

Abstract
Pneumococcal meningitis (PM) results in high mortality rates and long-lasting neurological deficits. Hippocampal apoptosis and cortical necrosis are histopathological correlates of neurofunctional sequelae in rodent models and are frequently observed in autopsy studies of patients who die of PM. In experimental PM, inhibition of matrix metalloproteinases (MMPs) and/or tumor necrosis factor (TNF)-converting enzyme (TACE) has been shown to reduce brain injury and the associated impairment of neurocognitive function. However, none of the compounds evaluated in these studies entered clinical development. Here, we evaluated two second-generation MMP and TACE inhibitors with higher selectivity and improved oral availability. Ro 32-3555 (Trocade, cipemastat) preferentially inhibits collagenases (MMP-1, -8, and -13) and gelatinase B (MMP-9), while Ro 32-7315 is an efficient inhibitor of TACE. PM was induced in infant rats by the intracisternal injection of live Streptococcus pneumoniae. Ro 32-3555 and Ro 32-7315 were injected intraperitoneally, starting at 3 h postinfection. Antibiotic (ceftriaxone) therapy was initiated at 18 h postinfection, and clinical parameters (weight, clinical score, mortality rate) were recorded. Myeloperoxidase activities, concentrations of cytokines and chemokines, concentrations of MMP-2 and MMP-9, and collagen concentrations were measured in the cerebrospinal fluid. Animals were sacrificed at 42 h postinfection, and their brains were assessed by histomorphometry for hippocampal apoptosis and cortical necrosis. Both compounds, while exhibiting disparate MMP and TACE inhibitory profiles, decreased hippocampal apoptosis and cortical injury. Ro 32-3555 reduced mortality rates and cerebrospinal fluid TNF, interleukin-1β (IL-1β) and collagen levels, while Ro 32-7315 reduced weight loss and cerebrospinal fluid TNF and IL-6 levels.
INTRODUCTION
Pneumococcal meningitis (PM) is characterized by a considerably high mortality rate and leads to long-lasting neurofunctional deficits such as epilepsy, hearing loss, and cerebral palsy, as well as behavioral problems and cognitive deficits (1–4). Neuronal apoptosis in the dentate gyrus (DG) of the hippocampus, a region critical for memory acquisition, and cortical necrosis are the neuropathological correlates of these functional symptoms of PM in both experimental models and human disease (5–7). The invading pathogens trigger an excessive inflammatory immune response with high levels of cytokine and chemokine release into the cerebrospinal fluid (CSF) space acting as the primary driver of the pathophysiology of PM (1). Tumor necrosis factor (TNF) is the most prominent of the early-response cytokines, which also include interleukin-1β (IL-1β) and IL-6, and triggers a cascade of proinflammatory mediators that are elevated in PM (8, 9). TNF is released by a metalloproteinase (TNF-converting enzyme [TACE], ADAM-17) closely related to the family of matrix metalloproteinases (MMPs) (10). MMPs are proteolytic enzymes and play a crucial role in inflammation and tissue degradation and remodeling. Moreover, they release and activate soluble cytokines and their receptors (11–13). In PM, these processes also occur at the level of the brain microvasculature, where MMPs promote blood-brain barrier (BBB) breakdown, enable leukocyte extravasation (1, 14, 15), and thereby contribute to the development of brain edema, hydrocephalus, vasculitis, intracranial bleeding, brain ischemia, and other major complications of PM (16). An imbalance of MMP and TACE activity, compared to that of their endogenous inhibitor (tissue inhibitor of metalloproteinase), is a molecular factor contributing to tissue damage (17, 18). Important in the clinical context is the finding that MMP-8 and MMP-9 are elevated in the CSF of children with bacterial meningitis, and a high concentration of MMP-9 has been identified as a risk factor for the development of neurological sequelae (19). MMP-9 is regarded as an inflammatory marker that correlates with the CSF cell count but not with BBB impairment (20). MMP-9 is also increased in other inflammatory diseases, including multiple sclerosis, while it is usually below the limit of detection in the CSF of healthy subjects (20, 21).
Originally, MMP inhibitors were developed for the treatment of cancer and chronic inflammatory diseases, mainly arthritis (22). Lack of oral bioavailability; long-term adverse effects, i.e., arthralgia and myalgia; and efficacy in human disease vis-à-vis animal models that remains below expectation have led the industry to halt virtually all development programs (23, 24). Part of the issues were addressed with the development of compounds with more selective activity profiles (25). Furthermore, long-term adverse effects would not occur during short-term use against acute diseases such as PM. There, MMP inhibitors, particularly in combination with TACE inhibitors (e.g., BB 1101, GM 6001, TNF 484, doxycycline), have been successful against pathophysiological aspects that are outside the reach of standard therapies and have attenuated at least one form of brain injury (14, 26–28).
Ro 32-3555 (Trocade, cipemastat) is an inhibitor of collagenases (MMP-1, -8, and -13) and gelatinase B (MMP-9) but is weakly active against TACE (29). In a rodent model of rheumatoid arthritis, Ro 32-3555 prevented articular cartilage and bone destruction (30). Tolerability of oral Ro 32-3555 application has been proven in a clinical phase II trial in patients with rheumatoid arthritis; however, further clinical development for the treatment of arthritis was not pursued because long-term progression of joint damage could not be altered (25, 30–32). Ro 32-7315 is a potent inhibitor of TACE (50% inhibitory concentration [IC50], 5 nM) with inhibitory activity against MMPs 5 to 10 times less than that of Ro 32-3555 (29, 33). Its clinical safety has been evaluated in a phase I trial with healthy human subjects (33). Because of their more selective activity profile and improved bioavailability than earlier compounds, in the present study, Ro 32-3555 and Ro 32-7315 were evaluated head to head in the infant rat model of PM with regard to their potential beneficial effects on structural brain damage and clinical and inflammatory parameters. The dosage used was adapted from earlier rodent studies (Roche Pharma, personal communication; 33–35).
MATERIALS AND METHODS
Infecting organism.
A clinical isolate of S. pneumoniae (serotype 3) was cultured overnight in brain heart infusion medium, diluted in fresh medium, and grown for 5 h to logarithmic phase as reported earlier (14). The culture broth was centrifuged for 10 min at 1,600 × g, pelleted, and resuspended in sterile, pyrogen-free saline to an optical density at 570 nm (OD570) of 0.013 to 0.015. The accuracy of the inoculum size was routinely confirmed by quantitative culture on sheep blood agar plates.
Infant rat model of PM.
All animal studies were approved by the Animal Care and Experimentation Committee of the Canton of Bern, Switzerland (license BE 100/11), and followed the Swiss national guidelines for the performance of animal experiments. A well-established infant rat model of PM was used as previously described (14). Litters of 12 nursing Wistar rats with their dams were obtained from Charles River (Sulzfeld, Germany) and acclimatized for 5 days prior to infection on day 11 by the intracisternal (i.c.) injection of 10 μl of 0.85% NaCl containing live S. pneumoniae bacteria at log10 6.05 ± 5.98 CFU/ml.
The effects of Ro 32-3555 at 2 × 75 mg kg−1 day−1 (n = 56) and Ro 32-7315 at 2 × 25 mg kg−1 day−1 on rats (n = 41) were compared to those of treatment with the vehicle alone, i.e., succinylated gelatin (Physiogel; Spitalpharmazie Inselspital, Bern University Hospital, Bern, Switzerland) on littermates (n = 92). Therefore, animals were randomly assigned to the treatment groups. The compounds were resuspended in the vehicle directly before use and administered by intraperitoneal injection at 3 h postinfection (hpi) to maximize their effect and then at 18 and 27 hpi (15 min before the injection of ceftriaxone). Vehicle-treated littermates served as controls (n = 92). PM was confirmed by quantitative analysis of bacterial titers in CSF samples when animals developed symptomatic disease at 18 hpi. These titers did not vary significantly between the treatment groups (Table 1).
