

Abstract
The virulence factors mediating Francisella pathogenesis are being investigated, with an emphasis on understanding how the organism evades innate immunity mechanisms. Francisella tularensis produces a lipopolysaccharide (LPS) that is essentially inert and a polysaccharide capsule that helps the organism to evade detection by components of innate immunity. Using an F. tularensis Schu S4 mutant library, we identified strains that are disrupted for capsule and O-antigen production. These serum-sensitive strains lack both capsule production and O-antigen laddering. Analysis of the predicted protein sequences for the disrupted genes (FTT1236 and FTT1238c) revealed similarity to those for waa (rfa) biosynthetic genes in other bacteria. Mass spectrometry further revealed that these proteins are involved in LPS core sugar biosynthesis and the ligation of O antigen to the LPS core sugars. The 50% lethal dose (LD50) values of these strains are increased 100- to 1,000-fold for mice. Histopathology revealed that the immune response to the F. tularensis mutant strains was significantly different from that observed with wild-type-infected mice. The lung tissue from mutant-infected mice had widespread necrotic debris, but the spleens lacked necrosis and displayed neutrophilia. In contrast, the lungs of wild-type-infected mice had nominal necrosis, but the spleens had widespread necrosis. These data indicate that murine death caused by wild-type strains occurs by a mechanism different from that by which the mutant strains kill mice. Mice immunized with these mutant strains displayed >10-fold protective effects against virulent type A F. tularensis challenge.
INTRODUCTION
Francisella tularensis is a small Gram-negative bacterium that causes tularemia in humans. This bacterial pathogen causes disease by diverse routes, including oral, intravenous, subcutaneous, and pneumonic routes; the respiratory route is of particular concern, because infection with 10 or fewer organisms is associated with mortality rates of 30 to 60% if left untreated (1, 2). The pathogen employs a virulence strategy in which it enters host cells, escapes the phagosome, and grows within the cytosol of the cell, apparently with diminished or delayed detection by host innate defenses (3). The interplay of bacteria with host macrophages is a significant focus of research efforts, as these interactions are critical to virulence (4). However, the organism also has important interactions with other cells, including epithelial and endothelial cells, hepatocytes, and neutrophils (5–9). Migration of infected leukocytes throughout the vasculature leads to seeding of sites in the lymphatic system, establishment of systemic disease, and, eventually, death by multiple-organ failure (10). Due to the extreme virulence and ease of aerosol dissemination of F. tularensis, the U.S. Centers for Disease Control and Prevention have classified this organism as a tier 1 select agent because of its potential for development into a bioweapon (11).
One important contributing factor to the high virulence of this pathogen is that its intracellular growth appears to be relatively unchecked because it fails to induce significant host immune responses (12). Lipid A of F. tularensis is highly unusual in that it is asymmetrical and tetra-acylated and has long-chain fatty acids (16 to 18 carbons). In addition, the phosphate at the 1 position on the diglucosamine backbone can be shielded by a galactosamine, while a phosphate group at the 4′ position of the sugar backbone has not been observed (13–15). This is in contrast to the lipid A structures of most Gram-negative bacteria, which contain six acyl chains of 12 to 14 carbons and phosphate groups that are available for interactions with Toll-like receptor 4 (TLR4), which can stimulate a strong proinflammatory response (16–18). In comparison, the endotoxin of F. tularensis does not bind to lipopolysaccharide-binding protein (LBP) and therefore does not activate TLR4 signaling pathways, rendering it inert compared to typical endotoxins (15, 19). Several research groups are working to understand how acylation, the length of fatty acid side chains, and shielding of the phosphates of the endotoxin contribute to the lack of bioactivity of F. tularensis lipopolysaccharide (LPS). In addition, F. tularensis LPS and capsule opsonize complement components that aid in the uptake of the bacterium into host cells while simultaneously protecting the organism from the killing activity of complement (20). Each of these areas of immune evasion continues to be an active area of research to understand the Francisella virulence strategy.
Our research group is actively focusing on understanding how the Francisella capsule contributes to the pathogenesis of this organism. Work published in 1977 described a crude “capsular” preparation that contained mannose, rhamnose, and dideoxy sugars (21). Sandstrom et al. created an acapsular Francisella strain and found that it was sensitive to antibody-mediated killing (22). Recent work using a monoclonal antibody directed against purified capsular material of Francisella has helped to characterize several aspects of this capsule (23). The capsular material was shown to range in size from 100 to 250 kDa, and the material could be separated from LPS by selective Triton X-114 treatment. Furthermore, the capsule was detected in all type A and B strains of Francisella tularensis that were examined (23). Immunization of mice with purified capsular material elicited circulating anti-capsule antibodies in the mice that protected them from challenge with F. tularensis LVS but did not protect from F. tularensis Schu S4 challenge (23). In this report, we extend our previous findings by characterizing in more detail how the FTT1236 and FTT1238c gene products participate in LPS core biosynthesis and examining how these structures contribute to the virulence of this pathogen in the murine host. Based on results presented here, we propose the gene names waaY, waaZ, and waaL for the F. tularensis FTT1236, FTT1237, and FTT1238c genes, respectively. We have chosen to use the “waa” nomenclature with these genes because the proteins produced are involved in LPS core biosynthesis as well as O-antigen ligation to the core of the LPS (24). However, the genes are not exact orthologues of the Escherichia coli or Salmonella enterica genes, so the last letter of each gene name is unique to Francisella to make this point. FTT1238c is the exception, in that the Francisella protein appears to have the same role as WaaL proteins in other organisms. Our data also demonstrate that these mutant strains can evoke protection and a delay to death after lethal Schu S4 challenge of mice.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Francisella tularensis subsp. tularensis Schu S4 was routinely cultured on modified Mueller-Hinton (MMH) plates (Acumedia, Lansing, MI) or in MMH broth. E. coli mutants were acquired from the Keio collection (National Bioresource Project, Japan), and the E. coli parent BW25113 was obtained from David Weiss (University of Iowa). The Salmonella enterica serovar Typhimurium SL1344 strain and E. coli strains were grown on L agar or in L broth supplemented with either 50 μg kanamycin or 50 μg chloramphenicol, as appropriate. All work with F. tularensis Schu S4 was performed within the Carver College of Medicine Biosafety Level 3 (BSL3) Core Facility, and experimental protocols were reviewed for safety by the BSL3 Oversight Committee of The University of Iowa Carver College of Medicine. Recombinant DNA work with F. tularensis Schu S4 was reviewed and approved by the University of Iowa Institutional Biosafety Committee.
We constructed LVS 0708::TrgTn and LVS 0706::TrgTn mutants by using previously constructed plasmids (25) and the Targetron system (Sigma). These mutations appeared to have effects on the lipid A core structures of the LVS mutants identical to those in the corresponding Schu S4 FTT1236::TrgTn and Schu S4 FTT1238::TrgTn mutants. The LVS mutant strains were used to purify sufficient quantities of LPS to determine the structures and compositions of the lipid A core structures.
Creation of a Tn5lacZ Francisella tularensis Schu S4 mutant library.
Tn5lacZ insertions were generated by introducing the temperature-sensitive transposon delivery plasmid pBB105 into Francisella tularensis Schu S4 via cryotransformation (26). Transformed Francisella organisms were selected on MMH plates containing kanamycin at 32°C to select for the presence of pBB105. A single kanamycin-resistant colony was cultured in 2 ml of MMH broth with kanamycin and grown overnight at 32°C to an optical density at 600 nm (OD600) of ∼1.1. The culture was serially diluted, plated onto chocolate agar, and grown at 40°C in 5% CO2 for 48 h to cure the temperature-sensitive delivery plasmid and select for Tn5lacZ chromosomal insertion mutants. Mutant colonies were picked, arrayed into 96-well plates containing 100 μl of MMH broth, and grown for 24 h at 37°C with 5% CO2. Approximately 7,500 mutants were created and isolated using this technique. Freezing medium was added to each well, at a 1:1 ratio, and plates were stored at −80°C for long-term storage.
Screening for F. tularensis Schu S4 capsule-deficient mutants by ELISA.
In preparation for screening of the transposon library, each mutant in the 96-well plate library was transferred from the master plate to a fresh plate containing 200 μl of MMH broth per well. Plates were incubated at 37°C for 48 h. Plates were read by measuring the OD600 in a microplate autoreader (Bio-Tek Instruments). A final concentration of 4% paraformaldehyde (PFA) was used to simultaneously fix and kill the organisms in each well. Wells were air dried to evaporate the broth-PFA mixture, and then each well was washed with phosphate-buffered saline (PBS) and blocked with 1% bovine serum albumin (BSA) in Tris-buffered saline (TBS). Next, the dead organisms were incubated with the primary antibody 11B7 (1:500 dilution), washed with PBS, incubated with a goat anti-mouse IgG secondary antibody (1:2,500; Jackson Immunoresearch), and washed with PBS again before adding the p-nitrophenyl phosphate substrate. Binding of antibody to capsular material was quantitated by correlation to the production of the p-nitrophenol product, which was measured by the OD405. Data were normalized to the value for the wild type (WT), which was arbitrarily set to 100%, and the OD405/600 ratio of each well was compared to that of the parent strain positive control. An arbitrary cutoff of 15% of the WT level was used to identify F. tularensis Schu S4 strains with reduced binding to the anti-capsule antibody. Additionally, mutants that met the 15% cutoff were characterized further by a second enzyme-linked immunosorbent assay (ELISA) using the monoclonal antibody FB11 (QED Biosciences) to determine if the LPS structure was also altered.
Identification of F. tularensis capsule mutants.
Mutants of interest were picked, plated onto MMH agar containing kanamycin, and grown at 37°C with 5% CO2 for 48 h. A small loopful of bacteria was collected from the agar plate to obtain genomic DNA by use of a DNeasy blood and tissue kit (Qiagen). The genomic DNA was digested with the restriction enzyme EcoRI or NdeI, ligated with T4 DNA ligase, and then transformed into E. coli λpir-1 cells before plating on LB plates with kanamycin. This approach clones the majority of the transposon sequence, including the kanamycin resistance gene and flanking DNA, since the Tn5lacZ transposon carries the R6K plasmid origin of replication. Inclusion of this plasmid origin on the transposon allows recovery of clones in E. coli host strains that express the Pir protein for replication. Single colonies were picked and grown in LB broth with kanamycin. Plasmid purification was done by using a plasmid miniprep kit (Qiagen). A Tn5 primer was used to sequence the genomic DNA flanking the transposon insertion at The University of Iowa School of Medicine DNA Core.
Protein similarity analysis and functional complementation experiments.
