

Abstract
Pathogen mutants arise during infections. Mechanisms of selection for pathogen variants are poorly understood. We tested whether neutrophils select mutations in the two-component regulatory system CovRS of group A Streptococcus (GAS) during infection using the lack of production of the protease SpeB (SpeB activity negative [SpeBA−]) as a marker. Depletion of neutrophils by antibodies RB6-8C5 and 1A8 reduced the percentage of SpeBA− variants (SpeBA−%) recovered from mice infected with GAS strain MGAS2221 by >76%. Neutrophil recruitment and SpeBA−% among recovered GAS were reduced by 95% and 92%, respectively, in subcutaneous MGAS2221 infection of CXCR2−/− mice compared with control mice. In air sac infection with MGAS2221, levels of neutrophils and macrophages in lavage fluid were reduced by 49% and increased by 287%, respectively, in CXCR2−/− mice compared with control mice, implying that macrophages play an insignificant role in the reduction of selection for SpeBA− variants in CXCR2−/− mice. One randomly chosen SpeBA− mutant outcompeted MGAS2221 in normal mice but was outcompeted by MGAS2221 in neutropenic mice and had enhancements in expression of virulence factors, innate immune evasion, skin invasion, and virulence. This and nine other SpeBA− variants from a mouse all had nonsynonymous covRS mutations that resulted in the SpeBA− phenotype and enhanced expression of the CovRS-controlled secreted streptococcal esterase (SsE). Our findings are consistent with a model that neutrophils select spontaneous covRS mutations that maximize the potential of GAS to evade neutrophil responses, resulting in variants with enhanced survival and virulence. To our knowledge, this is the first report of the critical contribution of neutrophils to the selection of pathogen variants.
INTRODUCTION
The human pathogen group A Streptococcus (GAS) causes both relatively mild pharyngitis and superficial skin infections and potentially lethal, severe invasive infections (1). Severe invasive infections are most frequently caused by GAS strains of serotypes M1, M3, and M12 among GAS strains of >200 M protein serotypes in the United States (2). In particular, a serotype M1T1 clone of M1 GAS that emerged in the 1980s has globally disseminated and has been associated with the resurgence of severe invasive GAS infections in the last 30 years (3–11). Clinical isolates from severe invasive infections usually possess hypervirulence and an enhanced capacity to invade soft tissues and evade neutrophil responses compared with pharyngitis isolates (12–14). Invasive GAS isolates frequently carry a mutation in the genes encoding the two-component regulatory system CovRS (also known as CsrRS) (15, 16), and covRS mutations are a common cause of their hypervirulence and enhancement of soft tissue invasion and innate immune evasion (12–14). CovRS negatively regulates many virulence factors, including most of those that are involved in innate immune evasion (17–20). As a result of CovRS mutations, the loss of the production of the protease SpeB and enhanced production of the hyaluronic acid capsule and secreted streptococcal esterase (SsE) contribute to the phenotype of hypervirulent isolates (14, 21–25).
The association of the M1T1 GAS clone with severe invasive infections appears to be linked to its proneness to the selection of covRS mutations during infection. A natural covS deletion in an invasive M1T1 isolate is responsible for its hypervirulence and enhanced innate immune evasion (14). Null covS mutations of M1T1 isolates arise in experimental invasive infection in mice (12, 26, 27). The lack of production of the protease SpeB (SpeBA−, for the SpeB activity-negative phenotype) has been used as a marker for GAS variants with covRS mutations (27, 28), although the validity of this approach has not been rigorously tested. In contrast, the first sequenced M1 GAS strain, SF370, rarely switches to the SpeBA− phenotype during experimental mouse infection (27). The DNase Sda1, encoded by a prophage, which is carried by some M1T1 isolates but not by SF370, plays a critical role in the selection of covRS mutations of M1T1 isolate 5448 during infection in mice (27). However, introduction of the Sda1-encoding prophage into SF370 does not facilitate the selection of SpeBA− mutants in vivo (29). Furthermore, hypervirulent variants with covS mutations arise in strains that lack Sda1 (30). Besides Sda1, the capsule synthetase gene hasA and the M protein gene emm are required for the selection of SpeBA− variants (28). Despite these considerable efforts and advancement, the exact basis for the selection of covRS mutants in vivo remains unknown.
In contrast to the active search for the basis on the pathogen side for the selection of covRS mutations, there has been no report on host factors that contribute to the selection of CovRS mutants. Here we report the first examination of the role of host factors in the selection of GAS covRS mutations. We found that neutrophils are the primary selection pressure for covRS mutants of M1T1 GAS strain MGAS2221. We also demonstrated that a randomly chosen isolate with a null covS mutation had an advantage in survival and an enhanced capacity to evade innate immune responses. These findings demonstrate that neutrophils critically contribute to the selection of M1T1 GAS covRS mutations that compromise neutrophil responses and enhance GAS survival and virulence.
MATERIALS AND METHODS
Declaration of ethical approval.
All animal experimental procedures were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals (31). The protocols for the experiments were approved by the Institutional Animal Care and Use Committee at Montana State University (permit number 2011-57).
Bacterial strains and growth.
Sequenced M1 isolate SF370 (32) and M1T1 strain MGAS2221 (12) were used in this study. SpeBA− strain 1 (SpeBA−1) was a randomly chosen mutant among isolates that were recovered from MGAS2221 skin infection sites in mice and had abolished SpeB production. This strain had a deletion of 5 bases at positions 132 to 136 of the covS gene. These GAS isolates and other testing strains were grown in Todd-Hewitt broth supplemented with 0.2% yeast extract (THY) at 37°C in 5% CO2. Tryptose agar with 5% sheep blood and THY agar were used as the solid media. GAS bacteria used for animal experiments were harvested at the mid-exponential growth phase (optical density at 600 nm [OD600] of 0.4) and washed three times with and resuspended in pyrogen-free Dulbecco's phosphate-buffered saline (DPBS) to the desired doses.
Neutrophil depletion.
Rat anti-murine Ly6G monoclonal antibodies (MAbs) RB6-8C5 and 1A8 and isotype control MAb 2A3 for 1A8 were purchased from Bio X Cell. To deplete and maintain the depletion of neutrophils, 250 μg RB6-8C5 or 1A8 in 0.5 ml DPBS was injected into the intraperitoneal cavity of each female C57BL/6 mouse (5 weeks old) at 24 h prior to and 24 h after GAS inoculation. Control mice were treated similarly with 2A3 or DPBS. The efficiency of neutrophil depletion was evaluated by flow cytometry analysis. Blood samples were collected into heparinized tubes from maxillary bleeds of mice 24 h after they had been treated with RB6-8C5, 1A8, or the control. The blood samples were incubated with an antibody cocktail of fluorescein isothiocyanate (FITC)-conjugated rat anti-mouse CD11b (BD Biosciences) and Alexa Fluor 647-conjugated rat anti-mouse Ly6G (BioLegend) at a 1:200 dilution at 37°C for 15 min. Red blood cells in the samples were then lysed by using the Whole Blood Lysing Reagent kit (Beckman Coulter) according to the manufacturer's protocol. White blood cells in the stained samples were analyzed on a BD LSR flow cytometer running FACSDiva software (BD Biosciences). Flow cytometry data were analyzed by using the FlowJo software program (TreeStar Inc.). Levels of neutrophils at skin infection sites 24 h after GAS inoculation were also determined, as described below, to confirm the efficiency of neutrophil depletion.
Mouse infections.
