2001. CD19(+) cells produce IFN-gamma in mice infected with Borrelia burgdorferi. Eur. J. Immunol. 31:3460–3468. [DOI] [PubMed] [Google Scholar]
- 7.Hastey CJ, Elsner RA, Barthold SW, Baumgarth N. 2012. Delays and diversions mark the development of B cell responses to Borrelia burgdorferi infection. J. Immunol. 188:5612–5622. 10.4049/jimmunol.1103735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Radolf JD, Caimano MJ, Stevenson B, Hu LT. 2012. Of ticks, mice and men: understanding the dual-host lifestyle of Lyme disease spirochaetes. Nat. Rev. Microbiol. 10:87–99. 10.1038/nrmicro2714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nowakowski J, Schwartz I, Nadelman RB, Liveris D, Aguero-Rosenfeld M, Wormser GP. 1997. Culture-confirmed infection and reinfection with Borrelia burgdorferi. Ann. Intern. Med. 127:130–132. 10.7326/0003-4819-127-2-199707150-00006 [DOI] [PubMed] [Google Scholar]
- 10.Piesman J, Dolan MC, Happ CM, Luft BJ, Rooney SE, Mather TN, Golde WT. 1997. Duration of immunity to reinfection with tick-transmitted Borrelia burgdorferi in naturally infected mice. Infect. Immun. 65:4043–4047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cyster JG. 2005. Chemokines, sphingosine-1-phosphate, and cell migration in secondary lymphoid organs. Annu. Rev. Immunol. 23:127–159. 10.1146/annurev.immunol.23.021704.115628 [DOI] [PubMed] [Google Scholar]
- 12.Forster R, Mattis AE, Kremmer E, Wolf E, Brem G, Lipp M. 1996. A putative chemokine receptor, BLR1, directs B cell migration to defined lymphoid organs and specific anatomic compartments of the spleen. Cell 87:1037–1047. 10.1016/S0092-8674(00)81798-5 [DOI] [PubMed] [Google Scholar]
- 13.Mori S, Nakano H, Aritomi K, Wang CR, Gunn MD, Kakiuchi T. 2001. Mice lacking expression of the chemokines CCL21-ser and CCL19 (plt mice) demonstrate delayed but enhanced T cell immune responses. J. Exp. Med. 193:207–218. 10.1084/jem.193.2.207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.St John AL, Abraham SN. 2009. Salmonella disrupts lymph node architecture by TLR4-mediated suppression of homeostatic chemokines. Nat. Med. 15:1259–1265. 10.1038/nm.2036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Narayan K, Dail D, Li L, Cadavid D, Amrute S, Fitzgerald-Bocarsly P, Pachner AR. 2005. The nervous system as ectopic germinal center: CXCL13 and IgG in lyme neuroborreliosis. Ann. Neurol. 57:813–823. 10.1002/ana.20486 [DOI] [PubMed] [Google Scholar]
- 16.Rupprecht TA, Kirschning CJ, Popp B, Kastenbauer S, Fingerle V, Pfister HW, Koedel U. 2007. Borrelia garinii induces CXCL13 production in human monocytes through Toll-like receptor 2. Infect. Immun. 75:4351–4356. 10.1128/IAI.01642-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McLachlan JB, Hart JP, Pizzo SV, Shelburne CP, Staats HF, Gunn MD, Abraham SN. 2003. Mast cell-derived tumor necrosis factor induces hypertrophy of draining lymph nodes during infection. Nat. Immunol. 4:1199–1205. 10.1038/ni1005 [DOI] [PubMed] [Google Scholar]
- 18.Barbour AG. 1984. Isolation and cultivation of Lyme disease spirochetes. Yale J. Biol. Med. 57:521–525 [PMC free article] [PubMed] [Google Scholar]
- 19.Rothaeusler K, Baumgarth N. 2006. Evaluation of intranuclear BrdU detection procedures for use in multicolor flow cytometry. Cytometry A 69:249–259. 10.1002/cyto.a.20252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duray PH, Steere AC. 1988. Clinical pathologic correlations of Lyme disease by stage. Ann. N. Y. Acad. Sci. 539:65–79. 10.1111/j.1749-6632.1988.tb31839.x [DOI] [PubMed] [Google Scholar]
- 21.Pachner AR, Dail D, Narayan K, Dutta K, Cadavid D. 2002. Increased expression of B-lymphocyte chemoattractant, but not pro-inflammatory cytokines, in muscle tissue in rhesus chronic Lyme borreliosis. Cytokine 19:297–307. 10.1006/cyto.2002.1973 [DOI] [PubMed] [Google Scholar]
- 22.Ljostad U, Mygland A. 2008. CSF B-lymphocyte chemoattractant (CXCL13) in the early diagnosis of acute Lyme neuroborreliosis. J. Neurol. 255:732–737. 10.1007/s00415-008-0785-y [DOI] [PubMed] [Google Scholar]
- 23.Puneet P, Yap CT, Wong L, Lam Y, Koh DR, Moochhala S, Pfeilschifter J, Huwiler A, Melendez AJ. 2010. SphK1 regulates proinflammatory responses associated with endotoxin and polymicrobial sepsis. Science 328:1290–1294. 10.1126/science.1188635 [DOI] [PubMed] [Google Scholar]
- 24.Salazar JC, Pope CD, Sellati TJ, Feder HM, Jr, Kiely TG, Dardick KR, Buckman RL, Moore MW, Caimano MJ, Pope JG, Krause PJ, Radolf JD. 2003. Coevolution of markers of innate and adaptive immunity in skin and peripheral blood of patients with erythema migrans. J. Immunol. 171:2660–2670 [DOI] [PubMed] [Google Scholar]