The F. tularensis Schu S4 FTT1236, FTT1237, and FTT1238c gene products were compared to proteins encoded in the E. coli MG1655 genome by using NCBI's position-specific iterated BLAST (PSI-BLAST) function. HMMTOP and TMRPres2D software was used to predict transmembrane helices and topology of the FTT1238c gene product. Clustal Omega was used to align sequences of WaaL proteins from E. coli and Salmonella with the translated FTT1238c sequence of F. tularensis. Plasmids carrying sequences for FTT1236 (pSL129), FTT1237 (pSL130), or FTT1238c (pSL149) were transformed into E. coli waa mutants (Keio strains) or into the S. enterica SL1344 ΔwaaL mutant constructed for this work and were selected on 50 μg spectinomycin in L agar. Colonies were picked and grown in L broth for 12 to 24 h at 37°C. One milliliter of broth culture was centrifuged at 8,000 × g for 2 min, the cell pellet was resuspended in buffer A, and organisms were lysed by heating at 65°C. Samples were treated with proteinase K (Sigma) at 37°C for 24 h. Samples were examined for carbohydrate content by a silver staining method. Briefly, samples separated by SDS-PAGE were incubated in a mixture of 30% ethanol and 10% acetic acid for 30 min. The solution was decanted before incubation of the gel in 10% ethanol for 15 min. The ethanol was removed, and 5% glutaraldehyde was added for 15 min. The gel was washed six times with double-distilled water (ddH2O) for 5 min each, and then the gel was bathed in a 5-μg/ml dithiothreitol (DTT) solution for 15 min. AgNO3 (0.1%) was added, and the gel was incubated for 15 min. The gel was then washed extensively with ddH2O and developed with 3% Na2CO3 and 0.02% formaldehyde until bands appeared. A 3% acetic acid solution was used to stop the reaction.
PAGE, immunoblotting, and emerald green staining of bacterial whole-cell lysates.
Francisella strains from newly streaked agar plates were inoculated into MMH broth and grown for 24 h before the OD600 was measured and recorded. One-milliliter broth cultures were centrifuged at 8,000 × g for 2 min before resuspension of each pellet in buffer A (6 mM Tris, 10 mM EDTA, and 2% [wt/vol] sodium dodecyl sulfate [pH 6.8]) and heating to 65°C to sterilize cultures. Bacterial lysates were then incubated with or without proteinase K (New England BioLabs, Ipswich, MA) at 37°C for 24 h before being lyophilized. Approximately 14 μg of bacterial material from each sample was mixed with NuPage (Life Technologies, Carlsbad, CA) sample reducing agent and buffer, boiled for 10 min, loaded into a 4 to 12% Bis-Tris NuPage gel, and electrophoresed using NuPage morpholineethanesulfonic acid (MES)-SDS running buffer (Life Technologies, Carlsbad, CA). For immunoblots, samples were transferred to nitrocellulose and probed with either the primary antibody 11B7 (to detect capsule) or FB11 (to detect LPS O antigen) (QED Bioscience, San Diego, CA). Bands were visualized using goat anti-mouse IgG(H+L) conjugated to horseradish peroxidase (Jackson ImmunoResearch, West Grove, PA) and SuperSignal West Pico chemiluminescence substrate (Pierce Biotechnology, Rockford, IL). ProQ-Emerald 300 stain was used to visualize carbohydrates according to the instructions of the manufacturer (Invitrogen).
LPS isolation.
LPS was isolated by the method of McLendon et al. (27). In brief, bacteria were grown for ∼16 h on chocolate agar with appropriate antibiotics at 37°C, collected in 6 mM Tris base, 10 mM EDTA, and 2.0% SDS (wt/vol), pH 6.8, containing 50 μg/ml proteinase K, and incubated at 65°C for 1 h and then overnight at 37°C. To remove SDS, samples were precipitated with 0.3 M sodium acetate and 3 volumes of cold 100% ethanol, flash cooled in a dry ice-ethanol bath, and incubated overnight at −20°C. Samples were centrifuged for 10 min at 12,000 × g at 4°C, and pellets were suspended in deionized water and precipitated a total of three times. Samples were suspended in water and treated with 80 U of micrococcal nuclease (Sigma, St. Louis, MO) for 2 h at 37°C. LPS samples and phenol were equilibrated to 65°C, and an equal volume of phenol was added to the lysates. Samples were mixed, incubated at 65°C for 30 min, cooled, and centrifuged at 3,000 rpm for 10 min at 4°C. The aqueous layer was collected, and the organic layer was back extracted with an equal volume of water. The final aqueous samples were precipitated with ethanol three times, and after the last precipitation, pellets were suspended in high-pressure liquid chromatography (HPLC)-grade water and lyophilized overnight.
Composition and linkage analyses of LPS.
Composition analysis of LPS samples was done by gas chromatography-mass spectrometry (GC-MS) analysis of trimethylsilyl (TMS) derivatives of methylglycosides as previously described (23). Briefly, LPS samples (100 μg) containing 5 μg of myo-inositol as an internal standard were methanolyzed using 1 M methanolic HCl at 80°C for 16 h, re-N-acetylated using CH3OH-pyridine-acetic anhydride (4:1:1 [vol/vol/vol]) at 100°C for 1 h, dried, and then silylated using Tri-Sil reagent at 80°C for 30 min. After removal of Tri-Sil, samples were extracted in hexane and analyzed by GC-MS using an Agilent GC-MS (Agilent Technologies 7820A GC attached to a 5975 series MSD) operating in the electron impact (EI) mode and equipped with an Rtx-5ms column (30 m × 0.25 mm × 0.25 μm; Restek). A temperature gradient of 80°C to 140°C at 10°C/min, 140°C to 220°C at 2°C/min (with a 1-min hold), and 220°C to 240°C at 5°C/min was used to separate silylated monosaccharides. Monosaccharides were identified and characterized by comparison to the retention times and fragmentation patterns of authentic standards.
Linkage analysis of the LPS samples was performed using GC-MS analysis of their partially methylated alditol acetate (PMAA) forms as previously reported, with some minor modifications (23). Briefly, 0.4 mg of each LPS sample was permethylated using a method modified from the work of Ciucanu (28) and hydrolyzed to partially O-methylated monosaccharides, followed by reduction and peracetylation with sodium borodeuteride and pyridine-acetic anhydride, respectively. After dissolving in dichloromethane, 1 μl of the final PMAA sample was injected in splitless mode and analyzed using the same GC-MS system and temperature gradient, with the exception that the final temperature was 230°C. The PMAA peaks on the GC-MS chromatograph were identified by comparison with some authentic standards and fragmentation patterns.
MALDI-MS analyses of LPS.
To generate LPS that was more amenable to mass spectrometric analyses, O-deacylated (O-LPS) samples were prepared by treating ∼100 μg of LPS with 50 μl of anhydrous hydrazine followed by acetone precipitation as described previously (29). Additionally, to remove contaminating free lipid A, some LPS samples were extracted with chloroform-methanol-water (10:5:6 [vol/vol/vol]). The chloroform phase was removed, and the aqueous phase and interface were dried down under a stream of dry nitrogen; these samples were then O-deacylated as described above. All samples were desalted by drop dialysis using 0.025-μm-pore-size nitrocellulose membranes (Millipore, Bedford, MA) and then lyophilized. Samples were reconstituted in 5 to 100 μl of HPLC-grade H2O; 1 μl was loaded onto the target, dried, and then overlaid with 1 μl of matrix, consisting of 50 mg/ml 2,5-dihydroxybenzoic acid in 70% acetonitrile. Samples were subsequently analyzed using matrix-assisted laser desorption ionization mass spectrometry (MALDI-MS) on an LTQ linear-ion-trap mass spectrometer coupled to a vMALDI ion source (MALDI-LIT; Thermo Fisher, Waltham, MA) operating with a nitrogen laser at 337 nm, a 3-ns pulse duration, and a 60-Hz repetition rate. Data were collected in the negative-ion mode, using the automated gain control and the automatic spectrum filter settings. Tandem mass spectrometry (MSn) data were collected using a precursor ion selection window of m/z 3 and a normalized collision energy of 35%.
Murine infections and organ dissemination.
BALB/c female mice of 6 to 8 weeks of age were purchased from the National Cancer Institute (NCI) for all sets of animal experiments. Groups of five mice were used for evaluation of virulence of Francisella strains or immunization studies of attenuated Francisella strains, and groups of three mice were used for experiments in which bacterial growth in murine organs was examined. Mice were infected intraperitoneally (i.p.) with 100-μl inocula or intranasally with 50-μl inocula and were monitored for up to 26 days postinfection. Doses, ranging from 102 to 108 CFU, were estimated from OD600 readings and confirmed by dilution and plating of the bacterial suspension. For determination of bacterial burdens in organs, mice were infected intranasally with 290 CFU, 2.2 × 106 CFU, or 2.4 × 106 CFU of F. tularensis Schu S4, F. tularensis Schu S4 waaY (FTT1236), or F. tularensis Schu S4 waaL (FTT1238c), respectively. Lungs, livers, and spleens were harvested on days 3, 6, 9, and 12. Data from Schu S4-infected mice were collected only until day 5 postinfection, since all mice infected with Schu S4 succumbed to infection after day 5. Organs were homogenized using 35-ml tissue grinders (Thermo Fisher Scientific) and 2 ml of 1% saponin (Acros). Organ homogenates were serially diluted in PBS and plated to enumerate the bacterial load per organ. The amount of growth for a strain in the lung was determined by taking the amount of bacterial growth on day 3, 6, 9, or 12 and dividing it by the initial inoculum. No statistical significance was found for these data by use of the paired two-tailed Student t test. All work with F. tularensis Schu S4 was performed within the Carver College of Medicine Biosafety Level 3 (BSL3) Core Facility, and all experimental protocols were reviewed for safety by the BSL3 Oversight Committee of the University of Iowa Carver College of Medicine. Recombinant DNA work with F. tularensis Schu S4 was approved by the Institutional Biosafety Committee.
Pathology of murine spleen, liver, and lungs.
Four BALB/c mice were infected with 20 CFU of F. tularensis Schu S4, and groups of eight mice were infected with 1.5 × 106 CFU of the F. tularensis Schu S4 waaY (FTT1236) mutant or the F. tularensis Schu S4 waaL (FTT1238c) mutant. Two mice from each group were sacrificed by CO2 inhalation, and organs (lungs, liver, and spleen) were harvested on days 3, 6 (day 5 for WT-infected mice), 9, and 12. Tissues were fixed in 10% formalin in nonbuffered saline. Fixed tissues (lungs, liver, and spleen) were submitted to the Comparative Pathology Laboratory (University of Iowa) for routine tissue processing, paraffin embedding, sectioning (∼4 μm), and hematoxylin and eosin (HE) staining. Tissue sections were examined, and digital images were collected using a high-resolution digital camera (DP71; Olympus) on a BX51 microscope (Olympus). Scoring of lung tissues was performed following the principles of histopathologic scoring (30). Groups of 5 BALB/c mice were infected with 33 CFU of Francisella tularensis Schu S4, 1.4 × 106 CFU of F. tularensis waaY::TrgTn, 1.4 × 106 CFU of F. tularensis waaL::TrgTn, 27 CFU of F. tularensis waaY::TrgTn/pSL129, or 45 CFU of F. tularensis waaL::TrgTn/pSL149. Organs were harvested and fixed as described above on day 4 postinfection. Scoring parameters for necrotic debris in airspaces were as follows: 0, no necrosis detected; 1, <25% of airspaces obstructed with necrotic cellular debris; 2, 25 to 50% of airspaces obstructed with necrotic cellular debris; 3, 51 to 75% of airspaces obstructed with necrotic cellular debris; and 4, >75% of airspaces obstructed with necrotic cellular debris. Statistical significance of semiquantitative scoring was determined using Prism v5 software, Kruskal-Wallis one-way analysis of variance (ANOVA), and the Dunn posttest. Significance was set at 0.05.
Immunization with capsule/O-antigen F. tularensis mutants.