Selection of SpeBA− variants was done by using 5-week-old female C57BL/6 neutropenic mice, male CXCR2 knockout (KO) (CXCR2−/−) mice in the BALB/c background, and BALB/c control mice for CXCR2−/− mice. These mice were bred at the Animal Resource Center at Montana State University using breeding pairs of mice from the Jackson Laboratory (Bar Harbor, ME). The CXCR2−/− mice [C.129S2(B6)-Cxcr2tm1Mwm/J] had a deficiency in chemokine (C-X-C) receptor 2 and impaired neutrophil migration (33). Each mouse was subcutaneously inoculated with 0.2 ml of an MGAS2221 or SF370 suspension at an OD600 of 0.9. In experiments using neutropenic mice, GAS was inoculated subcutaneously in mice 24 h after mice were injected intraperitoneally with 250 μg RB6-8C5 or 1A8, and the mice were injected with the antibodies again at 24 h after GAS infection. The infection site in the skin was collected at the indicated times after inoculation to measure the percentage of SpeBA− variants (SpeBA−%) of GAS bacteria isolated from these tissues.
Five-week-old female CD-1 mice from Charles River Laboratories were used to compare the SpeBA−1 strain with MGAS2221 in virulence, neutrophil recruitment, skin invasion, and systemic GAS dissemination. Groups of 15 mice were subcutaneously inoculated with 0.2 ml of MGAS2221 and SpeBA−1 suspensions in DPBS at an OD600 of 0.9. Five mice of each group were euthanized to collect skin samples for measurement of lesion size and neutrophil recruitment as described below, and the liver and spleen were also harvested to measure numbers of viable GAS bacteria. The other 10 mice of each group were monitored daily for 14 days to determine survival rates.
Quantification of SpeBA− isolates.
The skin and liver samples obtained as described above were homogenized in DPBS by using a Kontes pestle and plated at appropriate dilutions. Forty-eight colonies were randomly picked from each sample, inoculated in 200 μl THY in 96-well plates, and cultured overnight. Three microliters of 10% β-mercaptoethanol was added to each well, and the cultures in the plate were centrifuged at 3,500 rpm. The SpeB activity in the supernatant of GAS cultures grown overnight was detected by using a casein plate assay as described previously (34).
Cytospin analysis.
MGAS2221 (0.1 ml of bacterial suspension in DPBS with an OD600 of 1.1) was injected with 0. 9 ml air subcutaneously into 5 male BALB/c (control) or CXCR2−/− mice. Twelve hours later, the mice were euthanized, and the air sac of each mouse was lavaged with 1 ml of cold DPBS. An aliquot of the recovered lavage fluid was used to determine the total number of live cells by trypan blue exclusion counts. A second aliquot of the recovered lavage fluid at an appropriate dilution was used to prepare cytospin slides using a Shandon Cytospin cytocentrifuge. The slides were stained by using the Diff-Quik stain kit from Fisher Scientific. Neutrophils and macrophages among 150 host cells on each cytospin slide were counted to determine the percentages of neutrophils and macrophages, and the total numbers of neutrophils and macrophages were calculated from the percentage data and the total counts of live cells in the lavage samples.
GAS competitive growth/survival assay.
A 1:1 SpeBA−1-MGAS2221 mixture (0.2 ml) was injected with 0.8 ml air subcutaneously into 18 C57BL/6 or neutropenic mice obtained from C57BL/6 mice by treatment with MAb RB6-8C5. Nine C57BL/6 or neutropenic mice were euthanized at 1 h or 24 h after inoculation, and the air sac was lavaged with 1 ml PBS. The lavage samples were plated onto THY agar plates. The percentage of SpeBA−1 variants in GAS colonies for each lavage sample was determined by analyzing 48 colonies of each sample with an SpeB activity assay, as described above. SpeBA−1 and MGAS2221 showed negative and positive SpeB production, respectively.
Quantification of neutrophil infiltration.
The skin around the infection site was peeled off, and the skin lesion was recognized by the boundary of the inflammation area. The size of skin lesions was measured by analyzing the lesion pictures using the area measurement tool of the Adobe Acrobat 9 software program (Adobe Systems Inc.). The skin containing the infection area was excised for neutrophil measurement. Numbers of recruited neutrophils in the infected skin samples were determined by a myeloperoxidase assay, as described previously (35).
DNA sequencing.
A DNA fragment containing the covRS genes was amplified from test strains by using the primers 5′-TCGCTAGAAGACTATTTGAC-3′ and 5′-TTCATGTCATCCATCATTGC-3′ and the Phusion high-fidelity PCR kit (New England BioLabs). DNA sequencing of the amplified PCR products was performed by using the BigDye Terminator v3.1 cycle sequencing kit and an Applied Biosystems 3130 genetic analyzer. Primers used for sequencing were 5′-TCGCTAGAAGACTATTTGAC-3′, 5′-TTCATGTCATCCATCATTGC-3′, 5′-AACGGCTTCATCATATTTCC-3′, 5′-AAATCCACAAAACCGTTCAG-3′, 5′-TGATACACACGACCGATAG-3′, 5′-TTGATGACAGAAAGGGCAG-3′, 5′-TACGCGAACCATGTCTAAC-3′, and 5′-GTTGGGGTAAAGATGACAG-3′. Sequence data were analyzed by using Sequencer 5.1 software (Gene Codes Corporation).
Real time RT-PCR analysis.
MGAS2221 and SpeBA−1 were grown at 37°C (5% CO2) to an OD600 of 0.2 in THY. Total RNA was isolated from these GAS bacteria as described previously (24). High RNA quality was confirmed by using an Agilent 2100 Bioanalyzer and an RNA 6000 Lab Chip kit (Agilent Technologies). TaqMan quantitative reverse transcription-PCR (RT-PCR) assays were performed by using probes specific for emm, hasA, spyCEP, sse, and gyrA (control) and the ABI 7500 Fast system (Applied Biosystems Inc.), as described previously (24). Control reaction mixtures that did not contain reverse transcriptase revealed no contamination of genomic DNA in any RNA sample. All RNA samples were assayed in triplicate, and mRNA levels of genes were compared by using the ΔΔCT method with normalization to the mRNA levels of the gyrA gene, which were about the same in all samples.
Analyses for SsE production.
Relative levels of SsE production by MGAS2221 and its variants were determined by Western blotting and the 2-thio-PAF hydrolysis assay (36) for platelet-activating factor acetylhydrolase (PAF-AH) activity in culture supernatants. Western blotting to detect the presence of SsE and Spy0469 (control), a secreted protein, in culture supernatants was performed as described previously (24). In the 2-thio-PAF hydrolysis assay, 100 μl of the culture supernatant from the exponential growth phase (OD600 = ∼0.33) was mixed with 30 μl of a reactant solution containing 0.9 mM 2-thio-PAF and 1.3 mM 5,5′-dithiobis(2-nitrobenzoic acid) at room temperature in a well of a 96-well plate. The absorbance change at 414 nm (ΔA414), as a measure of SsE-catalyzed 2-thio-PAF hydrolysis, was recorded with time by using a SPECTRAMax 384 Plus spectrophotometer (Molecular Devices).
Complementation of CovRS mutants.
Plasmid pCovSC for in trans complementation and the vector control pDCBB were described previously (20). To construct the suicide plasmid pGRV-CovRS, a DNA fragment containing the covRS genes was amplified by using primers 5′-GGACAAGCTTTGAAATAGTCTAGGATATGAG-3′ and 5′-GCGGATCCTGGTAGATAGAGACCGCGTCA-3′, and the PCR product was cloned into pGRV (37) at the BglII and BamHI sites. SpeBA− variants with covS mutations were transformed with pCovSC or pDCBB (20). For complementing SpeBA− variants with covR mutations, pGRV-CovRS was integrated into the genome of the variants by a single crossover. All complement and vector control strains were selected with 10 mg/liter chloramphenicol.
Statistical analyses.
The Prism software program (Graph-Pad Software Inc.) was used for all statistical analyses. Survival data were analyzed by using the log-rank (Mantel-Cox) test. The data in Fig. 1B and 5A were analyzed by using one-way analysis of variance (ANOVA) with Tukey's multiple-comparison test. The P values other than that in the survival study were obtained by using the two-tailed Mann-Whitney t test.