TABLE 1.
Bacterial titers, clinical scores, weight changes, CSF MPO activity levels, and MMP-9 concentrations of infant rats with acute PM treated with ceftriaxone and Ro 32-3555, Ro 32-7315, or the vehicle
| Parameter | Mean value ± SD (no. of animals) |
||
|---|---|---|---|
| Vehicle | Ro 32-3555 | Ro 32-7315 | |
| CSF bacterial titer (log10 CFU/ml), 18 hpi | 8.73 ± 9.13 (91) | 8.83 ± 9.17 (55) | 8.16 ± 8.16 (41) |
| Clinical score at: | |||
| 18 hpi | 4.18 ± 0.37 (92) | 4.10 ± 0.32 (56) | 4.21 ± 0.46 (41) |
| 27 hpi | 3.60 ± 0.91 (69) | 3.92 ± 0.6 (51) | 3.48 ± 1.01 (22) |
| 42 hpi | 4.39 ± 0.78 (57) | 4.34 ± 0.79 (47) | 4.69 ± 0.48 (13) |
| Weight change (g), 0–42 hpi | −1.12 ± 1.49 (52) | −1.01 ± 1.06 (43) | −0.04 ± 1.86 (13)a |
| MPO activity (OD450)b at: | |||
| 18 hpi | 7.35 ± 9.94 (28) | 7.79 ± 6.65 (22) | 2.67 ± 2.68 (9) |
| 27 hpi | 13.7 ± 19.2 (18) | 20.1 ± 22.4 (24) | 4.87 ± 7.83 (3) |
| 42 hpi | 6.60 ± 10.5 (13) | 5.04 ± 7.28 (22) | 4.97 ± 5.43 (4) |
Significantly different (P < 0.05) from the value for vehicle-treated animals.
Arbitrary units.
To assess disease severity, animals were weighed and examined clinically at 0, 18, and 27 hpi and at the time of sacrifice (42 hpi). The severity of disease was scored as previously described, on the following scale: 1, coma; 2, does not stand upright after being turned on the back; 3, stands upright within 30 s; 4, minimal ambulatory activity, stands upright in <5 s; 5, normal (14). Animals reaching a score of ≤2 were sacrificed by an overdose of pentobarbital (Esconarkon; Streuli, Uznach, Switzerland; 100 mg kg−1 day−1 intraperitoneally) for ethical reasons; spontaneous deaths were documented. Antibiotic therapy with ceftriaxone (Rocephine; 2 × 100 mg kg of body weight−1 day−1 intraperitoneally; Roche Pharma, Basel, Switzerland) was started at 18 hpi. Punctures of the cisterna magna with a 30-gauge needle were performed to obtain CSF samples at 18, 27, and 42 hpi. CSF samples not used for bacterial titer determination or myeloperoxidase (MPO) assays were centrifuged (16,000 × g at 4°C for 10 min), and the supernatants were frozen at −80°C until further use for gelatin gel zymography (MMP-2 and MMP-9) or determination of cytokine or collagen concentrations. Animals were sacrificed with an overdose of pentobarbital at 42 hpi and perfused with 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) before their brains were removed and fixed in PFA for histological analysis.
Assessment of MPO activity.
MPO activity in CSF samples was measured as a parameter of inflammation and leukocyte pleocytosis as described earlier (9). Five microliters of noncentrifuged CSF was resuspended in 200 μl of HETAB solution (0.5% hexadecyltrimethylammonium bromide in 100 mM potassium phosphate buffer) and repeatedly submitted to three cycles of freeze-thawing, and sonication, followed by centrifugation for 5 min at 16,000 × g at 4°C. Supernatants were stored at 80°C until use. Assays were performed in triplicate by mixing 155 μl of HETAB buffer with 10 μl of sample and 10 μl of O-dianisidine dihydrochloride (20 mg/ml in H2O) in a 96-well plate. The reaction was initiated by the addition of 25 μl of 2 mM H2O2. Absorbance at 450 nm was measured every 30 s for 10 min at 37°C with a Thermomax microplate reader (Molecular Devices Inc., Sunnyvale, CA). The linear portion of the curve was used to determine the increase in absorbance. MPO activity was expressed as a change in OD per unit of time corresponding to Vmax as determined with SOFTmax PRO software version 3.1.2 (Molecular Devices Inc., Sunnyvale, CA).
Quantification of MMP-2 and MMP-9 by gelatin gel zymography.
MMP-9 and MMP-2 were quantified by gelatin gel zymography as described in detail previously (14). In brief, samples of 1.8 to 2.5 μl of CSF were diluted to a loading volume of 20 μl and electrophoresed in gels containing 10% polyacrylamide–0.1% sodium dodecyl sulfate (SDS) plus 1 mg/ml gelatin; purified human neutrophil MMP-9 and MMP-2 (Calbiochem, Darmstadt, Germany) were used as standards on each gel. After electrophoresis for 2 h at 100 V, MMPs were renatured by immersing the gel for 1 h in Triton X-100 (2% [vol/vol]) to remove SDS. The gels were then incubated in zymography buffer (10 mM CaCl2, 50 mM Tris, 50 mM NaCl [pH 7.65]) for 18 h at 37°C to allow proteolysis of the gelatin substrate and stained with Coomassie blue. Gels were scanned, and densities of substrate lysis zones at 86 kDa (active MMP-9) or 62 kDa (active MMP-2) were measured with the ImageJ software (NIH). The gelatinolytic activities of MMP-9 (gelatinase B, 92 kDa) and MMP-2 (gelatinase A, 72 kDa) were determined by quantification of gelatin lysis zones and expressed as absolute values for each sample (29).
CSF collagen content.
The collagen concentrations in CSF samples were assessed as an index of gelatinase and collagenase activity in the central nervous system (CNS) with the QuickZyme Total Collagen Assay (QuickZyme Biosciences, Leiden, Netherlands) according to the manufacturer's protocol with minor modifications. Samples of 10 or 20 μl CSF supernatant were diluted with HCl to a final concentration of 4 M HCl and a final volume of 50 μl. Hydrolysis was performed in screw-cap tubes for 20 h at 95°C with constant shaking. A 35-μl volume of the centrifuged, undiluted supernatant (16,000 × g for 10 min) was used for analysis and compared to a standard curve of hydrolyzed collagen (hydroxyproline) provided in the kit (1,200 μg/ml in 0.02 M acetic acid, dilution with 4 M HCl). A 75-μl volume of assay buffer was added to each well of the plate, and samples were incubated for 60 min at 60°C while shaking. Absorbance at 550 nm was measured after the plate cooled to room temperature.
Quantitative analysis of cytokines in CSF.
A panel of cytokines and chemokines known to be involved in the pathophysiology of bacterial meningitis was selected to document the inflammatory response in the infant rat model of PM (8): TNF-α, IL-6, IL-1β, IL-10, monocyte chemoattractant protein 1 (MCP-1), MIP-1α, and gamma interferon (IFN-γ) (36). The concentrations of these analytes in CSF were determined with microsphere-based multiplex assays (MILLIPLEX MAP kit, Rat Cytokine/Chemokine Magnetic Bead Panel, catalog number RECYTMAG-65K; Millipore Corporation, Billerica, MA). A 5-μl sample of CSF supernatant was diluted to a final volume of 25 μl with the assay buffer provided. A minimum of 50 beads per analyte were measured with a Bio-Plex 200 station (Bio-Rad Laboratories, Hercules, CA). Calibration curves from recombinant standards were calculated with Bio-Plex Manager software version 4.1.1 by five-parameter logistic curve fitting. If the sample concentration was below the limit of detection, the limit of detection provided by the manufacturer multiplied by the dilution factor was used for statistical analysis, i.e., TNF-α, 9.5 pg/ml; IL-6, 153.5 pg/ml; IL-1β, 14 pg/ml; IL-10, 13.5 pg/ml; MCP-1, 45 pg/ml; MIP-1α, 4.0 pg/ml; IFN-γ, 31 pg/ml. Interassay variations were corrected by normalization to an internal control with a known concentration.