Groups of five BALB/c mice were infected with 76 CFU of F. tularensis Schu S4 i.p. in 100 μl of PBS. Groups of five mice were infected i.p. with three different doses of the F. tularensis waaY (FTT1236) and waaL (FTT1238c) mutants. Mice infected with the F. tularensis waaY (FTT1236) mutant were given doses of 5.6 × 102, 5.6 × 103, and 5.6 × 104 CFU, while mice infected with the F. tularensis waaL (FTT1238c) mutant were given doses of 9.6 × 102, 9.6 × 103, and 9.6 × 104 CFU. At 14 days postinfection, mice were boosted i.p. with 5 × 105 CFU of the same strain that was originally used to challenge the mice. After an additional 28 days, the mice infected with the F. tularensis Schu S4 waaY (FTT1236) mutant were challenged i.p. with 20, 200, and 2,000 CFU of Schu S4, and the F. tularensis Schu S4 waaL (FTT1238c) mutant-infected mice were challenged intranasally with 20, 200, and 2,000 CFU of Schu S4. A group of seven mice was infected i.p. with F. tularensis Schu S4 as a positive control. Each group of mice was monitored for 14 days after challenge with the F. tularensis Schu S4 parent strain.
To examine the efficacy of vaccinating mice by the intranasal route (5), groups of five BALB/c mice were immunized intranasally with the sublethal dose of 675 CFU of the F. tularensis waaY::TrgTn mutant. Two of the groups of mice were boosted with 500 CFU intranasally at day 14, while the other three groups of mice were left unboosted. At 28 days postvaccination, the mice were challenged with various doses of F. tularensis Schu S4 to determine the level of protection provided by the F. tularensis waaY::TrgTn mutant.
RESULTS
Screening for capsule-deficient mutants in an F. tularensis Schu S4 Tn5 library.
To identify F. tularensis Schu S4 mutants lacking capsule production, we screened an F. tularensis Schu S4 Tn5 transposon insertion library, comprising ∼7,500 F. tularensis mutants (∼4-fold coverage of the Schu S4 genome), with the 11B7 monoclonal antibody to capsule (23). A total of 116 isolates with significantly reduced binding (<15% of the wild-type level) were initially identified as candidate capsule mutants for further characterization. Confirmatory screening (screening the 116 isolates again with 11B7) of these strains revealed that only 10 of these strains reproducibly had less than 15% of wild-type binding to the 11B7 antibody, and these were selected for further characterization. To identify disrupted genes, transposon elements (i.e., the kanamycin resistance gene and the R6K origin of plasmid replication) and flanking chromosomal DNA were rescued from these strains by restriction enzyme digestion and ligation to create plasmids that replicated within E. coli pir+ strains. The plasmids were sequenced across the transposon insertion site to determine which gene the transposon had disrupted. Sequence analysis revealed that these 10 strains represented insertions in seven different Francisella genes (Table 1), with three separate mutations in one gene (FTT1238c) and two identical insertions in another gene (FTT1236). In addition to finding capsule defects in each of these F. tularensis Schu S4 mutants, we also found LPS defects in these mutants. In previously published work, our laboratory identified the FTT1236, FTT1237, and FTT1238c genes as being important for intracellular growth in human monocyte-derived macrophages (MDMs) by screening of an F. tularensis Schu S4 TraSH library for genes required for intracellular survival (25). To confirm that mutations in these genes did significantly reduce capsule and LPS antibody binding, we examined our previously constructed mutants in FTT1236, FTT1237, and FTT1238c by ELISA for capsule production and found that the FTT1236 and FTT1238c mutants each had <15% reactivity, while the FTT1237 mutant had 74% reactivity to the anti-capsule 11B7 monoclonal antibody compared to the WT strain. In the ELISA, the F. tularensis FTT1236 mutant had 39% reactivity, the F. tularensis FTT1237 mutant had 50% reactivity, and the F. tularensis FTT1238c mutant had 33% reactivity to the anti-LPS FB11 monoclonal antibody compared to the reactivity for the F. tularensis Schu S4 parent strain (100% reactivity). These results are consistent with Western blot data showing that mutations in these genes abolish capsule production and significantly reduce LPS O-antigen laddering (25).
TABLE 1.
Identification of F. tularensis Schu S4 genes involved in capsule productiona
| Gene locus | Tn5 insertion site (nucleotide position in gene) | Predicted function of gene product |
|---|---|---|
| FTT0025c (fslE) | 137 | Iron acquisition protein |
| FTT0846 | 896 | Potential deoxyribodipyrimidine photolyase |
| FTT1138 (hemH) | 637 | Potential ferrochelatase |
| FTT1236 | 688 | Hypothetical protein |
| FTT1238c | 463, 681, 989 | Hypothetical protein |
| FTT1458c (wzy) | 593 | O-antigen polymerase |
| FTT1464c (wbtA) | 641 | dTDP-glucose 4,6-dehydratase |
Identified by reduced binding of the gene mutants to anti-capsule antibody; all mutants had binding that was less than 15% of the WT level.
Gene products of FTT1236, FTT1237, and FTT1238c are similar to E. coli Waa proteins and functionally complement waa mutants in E. coli or Salmonella.
The proteins produced by FTT1236, FTT1237, and FTT1238c are annotated as hypothetical proteins in Francisella genome sequences. In an effort to understand how mutation of these genes alters biosynthesis of capsule and LPS, we analyzed the predicted translational product of each gene. Using NCBI protein PSI-BLAST to search for homology, we discovered that these genes share some similarity with the Waa protein genes of Escherichia coli MG1655 (Fig. 1A). The FTT1236 gene product has regions of similarity (24% similarity) to E. coli WaaB (Fig. 1A). Analysis of FTT1237 revealed significant regions of similarity to the E. coli WaaI and WaaJ proteins (41% and 43% similarities, respectively). FTT1238c was compared to E. coli WaaL and S. enterica WaaL, and regions of similarity were observed for both (43% and 40% similarities, respectively). With the aid of Clustal Omega (31), the F. tularensis FTT1237 protein sequence was compared to the E. coli WaaI and WaaJ sequences, revealing a DxD motif, which is a characteristic of glycosyltransferases (Fig. 1B). Additionally, the F. tularensis FTT1238c protein sequence was compared to the E. coli WaaL and Salmonella WaaL sequences, revealing a conserved histidine residue and a conserved arginine residue that have been shown to be important for function in other organisms (32–34) (Fig. 1B). In E. coli as well as other Gram-negative organisms, Waa proteins are important for LPS core biosynthesis (i.e., core sugar assembly) and for ligation of the O-antigen repeating unit to the synthesized core (24). Using the programs HMMTOP (35, 36) and TMRPres2D (37) to predict membrane topology, we constructed a transmembrane model for the FTT1238c gene product (Fig. 1C). As for other WaaL proteins, the FTT1238c gene product was predicted to have 12 transmembrane segments, 5 cytoplasmic loops, 6 periplasmic loops, and localization of both the N and C termini in the bacterial cytoplasm. The large periplasmic loop between segments 9 and 10 contains the highly conserved residues R270 and H361.
FIG 1.

Similarity of F. tularensis Waa proteins to E. coli and S. enterica proteins. (A) Francisella proteins FTT1236, FTT1237, and FTT1238c are similar to E. coli (*) and S. enterica (‡) LPS biosynthetic proteins. (B) In the protein alignments, single dots denote semiconserved residues, double dots are conserved residues, and asterisks identify exact matches of amino acid residues. Individual alignments are separated by thick black lines. The FTT1237 protein has a DxD motif (shown in bold) that is conserved in both WaaI and WaaJ of E. coli. Alignment of the FTT1238c protein to the E. coli and S. enterica WaaL proteins reveals the presence of the highly conserved arginine and histidine amino acids (in bold) that are important for the function of the protein. (C) The membrane topology map of FTT1238c shows the 12 predicted transmembrane domains as well as a large periplasmic loop containing the conserved arginine (*; R270) and histidine (**; H361) residues.
To explore possible functions for the Francisella FTT1236, FTT1237, and FTT1238c genes, we obtained E. coli mutants in the waaB, waaI, and waaJ genes from the Keio collection to determine whether Francisella genes could functionally complement the LPS biosynthetic defect in these E. coli mutants. Since the E. coli parent strain (BW25113) also lacks the ability to synthesize O antigen, we could not use this strain to attempt to complement a waaL defect. Accordingly, we constructed a waaL mutant in S. enterica Typhimurium SL1344 to assay for functional complementation with a plasmid carrying the intact FTT1238c gene (pSL149). When the F. tularensis FTT1236 gene (pSL129) was introduced into the E. coli waaB mutant, the core sugar bands were observed to have a slight yet detectable shift in comparison to the uncomplemented mutant strain (Fig. 2A). When a plasmid with F. tularensis FTT1237 (pSL130) was introduced into the E. coli waaI or waaJ mutant, there was a slight yet detectable shifting of the carbohydrate bands in comparison to the uncomplemented mutant strain (Fig. 2A). We therefore observed partial complementation of E. coli LPS biosynthetic defects with both the Francisella FTT1236 and FTT1237 genes. As negative controls for functional complementation, E. coli mutants in waaG and waaQ did not show any complementation when complemented with either pSL129 or pSL130. Introduction of a plasmid with a functional copy of the FTT1238c gene (pSL149) into the Salmonella ΔwaaL mutant partially restored the O-antigen banding that was observed in the parent SL1344 strain (Fig. 2B). We observed no shift in carbohydrate band migration for strains carrying empty vectors as negative controls (data not shown). The presence of any complementation in these experiments is considered significant due to the species-specific requirements for these LPS biosynthetic enzymes. We believe that this is particularly true for Francisella, since the LPS of this organism is very different from those of the Enterobacteriaceae (13, 14) and the similarities that we have detected between the Francisella genes and E. coli/Salmonella genes are quite low.
FIG 2.
Functional complementation of E. coli and S. enterica mutants in waa genes with Francisella tularensis genes. (A) E. coli mutants in waaB, waaI, and waaJ from the Keio collection were complemented with a plasmid carrying either FTT1236 (pSL129) or FTT1237 (pSL30). Proteinase K-treated whole-cell lysates were prepared for each strain, separated by PAGE, and visualized by silver staining. *, upward mobility shift of lipid A-core molecules in each E. coli strain. (B) Whole-cell lysates (proteinase K treated) of S. enterica SL1344, the S. enterica waaL mutant, and the S. enterica waaL mutant complemented with FTT1238c (pSL149) were separated by PAGE, and O-acylated species were visualized by silver staining.
F. tularensis waa mutants have altered LPS core structures.
Previously, we showed that the FTT1236::TrgTn, FTT1237::TrgTn, and FTT1238c::TrgTn mutants were sensitive to complement (25), consistent with observations that waa mutants of other organisms also display significantly increased sensitivity (38–40). To examine other phenotypes of our mutants, we prepared whole-cell lysates of the FTT1236::TrgTn, FTT1237::TrgTn, and FTT1238c::TrgTn mutants to determine if core sugars were altered. Whole-cell lysates of each strain were separated by SDS-PAGE, and the samples were stained for carbohydrates (Fig. 3A). We observed that the FTT1236 mutant and the FTT1237 mutant each had core sugar defects and that the FTT1236 mutant strain was more truncated in its core than the FTT1237 mutant. These types of core sugar shifts were consistent with core sugar changes previously observed in E. coli waa mutants (40, 41). In contrast, the waaL mutant appeared to have a complete lipid A and core sugar structure but lacked O-antigen laddering as previously described (40). This result provides additional evidence that the FTT1238c gene product is similar to WaaL, as other bacterial strains mutated at waaL cannot ligate the O antigen to the LPS core and therefore also do not have O-antigen laddering.