FIG 1.
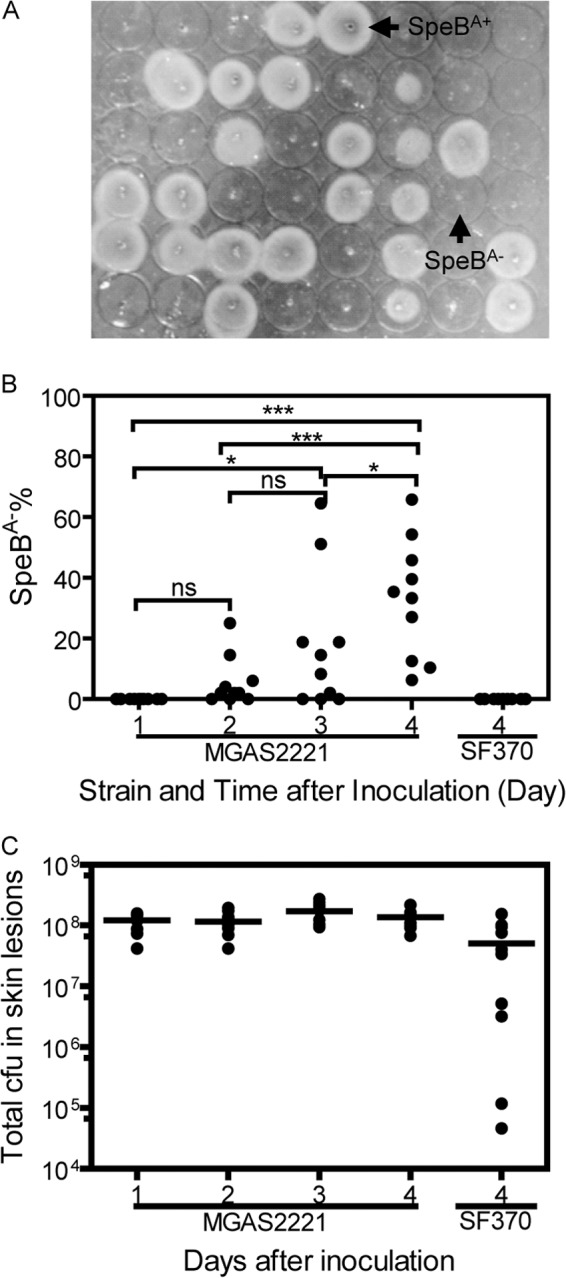
Time course of accumulation of SpeBA− variants and number of GAS bacteria at skin infection sites. (A) Representative picture of the casein SpeB protease activity assay showing the presence (SpeBA+) and absence (SpeBA−) of SpeB activity in the culture supernatants of 48 GAS isolates from a mouse at day 4 after subcutaneous MGAS2221 infection. (B) SpeBA−% among GAS bacteria recovered at skin infection sites of C57BL/6 mice at days 1 to 4 after subcutaneous inoculation with 2.5 × 107 CFU of MGAS2221 or at day 4 after inoculation with 2.0 × 107 CFU of SF370. ns, not significant. (C) Total numbers of viable GAS bacteria at the skin infection site of MGAS2221 as a function of time after inoculation in panel B. The data for SF370 at day 4 after infection are included in panels B and C for comparison. Asterisks indicate significance, with *** showing more significance than *. ns, not significant.
RESULTS
Time course of in vivo accumulation of MGAS2221 SpeBA− variants.
We hypothesize that GAS SpeBA− variants are selected for better survival against innate immunity during infection. If this hypothesis is correct, SpeBA− variants should accumulate with time. Thus, we measured the percentage of SpeBA− variants of MGAS2221 among GAS bacteria recovered from infection sites in the skin at different time points after inoculation by analyzing 48 colonies from each mouse using the casein hydrolysis assay for the SpeB protease activity in the supernatants of cultures grown overnight (Fig. 1A). All colonies analyzed were confirmed to be GAS by their beta-hemolytic activity on blood agar plates, and GAS isolates with the SpeBA− phenotype were analyzed twice and could be repeated. No SpeBA− isolates were detected at 24 h after inoculation of MGAS2221, an M1T1 isolate. On day 2, the percentage (mean ± standard deviation [SD]) of SpeBA− variants was 5.5% ± 8.1%, which increased to 17.9% ± 22.5% and 33.0% ± 19.5% on days 3 and 4, respectively (Fig. 1B). The data at one time point were significantly different from those 2 days apart. As reported previously (27), no SpeBA− variants of GAS strain SF370 were detected at day 4 after GAS inoculation in mice (Fig. 1B). The total CFU of GAS at the MGAS2221 infection site remained nearly constant, ranging from 1.1 × 108 to 1.7 × 108 during a period of 4 days (Fig. 1C). However, the data showing that one-third of GAS bacteria on day 4 were SpeBA− variants indicate active growth and clearance of GAS at infection sites.
Neutrophils exert pressure for selection of SpeBA− variants.
We next tested whether neutrophils exert primary pressure for selection of SpeBA− variants. We first used neutropenic mice to address this question. To obtain neutropenic mice, 250 μg of monoclonal antibody RB6-8C5, which was commonly used to deplete mice of neutrophils (38), was injected into the intraperitoneal cavity. The efficiency of neutrophil depletion was examined by fluorescence-activated cell sorting (FACS). Blood was collected 24 h after the RB6-8C5 treatment, and white blood cells were incubated with Alexa Fluor-labeled anti-Ly6G and FITC-labeled anti-CD11b antibodies. FACS analysis detected Ly6G+/CD11b+ neutrophils in the blood of DPBS-treated (control) but not RB6-8C5-treated mice (Fig. 2A and B). To determine whether the RB6-8C5 treatment reduces the levels of neutrophils at infections sites, RB6-8C5-treated and DPBS-treated mice were infected subcutaneously with MGAS2221 24 h after RB6-8C5 injection, and levels of neutrophils at skin infection sites were measured 24 h after GAS injection by using the myeloperoxidase assay (35). Because monocytes also produce myeloperoxidase at levels from 33% of those in neutrophils in human (39) to 1.8% of those in neutrophils in rats (35), we first estimated the contribution of inflammatory macrophages to levels of myeloperoxidase at the skin infection sites 24 h after MGAS2221 inoculation. Percentages of neutrophils and macrophages among 150 cells at hematoxylin and eosin (H&E)-stained skin infection sites were 80% and 20%, respectively (Fig. 2E). Using the relative myeloperoxidase contents in human monocytes compared to those in neutrophils, 33%, for the relative myeloperoxidase content in mouse monocytes relative to that in mouse neutrophils, the contribution of monocytes to myeloperoxidase levels at the MGAS2221 skin infection site would be about 7%. Since mouse is more closely related to rat than human, mouse monocytes might contribute <7% of myeloperoxidase at MGAS2221 skin infection sites. Thus, the myeloperoxidase assay is a reasonably valid approach to estimate the levels of neutrophils at the skin infection site for infection of mice with MGAS2221. Using this assay, the infection site in the control mice had (4.5 ± 1.6) × 105 neutrophils/mm2, whereas the infection site in RB6-8C5-treated mice had (1.8 ± 0.6) × 104 neutrophils/mm2, representing a 96% reduction in recruited neutrophils at infection sites in the RB6-8C5-treated mice (Fig. 2F). The neutrophil depletion was maintained during the 5-day experiment by a second injection of RB6-8C5 at 24 h after GAS inoculation.
FIG 2.