Histomorphometric analysis.
Brain damage was quantified as previously described in animals sacrificed at 42 hpi (37). Brains were fixed in PFA and cryopreserved in 18% sucrose in PBS at 4°C overnight. Coronal brain cryosections (45 μm thick) obtained by systematic uniform random sampling were stained for Nissl substance with cresyl violet. Cortical damage was defined as areas of decreased neuronal density or frank cortical necrosis by simultaneous bright-field microscopy and scanned digitized images with the software ImageJ 1.45l (National Institutes of Health, http://imagej.nih.gov/ij). The volume of cortical brain damage was expressed as a percentage of the total cortical volume determined by the Cavalieri principle by investigating at least 16 brain sections per animal as described in detail previously (38).
Histological features of apoptosis (condensed, fragmented, dark nuclei, apoptotic bodies) were counted in four different slices spanning the hippocampus of both hemispheres by a person blinded to the experimental grouping. Cells were counted in three visual fields in each of the two blades of the DG, and a mean value per animal (a total of 48 fields) was calculated.
Statistical analyses.
Statistical analyses were performed with GraphPad Prism software (Prism 6 for Windows; GraphPad Software Inc., San Diego, CA). If not stated otherwise, results are presented as mean values ± standard deviations. The D'Agostino-Pearson omnibus normality test was used to discriminate between parametric and nonparametric values. To compare data between two groups, an unpaired Student t test was used for parametric data; otherwise, the Mann-Whitney test was used. To compare three groups, one-way analysis of variance was used for parametric data and the Kruskal-Wallis test was used for nonparametric data together with Dunn's multiple-comparison test. Mortality rates were calculated by using the log rank (Mantel-Cox) test for significance based on all infected animals and numbers of animals sacrificed for ethical reasons (clinical score of ≤2) or dying spontaneously. A cutoff finder using the R statistical engine was used as previously described (http://molpath.charite.de/cutoff/load.jsp) (39). A chi-square test was used to compare animals with clinical scores of <4 (but not in a coma or dead) with those with scores of 4 to 5. A two-tailed P value of <0.05 was considered statistically significant.
RESULTS
Clinical scores and mortality rates.
A total of 194 animals weighing 25.0 ± 2.9 g were infected. All animals had meningitis, as indicated by positive bacterial titers in CSF samples and disease symptoms (weight loss, a clinical score of <5), except for five animals, which were consequently excluded from all analyses. Clinical scores and weights were recorded at infection and 18, 27, and 42 hpi.
Ro 32-3555 improved the clinical status at 27 hpi, where only 9 (17.6%) of 51 animals had scores of <4 compared to 28 (40.6%) of 69 vehicle-treated animals that had a low score (P = 0.0269). Conversely, the influence of Ro 32-7315 was weak (11 [50%] of 22 animals had scores of <4 [P = 0.7392]). The lowest clinical score in the control group was recorded at 27 hpi (Table 1). Comparably, the mortality rate was highest during the acute disease phase, between 21 and 29 hpi (Fig. 1A). Adjunctive treatment (in addition to antibiotics) with Ro 32-3555 improved survival significantly from 62.9% (vehicle; n = 92) to 85.5% (n = 56; P = 0.0043; Fig. 1A), while the survival rate of animals treated with Ro 32-7315 tended to be lower; however, this effect remained below the level of statistical significance (43.7%; n = 41, P = 0.0533).
FIG 1.
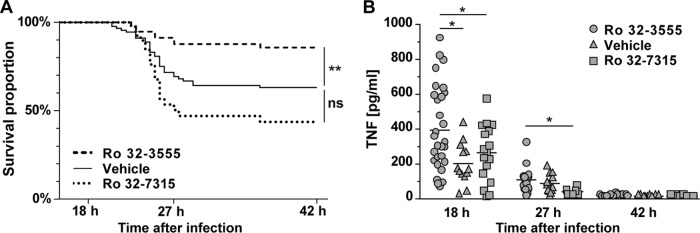
(A) Kaplan-Meier curves showing the survival proportions of animals with PM treated with antibiotics and MMP and/or TACE inhibitors or the vehicle (succinylated gelatin). Results represent spontaneous deaths and killing for ethical reasons (severe disease with a clinical score of <2). Ro 32-3555 reduced the mortality rate significantly (**, P < 0.005) to 14.5% compared to that of control littermates (37.1%), while Ro 32-7315 did not alter the mortality rate significantly (56.3%, P < 0.05). (B) Ro 32-3555 reduced the concentrations of TNF in CSF significantly at 18 hpi, while Ro 32-7315 reduced TNF levels at 18 and 27 hpi compared to those of littermates treated with the vehicle. At 42 hpi TNF values were close to the limit of detection (*, P < 0.05).
Weight loss.
The comparison of weight changes at the time of infection versus 42 hpi showed less weight loss in animals treated with Ro 32-7315 than in animals treated with the vehicle (P = 0.0106, but not versus Ro 32-3555) (Table 1).
MPO activity in CSF samples.
MPO activity measured as an index of the leukocyte concentration peaked at 27 hpi in the control group (Table 1). None of the compounds had a significant effect on MPO activity.
Cytokines and chemokines in CSF samples.
Cytokine and chemokine levels in CSF were measured at 18, 27, and 42 hpi (Table 2). TNF expression was most accentuated at 18 hpi, less accentuated at 27 hpi, and close to the limit of detection at 42 hpi (Ro 32-3555, 12.8 ± 5.9 pg/ml, n = 7; vehicle, 11.5 ± 1.9 pg/ml, n = 9 [no statistically significant difference]). IL-6, IL-1β, MCP-1, and MIP-1α peaked at 18 hpi; IFN-γ and IL-10 peaked at 27 hpi (Table 2). TNF was significantly reduced by both MMP and TACE inhibitors at 18 hpi and by Ro 32-7315 at 27 hpi (Table 2; Fig. 1B). Concentrations of TNF in CSF correlated with MMP-9 levels (P = 0.0212, r = 0.32, n = 52 [Spearman correlation coefficient]). Animals with a low TNF concentration in their CSF (<128 pg/ml), independently of treatment, showed significantly increased survival rates compared to those of littermates with high TNF concentrations (Fisher's exact test, P = 0.0121). IL-1β concentration was reduced by Ro 32-3555 but not by Ro 32-7315 compared to that in control littermates at 18 hpi, while Ro 32-7315 reduced IL-6 at 27 hpi. A nonsignificant trend toward decreased levels of IFN-γ was observed at 27 hpi for Ro 32-3555 and Ro 32-7315 (P = 0.4371 and P = 0.8653; Table 2).
TABLE 2.