FIG 3.

Examination of the lipopolysaccharide core structures of O-antigen mutants by staining of carbohydrates and negative-ion multistage MALDI-MS. (A) Consistent with previously published data (58–60), the bands in the purified LPS lane shown in the PAGE gel and stained by emerald green are labeled lipid A–core–O-antigen (one unit), lipid A-core (no O-antigen subunit), and lipid A and are indicated by black arrows. (B) MALDI-MS analysis of O-LPS derived from the LVS 0708 mutant. MSn analyses were performed to sequentially determine the components of the O-LPS. (C) MS/MS analysis of the precursor ion observed at m/z 1,747. (D) Subsequent MS3 analysis performed on the fragment ion observed at m/z 1,527. Peaks marked with asterisks designate major masses minus water or CO2. (E) Completed core structure for LVS 0708::TrgTn (predicted for the Schu S4 1236::TrgTn strain as well) and predicted structure for LVS 0706::TrgTn (as well as the Schu S4 1238::TrgTn strain).
Since previous work indicated that the FTT1237::TrgTn strain has a less attenuated phenotype than either the FTT1236 or FTT1238c mutant, we focused our remaining work on the phenotypes of the latter two mutants. LPS species from the Schu S4 FTT1236 and FTT1238c mutants were isolated and subsequently O-deacylated to make the LPS more amenable to mass spectrometric analyses. MALDI-MS analyses of these samples showed abundant peaks corresponding to O-deacylated lipid A, with no additional carbohydrate component detected (data not shown). Multiple samples were tested using our standard protocols, and each time only O-deacylated lipid A was detected at m/z 983 and 1,144.
Since we were unable to successfully detect O-LPS components from the Schu S4 strains, we created mutations in the analogous LVS genes and used LPS isolated from the LVS mutant strains. We have shown previously that the lipid A-core sugar bands from these strains appear identical to those from the Schu S4 mutants (data not shown). The analogous LVS strain for the Schu S4 1236::TrgTn mutant is LVS 0708::TrgTn, and the corresponding LVS strain for the Schu S4 1238c::TrgTn mutant is LVS 0706::TrgTn. The use of these F. tularensis LVS mutants allowed us to use larger starter culture volumes to generate larger quantities of LPS for analyses, which were isolated as previously detailed. As described above, the LPS samples were O-deacylated and subsequently analyzed by MALDI-MS. Initial analyses of these O-deacylated LPS preparations clearly demonstrated that we were able to detect O-LPS components in the LVS 0708::TrgTn strain, but the most abundant peak detected was that corresponding to the O-deacylated lipid A (data not shown). To decrease the amount of free lipid A present in the LPS samples, intact LPS was extracted with chloroform-methanol-water, and the aqueous and interface layers were isolated, O-deacylated, and subsequently analyzed by MALDI-MS. Indeed, the most intense peak was then observed at m/z 1,747.55, with less abundant peaks at m/z 1,649.45, 1,585.55, 1,527.55, 1,365.55, and 983.55. The mass observed at m/z 983.55 corresponds to O-deacylated lipid A containing one phosphate group and agrees with the previously predicted lipid A structure (13, 14).
To determine the individual components of the O-LPS structure, multistage mass spectrometric analyses (MSn) were performed (Fig. 3B). MS/MS analyses of the major peak at m/z 1,747.55 showed the generation of two major peaks, at m/z 1,527.5 and 983.58 (Fig. 3C). The mass difference between the precursor ion, m/z 1,747, and the fragment ion at m/z 1,527 is 220 Da, corresponding to the loss of one 2-keto-3-deoxyoctulosonic acid (KDO) moiety. Further fragmentation (MS3) of the peak at m/z 1,527 generated one major peak, at m/z 983.58, which is O-deacylated lipid A (Fig. 3D). The mass difference between the precursor ion peak at m/z 1,527.5 and the ion at m/z 983.58 is 544 Da, which could nominally be attributed to two hexoses and one KDO. Composition analyses of LVS 0708::TrgTn showed the presence of mannose (Man), glucose (Glc), and KDO, suggesting that the 544 Da consisted of one KDO, Glc, and Man. MS analyses demonstrated that the loss of one KDO or one hexose could readily be observed as one of the first losses from the O-LPS, confirming that both of these sugars are at terminal positions. Linkage analysis confirmed the presence of both Glc and Man as terminal sugars in the LVS 0708::TrgTn LPS structure (Fig. 3E). Additional peaks were observed in the spectrum, including peaks that corresponded to components of the lipid A and to probable KDO degradation products and some peaks which could not be attributed to the known LPS structure. The previously published wild-type LVS LPS structure identified only one KDO moiety in the core structure (14); however, our data clearly demonstrate the presence of a second KDO moiety as part of the core structure, which is most likely linked to the other core KDO moiety.
MALDI-MS analyses were also performed on the O-LPS from the LVS waaL mutant LVS706. Initial MS data from the LVS 0706::TrgTn O-LPS once again showed only the presence of O-deacylated lipid A; therefore, a chloroform-methanol-water extraction was performed on the intact LVS 0706::TrgTn LPS to reduce the free lipid A components in this preparation. As expected, the intensity of the O-deacylated lipid A peak decreased dramatically after this extraction; however, a higher-mass substituted O-LPS was not detected. To gain further insight into the LVS 0706::TrgTn LPS structure, composition analyses were performed. These data showed the presence of Man, Glc, KDO, GalNAc, and GlcNAc in the LVS 0706::TrgTn strain. The GlcNAc was seen in both the LVS 0706::TrgTn and LVS 0708::TrgTn LPS samples and was likely derived from lipid A. The remaining constituents are components of the LPS core. Comparisons of the composition analyses of the LVS 0706::TrgTn and LVS 0708::TrgTn LPS species showed that the LVS 0706::TrgTn LPS contained higher levels of Man and Glc and contained one GalNAc, whereas GalNAc was not detected in the LVS 0708::TrgTn LPS sample. Linkage analyses confirmed the presence of terminal Glc and GalNAc as well as 2,3-Man and 2,4-Man, correlating with the previously published LPS structure. Additional peaks were observed which corresponded to the GlcNAc from lipid A and to probable KDO degradation products, with some peaks which did not correlate with the published LPS structure. These data combined with the previously published LVS core structure indicate that the LVS 0706::TrgTn LPS consists of a full core structure like that of the wild type (14, 42), including an additional KDO moiety linked to the other core KDO residue (Fig. 3E). Due to these results, we refer to the FTT1236 gene as waaY, the FTT1237 gene as waaZ, and the FTT1238c gene as waaL.
Determination of virulence and organ dissemination in murine infections.
We previously demonstrated that when human MDMs were infected with either the waaY or waaL mutant, the organisms gained entry into the cells in larger numbers and caused cell death of MDMs 30 h sooner than the WT strain (25). We wanted to extend these observations by examining the virulence of these strains in a murine model of disease. We infected mice with F. tularensis Schu S4 (12 CFU), the waaY mutant, or the waaL mutant at doses ranging from 102 to 108 CFU via the intraperitoneal (i.p.) or intranasal (i.n.) route of infection. The i.p. infection experiments revealed that the F. tularensis mutants were significantly attenuated for mouse virulence when inoculated into the peritoneal cavity. The accepted 50% lethal dose (LD50) of F. tularensis Schu S4 for BALB/c mice is <10 CFU, and the group of mice infected with 12 CFU of Schu S4 all died within 5 days. However, the i.p. LD50 for the F. tularensis waaY mutant was calculated (43) to be 7.6 × 106 CFU, and the i.p. LD50 for the F. tularensis waaL mutant was calculated to be 3.8 × 105 CFU, which are about 105 and 104 reductions in virulence, respectively, for these strains. This result was not completely surprising, since we have previously reported that these strains are highly sensitive to complement killing (25).
For mice that were challenged intranasally, we used 10-fold dilution doses from ∼103 to 108 CFU and observed the animals for signs of disease for up to 26 days postinfection (Table 2). Regardless of the infectious dose, mice that were infected with each of the mutants appeared sick, as the fur was ruffled for 2 days after the infection. Minimal conjunctivitis was observed on days 4 through 6 postinfection. Typically, a day or two before becoming moribund (for all doses), the mice intranasally infected with these mutant strains had moderately severe conjunctivitis and began to have labored breathing which worsened significantly until the mice succumbed. Using the calculation method of Reed and Muench (43), we determined that the LD50 for i.n. infection of mice with the waaY mutant was 1.3 × 104 CFU, and that for the waaL mutant was 3 × 103 CFU. To determine the bacterial burdens in organs, we intranasally infected groups of BALB/c mice with ∼300 CFU of Schu S4 and 2 × 106 CFU of the waaY or waaL mutant strain. At these doses, wild-type-infected mice died at about day 5, and F. tularensis mutant-infected mice died at about day 13. On days 3, 6, 9, and 12 postinfection, we harvested the lungs, liver, and spleen and plated dilutions of the organ homogenates to determine the bacterial burden carried in each organ. Organ burdens in WT-infected mice were evaluated only on day 3 postinfection, as all mice had succumbed to the infection by day 6 (Fig. 4A). Regardless of when organ burdens were examined, mice infected with the mutant strains did not clear the infections from the lungs, liver, or spleen. During early stages of infection, these strains persisted at low levels in the liver and spleen, although by day 12 the levels of growth were comparable to those observed for the virulent WT Schu S4 strain at day 3 postinfection. In contrast, the lungs of mice infected with either the waaY or waaL mutant had significantly higher levels of bacterial growth than the WT strain (106 CFU/organ) at 3 days postinfection, with the burden in the lungs continuing to increase exponentially, to 1011 CFU/organ by day 12 postinfection. In comparing the growth on day 3 postinfection, the trend points to the mutants having slower growth kinetics in vivo than that of the WT (Fig. 4B). By day 12 postinfection, the mutants had relatively similar growth to that of the WT on day 3 postinfection. These data suggest that the organ lung burdens were similar for the mutants and the WT prior to mouse death (WT-infected mice died on day 5 or 6 postinfection, whereas mutant-infected mice, at the dose given, died on days 13 to 15 postinfection). Another significant observation from these murine infection studies is that highly serum-sensitive F. tularensis mutant strains are still able to colonize, grow within mice, and cause fatality at relatively low doses when delivered intranasally. This suggests that complement is low in abundance in the lungs or that the organisms are able to escape its killing action by an unknown and unanticipated mechanism.
TABLE 2.