- 25.Miller JC, Ma Y, Bian J, Sheehan KC, Zachary JF, Weis JH, Schreiber RD, Weis JJ. 2008. A critical role for type I IFN in arthritis development following Borrelia burgdorferi infection of mice. J. Immunol. 181:8492–8503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Salazar JC, Duhnam-Ems S, La Vake C, Cruz AR, Moore MW, Caimano MJ, Velez-Climent L, Shupe J, Krueger W, Radolf JD. 2009. Activation of human monocytes by live Borrelia burgdorferi generates TLR2-dependent and -independent responses which include induction of IFN-beta. PLoS Pathog. 5:e1000444. 10.1371/journal.ppat.1000444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller JC, Maylor-Hagen H, Ma Y, Weis JH, Weis JJ. 2010. The Lyme disease spirochete Borrelia burgdorferi utilizes multiple ligands, including RNA, for interferon regulatory factor 3-dependent induction of type I interferon-responsive genes. Infect. Immun. 78:3144–3153. 10.1128/IAI.01070-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Colonna M, Trinchieri G, Liu YJ. 2004. Plasmacytoid dendritic cells in immunity. Nat. Immunol. 5:1219–1226. 10.1038/ni1141 [DOI] [PubMed] [Google Scholar]
- 29.de Souza MS, Fikrig E, Smith AL, Flavell RA, Barthold SW. 1992. Nonspecific proliferative responses of murine lymphocytes to Borrelia burgdorferi antigens. J. Infect. Dis. 165:471–478. 10.1093/infdis/165.3.471 [DOI] [PubMed] [Google Scholar]
- 30.Yang L, Ma Y, Schoenfeld R, Griffiths M, Eichwald E, Araneo B, Weis JJ. 1992. Evidence for B-lymphocyte mitogen activity in Borrelia burgdorferi-infected mice. Infect. Immun. 60:3033–3041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shiow LR, Rosen DB, Brdickova N, Xu Y, An J, Lanier LL, Cyster JG, Matloubian M. 2006. CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature 440:540–544. 10.1038/nature04606 [DOI] [PubMed] [Google Scholar]
- 32.Rickert RC, Roes J, Rajewsky K. 1997. B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nucleic Acids Res. 25:1317–1318. 10.1093/nar/25.6.1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Golde WT, Robinson-Dunn B, Stobierski MG, Dykhuizen D, Wang IN, Carlson V, Stiefel H, Shiflett S, Campbell GL. 1998. Culture-confirmed reinfection of a person with different strains of Borrelia burgdorferi sensu stricto. J. Clin. Microbiol. 36:1015–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schwab SR, Pereira JP, Matloubian M, Xu Y, Huang Y, Cyster JG. 2005. Lymphocyte sequestration through S1P lyase inhibition and disruption of S1P gradients. Science 309:1735–1739. 10.1126/science.1113640 [DOI] [PubMed] [Google Scholar]
- 35.Pham TH, Baluk P, Xu Y, Grigorova I, Bankovich AJ, Pappu R, Coughlin SR, McDonald DM, Schwab SR, Cyster JG. 2010. Lymphatic endothelial cell sphingosine kinase activity is required for lymphocyte egress and lymphatic patterning. J. Exp. Med. 207:17–27. 10.1084/jem.20091619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cyster JG, Schwab SR. 2012. Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu. Rev. Immunol. 30:69–94. 10.1146/annurev-immunol-020711-075011 [DOI] [PubMed] [Google Scholar]
- 37.Pappu R, Schwab SR, Cornelissen I, Pereira JP, Regard JB, Xu Y, Camerer E, Zheng YW, Huang Y, Cyster JG, Coughlin SR. 2007. Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science 316:295–298. 10.1126/science.1139221 [DOI] [PubMed] [Google Scholar]
- 38.Trinchieri G. 2010. Type I interferon: friend or foe? J. Exp. Med. 207:2053–2063. 10.1084/jem.20101664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parker D, Prince A. 2011. Type I interferon response to extracellular bacteria in the airway epithelium. Trends Immunol. 32:582–588. 10.1016/j.it.2011.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petnicki-Ocwieja T, Chung E, Acosta DI, Ramos LT, Shin OS, Ghosh S, Kobzik L, Li X, Hu LT. 2013. TRIF mediates Toll-like receptor 2-dependent inflammatory responses to Borrelia burgdorferi. Infect. Immun. 81:402–410. 10.1128/IAI.00890-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Petnicki-Ocwieja T, DeFrancesco AS, Chung E, Darcy CT, Bronson RT, Kobayashi KS, Hu LT. 2011. Nod2 suppresses Borrelia burgdorferi mediated murine Lyme arthritis and carditis through the induction of tolerance. PLoS One 6:e17414. 10.1371/journal.pone.0017414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Petzke MM, Brooks A, Krupna MA, Mordue D, Schwartz I. 2009. Recognition of Borrelia burgdorferi, the Lyme disease spirochete, by TLR7 and TLR9 induces a type I IFN response by human immune cells. J. Immunol. 183:5279–5292. 10.4049/jimmunol.0901390 [DOI] [PubMed] [Google Scholar]
- 43.Cervantes JL, Dunham-Ems SM, La Vake CJ, Petzke MM, Sahay B, Sellati TJ, Radolf JD, Salazar JC. 2011. Phagosomal signaling by Borrelia burgdorferi in human monocytes involves Toll-like receptor (TLR) 2 and TLR8 cooperativity and TLR8-mediated induction of IFN-beta. Proc. Natl. Acad. Sci. U. S. A. 108:3683–3688. 10.1073/pnas.1013776108 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.