Assessment of the efficiency of neutrophil depletion in mice treated with monoclonal antibody RB6-8C5 or 1A8. Neutrophil depletion and sample preparation are described in Materials and Methods. (A to D) FACS analyses of Ly6G+/CD11b+ neutrophils among all white cells from mice that were treated with DPBS (A), RB6-8C5 (B), 1A8 (C), or 2A3 (D). (E) Percentages of neutrophils and macrophages at the skin infection site of MGAS2221 at 24 h after inoculation. The numbers of neutrophils and macrophages were counted among 150 cells by examining H&E-stained skin samples under a microscope. (F) Levels of infiltrated neutrophils at the infection site in the skin at 24 h after inoculation of 9.4 × 107 CFU MGAS2221 in C57BL/6 mice. The mice were intraperitoneally injected with DPBS, RB6-8C5, or 1A8 at 24 h prior to MGAS2221 infection. PMN, polymorphonuclear leukocytes.
MGAS2221 bacteria were recovered from the skin infection sites on day 4 after inoculation and were analyzed to determine whether they produced SpeB by the SpeB protease activity assay (34). Dead mice were excluded from the SpeB activity assay. A total of (1.5 ± 2.0)% of GAS bacteria recovered from RB6-8C5-treated mice did not produce SpeB, whereas (53 ± 26)% of GAS bacteria recovered from DPBS-treated mice had the SpeBA− phenotype (P = 0.0009) (Fig. 3A). The treatment of mice with RB6-8C5 nearly abolished the selection of SpeBA− variants during infection.
FIG 3.
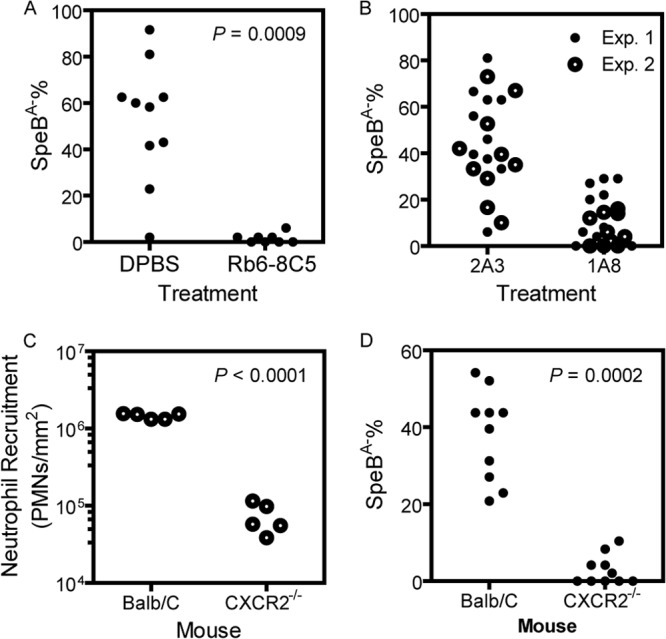
Neutrophils select SpeBA− variants. (A and B) SpeBA−% of GAS isolates recovered from the skin infection site of MGAS2221 in C57BL/6 mice with intraperitoneal treatment with DPBS or RB6-8C5 (A) and 2A3 (subtype MAb control) or 1A8 (B) at day 4 post-subcutaneous infection with 5 × 107 CFU of MGAS2221. (C) Levels of neutrophil infiltration at the skin infection site of MGAS2221 in BALB/c and CXCR2−/− mice at 24 h postinoculation. (D) SpeBA−% of GAS isolates recovered from the skin infection site in BALB/c and CXCR2−/− mice at day 4 post-subcutaneous infection with MGAS2221.
Because RB6-8C5 also depletes a subset of monocytes that express granulocyte receptor 1 (Gr1) (38), the results of the RB6-8C5 experiment would indicate a role of neutrophils and/or inflammatory macrophages in the selection of SpeBA− variants during infection. We performed two additional tests to further examine the role of neutrophils in SpeBA− selection. First, the anti-Ly6G-specific MAb 1A8, which targets only neutrophils (38), was used to deplete neutrophils. FACS analyses showed that treatment with 1A8 (Fig. 2E), but not 2A3 (subtype control MAb) (Fig. 2D), efficiently depleted neutrophils. The myeloperoxidase assay showed that 1A8 reduced neutrophil levels at MGAS2221 infection sites by 92% compared with DPBS-treated mice (Fig. 2F), and this reduction efficiency was slightly lower than that for the RB6-8C5 treatment. The percentages of SpeBA− isolates among GAS bacteria recovered from 1A8-treated mice in two independent experiments (experiment 1, 14% ± 12%; experiment 2, 7% ± 7%; combined, 11% ± 10%) were significantly lower than those recovered from mice treated with 2A3 (experiment 1, 49% ± 21%; experiment 2, 40% ± 20%; combined, 46% ± 21%) (P values of 0.0011 [experiment 1], 0.0001 [experiment 2], and <0.0001 [combined]) (Fig. 3B). The results indicate a major role of neutrophils in the selection of SpeBA− variants.
The second experiment used CXCR2−/− mice. These mice have impaired neutrophil migration (33). MGAS2221 induced (7.3 ± 3.2) × 104 neutrophils/mm2 at subcutaneous infection sites in CXCR2−/− mice, which was only 5% of that at infection sites in BALB/c mice ([1.45 ± 0.12] × 106 neutrophils/mm2) (P < 0.0001) (Fig. 3C). The SpeBA−% for GAS bacteria recovered from skin infection sites of MGAS2221 in CXCR2−/− mice was 2.9% ± 3.8%, which was only 7.6% of that for GAS bacteria from BALB/c mice (38.0% ± 12%) (P = 0.0002) (Fig. 3D). It is possible that CXCR2−/− mice reduced the recruitment of inflammatory macrophages due to the impaired neutrophil infiltration during MGAS2221 infection, resulting in a reduction in the selection of SpeBA− variants. To examine this possibility, we quantified the numbers of neutrophils and macrophages in the lavage fluid of the skin air sac containing MGAS2221 at 12 h after inoculation in CXCR2−/− and BALB/c (control) mice. More than 90% of host cells were neutrophils in the lavage fluid from BALB/c mice (Fig. 4A and C), whereas macrophages constituted 40% of host cells in the lavage fluid from CXCR2−/− mice (Fig. 4B and C). The CXCR2−/− mouse lavage fluid had a 67% reduction in neutrophil numbers (P = 0.0079) and a 290% increase in macrophage numbers (P = 0.0159) compared to the BALB/c lavage fluid (Fig. 4C). If inflammatory macrophages play a critical role in the selection of SpeBA− variants, CXCR2−/− mice should have lower macrophage infiltration during MGAS2221 infection since the SpeBA−% of GAS isolates recovered from CXCR2−/− mice was lower than that from control mice. Thus, the increase in macrophage infiltration in infection of CXCR2−/− mice rules out a major role of inflammatory macrophages in the selection of SpeBA− variants of MGAS2221. Taken together, all the results of the three experiments indicate that neutrophils play a dominant role in the selection of MGAS2221 SpeBA− variants during infection.
FIG 4.