Murine virulence studies of F. tularensis Schu S4, F. tularensis waaB::TrgTn, and F. tularensis waaL::TrgTn strains
| Strain and infection route | Infection dose (CFU) | Survival ratio (no. of survivors/no. of mice in group) | Mean time to death (days) |
|---|---|---|---|
| i.p. infection | |||
| Schu S4 | 12 | 0/5 | 5 |
| waaY::TrgTn | 1.8 × 104 | 5/5 | >14 |
| 1.8 × 105 | 5/5 | >14 | |
| 1.8 × 106 | 5/5 | >14 | |
| 2.2 × 107 | 1/5 | 3.75, >14 | |
| 2.2 × 108 | 0/5 | 3.2 | |
| waaL::TrgTn | 1.2 × 104 | 5/5 | >14 |
| 1.2 × 105 | 5/5 | >14 | |
| 1.2 × 106 | 0/5 | 5 | |
| 2.4 × 107 | 1/5 | 3.75, >14 | |
| 2.4 × 108 | 0/5 | 2.8 | |
| i.n. infection | |||
| Schu S4 | 16 | 0/5 | 6 |
| waaY::TrgTn | 1.3 × 103 | 5/5 | >26 |
| 1.3 × 104 | 2/5 | 19.3, >26 | |
| 1.3 × 105 | 1/4 | 16.6, >26 | |
| 1.3 × 106 | 0/5 | 18 | |
| 1.3 × 107 | 0/5 | 19.8 | |
| 1.3 × 108 | 0/5 | 15.8 | |
| waaL::TrgTn | 7 × 102 | 5/5 | >26 |
| 2.4 × 104 | 0/5 | 16.2 | |
| 7 × 104 | 1/5 | 19, >26 | |
| 7 × 105 | 0/5 | 12 | |
| 7 × 106 | 1/5 | 15, >26 | |
| 7 × 107 | 0/5 | 11.6 |
FIG 4.

F. tularensis O-antigen mutants in the waaY or waaL gene are able to disseminate to and replicate in the mouse liver and spleen. (A) BALB/c mice were intranasally infected with 300 CFU of F. tularensis Schu S4, 2.2 × 106 CFU of F. tularensis waaY::TrgTn, or 2.4 × 106 CFU of F. tularensis waaL::TrgTn. Homogenates of the lungs, liver, and spleen were plated from mice infected for 3, 6, 9, or 12 days to determine bacterial counts per organ. Horizontal lines represent the means of the organ counts for each strain at each time point. (B) From the initial inoculum to day 3 postinfection, fold growth of bacteria in the lung was calculated for Schu S4 and mutants. Fold growth was also calculated for mice infected with the mutants after day 3 postinfection. Comparison of fold growth after day 3 postinfection in mice infected with mutants could not be compared to that in Schu S4-infected mice because Schu S4-infected mice died before day 6 postinfection.
Pathology of murine tissues infected with F. tularensis Schu S4 or the waaY or waaL mutant. (i) Spleen.
Three days after intranasal inoculation, mice inoculated with WT bacteria lacked lesions in the spleen (Fig. 5a), but by day 5 postinfection, the splenic red pulp had become effaced by coalescing to diffuse caseous necrosis that extended into the periphery of the white pulp (Fig. 5b). In contrast, on day 3 postinfection, mice inoculated with either the waaY or waaL mutant had increased numbers of neutrophils in the red pulp (Fig. 5c). At day 6 postinfection, the red pulp continued to have neutrophilia, with detectable small islands of granulocytic/myeloid-predominant extramedullary hematopoiesis and rare thrombi (Fig. 5d and e). At day 12 postinfection, mice inoculated with the mutant strains had a prominent lymphoid hyperplasia characterized by germinal centers in the splenic white pulp, while the red pulp commonly had extramedullary hematopoiesis with neutrophilia and an absence of thrombi or necrosis (Fig. 5f).
FIG 5.

Pathology of spleens infected with F. tularensis Schu S4, the F. tularensis waaY mutant, and the F. tularensis waaL mutant. Sections of spleens are shown for mice infected with Schu S4 for 3 and 5 days (a and b) or with F. tularensis mutant strains for 3, 6, and 12 days (c to f). The lesions observed in samples from F. tularensis waaY and F. tularensis waaL mutant infections were indistinguishable from one another. (a) Spleen section from F. tularensis Schu S4-infected mouse at 3 days postinfection. (b) Spleen section from F. tularensis Schu S4-infected mouse at 5 days postinfection. The white asterisks denote areas of diffuse to coalescing necrosis in the red pulp. (c and d) Spleen sections from F. tularensis mutant-infected mouse at 3 days postinfection (c) and 6 days postinfection (d). The spleen had increased neutrophils (arrows) in the red pulp. (e) Spleen section from F. tularensis mutant-infected mouse at 6 days postinfection. The spleen had rare thrombi in red pulp (black asterisk) and lacked detectable necrosis. (f) Tissue section from F. tularensis mutant-infected mouse at 12 days postinfection. The spleen had white pulp with large germinal centers (white asterisk) and red pulp with granulocytic/myeloid-predominant extramedullary hematopoiesis (arrows). The scale bar applies to each panel shown.
(ii) Liver.
At day 3 postinfection, both inoculation groups had similar liver changes, characterized by sporadic focal neutrophil and macrophagic inflammation (pyogranulomas). At day 5 postinfection, mice from the WT inoculation group had multifocal hepatic parenchymal necrosis, with numerous Kupffer cells distended by bacteria. From days 6 to 12 postinfection, livers from mice infected with the F. tularensis waaY and waaL mutants had multifocal pyogranulomas similar to those seen on day 3 postinfection, but they also had multifocal evidence of extramedullary hematopoiesis, with rare necrosis, thrombi, and multifocal extramedullary hematopoiesis that did not produce obvious temporal group changes.
(iii) Lungs.
By day 3 postinfection, mice inoculated with WT bacteria lacked overt lung inflammation or lesions (Fig. 6a). By day 5 postinfection, WT groups had multifocal caseous necrosis that was often centered on vessel walls (Fig. 6b). At times, the necrosis resulted in complete obliteration of the vessel and adjacent tissue. In contrast, at day 3 postinfection, mice inoculated with mutant bacteria had multifocal airspaces in the lungs filled with necrotic cellular debris and inflammation that was often located in peribronchiolar/perivascular regions of small airways (Fig. 6c). At day 6 postinfection and through the end of the time course, these multifocal lesions continued to expand (Fig. 6d), and by day 12 postinfection, they effaced the majority of lung airspaces and airways with necrotic cellular debris, inflammatory cells (neutrophils and macrophages), and edema (Fig. 6e and f).
FIG 6.

Pathology of lungs infected with Schu S4 and the waaY and waaL mutants. Lung sections are shown for mice infected for 3 and 5 days with F. tularensis Schu S4 (a and b) or for 3, 6, and 12 days with F. tularensis mutant strains (c to f). The lesions observed in samples from F. tularensis waaY and F. tularensis waaL mutant infections were indistinguishable from one another. (a) Lung section from F. tularensis Schu S4-infected mouse at 3 days postinfection. Lungs lacked overt lesions. (b) Lung section from F. tularensis Schu S4-infected mouse at 5 days postinfection. Multifocal vessels (large and small) had segmental to circumferential necrosis of the vascular wall (arrows), without overt inflammation in airways or lumens (asterisk). (c and d) Lung sections from F. tularensis mutant-infected mouse at 3 days postinfection (c) and 6 days postinfection (d). Lungs (day 3) had necrotic cellular debris and inflammation (neutrophils and lesser macrophages) that localized near distal airways/vessels, with extension into the adjacent parenchyma (c; arrows). (d) Lungs had perivascular inflammation (arrow) with progressive expansion of the lesions (necrotic debris, neutrophils, macrophages, and some edema) into adjacent parenchyma and airway lumens (asterisk). (e and f) By day 12 postinfection, pathology in the lungs of mice infected with F. tularensis mutant strains had progressed to diffuse airspace consolidation, with filling of most airways (e; asterisks) by edema, necropurulent debris (f; asterisks), and macrophages (f; arrows). The scale bar in panel a applies to panels a to e.
The lung lesions were semiquantitatively measured by scoring the necrotic cellular debris (determining the percentage of pulmonary airspaces that were obstructed by necrotic debris). F. tularensis Schu S4, waaY::TrgTn, and waaL::TrgTn and the complemented mutant strains were scored for lung inflammation from 5 sets of murine lungs on day 4 postinfection. F. tularensis Schu S4-infected mice were given a ranking of only 1, even though the animals were moribund on day 5 (Fig. 7). For the mice infected with the F. tularensis mutants, the lung scores were similar for the waaY::TrgTn and waaL::TrgTn mutants, and these were both significantly increased compared to those for the wild type. Infections with the complemented mutant strains appeared like WT infection and were not significantly different from WT infection. It is interesting that when mice were infected with the complemented mutants at ∼20 to 40 CFU, they succumbed to infection, albeit with slightly delayed timing relative to that for typical Schu S4 infections at these doses (data not shown). Taken together, these data provide additional support for the gross inflammation that these F. tularensis mutants elicit in the lungs of infected mice, in contrast to the lack of inflammation within the lungs of wild-type-infected mice, even at the point of death.
FIG 7.
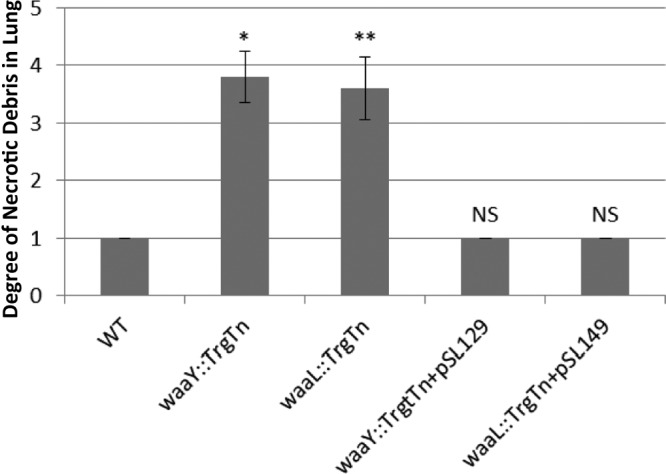
Comparison of necrotic debris for Schu S4, Francisella tularensis waaY and waaL mutants, and complemented mutants. Groups of 5 BALB/c mice were infected with 33 CFU of Francisella tularensis Schu S4, 1.4 × 106 CFU of F. tularensis waaY::TrgTn, 1.4 × 106 CFU of F. tularensis waaL::TrgTn, 27 CFU of F. tularensis waaY::TrgTn plus pSL129, or 45 CFU of F. tularensis waaL::TrgTn plus pSL149. On day 4 postinfection, lungs were scored for percent airspace filled with necrotic debris. 0, no detected necropurulent debris in the airspaces; 1, 0 to 25% of airspaces partially to fully obstructed by necrotic cellular debris; 2, 25 to 50% obstructed; 3, 50 to 75% obstructed; 4, >75% obstructed. *, P < 0.01; **, P < 0.05; NS, not significant. P values were determined using Prism v5 software, Kruskal-Wallis one-way ANOVA, and the Dunn posttest.
Immunization with F. tularensis mutants and subsequent challenge with Schu S4.