Cyto- and chemokine levels in CSF samples of infant rats with acute PM at 18 and 27 hpi
| Cyto- or chemokine | Mean concn (pg/ml) in CSF ± SD after treatment with: |
|||||
|---|---|---|---|---|---|---|
| Vehicle, 18 hpi (n = 30) | Ro 32-3555, 18 hpi (n = 13) | Ro 32-7315, 18 hpi (n = 18) | Vehicle, 27 hpi (n = 18) | Ro 32-3555, 27 hpi (n = 14) | Ro 32-7315, 27 hpi (n = 6) | |
| TNF | 382.7 ± 246.9 | 190.1 ± 118.3a | 242.9 ± 161.7a | 101.7 ± 109.7 | 85.86 ± 61.99 | 29.54 ± 21.25a |
| IL-6 | 61,183 ± 32,192 | 53,491 ± 24,250 | 51,725 ± 32,281 | 41,295 ± 31,248 | 38,401 ± 23,327 | 17,605 ± 23,004a |
| IL-1β | 1,363 ± 967.5 | 678.3 ± 416.7a | 1,338 ± 969.0 | 692.4 ± 636.1 | 347.7 ± 208.1 | 500.2 ± 598.9 |
| MIP-1α | 2,081 ± 1,683 | 1,270 ± 771.7 | 2,297 ± 2,137 | 1,000 ± 931.1 | 1,355 ± 1,298 | 528.9 ± 839.9 |
| MCP | 85,276 ± 38,771 | 61,103 ± 23,455 | 151,935 ± 244,055 | 52,478 ± 45,868 | 70,716 ± 72,173 | 24,588 ± 14,139 |
| IL-10 | 432.3 ± 236.5 | 273.5 ± 178.1 | 425.1 ± 275.4 | 624.5 ± 376.9 | 868.4 ± 776.2 | 411.4 ± 360.4 |
| IFN-γ | 7,776 ± 7,095 | 3,426 ± 3,313 | 8,386 ± 7,272 | 8,933 ± 7,421 | 4,887 ± 2,703 | 4,867 ± 2,144 |
Significantly different (P < 0.05) from the value for vehicle-treated animals.
Quantification of MMP-9 and MMP-2 in CSF samples.
Levels of MMP-9 and MMP-2 in CSF samples were measured at 18, 27, and 42 hpi. In the control group, the highest values were measured at 18 hpi (Table 3). Neither Ro 32-3555 nor Ro 32-7315 significantly influenced the amount of MMP-9. Accordingly, MMP-2 was not altered by either treatment and remained stable at different time points (no statistically significant difference; data not shown). Consequently, the ratio of MMP-9 to MMP-2 was changed only marginally over time (Table 3; Fig. 2A).
TABLE 3.
Concentrations of MMP-2 and MMP-9 in CSF samples from infant rats treated with MMP and/or TACE inhibitors during the acute phase of PM
| Parameter | Mean value ± SD (no. of animals) |
||
|---|---|---|---|
| Vehicle | Ro 32-3555 | Ro 32-7315 | |
| MMP-9 concn (ng/ml) at: | |||
| 18 hpi | 1,468 ± 2,689 (32) | 778 ± 1,168 (20) | 262 ± 280 (20) |
| 27 hpi | 377 ± 334 (20) | 200 ± 112 (21) | 235 ± 155 (4) |
| 42 hpi | 238 ± 225 (7) | 352 ± 420 (12) | 39.2 ± 13 (4) |
| MMP-9/MMP-2 ratio at: | |||
| 18 hpi | 2.80 ± 1.70 (32) | 2.19 ± 1.41 (20) | 2.98 ± 1.96 (20) |
| 27 hpi | 1.07 ± 0.47 (25) | 0.84 ± 0.41 (21) | 1.06 ± 0.49 (7) |
| 42 hpi | 0.54 ± 0.19 (7) | 0.55 ± 0.33 (12) | 033 ± 0.08 (4) |
| Collagen concn (μg/ml) at: | |||
| 18 hpi | 40.9 ± 12.5 (27) | 33.1 ± 5.0 (12)a | 44.1 ± 8.7 (22) |
| 27 hpi | 60.1 ± 17.3 (7) | 58.7 ± 17.3 (5) | n = 0 |
Significantly different (P < 0.05) from the value for vehicle-treated animals.
FIG 2.
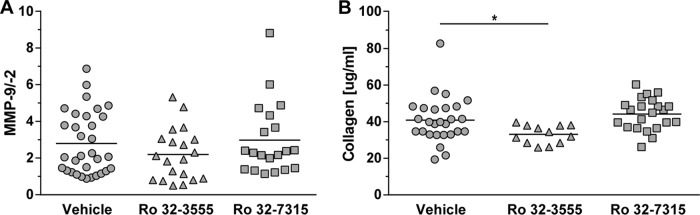
(A) The effect of MMP and/or TACE inhibition on MMP-9/MMP-2 ratios measured in samples of CSF at 18 hpi remained below the level of statistical significance. (B) The collagen content of samples of CSF at 18 hpi was reduced by treatment with the collagenase and gelatinase inhibitor Ro 32-3555 but not by treatment with the TACE inhibitor Ro 32-7315 (*, P < 0.05).
Collagen contents of CSF samples.
The concentration of collagen was assessed as an index of gelatinase and collagenase activity in the CNS. CSF samples obtained at 18 and 27 hpi were hydrolyzed, and the substrate hydroxyproline was quantified by spectrophotometry. At 18 hpi, the collagen concentration in the CSF of animals treated with the collagenase and gelatinase inhibitor Ro 32-3555 was significantly lower than that in control rat CSF (P = 0.0312), while the TACE inhibitor Ro 32-7315 had no effect (no statistically significant difference; Table 3; Fig. 2B).
Hippocampal injury and cortical damage.
Histomorphometric analysis of the brains of animals sacrificed at 42 hpi was performed. The two forms of brain injury characteristic of PM, i.e., hippocampal apoptosis and cortical injury, were found in the infected animals and were quantified (Fig. 3). Treatment with the collagenase and gelatinase inhibitor Ro 32-3555 resulted in significantly less apoptotic brain injury in the DG of the hippocampus than treatment with the vehicle (Ro 32-3555, 3.63 ± 7.89 apoptotic cells per visual field [c/f], n = 43; vehicle, 6.10 ± 8.05 c/f, n = 52 [P = 0.0455]) (Fig. 4A). Treatment with the TACE inhibitor Ro 32-7315 led to an even more pronounced reduction of hippocampal apoptosis (to 0.92 ± 1.47 c/f [n = 13, P = 0.0041]) compared to that of controls.
FIG 3.

Representative images (Nissl staining) of cryosections prepared 42 h after the induction of PM in infant rats. (A) Apoptotic neurons (selected cells are marked with arrowheads; inset) were found throughout the hippocampal DG of animals treated with the vehicle and ceftriaxone, whereas apoptotic cells were found only sporadically in animals treated with Ro 32-7315 and ceftriaxone (B). (C) A sharply demarcated necrotic zone in the cortex (arrows) with meningeal leukocyte infiltration (asterisks) is visible in an animal treated with the vehicle and ceftriaxone, as opposed to the intact cortex with slight meningeal leukocyte infiltration (asterisks) in an animal treated with Ro 32-7315 (D).
FIG 4.
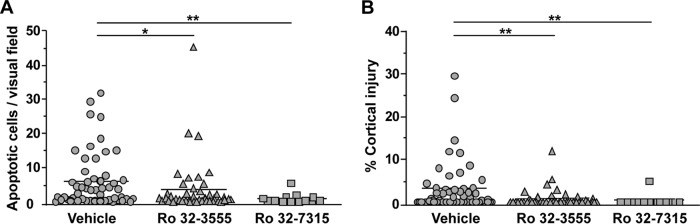
Histomorphometric analysis of the two characteristic forms of brain damage due to PM in cryosections of animals sacrificed at 42 hpi. (A) Rats treated with Ro 32-3555 or Ro 32-7315 showed significantly less hippocampal apoptosis than vehicle-treated littermates (*, P < 0.05; **, P < 0.01). (B) Infant rats treated with Ro 32-3555 or Ro 32-7315 showed significantly less cortical necrosis than vehicle-treated littermates (**, P < 0.01).