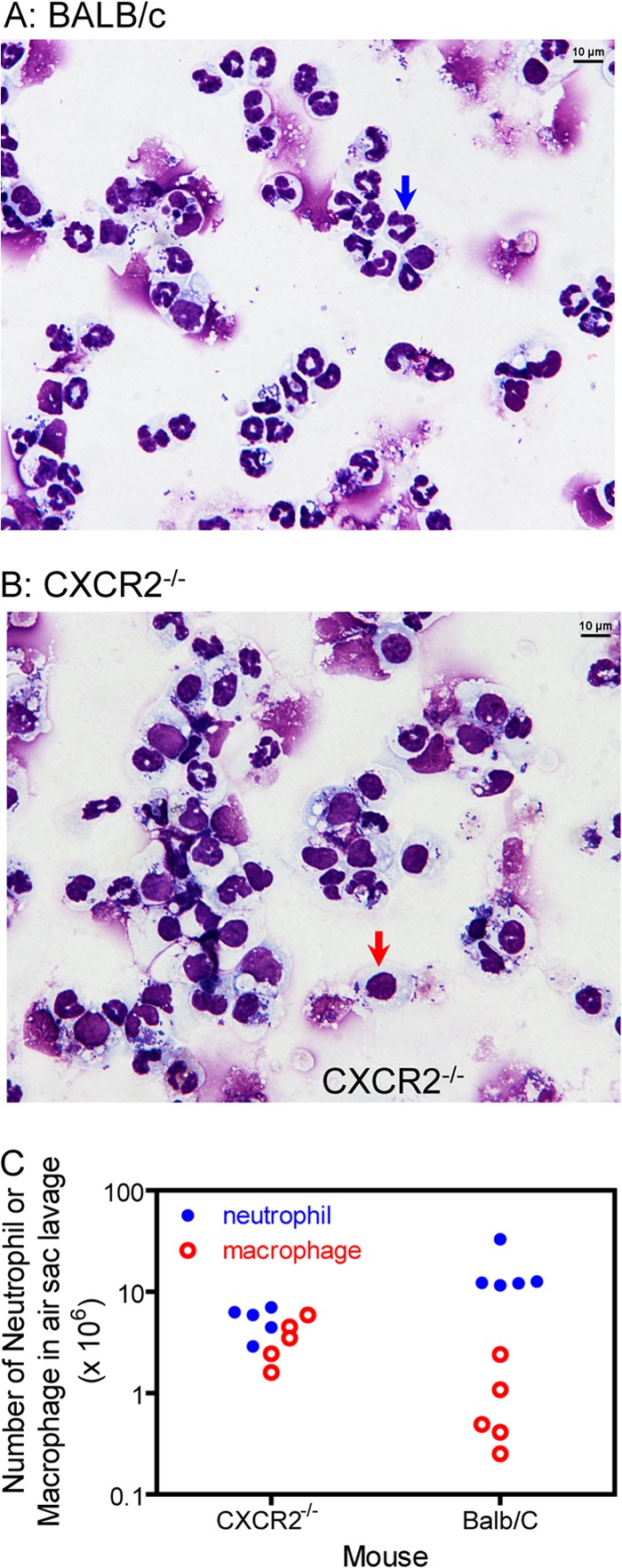
Comparison of neutrophil and macrophage infiltration in air sac MGAS2221 infection of CXCR2−/− and BALB/c mice by cytospin analysis. Air sacs containing MGAS2221 were lavaged at 12 h after GAS inoculation, and neutrophils and macrophages in the lavage fluid were quantified as described in Materials and Methods. (A and B) Representative pictures of cytospin showing neutrophils (blue arrow) and macrophages (red arrow) for the lavage samples from BALB/c (A) and CXCR2−/− (B) mice. (C) Numbers of neutrophils and macrophages in the lavage samples from BALB/c and CXCR2−/− mice.
Advantage in survival and disadvantage in growth of the SpeBA−1 strain compared to the parent strain.
The role of neutrophils in the selection of SpeBA− variants suggests that the mutants may be more resistant to neutrophil-mediated clearance than MGAS2221. To test this idea, we performed a competitive growth/clearance assay in normal and neutropenic mice. The neutropenic mice were prepared by depletion of neutrophils with RB6-8C5, as described above. RB6-8C5 was used for this test was because the test was performed immediately after we observed the detrimental effect of the RB6-8C5 treatment of mice on the selection for SpeBA− variants of MGAS2221 during infection and before 1A8 was used later for additional tests. In this competitive assay, an approximate 1:1 mixture of SpeBA−1, a randomly chosen SpeBA− mutant, and MGAS2221 was subcutaneously inoculated into C57BL/6 mice without (normal mice) or with (neutropenic mice) depletion of neutrophils, and 24 h later, viable bacteria were recovered from infection sites to determine SpeBA−% in the recovered SpeBA−1-MGAS2221 mixture. The number of SpeBA− variants derived from MGAS2221 at 24 h after inoculation should be negligible according to the data in Fig. 1B. Indeed, sequencing of the covRS genes in 10 randomly picked SpeBA− colonies from this competitive assay found that they had the same covS mutation as SpeBA−1. Thus, the SpeBA−% data in this test reflected the relative growth and survival of SpeBA−1 and MGAS2221 in both normal and neutropenic mice. The percentage of SpeBA−1 variants in GAS in neutropenic mice significantly decreased from 66% ± 8% at 1 h after inoculation to 55% ± 8% at 24 h after inoculation (P = 0.0213) (Fig. 5A). These data indicate that the relative population of SpeBA−1 decreased with time in neutropenic mice and that SpeBA−1 grew more slowly than MGAS2221 in vivo. The growth rate of SpeBA−1 in THY was 75% of that of MGAS2221 according to the slope of the growth curves in the exponential growth phase (Fig. 5B). In contrast, the percentage of SpeBA−1 bacteria in GAS from normal mice significantly increased from 70% ± 6% at 1 h after inoculation to 83% ± 13% at 24 h after inoculation (P = 0.0133) (Fig. 5A). The SpeBA−1% value at 24 h after inoculation in normal mice was 51% higher than that in neutropenic mice (P = 0.0006). The data show that the relative population of SpeBA−1 bacteria increased with time in normal mice. Thus, even though SpeBA−1 had a disadvantage in growth in vivo compared to MGAS2221, it had a survival advantage over the parent strain against innate immune responses. The data suggest that SpeBA− mutants are selected because of their advantage for survival against innate immunity over the parent strain.
FIG 5.
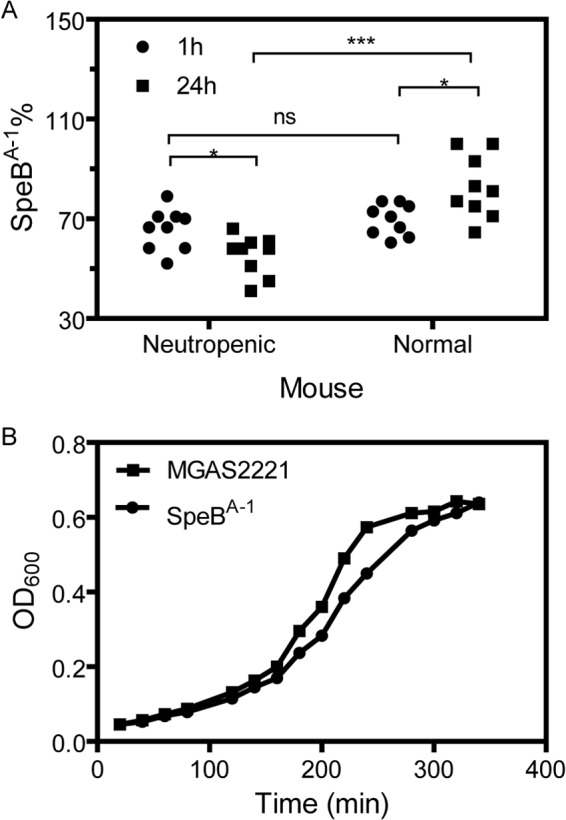
Survival advantage and growth disadvantage of SpeBA−1 compared to wild-type MGAS2221. (A) Percentage of SpeBA−1 variants at 1 h and 24 h after inoculation of an SpeBA−1-MGAS2221 mixture in a mouse model of air sac infection. The data are for mice (9 mice per group) treated with RB6-8C5 (neutropenic mice) and DPBS (normal mice). (B) Growth curves of MGAS2221 and SpeBA−1 in THY. Asterisks indicate significance, with *** showing more significance than *. ns, not significant.
Enhanced innate immune evasion by SpeBA−1.