In previous studies, we immunized mice with Francisella capsular material and demonstrated that this immunization elicited an antibody response against capsule. However, an ensuing challenge of the immunized mice with Schu S4 yielded no protection, although the mice were protected significantly against challenge with F. tularensis LVS (23). With our current study of O-antigen/capsule mutants, we explored the potential of using F. tularensis waaY and waaL mutant strains as a vehicle to provide protection against a lethal Schu S4 challenge. A preliminary vaccine experiment was performed using mice that had been exposed to either F. tularensis waaY::TrgTn or F. tularensis waaL::TrgTn in the virulence study. In this experiment, mice that survived intraperitoneal challenge with 560, 5,600, or 56,000 CFU of the waaY::TrgTn mutant or 960, 9,600, or 96,000 CFU of the waaL::TrgTn mutant were challenged with 5 × 105 CFU of the strain that was originally used to infect the mice. Twenty-eight days later, the waaY mutant-infected mice were reinfected i.p. with 20, 200, or 2,000 CFU of Schu S4 (Fig. 8A), while the waaL mutant-infected mice were challenged i.n. with 20, 200, or 2,000 CFU of Schu S4 (Fig. 8B). Control mice were given a dose of 20 CFU i.p., and all succumbed to infection by 6 days postinfection. Of the waaY mutant-immunized mice, 4 of 5 mice (80%), 3 of 5 mice (60%), and 1 of 5 mice (20%) survived the Schu S4 doses of 20, 200, and 2,000 CFU, respectively. Interestingly, 5 of 5 (100%) of the waaL mutant-immunized mice that were challenged with either 20 or 200 CFU of Schu S4 i.n. survived, while 4 of 5 (80%) of the waaL mutant-immunized mice survived challenge with 2,000 CFU of Schu S4 i.n. This preliminary experiment provides strong evidence that mice challenged intraperitoneally with sublethal doses of either of the two mutant strains can elicit a protective immune response against challenge with up to 2,000 CFU of Schu S4. It is worth noting that in our laboratory, every naive mouse challenge experiment with wild-type F. tularensis Schu S4 has resulted in death of the mouse, suggesting that a single organism may always be lethal for mice. The protection that we observed in these experiments is of high interest, even though the experimental protocol had many variables.
FIG 8.

Vaccination with Francisella tularensis waaY or waaL mutant provides protection against challenge with F. tularensis Schu S4. Groups of 5 BALB/c mice were initially infected with three doses, from 102 to 104 CFU, of F. tularensis waaY::TrgTn or F. tularensis waaL::TrgTn. At 14 days postinfection (DPI), mice were rechallenged with 5 × 105 CFU of the same strain. Twenty-eight days later, F. tularensis waaY::TrgTn-infected mice were challenged intraperitoneally with F. tularensis Schu S4 (A), and F. tularensis waaL::TrgTn-infected mice were challenged intranasally with F. tularensis Schu S4 (B). Mice were tracked for 14 days for signs of disease.
Since the previous infection experiment had differing challenge doses, we performed another experiment with the F. tularensis waaY::TrgTn strain. Five groups of five BALB/c mice were challenged intranasally with 675 CFU of the waaY::TrgTn strain, which is a sublethal dose. Two groups of five mice were boosted intranasally with 500 CFU of the waaY::TrgTn strain, while the other three groups of five mice were not boosted. The unboosted mice were challenged 28 days later with either 19, 190, or 1,900 CFU of F. tularensis Schu S4 (Fig. 9A), and the boosted mice were challenged with either 190 or 1,900 CFU of F. tularensis Schu S4 (Fig. 9B). As can be seen in Fig. 9A and B, a single intranasal dose with the waaY::TrgTn strain protected 1 of 5 mice challenged i.n. with 1,900 CFU of Schu S4, 1 of 5 mice challenged with 190 CFU of Schu S4, and 4 of 5 mice challenged with 19 CFU of Schu S4. While this protection is significant considering the extreme lethality of F. tularensis Schu S4 for mice, boosting the mice with a second dose of the waaY::TrgTn mutant significantly increased the ability of the strain to elicit a protective immune response to intranasal challenge with Schu S4. The boosting of mice protected 2 of 5 mice (40%) challenged i.n. with 190 CFU of Schu S4 and 3 of 5 mice (60%) challenged i.n. with 1,900 CFU of Schu S4. These levels of protection are comparable to those observed in the experiment described for Fig. 8 and provide evidence that mice may be able to develop a memory immune response when infected with these F. tularensis mutant strains. The response that is elicited provides protection against virulent F. tularensis Schu S4 challenge, which is a significant step forward both in understanding the host response to Francisella infection and in efforts to develop effective biotherapeutics.
FIG 9.

Intranasal vaccination and boosting of BALB/c mice with F. tularensis waaY::TrgTn provide significant protection against challenge with F. tularensis Schu S4. (A and B) Five groups of BALB/c mice were inoculated intranasally with 675 CFU of F. tularensis waaY::TrgTn. (B) Two groups of mice were boosted with 500 CFU of F. tularensis waaY::TrgTn 14 days after the initial infection. Twenty-eight days later, the boosted and unboosted mice were challenged intranasally with F. tularensis Schu S4, and the mice were tracked for 14 days for signs of disease. A group of naive mice were infected with 19 CFU of F. tularensis Schu S4 as an infection control.
DISCUSSION
In this work, we identified F. tularensis Schu S4 mutants that are defective in capsule production with the goal of elucidating the role of the Francisella capsule in the virulence strategy of this organism. This genetic screen identified 10 strains that represent 7 different genes that are defective for capsule production and have altered LPS structures as well. Since two of these capsule-deficient mutants were previously identified by our research group as defective for growth in human macrophages, we carried out more extensive characterization of the F. tularensis Schu S4 FTT1236 and FTT1238c genes, as well as the FTT1237 gene, which compose a locus on the chromosome. Analysis of the translation products of these genes revealed that they are similar to Waa gene products that are involved in LPS biosynthesis in Gram-negative bacteria by performing the specific functions of adding sugars to the core structure of LPS (WaaY and WaaZ) or adding O-antigen subunits to the lipid A-core structure (WaaL). Interestingly, a fourth gene in the same region, FTT1235c, encodes a product that is similar to the mannosyltransferase LpcC as well as the glycosyltransferase WaaG. This protein is predicted to add the first mannose of the core to 3-deoxy-d-manno-octulosonic acid (Kdo). As we previously reported, we have been unable to make a mutant in FTT1235c, as the mutation appears to be lethal (25). The specific functions of WaaB, WaaI, and WaaJ are known for E. coli and Salmonella species. In those species, WaaB adds a galactose as a branch sugar to a core backbone glucose, while WaaI and WaaJ are responsible for adding a single hexose to the backbone, in concert (40). Interestingly, Francisella tularensis does have a galactose derivative (galactosamine) as a branch core sugar (13). Additionally, as mentioned previously, WaaL ligates polymerized O-antigen units to the complete core of lipid A (40).
Since our sequence analysis indicated that the mutated Francisella genes were likely involved in LPS biosynthesis, we performed functional complementation experiments to examine whether the F. tularensis waaY (FTT1236), waaZ (FTT1237), or waaL (FTT1238c) gene had the ability to complement LPS biosynthesis defects in E. coli strains (E. coli waaB or E. coli waaJ mutant) or a Salmonella strain (S. enterica waaL mutant). Due to the low sequence similarity between these Francisella genes and the E. coli and Salmonella genes, as well as the specific sugar reactions catalyzed by these enzymes, there seemed to be a low probability that we would observe any functional complementation in these experiments. Surprisingly, however, each of the Francisella genes of interest did modify core structures of E. coli or Salmonella waa mutant strains, although the complementation was only partial in each instance.
As another approach to investigate the functions of these Francisella genes, we examined the core structures and O-antigen laddering of the LPS species of the mutants following carbohydrate staining of electrophoretically separated preparations. These experiments revealed that each of the mutants had detectable alterations in LPS core biosynthesis compared to the F. tularensis Schu S4 purified LPS. The F. tularensis waaY (FTT1236) mutant had the most truncated core, and the core of the F. tularensis waaZ (FTT1237) mutant was slightly larger than the core of the waaY mutant. The F. tularensis waaL (FTT1238c) mutant appeared to have a core structure similar in size to that of the F. tularensis Schu S4 parent but lacked the O-antigen repeating units.
Mass spectrometric analyses further confirmed that the waaL and waaY mutants had truncated LPS structures. The waaY mutant expressed an LPS with a truncated core structure consisting of 1 mannose (Man), 1 glucose (Glc), and 2 KDO residues. Composition and mass spectrometric analyses suggested that the waaL mutant expressed a full core structure consisting of GalNAc, 2 Man residues, 2 Glc residues, and 2 KDO residues. Although our predicted Francisella core structure contains two KDO sugars instead of one KDO as previously reported by Vinogradov et al. (14, 42), ketosidic bonds are known to be labile, and this discrepancy is most likely due to the isolation and processing of the LPS prior to analyses. Additionally, the previously published lipid A structure from this group identified a lipid A which lacked the addition of any phosphate groups (14). Both in the present study and in previous studies by us and others, the predominant Francisella lipid A structure has been identified as being monophosphorylated. This phosphate group can also be substituted further with a galactosamine (13, 44). In the previous study by Vinogradov et al., neither of these additions to the lipid A structure were identified, lending further support to the possibility of the loss of labile groups in the processing of LPS (14). In fact, this group even suggested the possibility of the loss of some constituents due to the harsh conditions used for deacylation of the structure (14).
The presence of free lipid A in the LPS samples made analyses of the LPS more challenging. The chloroform-methanol-water extraction of the LPS samples improved the signal strength of the LPS from LVS 0708::TrgTn but was not enough to improve the MS signal intensity of LVS 0706::TrgTn LPS to detectable levels. In a previous study by Wang et al., it was estimated that less than 5% of the total lipid A is linked to LPS (44). The presence of such a large amount of free lipid A most likely contaminated the LPS samples, making characterization of the structures significantly more difficult.
The predominant Francisella LPS structure observed in vitro in the virulent 1547-57 strain lacks free phosphate moieties and produces LPS with two 16- and two 18-carbon acyl groups instead of six 14-carbon acyl groups (13). Importantly, some or all of these differences produce LPS with a very low endotoxic activity. Synthesis of LPS begins with the Lpx proteins, which synthesize and assemble the lipid A molecule (45), and initial transport of lipid A is carried out by LptB and ValA (46, 47). Completion of lipid A transport to the outer membrane is performed by LptA, LptD, and RplB (48–50). Initial E. coli core sugar assembly mutants were designated rfa (rough A) because the mutants grew with rough colony phenotypes (40). We noticed in the course of working with our F. tularensis mutants in FTT1236, FTT1237, and FTT1238c that each strain produced colonies on agar that were significantly rougher than those of the parental Schu S4 strain and that possessed altered colony consistency. In addition, electron microscopy of individual organisms indicated a significantly altered appearance (see Fig. S1 in the supplemental material). Together, these observations provide additional evidence of the role of these genes in LPS core assembly. As shown in Fig. 10, the products of this biosynthetic pathway are ligated to polymerized O-antigen subunits (51), which are synthesized by several proteins, including WbaP/PNPTs, Wzx, and Wzy, which carry out the functions of sugar synthesis, flipping, and polymerization of reaction intermediates to make the final O-antigen subunits available to be added to the lipid A-core sugar structure (52–55) (Fig. 10).
FIG 10.