The volume of injured cortex was significantly reduced by Ro 32-3555 from 3.16% ± 5.89% (n = 52) to 0.78% ± 2.09% (n = 43, P = 0.0050, Fig. 4B), and Ro 32-7315 significantly reduced the volume of injured cortex to 0.37% ± 1.34% (n = 13, P = 0.0032). In the control group, 28 (46.2%) of 52 animals showed relevant injury of the cortex, as defined by damage to >1% of the total cortical volume. The percentage of animals with cortical injury was significantly lowered by treatment with Ro 32-3555 (18.6%, 8 of 43 brains [P = 0.0183]) or Ro 32-7315 (7.7%,1 of 13 brains [P = 0.0388]).
DISCUSSION
In the present study, Ro 32-3555 and Ro 32-7315 were evaluated for their effectiveness in reducing brain injury and inflammatory parameters and improving clinical outcome. This is the first head-to-head comparison of two MMP and/or TACE inhibitors with distinct inhibitory profiles that target different pathogenetic pathways of PM. Ro 32-3555 is an inhibitor of collagenases with no effect on TACE, while Ro 32-7315 is a potent TACE inhibitor (29, 33). Therefore, we hypothesized that they might differ in their impact on neuropathological and clinical outcomes.
Brain injury in PM develops during an overshooting immune response. It involves an increase in MMP activity, even though the detailed mechanisms may be dissimilar in different forms of brain injury (40). Foci of cortical necrosis formation start appearing at 18 hpi; are probably a consequence of focal ischemia, vasculitis, and venous thrombosis; and are associated with brain edema and increased intracranial pressure (1, 41, 42). A marked increase in the number of apoptotic cells in the hippocampal DG, caused by an overwhelming immune response and bacterial toxins without direct exposure of the DG to pneumococci, is first observed between 18 and 24 hpi (37, 40, 43). In experimental PM, the mortality rate is highest between 21 and 29 hpi, when inflammation peaks because of bacteriolysis after the application of ceftriaxone, among other factors (28, 44). Therefore, potential pharmacologic interventions, including MMP and TACE inhibition, have to fall before or within this time window to be effective. In the present proof-of-concept study, this issue was addressed by maximizing the treatment effect by starting the adjunctive treatment at 3 hpi.
In rodent models of PM, MMPs contribute to brain injury via different mechanisms, including those located in the brain microvasculature. MMP-9 levels can be detected as early as 15 min after infection and peak at 12 hpi, which indicates a release from preformed stores by resident brain parenchymal cells (26). MMP-9 exerts direct effects on the BBB by degrading collagen, a main component of the basal lamina (11, 14). Thus, MMP-9 facilitates leukocyte extravasation and BBB leakage, which eventually lead to edema formation and a rise in intracranial pressure (18). Furthermore, MMPs activate cytokines and chemokines and their receptors by acting as sheddases and convertases and by initiating and sustaining a vicious circle of proinflammatory mechanisms that eventually contribute to the development of cortical necrosis, as well as of hippocampal apoptosis (15). Therefore, pharmacological inhibition of MMPs may indirectly downregulate inflammatory mediators, including TNF (1, 15, 45). No significant changes in MMP-2 concentrations were observed, since MMP-2 is constitutively expressed (26, 33), while MMP-9 is upregulated during the acute phase of PM (18).
Ro 32-3555 had a beneficial effect on the mortality rate and was neuroprotective by reducing hippocampal apoptosis and cortical necrosis, although this effect was less pronounced than that of Ro 32-7315, especially regarding hippocampal apoptosis (discussed below). To have comparable time points, we used only animals that survived until the end of the experiment (42 hpi) for histomorphometric analyses. This could have led to a selection bias for a population of healthier animals, especially in the control group, which is characterized by a higher mortality rate. Therefore, the effective brain injury level might have been higher in this group, leading to an underestimation of the neuroprotective effect of Ro 32-3555.
Concentrations of MMP-9 and MMP-2 in CSF were measured by gel zymography. MMP-9 was elevated at 18 hpi, while MMP-2 was stable over time and only the ratio of MMP-9 to MMP-2 was lowered by treatment with Ro 32-3555, without reaching significance. However, the determination of MMP levels in CSF by gel zymography does not reflect in vivo collagenolytic activity (46), which might explain why MMP-9 levels were not significantly affected by MMP and TACE inhibition in the present study. The concentration of collagen in CSF was measured as an index of MMP-mediated collagenolysis in the brain compartment. The concentrations of collagen and its constituent hydroxyproline in CSF are strongly increased during PM as a biomarker of extracellular matrix catabolism (18, 27). Ro 32-3555 reduced the collagen concentration at 18 hpi. As it has only a weak impact on the intrathecal MMP-9 concentration, it effectively inhibits its enzymatic activity and likely that of other MMPs in the CNS. In contrast, the TACE inhibitor Ro 32-7315, which has only limited inhibitory activity on gelatinases and collagenases, had no effect on collagen turnover (see below).
Concentrations of MMP-9 and TNF in CSF correlated in the present study, as described previously (14, 26). We hypothesize that early inhibition of collagenase and gelatinase activities by Ro 32-3555 impairs BBB breakdown and reduces proinflammatory processes; this is reflected by the decreased production and release of TNF and IL-1β by activated immune cells. These cytokines are known as markers of acute inflammatory processes and inflammation-related complications of PM (8). Ro 32-3555 reduced IFN-γ as a proinflammatory cytokine and IL-10 as an anti-inflammatory cytokine inhibiting TNF production (47, 48), indicating lower inflammatory activity than in vehicle-treated animals, although this effect did not reach statistical significance. In contrast, IFN-γ and IL-10 were not influenced by TACE inhibition. Thus, reduced cytokine and chemokine production and release could indirectly protect vulnerable neurons in the DG from apoptosis and reduce cerebrovascular complications that lead to necrosis in the cortex (40, 49). In summary, Ro 32-3555 may indirectly reduce the inflammatory reaction during the acute phase of PM without altering the CSF cell count and MMP-9 concentration by reducing the sheddase and convertase activities of MMPs and hence protecting the integrity of the BBB.
Ro 32-7315 prevented cortical injury and showed a profound antiapoptotic effect. The combined MMP and TACE inhibitor BB-1101 showed similar protective effects on cortical necrosis and hippocampal apoptosis in a previous study (26). As opposed to the effect of Ro 32-3555, which may limit brain injury by acting on the BBB and a generalized reduction of inflammatory activities, we propose that the neuroprotective effect of the TACE inhibitor Ro 32-7315 is mediated primarily by direct TNF reduction in the CNS. Here, the MMP-9/MMP-2 ratio and collagenase concentrations in CSF were not altered by TACE inhibition. The decreased TNF concentration at 18 and 27 hpi in animals treated with Ro 32-7315 indicates activity of the compound in the brain compartment. In accordance, no relevant changes in other cytokines were observed in CSF at 18 hpi. At 27 hpi, only IL-6, which is synthesized in response to TNF (8), was less upregulated than in vehicle-treated animals.