SpeBA− variants must have enhanced innate immune evasion to have an advantage in survival over the parent strain. To test this possibility, a mouse model of subcutaneous infection was used to compare SpeBA−1 to MGAS2221 in levels of neutrophil infiltration, skin invasion, systemic dissemination, and virulence. Levels of neutrophils at the MGAS2221 infection site at 24 h postinoculation (mean value ± SD of [1.1 ± 0.3] × 105 neutrophils/mm2) were 5.6-fold higher than those at the SpeBA−1 infection site ([1.9 ± 0.8] × 104 neutrophils/mm2) (P = 0.0004) (Fig. 6A). The lesion size caused by SpeBA−1 infection (406 ± 155 mm2) was 3-fold greater than that caused by MGAS2221 (131 ± 28 mm2) (P = 0.0045) (Fig. 6B). Numbers of viable GAS bacteria in the liver and spleen of mice infected with SpeBA−1 at 24 h after inoculation were (1.2 ± 1.6) × 105 CFU/g and (6.5 ± 12) × 107 CFU/g, respectively, whereas very few GAS bacteria were detected in the liver and spleen of mice infected by MGAS2221 (Fig. 6C and D). In the survival test, all mice infected with SpeBA−1 did not survive, but all mice infected with MGAS2221 did (P < 0.0001) (Fig. 6E). Thus, SpeBA−1 had a higher capacity than MGAS2221 to evade neutrophil responses, invade the skin tissue, and disseminate, resulting in the hypervirulence of the mutant.
FIG 6.

The SpeBA−1 isolate displays enhancement in innate immune evasion, skin evasion, systemic dissemination, virulence, and expression of virulence factors. (A to D) Neutrophil recruitment (A), lesion size (B), and numbers of viable GAS bacteria in the liver (C) and spleen (D) at 24 h after subcutaneous inoculation of 9.0 × 107 CFU MGAS2221 or 8.5 × 107 CFU SpeBA−1 in female CD1 mice. (E) Survival curves of CD1 mice after subcutaneous infection with the same dose of MGAS2221 and SpeBA−1. (F) Relative mRNA levels of the emm, hasA, spyCEP, and sse genes in SpeBA−1 at an OD600 of 0.2 compared with MGAS2221.
SpeB downregulation and SsE upregulation in SpeBA− variants were caused by covRS mutations.
The phenotype of SpeBA−1 described above was similar to that of GAS isolates from human patients and mice that have null covS mutations (12–14). Indeed, DNA sequencing of the covRS genes revealed that SpeBA−1 had a deletion of bases 132 to 136 of the covS gene (5′-TTTCT-3′). Transcription of the innate immunity-evading genes hasA, spyCEP, and sse in SpeBA−1 was enhanced by >35-fold compared with MGAS2221 (Fig. 6F). These results are consistent with previous findings on the effect of null covS deletion on expression of these virulence genes (14, 24).
To determine whether covRS mutations were common among SpeBA− variants of MGAS2221 recovered from mice, we sequenced the covRS genes of all 11 SpeBA− isolates and 2 randomly chosen SpeBA+ colonies among 48 tested GAS colonies from a mouse. All 11 SpeBA− variants had covRS mutations: 6 of them with a deletion, insertion, or nonsense mutation that resulted in the truncation of CovS; 3 variants with an Ala-to-Asp missense mutation at position 388 of CovS (CovSAla388Asp); and 2 variants with CovRIle205Phe mutations (Table 1). In contrast, both SpeBA+ isolates had the wild-type covRS sequence. In addition, there were no synonymous mutations in the covRS genes of the 11 SpeBA− variants, indicating that the covRS mutations were selected.
TABLE 1.
CovRS mutations and in vitro SsE expression of SpeBA− variants of MGAS2221 isolated from subcutaneous mouse infectionc
| Straina | covRS mutation | Mutated CovS or CovR | SpeB production | In vitro SsEb production |
|---|---|---|---|---|
| 1 | 132TTTCT136 deletion in covS | Truncated CovS | − | + |
| 2 | 223G to T in covS | Truncated CovS | − | + |
| 3 | 1166C to A in covS | CovSAla388Asp | − | + |
| 4 | 481C to T in covS | Truncated CovS | − | + |
| 5 | Insertion of ATTTTCTCTGC at nucleotide 99 in covS | Truncated CovS | − | + |
| 6 | 616A to T in covR | CovRIle205Phe | − | + |
| 7 | 616A to T in covR | CovRIle205Phe | − | + |
| 8 | 1166C to A in covS | CovSAla388Asp | − | + |
| 9 | 1363C to T in covS | Truncated CovS | − | + |
| 10 | 1363C to T in covS | Truncated CovS | − | + |
| 11 | 1166C to A in covS | CovSAla388Asp | − | + |
| 12 | None | wt | + | − |
| 13 | None | wt | + | − |
| 14 | wt | wt | + | − |
Strains 1 through 11 were all SpeBA− variants detected among 48 randomly chosen colonies isolated from a mouse infected with MGAS2221, and strains 12 and 13 were randomly chosen from 37 SpeBA+ colonies in the same test. Strain 14 was MGAS2221.
The SsE production status was assigned according to the data shown in Fig. 7.
wt, wild type.
We next checked the SsE expression of the 11 SpeBA− variants as an indicator of whether these covRS mutants enhanced expression of virulence genes. SsE solely confers the enzymatic activity of hydrolyzing platelet-activating factor (PAF) in GAS culture supernatants, and the PAF hydrolysis activity increases when the covS gene is deleted or has a null mutation (14, 25, 36). The PAF hydrolysis activity of SsE was very low in the culture supernatants of MGAS2221 and two SpeBA+ isolates at the mid-log growth phase (designated the SsEA− phenotype) but substantially increased in the culture supernatants of the SpeBA− variants (designated the SsEA+ phenotype) (Fig. 7A and D). These results were confirmed by the presence and absence of SsE in the culture supernatants of SpeBA− and SpeBA+ isolates, respectively, by Western blotting, whereas Spy0469 (control) was detected in all the culture supernatants (Fig. 7B). Thus, the SpeBA− variants with the covRS mutations all had enhanced SsE expression.
FIG 7.

The SpeBA−/SsEA+ phenotype of in vivo-selected MGAS2221 variants is caused by covRS mutations. (A) SsE PAF acetylhydrolase activity in the culture supernatant of SpeBA− variants with covS mutations and SpeBA+ isolates from MGAS2221-infected mice. (B) Western blots for SsE and Spy0469 (control) in culture supernatants showing positive detection of SsE in SpeBA− variants (strains 1 through 11) but not in SpeBA+ isolates (strains 12 and 13) and MGAS2221 (strain 14). (C) Complementation of SpeBA− variants with pGRV-CovRS (strains 6 and 7) and pCovSC (the other strains) converted the SpeBA− phenotype of the mutants into the SpeBA+ phenotype, and pDCBB controls for pCovSC did not change the status of SpeB production. Strain numbers correspond to those in Table 1. (D) SsE PAF-AH activity in the culture supernatant of pDCBB (control)- and pCovSC-transformed SpeBA− variants with covS mutations listed in Table 1. (E) SsE PAF-AH activity in culture supernatants of the two CovR mutants and their complemented strains with integrated pGRV-CovRS.
To determine whether the covRS mutations caused the SpeBA−/SsEA+ phenotype of these SpeBA− variants, these mutants were complemented with wild-type covS or covRS. The strains with the covS mutations were transformed with plasmid pCovSC, whereas the suicide plasmid pGRV-CovRS was integrated into the genomes of the two isolates with the CovRIle205Phe mutation. The complemented strains of these covRS mutants all restored SpeB production, whereas the vector controls still had the SpeA− phenotype (Fig. 7C). The complementation of the nine SpeBA− variants with covS mutations reduced SsE activity in the culture supernatants to the levels of MGAS2221 (Fig. 7D). The complemented covR mutants reduced SsE production but still had significantly higher SsE activity than MGAS2221 (Fig. 7E), which might be caused by the presence of a single copy of both the wild-type and mutated covR alleles in the complement strains. These results indicate that the observed covRS mutations are responsible for the SpeBA−/SsEA+ phenotype.