Model for the roles of the F. tularensis WaaY, WaaZ, and WaaL proteins in LPS biosynthesis. In general, LPS is synthesized as two separate components: (i) lipid A-core sugars and (ii) polymers of O-antigen subunits. O-antigen sugars are synthesized in the cytoplasm and assembled onto a lipid carrier, i.e., the undecaprenol pyrophosphate that is associated with WbaP or PNPT, polyisoprenyl-phosphate hexose-1-phosphate transferase, or N-acetylhexosamine-1-phosphate transferase. The Wzx protein flips the charged lipid carrier, with the assembled O-antigen unit, through the inner membrane from the cytoplasm to the periplasmic space. Repeating O-antigen subunits are polymerized on the lipid carrier by the Wzy protein. The lipid A portion of LPS is synthesized by a number of Lpt and Lpx proteins on the inner leaflet of the inner membrane. Waa proteins (WaaY and WaaZ) transfer or modify core sugars to the lipid A molecule as they help to assemble the core structure of LPS. ValA and LptB transport the lipid A-core sugars to the outer leaflet of the inner membrane, where O-antigen subunits are added by the O-antigen ligase, WaaL, and then they are chaperoned to the outer membrane by LptA and LptD/RlpB (55). F. tularensis mutants that are disrupted for WaaY and WaaZ production are depicted with truncated cores, while the lipid A-core sugar structure of an F. tularensis waaL mutant would be the same as that of wild-type Schu S4 but lacking O-antigen subunits, since the WaaL protein performs this ligation function.
Our results provide evidence that the Francisella gene products of waaY, waaZ, and waaL perform functions similar to those of the waa gene products of other organisms (i.e., E. coli and Salmonella). We found that an F. tularensis strain carrying a waaL mutation has lipid A and a complete core but lacks O antigens. We also present data showing that mutation of waaY results in the most truncated lipid A-core structure, while mutation of waaZ results in an intermediary structure that is larger than that of waaY but smaller than the intact core structure observed in the waaL mutant strain. These data establish that the proper biosynthesis of the Francisella LPS core is critical for the addition of O-antigen polymerized subunits to lipid A-core structures as well as for proper assembly of the capsule structure. This suggests that full-length LPS functions as a scaffold for capsule or that there is an interaction between LPS and capsule that is necessary for outer membrane capsular presence.
We have demonstrated that the Francisella waaY and waaL genes contribute to the interactions of F. tularensis with the murine host. For mouse infection by the intraperitoneal route, the LD50 of the waaY mutant was 7.6 × 106 CFU, and the LD50 of the waaL mutant was 3.8 × 105 CFU, in contrast to the LD50 of Schu S4, which is ∼1 CFU. We believe that this attenuation of i.p. virulence is most likely due to the serum sensitivity of these mutant strains (25). The difference in the LD50 values of the mutants (1.3 × 104 CFU for the waaY mutant and 3 × 103 CFU for the waaL mutant) and the Schu S4 parent (<10 CFU) for the intranasal route of infection was significantly smaller than that observed i.p. A precise explanation for this disparity is unknown, but it seems likely to be due to either lower effective concentrations of serum in the lung or a highly efficient mechanism possessed by Francisella to avoid the killing action of serum in the lung in order to find a favorable intracellular growth niche, perhaps within alveolar macrophages and epithelial and/or endothelial lung cells. The organ burden data acquired for the mutant strains, as a function of time postinfection, indicate that these strains can disseminate and grow within the murine host, although their growth rate is significantly reduced compared to that of Schu S4. A possible explanation for this difference may be the serum sensitivity of the mutants or that induction of early death of the host cell limits growth (25). In summary, the results from our virulence studies with mice demonstrate that the activities of the products of the waaY and waaL genes contribute to both the immune avoidance and high virulence observed in the F. tularensis Schu S4 strain, as these mutants are clearly more inflammatory in mice, based upon pathological evidence, and have reduced virulence by both intraperitoneal and intranasal routes of infection.
Other researchers have looked at the value of using bacterial mutants in rfaJ (waaJ) or rfaL (waaL) as potential vaccine strains. BALB/c mice infected with either a Salmonella enterica rfaJ or rfaL mutant showed significant protection from lethal challenge with a virulent Salmonella strain (56). Billips et al. also demonstrated that a waaL mutant in a uropathogenic strain of E. coli was able to provide protection against a broad range of uropathogenic isolates of E. coli (57). In this work, we present data indicating that mice infected intraperitoneally with the F. tularensis Schu S4 waaL mutant show protection against a lethal challenge with WT Schu S4. We showed that 80% of mice survived an intranasal challenge of 2,000 CFU, while all mice survived a challenge of 200 CFU. Additionally, mice infected either intraperitoneally or intranasally with the F. tularensis Schu S4 waaY mutant showed partial protection against lethal intraperitoneal and intranasal challenges with WT Schu S4, and a boost with the mutant essentially increased the protection observed 10-fold. Recently, another group also demonstrated that a Francisella strain that has modified LPS is able to provide protection against a lethal Schu S4 challenge (58). These data are very encouraging and provide the basis for future experiments to explore the possibility of creating a useful therapeutic against virulent strains of F. tularensis.
In summary, we have further characterized F. tularensis mutants that we first demonstrated were unable to grow in human MDMs (25). We confirmed that they do not produce capsule and that they have altered LPS structures. We have provided experimental data indicating that these mutations alter the synthesis of the core sugar structures (waaY and waaZ) or the addition of O antigen (waaL). Virulence studies in mice demonstrated significant attenuation of virulence by both the i.p. and i.n. routes of infection, and pathology studies indicated that the murine response to infection with these mutants is significantly different from that to the parent F. tularensis Schu S4 strain. We believe that these studies provide compelling evidence to investigate the immune response to these attenuated strains as part of our larger effort to understand how virulent Francisella strains avoid immune detection in a mammalian host and to understand in more detail the unique virulence strategy of these highly pathogenic bacteria.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful for use of the University of Iowa Carver College of Medicine Biosafety Level 3 Core Facility and thank the DNA, Central Microscopy Research, and Proteomics Core Facilities for their instruction, aid, and analysis. We also thank the Protein Facility at Iowa State University. We acknowledge the expert technical assistance of Lorri Reinders with the analysis of Francisella LPS. LPS composition and linkage analyses were performed by the University of California at San Diego Glycotechnology Core. We thank Matt Faron and Joshua Fletcher for critical reviews of the manuscript and Dana Ries for BSL3 assistance.
Financial support for this work was provided by NIH grants 2PO1 AI044642 (B.D.J., M.A.A., and B.W.G.) and 2U54 AI057160 and by project 14 of the Midwest Regional Center of Excellence (MRCE) for Biodefense and Emerging Infectious Disease Research to B.D.J.
Footnotes
Published ahead of print 22 January 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01640-13.
REFERENCES
- 1.McLendon MK, Apicella MA, Allen LA. 2006. Francisella tularensis: taxonomy, genetics, and immunopathogenesis of a potential agent of biowarfare. Annu. Rev. Microbiol. 60:167–185. 10.1146/annurev.micro.60.080805.142126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tarnvik A, Berglund L. 2003. Tularaemia. Eur. Respir. J. 21:361–373. 10.1183/09031936.03.00088903 [DOI] [PubMed] [Google Scholar]
- 3.Jones CL, Napier BA, Sampson TR, Llewellyn AC, Schroeder MR, Weiss DS. 2012. Subversion of host recognition and defense systems by Francisella spp. Microbiol. Mol. Biol. Rev. 76:383–404. 10.1128/MMBR.05027-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fortier AH, Green SJ, Polsinelli T, Jones TR, Crawford RM, Leiby DA, Elkins KL, Meltzer MS, Nacy CA. 1994. Life and death of an intracellular pathogen: Francisella tularensis and the macrophage. Immunol. Ser. 60:349–361 [PubMed] [Google Scholar]
- 5.Conlan JW, North RJ. 1992. Early pathogenesis of infection in the liver with the facultative intracellular bacteria Listeria monocytogenes, Francisella tularensis, and Salmonella typhimurium involves lysis of infected hepatocytes by leukocytes. Infect. Immun. 60:5164–5171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Forestal CA, Benach JL, Carbonara C, Italo JK, Lisinski TJ, Furie MB. 2003. Francisella tularensis selectively induces proinflammatory changes in endothelial cells. J. Immunol. 171:2563–2570 [DOI] [PubMed] [Google Scholar]
- 7.Horzempa J, O'Dee DM, Shanks RM, Nau GJ. 2010. Francisella tularensis ΔpyrF mutants show that replication in nonmacrophages is sufficient for pathogenesis in vivo. Infect. Immun. 78:2607–2619. 10.1128/IAI.00134-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lindemann SR, McLendon MK, Apicella MA, Jones BD. 2007. An in vitro model system used to study adherence and invasion of Francisella tularensis live vaccine strain in nonphagocytic cells. Infect. Immun. 75:3178–3182. 10.1128/IAI.01811-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schulert GS, McCaffrey RL, Buchan BW, Lindemann SR, Hollenback C, Jones BD, Allen LA. 2009. Francisella tularensis genes required for inhibition of the neutrophil respiratory burst and intramacrophage growth identified by random transposon mutagenesis of strain LVS. Infect. Immun. 77:1324–1336. 10.1128/IAI.01318-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ojeda SS, Wang ZJ, Mares CA, Chang TA, Li Q, Morris EG, Jerabek PA, Teale JM. 2008. Rapid dissemination of Francisella tularensis and the effect of route of infection. BMC Microbiol. 8:215. 10.1186/1471-2180-8-215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dennis DT, Inglesby TV, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Fine AD, Friedlander AM, Hauer J, Layton M, Lillibridge SR, McDade JE, Osterholm MT, O'Toole T, Parker G, Perl TM, Russell PK, Tonat K, Working Group on Civilian Biodefense 2001. Tularemia as a biological weapon: medical and public health management. JAMA 285:2763–2773. 10.1001/jama.285.21.2763 [DOI] [PubMed] [Google Scholar]
- 12.Bosio CM. 2011. The subversion of the immune system by Francisella tularensis. Front. Microbiol. 2:9. 10.3389/fmicb.2011.00009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Phillips NJ, Schilling B, McLendon MK, Apicella MA, Gibson BW. 2004. Novel modification of lipid A of Francisella tularensis. Infect. Immun. 72:5340–5348. 10.1128/IAI.72.9.5340-5348.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vinogradov E, Perry MB, Conlan JW. 2002. Structural analysis of Francisella tularensis lipopolysaccharide. Eur. J. Biochem. 269:6112–6118. 10.1046/j.1432-1033.2002.03321.x [DOI] [PubMed] [Google Scholar]