Ro 32-7315 inhibits TACE more efficiently and more selectively than BB-1101 (IC50, 5 nM versus 550 nM) and shows improved in vivo availability (29, 33). By inhibiting TNF release, Ro 32-7315 might contribute to the reduction of cortical injury indirectly. Earlier reports showed that i.c. TNF administration resulted in CSF leukocytosis, MMP activation, and BBB disruption (50, 51). Cortical injury has been prevented by different compounds that inhibit gelatinases and collagenases to various degrees (i.e., BB 1101, GM 6001, TNF 484), a concept confirmed in this study (14, 26, 27). Hippocampal apoptosis was reduced mainly by compounds that inhibit TACE (Ro 32-7315, TNF 484), and was only slightly reduced by Ro 32-3555, which is inactive against TACE (26). Similarly, a monoclonal antibody against TNF reduced hippocampal damage but had no effect on cortical injury and CSF inflammation earlier (52). In contrast, no antiapoptotic effect of GM 6001, which is inactive against TACE, was observed or of doxycycline, which inhibits TACE and MMPs at micromolar concentrations rather than the nanomolar concentrations of other MMP inhibitors (14, 28). In a previous study, a low dose of TNF 484, an inhibitor that targets TACE and other MMPs, reduced cortical injury, but not hippocampal apoptosis (27, 53). Altogether, this adds to the concept that inhibition of MMPs and TACE might be required for attenuation of hippocampal apoptosis (54). On the other side, the TACE inhibitor evaluated here showed a trend toward reduced survival. Increased mortality rates after TACE inhibition have already been observed earlier in models of PM when high doses of TNF 484 or TNF-deficient mice were used (27, 55). We hypothesize that inhibition of TNF release may reduce brain injury but also regulates other processes relevant to survival. Systemic effects, e.g., sepsis, may be associated more directly with the mortality rate, while brain injury predisposes to sequelae such as hearing loss, cognitive difficulties, and epilepsy, as there is no clear association of death and brain injury in the present study and in earlier studies of experimental bacterial meningitis (1, 40). Ro 32-7315 reduced weight loss, which is influenced by systemic effects, including capillary leakage, which leads to tissue edema. Further investigation of the influence of systemic processes, e.g., systemic TNF levels, was not the aim of this study but should be done.
MPO activity was used as an index of the leukocyte concentration in CSF samples (9, 56). MMP and/or TACE inhibition had little effect on the levels of MPO and the chemoattractants MIP-1α and MCP-1, which contribute to leukocyte extravasation and were detected in patients with bacterial meningitis in earlier studies (1, 57). Thus, the effect of MMP and/or TACE inhibition was probably too weak to influence neutrophil invasion significantly. Accordingly, the broad-spectrum MMP inhibitor batimastat (BB-94), which is inactive against TACE, reduced BBB disruption but not CSF white blood cell counts after intracranial injection of heat-killed meningococci (13). In summary, TACE inhibition by Ro 32-7315 may be sufficient to reduce brain injury but may increase the mortality rate because systemic inflammation is not reduced sufficiently by this selective approach.
Conclusion.
A better knowledge of how the timing and involvement of the different MMPs contribute to the different forms of brain damage is necessary for the development and evaluation of new metalloproteinase inhibitors that selectively target disease-relevant MMPs and TACE. An alternative approach is the combination of the two compounds presented in this study to create an optimal inhibition profile. Thereby, studies evaluating pharmacokinetics, including the mode of application, dosage, and intracerebral concentrations, should be performed. Beyond infectious brain diseases, this approach may be applicable as well to other neuroimmunological diseases such as neuromyelitis optica and optic neuritis, where excessive metalloproteinase activity is involved in neuronal injury (58).
ACKNOWLEDGMENTS
We acknowledge Roche Pharma, Basel, Switzerland, for providing Ro 32-3555 and Ro 32-7315. We thank Franziska Simon and Angela Bühlmann for excellent technical support.
This work was supported by grants from the Novartis Foundation for Medical-Biological Research (project 11A18), the Gottfried und Julia Bangerter-Rhyner Stiftung, and the Swiss National Science Foundation (grant 138094).
Footnotes
Published ahead of print 3 February 2014
REFERENCES
- 1.Koedel U, Scheld WM, Pfister HW. 2002. Pathogenesis and pathophysiology of pneumococcal meningitis. Lancet Infect. Dis. 2:721–736. 10.1016/S1473-3099(02)00450-4 [DOI] [PubMed] [Google Scholar]
- 2.Brouwer MC, Heckenberg SG, de Gans J, Spanjaard L, Reitsma JB, van de Beek D. 2010. Nationwide implementation of adjunctive dexamethasone therapy for pneumococcal meningitis. Neurology 75:1533–1539. 10.1212/WNL.0b013e3181f96297 [DOI] [PubMed] [Google Scholar]
- 3.Edmond K, Clark A, Korczak VS, Sanderson C, Griffiths UK, Rudan I. 2010. Global and regional risk of disabling sequelae from bacterial meningitis: a systematic review and meta-analysis. Lancet Infect. Dis. 10:317–328. 10.1016/S1473-3099(10)70048-7 [DOI] [PubMed] [Google Scholar]
- 4.Chandran A, Herbert H, Misurski D, Santosham M. 2011. Long-term sequelae of childhood bacterial meningitis: an underappreciated problem. Pediatr. Infect. Dis. J. 30:3–6. 10.1097/INF.0b013e3181ef25f7 [DOI] [PubMed] [Google Scholar]
- 5.Grandgirard D, Steiner O, Tauber MG, Leib SL. 2007. An infant mouse model of brain damage in pneumococcal meningitis. Acta Neuropathol. 114:609–617. 10.1007/s00401-007-0304-8 [DOI] [PubMed] [Google Scholar]
- 6.Bifrare YD, Gianinazzi C, Imboden H, Leib SL, Tauber MG. 2003. Bacterial meningitis causes two distinct forms of cellular damage in the hippocampal dentate gyrus in infant rats. Hippocampus 13:481–488. 10.1002/hipo.10142 [DOI] [PubMed] [Google Scholar]
- 7.Nau R, Soto A, Bruck W. 1999. Apoptosis of neurons in the dentate gyrus in humans suffering from bacterial meningitis. J. Neuropathol Exp. Neurol. 58:265–274. 10.1097/00005072-199903000-00006 [DOI] [PubMed] [Google Scholar]