DISCUSSION
This report presents two findings on the role of neutrophils in the selection of hypervirulent CovRS mutants of an M1T1 isolate during subcutaneous infection of mice. First, neutrophils play a critical role in the selection of CovRS mutants. Second, CovRS mutants of MGAS2221 are selected for better survival, even though they have a disadvantage in growth in vivo compared with wild-type MGAS2221. The significance of these findings is 2-fold. First, these findings are consistent with a model in which neutrophil responses mount pressure for the selection of spontaneous CovRS mutations that enhance innate immune evasion and consequently confer to GAS better survival under the attack of neutrophils. Second, these findings will facilitate the elucidation of the genetic variations among GAS isolates that dictate whether SpeBA− mutants are selected during soft tissue infections.
Null covS mutation-carrying M1T1 strains from severe invasive infection or mouse passage are SpeBA− strains (12–14, 20). The SpeBA− phenotype of such a strain has been shown to be caused by a null covS mutation (14). Conversely, characterized M1T1 SpeBA− variants from mouse passage have null covS mutations (13, 23, 27). SpeBA− variants have been used to quantify covRS mutations in mouse passage (27, 28), which was apparently based on the assumption that the selected SpeBA− phenotype is caused by covRS mutations. All 11 SpeBA− variants obtained from the same mouse had covRS mutations, and the SpeBA−/SsEA+ phenotype of these variants was caused by the covS or covR mutations. Thus, our data appear to validate the approach that uses the SpeBA− phenotype as a marker to screen for and quantify covRS mutations of M1T1 isolates during passage in mice. Our data also show that, besides covS truncation mutations, covS and covR missense mutations can lead to the SpeBA−/SsEA+ phenotype. Certain covS missense mutations have been shown to enhance the production of NADase and reduce SpeB production (40), and the CovSAla388Asp mutation that causes the SpeBA−/SsEA+ phenotype has not been reported. In addition, the CovRIle205Phe mutation has not been reported but is close to the known Arg203Ser and Gln216Pro mutations in the DNA-binding domain of CovR (41, 42). CovRArg119His confers a SpeBA− phenotype (20), and CovRIle205Phe is the second CovR mutation that has been shown to cause the SpeBA− phenotype.
A novel finding of this study is that neutrophils are required for the selection of CovRS mutations. Recruited macrophages play a role in clearance of GAS (43). However, macrophages appear not to play a critical role in the selection of covRS mutations in MGAS2221, since the decrease in the selection of SpeBA− variants of MGAS2221 in CXCR2−/− mice is correlated with the impaired neutrophil recruitment but not with the increased levels of macrophages. These results imply that CovRS mutations confer advantages against the clearance of GAS by neutrophils but not by macrophages. This implication is supported by the fact that most of the known innate immunity-evading factors of GAS target neutrophils. Neutrophil infiltration is reduced by interleukin-8 (IL-8)/CXC chemokine peptidase SpyCEP (44–46), C5a peptidase ScpA (47), and platelet-activating factor acetylhydrolase SsE (14, 25). GAS produces the hyaluronic acid capsule and the major surface protein M to resist phagocytosis by neutrophils (48, 49). Neutrophils can also be destroyed by streptolysins S and O (50–52). Specific antibody-mediated opsonophagocytosis is inhibited by the secreted protein Mac (53). GAS also produces DNases to help it escape neutrophils' DNA-derived extracellular traps (54). All of these virulence factors except for the M protein and ScpA are regulated by CovRS. All these studies suggest that neutrophils mount critical innate immune responses against GAS, which is supported by a number of studies examining GAS killing by neutrophils (55–59).
The SpeBA−1 mutant outcompetes wild-type bacteria in mice with normal innate immune responses but is outcompeted by wild-type bacteria when neutrophils and inflammatory macrophages are depleted. These observations suggest that the accumulation of SpeBA− mutants with time during infection is due to their advantage in survival against innate immune responses over wild-type bacteria, even though the mutants have a disadvantage in growth. Previous findings on the role of Sda1 in and the requirement of the hasA and emm gene for the selection of SpeBA− mutants imply a survival advantage of SpeBA− mutants in vivo (27, 28). Our findings explicitly demonstrate that covS mutants can survive better than the parent GAS strain in the presence of neutrophils and inflammatory macrophages. In addition, the ability of GAS to survive against neutrophil killing is more critical than the capacity of GAS to grow faster for the selection of CovRS mutants.
The survival advantage of CovRS mutants over wild-type GAS is apparently due to the enhanced innate immune evasion by the mutants. This advantage appears to be the reason for the in vivo accumulation of SpeBA− variants of MGAS2221 with time, which is also observed for M1T1 strain 5448 (60). Consistent with previous findings (14, 61), SpeBA−1 had enhanced expression of IL-8/CXC chemokine peptidase SpyCEP and platelet-activating factor acetylhydrolase SsE as a result of the covS deletion. A null covS deletion not only reduces neutrophil recruitment but also keeps neutrophils away from GAS, and SsE critically contributes to this covS deletion-caused severe innate immune evasion (14, 25). It is possible that CovRS mutants could generate a local neutrophil-free zone through the action of SsE and SpyCEP. The enhanced production of the capsule and Sda1 can also contribute to the survival advantage by enhancing the resistance of GAS to phagocytosis and help mutants escape neutrophil extracellular trap-enhanced clearance, respectively (27).
The covRS mutations in the analyzed SpeBA− variants were all nonsynonymous mutations and caused the SpeBA−/SsEA+ phenotype. No covRS mutations were found in the sequenced SpeBA+ isolates. These observations indicate that the covRS mutations are selected. Our findings support a model for the occurrence of SpeBA−/SsEA+ covRS mutants of M1T1 GAS strains during infection in which spontaneous covR or covS mutations are selected by neutrophils in soft tissue infections for an advantage against the innate immune system over parent strains with functional CovRS (Fig. 8). The new information in this study extends our current understanding that covRS mutations occur in clinical isolates and experimental animal infections and enhance innate immune evasion, skin invasion, and virulence.
FIG 8.
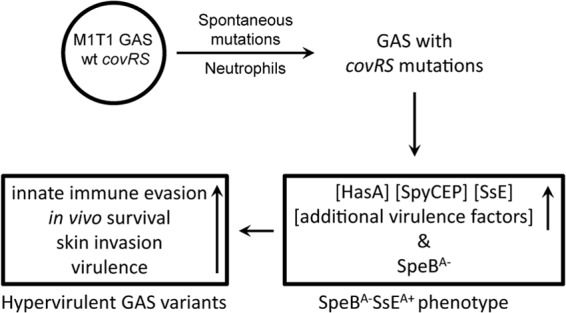
Model for in vivo selection of hypervirulent M1T1 GAS CovRS mutants. Neutrophils select spontaneous covRS mutations that enhance expression of multiple virulence genes and abolish SpeB production, resulting in mutants with enhanced innate evasion, in vivo survival, skin invasion, and virulence.