- 15.Gunn JS, Ernst RK. 2007. The structure and function of Francisella lipopolysaccharide. Ann. N. Y. Acad. Sci. 1105:202–218. 10.1196/annals.1409.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raetz CR. 1990. Biochemistry of endotoxins. Annu. Rev. Biochem. 59:129–170. 10.1146/annurev.bi.59.070190.001021 [DOI] [PubMed] [Google Scholar]
- 17.Poltorak A, Smirnova I, He X, Liu MY, Van Huffel C, McNally O, Birdwell D, Alejos E, Silva M, Du X, Thompson P, Chan EK, Ledesma J, Roe B, Clifton S, Vogel SN, Beutler B. 1998. Genetic and physical mapping of the Lps locus: identification of the Toll-4 receptor as a candidate gene in the critical region. Blood Cells Mol. Dis. 24:340–355. 10.1006/bcmd.1998.0201 [DOI] [PubMed] [Google Scholar]
- 18.Beutler B, Poltorak A. 2000. Positional cloning of Lps, and the general role of Toll-like receptors in the innate immune response. Eur. Cytokine Netw. 11:143–152 [PubMed] [Google Scholar]
- 19.Hajjar AM, Harvey MD, Shaffer SA, Goodlett DR, Sjostedt A, Edebro H, Forsman M, Bystrom M, Pelletier M, Wilson CB, Miller SI, Skerrett SJ, Ernst RK. 2006. Lack of in vitro and in vivo recognition of Francisella tularensis subspecies lipopolysaccharide by Toll-like receptors. Infect. Immun. 74:6730–6738. 10.1128/IAI.00934-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clay CD, Soni S, Gunn JS, Schlesinger LS. 2008. Evasion of complement-mediated lysis and complement C3 deposition are regulated by Francisella tularensis lipopolysaccharide O antigen. J. Immunol. 181:5568–5578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hood AM. 1977. Virulence factors of Francisella tularensis. J. Hyg. 79:47–60. 10.1017/S0022172400052840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sandstrom G, Lofgren S, Tarnvik A. 1988. A capsule-deficient mutant of Francisella tularensis LVS exhibits enhanced sensitivity to killing by serum but diminished sensitivity to killing by polymorphonuclear leukocytes. Infect. Immun. 56:1194–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Apicella MA, Post DM, Fowler AC, Jones BD, Rasmussen JA, Hunt JR, Imagawa S, Choudhury B, Inzana TJ, Maier TM, Frank DW, Zahrt TC, Chaloner K, Jennings MP, McLendon MK, Gibson BW. 2010. Identification, characterization and immunogenicity of an O-antigen capsular polysaccharide of Francisella tularensis. PLoS One 5:e11060. 10.1371/journal.pone.0011060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Whitfield C, Kaniuk N, Frirdich E. 2003. Molecular insights into the assembly and diversity of the outer core oligosaccharide in lipopolysaccharides from Escherichia coli and Salmonella. J. Endotoxin Res. 9:244–249. 10.1177/09680519030090040501 [DOI] [PubMed] [Google Scholar]
- 25.Lindemann SR, Peng K, Long ME, Hunt JR, Apicella MA, Monack DM, Allen LA, Jones BD. 2011. Francisella tularensis Schu S4 O-antigen and capsule biosynthesis gene mutants induce early cell death in human macrophages. Infect. Immun. 79:581–594. 10.1128/IAI.00863-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pavlov VM, Mokrievich AN, Volkovoy K. 1996. Cryptic plasmid pFNL10 from Francisella novicida-like F6168: the base of plasmid vectors for Francisella tularensis. FEMS Immunol. Med. Microbiol. 13:253–256. 10.1111/j.1574-695X.1996.tb00247.x [DOI] [PubMed] [Google Scholar]
- 27.McLendon MK, Schilling B, Hunt JR, Apicella MA, Gibson BW. 2007. Identification of LpxL, a late acyltransferase of Francisella tularensis. Infect. Immun. 75:5518–5531. 10.1128/IAI.01288-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ciucanu I. 2006. Per-O-methylation reaction for structural analysis of carbohydrates by mass spectrometry. Anal. Chim. Acta 576:147–155. 10.1016/j.aca.2006.06.009 [DOI] [PubMed] [Google Scholar]
- 29.Allen S, Zaleski A, Johnston JW, Gibson BW, Apicella MA. 2005. Novel sialic acid transporter of Haemophilus influenzae. Infect. Immun. 73:5291–5300. 10.1128/IAI.73.9.5291-5300.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gibson-Corley KN, Olivier AK, Meyerholz DK. 2013. Principles for valid histopathologic scoring in research. Vet. Pathol. 50:1007–1015. 10.1177/0300985813485099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD, Higgins DG. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7:539. 10.1038/msb.2011.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schild S, Lamprecht AK, Reidl J. 2005. Molecular and functional characterization of O antigen transfer in Vibrio cholerae. J. Biol. Chem. 280:25936–25947. 10.1074/jbc.M501259200 [DOI] [PubMed] [Google Scholar]
- 33.Perez JM, McGarry MA, Marolda CL, Valvano MA. 2008. Functional analysis of the large periplasmic loop of the Escherichia coli K-12 WaaL O-antigen ligase. Mol. Microbiol. 70:1424–1440. 10.1111/j.1365-2958.2008.06490.x [DOI] [PubMed] [Google Scholar]
- 34.Islam ST, Taylor VL, Qi M, Lam JS. 2010. Membrane topology mapping of the O-antigen flippase (Wzx), polymerase (Wzy), and ligase (WaaL) from Pseudomonas aeruginosa PAO1 reveals novel domain architectures. mBio 1:e00189–10. 10.1128/mBio.00189-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tusnady GE, Simon I. 1998. Principles governing amino acid composition of integral membrane proteins: application to topology prediction. J. Mol. Biol. 283:489–506. 10.1006/jmbi.1998.2107 [DOI] [PubMed] [Google Scholar]
- 36.Tusnady GE, Simon I. 2001. The HMMTOP transmembrane topology prediction server. Bioinformatics 17:849–850. 10.1093/bioinformatics/17.9.849 [DOI] [PubMed] [Google Scholar]
- 37.Spyropoulos IC, Liakopoulos TD, Bagos PG, Hamodrakas SJ. 2004. TMRPres2D: high quality visual representation of transmembrane protein models. Bioinformatics 20:3258–3260. 10.1093/bioinformatics/bth358 [DOI] [PubMed] [Google Scholar]
- 38.Hong M, Payne SM. 1997. Effect of mutations in Shigella flexneri chromosomal and plasmid-encoded lipopolysaccharide genes on invasion and serum resistance. Mol. Microbiol. 24:779–791. 10.1046/j.1365-2958.1997.3731744.x [DOI] [PubMed] [Google Scholar]
- 39.Ohno A, Isii Y, Tateda K, Matumoto T, Miyazaki S, Yokota S, Yamaguchi K. 1995. Role of LPS length in clearance rate of bacteria from the bloodstream in mice. Microbiology 141:2749–2756. 10.1099/13500872-141-10-2749 [DOI] [PubMed] [Google Scholar]
- 40.Rietschel ET. 1984. Chemistry of endotoxin. Elsevier, New York, NY [Google Scholar]
- 41.Pradel E, Parker CT, Schnaitman CA. 1992. Structures of the rfaB, rfaI, rfaJ, and rfaS genes of Escherichia coli K-12 and their roles in assembly of the lipopolysaccharide core. J. Bacteriol. 174:4736–4745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vinogradov E, Conlan WJ, Gunn JS, Perry MB. 2004. Characterization of the lipopolysaccharide O-antigen of Francisella novicida (U112). Carbohydr. Res. 339:649–654. 10.1016/j.carres.2003.12.013 [DOI] [PubMed] [Google Scholar]
- 43.Reed LJ, Muench H. 1938. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 27:493–497 [Google Scholar]
- 44.Wang X, Ribeiro AA, Guan Z, McGrath SC, Cotter RJ, Raetz CR. 2006. Structure and biosynthesis of free lipid A molecules that replace lipopolysaccharide in Francisella tularensis subsp. novicida. Biochemistry 45:14427–14440. 10.1021/bi061767s [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raetz CR, Guan Z, Ingram BO, Six DA, Song F, Wang X, Zhao J. 2009. Discovery of new biosynthetic pathways: the lipid A story. J. Lipid Res. 50(Suppl):S103–S108. 10.1194/jlr.R800060-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Raetz CR, Reynolds CM, Trent MS, Bishop RE. 2007. Lipid A modification systems in gram-negative bacteria. Annu. Rev. Biochem. 76:295–329. 10.1146/annurev.biochem.76.010307.145803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McDonald MK, Cowley SC, Nano FE. 1997. Temperature-sensitive lesions in the Francisella novicida valA gene cloned into an Escherichia coli msbA lpxK mutant affecting deoxycholate resistance and lipopolysaccharide assembly at the restrictive temperature. J. Bacteriol. 179:7638–7643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bos MP, Tefsen B, Geurtsen J, Tommassen J. 2004. Identification of an outer membrane protein required for the transport of lipopolysaccharide to the bacterial cell surface. Proc. Natl. Acad. Sci. U. S. A. 101:9417–9422. 10.1073/pnas.0402340101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Genevrois S, Steeghs L, Roholl P, Letesson JJ, van der Ley P. 2003. The Omp85 protein of Neisseria meningitidis is required for lipid export to the outer membrane. EMBO J. 22:1780–1789. 10.1093/emboj/cdg174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tran AX, Trent MS, Whitfield C. 2008. The LptA protein of Escherichia coli is a periplasmic lipid A-binding protein involved in the lipopolysaccharide export pathway. J. Biol. Chem. 283:20342–20349. 10.1074/jbc.M802503200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mulford CA, Osborn MJ. 1983. An intermediate step in translocation of lipopolysaccharide to the outer membrane of Salmonella typhimurium. Proc. Natl. Acad. Sci. U. S. A. 80:1159–1163. 10.1073/pnas.80.5.1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alexander DC, Valvano MA. 1994. Role of the rfe gene in the biosynthesis of the Escherichia coli O7-specific lipopolysaccharide and other O-specific polysaccharides containing N-acetylglucosamine. J. Bacteriol. 176:7079–7084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Keenleyside WJ, Whitfield C. 1996. A novel pathway for O-polysaccharide biosynthesis in Salmonella enterica serovar Borreze. J. Biol. Chem. 271:28581–28592. 10.1074/jbc.271.45.28581 [DOI] [PubMed] [Google Scholar]
- 54.Lehrer J, Vigeant KA, Tatar LD, Valvano MA. 2007. Functional characterization and membrane topology of Escherichia coli WecA, a sugar-phosphate transferase initiating the biosynthesis of enterobacterial common antigen and O-antigen lipopolysaccharide. J. Bacteriol. 189:2618–2628. 10.1128/JB.01905-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim TH, Sebastian S, Pinkham JT, Ross RA, Blalock LT, Kasper DL. 2010. Characterization of the O-antigen polymerase (Wzy) of Francisella tularensis. J. Biol. Chem. 285:27839–27849. 10.1074/jbc.M110.143859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leyman B, Boyen F, Van Parys A, Verbrugghe E, Haesebrouck F, Pasmans F. 2011. Salmonella Typhimurium LPS mutations for use in vaccines allowing differentiation of infected and vaccinated pigs. Vaccine 29:3679–3685. 10.1016/j.vaccine.2011.03.004 [DOI] [PubMed] [Google Scholar]
- 57.Billips BK, Yaggie RE, Cashy JP, Schaeffer AJ, Klumpp DJ. 2009. A live-attenuated vaccine for the treatment of urinary tract infection by uropathogenic Escherichia coli. J. Infect. Dis. 200:263–272. 10.1086/599839 [DOI] [PubMed] [Google Scholar]
- 58.Okan NA, Chalabaev S, Kim TH, Fink A, Ross RA, Kasper DL. 2013. Kdo hydrolase is required for Francisella tularensis virulence and evasion of TLR2-mediated innate immunity. mBio 4:e00638–12. 10.1128/mBio.00638-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chalabaev S, Kim TH, Ross R, Derian A, Kasper DL. 2010. 3-Deoxy-d-manno-octulosonic acid (Kdo) hydrolase identified in Francisella tularensis, Helicobacter pylori, and Legionella pneumophila. J. Biol. Chem. 285:34330–34336. 10.1074/jbc.M110.166314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zarrella TM, Singh A, Bitsaktsis C, Rahman T, Sahay B, Feustel PJ, Gosselin EJ, Sellati TJ, Hazlett KRO. 2011. Host-adaptation of Francisella tularensis alters the bacterium's surface-carbohydrates to hinder effectors of innate and adaptive immunity. PLoS One 6:e22335. 10.1371/journal.pone.0022335 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.