- 8.van Furth AM, Roord JJ, van Furth R. 1996. Roles of proinflammatory and anti-inflammatory cytokines in pathophysiology of bacterial meningitis and effect of adjunctive therapy. Infect. Immun. 64:4883–4890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grandgirard D, Burri M, Agyeman P, Leib SL. 2012. Adjunctive daptomycin attenuates brain damage and hearing loss more efficiently than rifampin in infant rat pneumococcal meningitis. Antimicrob. Agents Chemother. 56:4289–4295. 10.1128/AAC.00674-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maskos K, Fernandez-Catalan C, Huber R, Bourenkov GP, Bartunik H, Ellestad GA, Reddy P, Wolfson MF, Rauch CT, Castner BJ, Davis R, Clarke HR, Petersen M, Fitzner JN, Cerretti DP, March CJ, Paxton RJ, Black RA, Bode W. 1998. Crystal structure of the catalytic domain of human tumor necrosis factor-alpha-converting enzyme. Proc. Natl. Acad. Sci. U. S. A. 95:3408–3412. 10.1073/pnas.95.7.3408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rosenberg GA. 2002. Matrix metalloproteinases in neuroinflammation. Glia 39:279–291. 10.1002/glia.10108 [DOI] [PubMed] [Google Scholar]
- 12.Grandgirard D, Leib SL. 2006. Strategies to prevent neuronal damage in paediatric bacterial meningitis. Curr. Opin. Pediatr. 18:112–118. 10.1097/01.mop.0000193292.09894.b7 [DOI] [PubMed] [Google Scholar]
- 13.Paul R, Lorenzl S, Koedel U, Sporer B, Vogel U, Frosch M, Pfister HW. 1998. Matrix metalloproteinases contribute to the blood-brain barrier disruption during bacterial meningitis. Ann. Neurol. 44:592–600. 10.1002/ana.410440404 [DOI] [PubMed] [Google Scholar]
- 14.Leib SL, Leppert D, Clements J, Täuber MG. 2000. Matrix metalloproteinases contribute to brain damage in experimental pneumococcal meningitis. Infect. Immun. 68:615–620. 10.1128/IAI.68.2.615-620.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leppert D, Lindberg RL, Kappos L, Leib SL. 2001. Matrix metalloproteinases: multifunctional effectors of inflammation in multiple sclerosis and bacterial meningitis. Brain Res. Brain Res. Rev. 36:249–257. 10.1016/S0165-0173(01)00101-1 [DOI] [PubMed] [Google Scholar]
- 16.Kastenbauer S, Pfister HW. 2003. Pneumococcal meningitis in adults: spectrum of complications and prognostic factors in a series of 87 cases. Brain 126:1015–1025. 10.1093/brain/awg113 [DOI] [PubMed] [Google Scholar]
- 17.Amour A, Knight CG, Webster A, Slocombe PM, Stephens PE, Knauper V, Docherty AJ, Murphy G. 2000. The in vitro activity of ADAM-10 is inhibited by TIMP-1 and TIMP-3. FEBS Lett. 473:275–279. 10.1016/S0014-5793(00)01528-3 [DOI] [PubMed] [Google Scholar]
- 18.Sellner J, Leib SL. 2006. In bacterial meningitis cortical brain damage is associated with changes in parenchymal MMP-9/TIMP-1 ratio and increased collagen type IV degradation. Neurobiol Dis. 21:647–656. 10.1016/j.nbd.2005.09.007 [DOI] [PubMed] [Google Scholar]
- 19.Leppert D, Leib SL, Grygar C, Miller KM, Schaad UB, Hollander GA. 2000. Matrix metalloproteinase (MMP)-8 and MMP-9 in cerebrospinal fluid during bacterial meningitis: association with blood-brain barrier damage and neurological sequelae. Clin. Infect. Dis. 31:80–84. 10.1086/313922 [DOI] [PubMed] [Google Scholar]
- 20.Yushchenko M, Weber F, Mader M, Scholl U, Maliszewska M, Tumani H, Felgenhauer K, Beuche W. 2000. Matrix metalloproteinase-9 (MMP-9) in human cerebrospinal fluid (CSF): elevated levels are primarily related to CSF cell count. J. Neuroimmunol. 110:244–251. 10.1016/S0165-5728(00)00339-8 [DOI] [PubMed] [Google Scholar]
- 21.Tumani H, Hartung HP, Hemmer B, Teunissen C, Deisenhammer F, Giovannoni G, Zettl UK, BioMS Study Group 2009. Cerebrospinal fluid biomarkers in multiple sclerosis. Neurobiol Dis. 35:117–127. 10.1016/j.nbd.2009.04.010 [DOI] [PubMed] [Google Scholar]
- 22.Brinckerhoff CE, Matrisian LM. 2002. Matrix metalloproteinases: a tail of a frog that became a prince. Nat. Rev. Mol. Cell Biol. 3:207–214. 10.1038/nrm763 [DOI] [PubMed] [Google Scholar]
- 23.Catterall JB, Cawston TE. 2003. Assays of matrix metalloproteinases (MMPs) and MMP inhibitors: bioassays and immunoassays applicable to cell culture medium, serum, and synovial fluid. Methods Mol. Biol. 225:353–364. 10.1385/1-59259-374-7:353 [DOI] [PubMed] [Google Scholar]
- 24.Dormán G, Cseh S, Hajdu I, Barna L, Konya D, Kupai K, Kovacs L, Ferdinandy P. 2010. Matrix metalloproteinase inhibitors: a critical appraisal of design principles and proposed therapeutic utility. Drugs 70:949–964. 10.2165/11318390-000000000-00000 [DOI] [PubMed] [Google Scholar]
- 25.Jackson C, Nguyen M, Arkell J, Sambrook P. 2001. Selective matrix metalloproteinase (MMP) inhibition in rheumatoid arthritis-targetting gelatinase A activation. Inflamm. Res. 50:183–186. 10.1007/s000110050743 [DOI] [PubMed] [Google Scholar]
- 26.Leib SL, Clements JM, Lindberg RL, Heimgartner C, Loeffler JM, Pfister LA, Tauber MG, Leppert D. 2001. Inhibition of matrix metalloproteinases and tumour necrosis factor alpha converting enzyme as adjuvant therapy in pneumococcal meningitis. Brain 124:1734–1742. 10.1093/brain/124.9.1734 [DOI] [PubMed] [Google Scholar]
- 27.Meli DN, Loeffler JM, Baumann P, Neumann U, Buhl T, Leppert D, Leib SL. 2004. In pneumococcal meningitis a novel water-soluble inhibitor of matrix metalloproteinases and TNF-alpha converting enzyme attenuates seizures and injury of the cerebral cortex. J. Neuroimmunol. 151:6–11. 10.1016/j.jneuroim.2004.01.026 [DOI] [PubMed] [Google Scholar]
- 28.Meli DN, Coimbra RS, Erhart DG, Loquet G, Bellac CL, Täuber MG, Neumann U, Leib SL. 2006. Doxycycline reduces mortality and injury to the brain and cochlea in experimental pneumococcal meningitis. Infect. Immun. 74:3890–3896. 10.1128/IAI.01949-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Whittaker M, Floyd CD, Brown P, Gearing AJ. 1999. Design and therapeutic application of matrix metalloproteinase inhibitors. Chem. Rev. 99:2735–2776. 10.1021/cr9804543 [DOI] [PubMed] [Google Scholar]
- 30.Close DR. 2001. Matrix metalloproteinase inhibitors in rheumatic diseases. Ann. Rheum. Dis. 60(Suppl. 3):iii62–iii67. 10.1136/ard.60.90003.iii62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brewster M, Lewis EJ, Wilson KL, Greenham AK, Bottomley KM. 1998. Ro 32-3555, an orally active collagenase selective inhibitor, prevents structural damage in the STR/ORT mouse model of osteoarthritis. Arthritis Rheum. 41:1639–1644. [DOI] [PubMed] [Google Scholar]
- 32.Hemmings FJ, Farhan M, Rowland J, Banken L, Jain R. 2001. Tolerability and pharmacokinetics of the collagenase-selective inhibitor Trocade in patients with rheumatoid arthritis. Rheumatology (Oxford, England) 40:537–543. 10.1093/rheumatology/40.5.537 [DOI] [PubMed] [Google Scholar]
- 33.Beck G, Bottomley G, Bradshaw D, Brewster M, Broadhurst M, Devos R, Hill C, Johnson W, Kim HJ, Kirtland S, Kneer J, Lad N, Mackenzie R, Martin R, Nixon J, Price G, Rodwell A, Rose F, Tang JP, Walter DS, Wilson K, Worth E. 2002. (E)-2(R)-[1(S)-(Hydroxycarbamoyl)-4-phenyl-3-butenyl]-2′-isobutyl-2′-(methanesulfonyl)-4-methylvalerohydrazide (Ro 32-7315), a selective and orally active inhibitor of tumor necrosis factor-alpha convertase. J. Pharmacol. Exp. Ther. 302:390–396. 10.1124/jpet.302.1.390 [DOI] [PubMed] [Google Scholar]
- 34.Lewis EJ, Bishop J, Bottomley KM, Bradshaw D, Brewster M, Broadhurst MJ, Brown PA, Budd JM, Elliott L, Greenham AK, Johnson WH, Nixon JS, Rose F, Sutton B, Wilson K. 1997. Ro 32-3555, an orally active collagenase inhibitor, prevents cartilage breakdown in vitro and in vivo. Br. J. Pharmacol. 121:540–546. 10.1038/sj.bjp.0701150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hilpert H. 2001. Practical approaches to the matrix metalloproteinase inhibitor Trocade® (Ro 32-3555) and to the TNF-α converting enzyme inhibitor Ro 32-7315. Tetrahedron 57:7675–7683. 10.1016/S0040-4020(01)00720-7 [DOI] [Google Scholar]
- 36.Blaser C, Wittwer M, Grandgirard D, Leib SL. 2011. Adjunctive dexamethasone affects the expression of genes related to inflammation, neurogenesis and apoptosis in infant rat pneumococcal meningitis. PLoS One 6:e17840. 10.1371/journal.pone.0017840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gianinazzi C, Grandgirard D, Imboden H, Egger L, Meli DN, Bifrare DY, Joss PC, Täuber MG, Borner C, Leib SL. 2003. Caspase-3 mediates hippocampal apoptosis in pneumococcal meningitis. Acta Neuropathol. 105:499–507. 10.1007/s00401-003-0672-7 [DOI] [PubMed] [Google Scholar]
- 38.Gehre F, Leib SL, Grandgirard D, Kummer J, Buhlmann A, Simon F, Gaumann R, Kharat AS, Tauber MG, Tomasz A. 2008. Essential role of choline for pneumococcal virulence in an experimental model of meningitis. J. Intern. Med. 264:143–154. 10.1111/j.1365-2796.2008.01930.x [DOI] [PubMed] [Google Scholar]
- 39.Budczies J, Klauschen F, Sinn BV, Gyorffy B, Schmitt WD, Darb-Esfahani S, Denkert C. 2012. Cutoff Finder: a comprehensive and straightforward web application enabling rapid biomarker cutoff optimization. PLoS One 7:e51862. 10.1371/journal.pone.0051862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mook-Kanamori BB, Geldhoff M, van der Poll T, van de Beek D. 2011. Pathogenesis and pathophysiology of pneumococcal meningitis. Clin. Microbiol. Rev. 24:557–591. 10.1128/CMR.00008-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gerber J, Nau R. 2010. Mechanisms of injury in bacterial meningitis. Curr. Opin. Neurol. 23:312–318. 10.1097/WCO.0b013e32833950dd [DOI] [PubMed] [Google Scholar]
- 42.Leib SL, Kim YS, Chow LL, Sheldon RA, Tauber MG. 1996. Reactive oxygen intermediates contribute to necrotic and apoptotic neuronal injury in an infant rat model of bacterial meningitis due to group B streptococci. J. Clin. Invest. 98:2632–2639. 10.1172/JCI119084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mitchell L, Smith SH, Braun JS, Herzog KH, Weber JR, Tuomanen EI. 2004. Dual phases of apoptosis in pneumococcal meningitis. J. Infect. Dis. 190:2039–2046. 10.1086/425520 [DOI] [PubMed] [Google Scholar]
- 44.Grandgirard D, Leib SL. 2010. Meningitis in neonates: bench to bedside. Clin. Perinatol. 37:655–676. 10.1016/j.clp.2010.05.004 [DOI] [PubMed] [Google Scholar]
- 45.Liedtke W, Cannella B, Mazzaccaro RJ, Clements JM, Miller KM, Wucherpfennig KW, Gearing AJ, Raine CS. 1998. Effective treatment of models of multiple sclerosis by matrix metalloproteinase inhibitors. Ann. Neurol. 44:35–46. 10.1002/ana.410440110 [DOI] [PubMed] [Google Scholar]
- 46.Snoek-van Beurden PA, Von den Hoff JW. 2005. Zymographic techniques for the analysis of matrix metalloproteinases and their inhibitors. Biotechniques 38:73–83. 10.2144/05381RV01 [DOI] [PubMed] [Google Scholar]
- 47.Mitchell AJ, Yau B, McQuillan JA, Ball HJ, Too LK, Abtin A, Hertzog P, Leib SL, Jones CA, Gerega SK, Weninger W, Hunt NH. 2012. Inflammasome-dependent IFN-gamma drives pathogenesis in Streptococcus pneumoniae meningitis. J. Immunol. 189:4970–4980. 10.4049/jimmunol.1201687 [DOI] [PubMed] [Google Scholar]
- 48.Kornelisse RF, Savelkoul HF, Mulder PH, Suur MH, van der Straaten PJ, van der Heijden AJ, Sukhai RN, Hahlen K, Neijens HJ, de Groot R. 1996. Interleukin-10 and soluble tumor necrosis factor receptors in cerebrospinal fluid of children with bacterial meningitis. J. Infect. Dis. 173:1498–1502. 10.1093/infdis/173.6.1498 [DOI] [PubMed] [Google Scholar]
- 49.Grandgirard D, Bifrare YD, Pleasure SJ, Kummer J, Leib SL, Tauber MG. 2007. Pneumococcal meningitis induces apoptosis in recently postmitotic immature neurons in the dentate gyrus of neonatal rats. Dev. Neurosci. 29:134–142. 10.1159/000096218 [DOI] [PubMed] [Google Scholar]
- 50.Ramilo O, Mustafa MM, Porter J, Saez-Llorens X, Mertsola J, Olsen KD, Luby JP, Beutler B, McCracken GH., Jr 1990. Detection of interleukin 1 beta but not tumor necrosis factor-alpha in cerebrospinal fluid of children with aseptic meningitis. Am. J. Dis. Child. 144:349–352 [DOI] [PubMed] [Google Scholar]
- 51.Rosenberg GA, Estrada EY, Dencoff JE, Stetler-Stevenson WG. 1995. Tumor necrosis factor-alpha-induced gelatinase B causes delayed opening of the blood-brain barrier: an expanded therapeutic window. Brain Res. 703:151–155. 10.1016/0006-8993(95)01089-0 [DOI] [PubMed] [Google Scholar]
- 52.Bogdan I, Leib SL, Bergeron M, Chow L, Tauber MG. 1997. Tumor necrosis factor-alpha contributes to apoptosis in hippocampal neurons during experimental group B streptococcal meningitis. J. Infect. Dis. 176:693–697. 10.1086/514092 [DOI] [PubMed] [Google Scholar]
- 53.Janser P, Neumann U, Miltz W, Feifel R, Buhl T. 2006. A cassette-dosing approach for improvement of oral bioavailability of dual TACE/MMP inhibitors. Bioorg. Med. Chem. Lett. 16:2632–2636. 10.1016/j.bmcl.2006.02.042 [DOI] [PubMed] [Google Scholar]
- 54.Kim KS. 2003. Pathogenesis of bacterial meningitis: from bacteraemia to neuronal injury. Nat. Rev. Neurosci. 4:376–385. 10.1038/nrn1103 [DOI] [PubMed] [Google Scholar]
- 55.Gerber J, Bottcher T, Hahn M, Siemer A, Bunkowski S, Nau R. 2004. Increased mortality and spatial memory deficits in TNF-alpha-deficient mice in ceftriaxone-treated experimental pneumococcal meningitis. Neurobiol. Dis. 16:133–138. 10.1016/j.nbd.2004.01.013 [DOI] [PubMed] [Google Scholar]
- 56.Klebanoff SJ. 2005. Myeloperoxidase: friend and foe. J. Leukoc. Biol. 77:598–625. 10.1189/jlb.1204697 [DOI] [PubMed] [Google Scholar]
- 57.Lahrtz F, Piali L, Spanaus KS, Seebach J, Fontana A. 1998. Chemokines and chemotaxis of leukocytes in infectious meningitis. J. Neuroimmunol. 85:33–43. 10.1016/S0165-5728(97)00267-1 [DOI] [PubMed] [Google Scholar]
- 58.Hosokawa T, Nakajima H, Doi Y, Sugino M, Kimura F, Hanafusa T, Takahashi T. 2011. Increased serum matrix metalloproteinase-9 in neuromyelitis optica: implication of disruption of blood-brain barrier. J. Neuroimmunol. 236:81-86. 10.1016/j.jneuroim.2011.04.009 [DOI] [PubMed] [Google Scholar]