The selection power for MGAS2221 covRS mutations in murine skin infections is striking. Our SpeBA− data are consistent with data reported by Sumby et al. for mucoid isolates obtained from MGAS2221 in murine skin infections (12). Another M1T1 strain, 5448, readily switches to the SpeBA− phenotype in skin infection as well (21, 27). These data for animal models of GAS infections appear to correlate with the fact that invasive isolates frequently have covRS mutations (15, 16). For example, the invasive M1T1 isolate MGAS5005 has a single-base deletion at position 83 in covS, and this 1-base deletion causes the hypervirulence and SpeBA− phenotype of MGAS5005 (14). The sequenced MGAS315 and MGAS6180 genomes have CovSG457V and CovSG226E mutations, respectively, compared with CovS of the sequenced M1 strain SF370 (32, 62, 63). In addition, the well-known virulent strains NZ131 and CS101 are also SpeBA− strains (data not shown). However, not all GAS isolates switch to the SpeBA− phenotype, and the in vivo selection of SpeBA− variants appears to be associated with GAS strains that are linked to severe invasive infections. This would explain why we frequently obtain SpeBA+ isolates from human infections. MGAS2221 was isolated from a patient with scarlet fever and presumably from tonsil or pharynx (20). If true, the isolation of the SpeBA+ MGAS2221 strain would suggest that strains of the M1T1 clone associated with severe invasive infections may be subject to less selection pressure for SpeBA− variants at the pharynx and tonsil than in soft tissues. As for the carriage stage, strains with wild-type covRS may eventually prevail because they do not divert a significant amount of energy to synthesize high levels of virulence factors that should not be needed when acute inflammation is gone. Indeed, the SpeBA−1 isolate with the null covS mutation grows slower both in vivo and in vitro than MGAS2221. MGAS5005, which has a natural covS deletion and SpeBA− phenotype, grows slower in vitro than MGAS2221 (14). Tatsuno et al. recently also reported slower growth of covS mutants than strains with the wild-type covS gene (40). The slowdown in growth of covS mutants in comparison with wild-type strains could be the basis for the overgrowth of wild-type bacteria over covS mutants in saliva (20). Diversion of energy to the enhanced expression of CovRS-regulated virulence factors is most likely responsible for the slowdown in growth of SpeBA− mutants both in vitro and in vivo.
Sda1 has been shown to provide selection pressure for SpeBA− variants of M1T1 isolate 5448 (27). However, recent studies suggest that Sda1 does not play a direct and essential role in the selection of SpeBA− variants. M1 isolate SF370 does not switch to an SpeBA− phenotype (27), and the introduction of the Sda1-encoding phage into SF370 does not result in the selection of SpeBA− variants during infection (29). Furthermore, serotype M98 GAS apparently undergoes SpeBA− mutations in patients even though it lacks prophage-encoded Sda1 (30). Besides Sda1, the hasA and emm genes are also required for the selection of SpeBA− variants of the M1T1 isolate (28). Both the hyaluronic acid capsule and M protein are required for resistance of GAS to phagocytosis by neutrophils. The loss of hasA or emm may cause a loss of the survival advantage conferred by covRS mutations. SF370 has functioning hasA and emm genes. Therefore, hasA and emm are required for but are not the direct cause of the selection of SpeBA− variants. Thus, the exact basis for why covRS mutations are selected in strains of the M1T1 GAS clone associated with severe invasive infections but not the SF370 M1 isolate remains unknown. Since neutrophils exert pressure for the selection of CovRS mutants, identifying molecular events in the M1T1 clone that may lead to a disadvantage against neutrophil responses may be a good strategy to elucidate the basis for in vivo selection of covRS mutations.
ACKNOWLEDGMENTS
This work was supported in part by NIH grants AI095704 and AI097703 from the National Institute of Allergy and Infectious Diseases and GM103500-09 from the National Institute of General Medical Sciences, the USDA Formula Fund, and the Montana State University Agricultural Experimental Station. J.L. was supported by a Ph.D. student exchange scholarship of the Ministry of Education, China.
Footnotes
Published ahead of print 22 January 2014
REFERENCES
- 1.Carapetis JR, Steer AC, Mulholland EK, Weber M. 2005. The global burden of group A streptococcal diseases. Lancet Infect. Dis. 5:685–694. 10.1016/S1473-3099(05)70267-X [DOI] [PubMed] [Google Scholar]
- 2.O'Loughlin RE, Roberson A, Cieslak PR, Lynfield R, Gershman K, Craig A, Albanese BA, Farley MM, Barrett NL, Spina NL, Beall B, Harrison LH, Reingold A, Van Beneden C, Active Bacterial Core Surveillance Team 2007. The epidemiology of invasive group A streptococcal infection and potential vaccine implications: United States, 2000-2004. Clin. Infect. Dis. 45:853–862. 10.1086/521264 [DOI] [PubMed] [Google Scholar]
- 3.Musser JM, Hauser AR, Kim MH, Schlievert PM, Nelson K, Selander RK. 1991. Streptococcus pyogenes causing toxic-shock-like syndrome and other invasive diseases: clonal diversity and pyrogenic exotoxin expression. Proc. Natl. Acad. Sci. U. S. A. 88:2668–2672. 10.1073/pnas.88.7.2668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cleary PP, Kaplan EL, Handley JP, Wlazlo A, Kim MH, Hauser AR, Schlievert PM. 1992. Clonal basis for resurgence of serious Streptococcus pyogenes disease in the 1980s. Lancet 339:518–521. 10.1016/0140-6736(92)90339-5 [DOI] [PubMed] [Google Scholar]
- 5.Musser JM, Kapur V, Kanjilal S, Shah U, Musher DM, Barg NL, Johnston KH, Schlievert PM, Henrichsen J, Gerlach D, Rakita RM, Tanna A, Cookson BD, Huang JC. 1993. Geographic and temporal distribution and molecular characterization of two highly pathogenic clones of Streptococcus pyogenes expressing allelic variants of pyrogenic exotoxin A (scarlet fever toxin). J. Infect. Dis. 167:337–346. 10.1093/infdis/167.2.337 [DOI] [PubMed] [Google Scholar]
- 6.Martin DR, Single LA. 1993. Molecular epidemiology of group A streptococcus M type 1 infections. J. Infect. Dis. 167:1112–1117. 10.1093/infdis/167.5.1112 [DOI] [PubMed] [Google Scholar]
- 7.Seppälä H, Vuopio-Varkila J, Osterblad M, Jahkola M, Rummukainen M, Holm SE, Huovinen P. 1994. Evaluation of methods for epidemiologic typing of group A streptococci. J. Infect. Dis. 169:519–525. 10.1093/infdis/169.3.519 [DOI] [PubMed] [Google Scholar]
- 8.Musser JM, Kapur V, Szeto J, Pan X, Swanson DS, Martin DR. 1995. Genetic diversity and relationships among Streptococcus pyogenes strains expressing serotype M1 protein: recent intercontinental spread of a subclone causing episodes of invasive disease. Infect. Immun. 63:994–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chatellier S, Ihendyane N, Kansal RG, Khambaty F, Basma H, Norrby-Teglund A, Low DE, McGeer A, Kotb M. 2000. Genetic relatedness and superantigen expression in group A streptococcus serotype M1 isolates from patients with severe and nonsevere invasive diseases. Infect. Immun. 68:3523–3534. 10.1128/IAI.68.6.3523-3534.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sumby P, Porcella SF, Madrigal AG, Barbian KD, Virtaneva K, Ricklefs SM, Sturdevant DE, Graham MR, Vuopio-Varkila J, Hoe NP, Musser JM. 2005. Evolutionary origin and emergence of a highly successful clone of serotype M1 group A Streptococcus involved multiple horizontal gene transfer events. J. Infect. Dis. 192:771–782. 10.1086/432514 [DOI] [PubMed] [Google Scholar]
- 11.Aziz RK, Kotb MZ. 2008. Rise and persistence of global M1T1 clone of Streptococcus pyogenes. Emerg. Infect. Dis. 14:1511–1517. 10.3201/eid1410.071660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sumby P, Whitney AR, Graviss EA, DeLeo FR, Musser JM. 2006. Genome-wide analysis of group A streptococci reveals a mutation that modulates global phenotype and disease specificity. PLoS Pathog. 2:e5. 10.1371/journal.ppat.0020005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kansal RG, Datta V, Aziz RK, Abdeltawab NF, Rowe S, Kotb M. 2010. Dissection of the molecular basis for hypervirulence of an in vivo-selected phenotype of the widely disseminated M1T1 strain of group A Streptococcus bacteria. J. Infect. Dis. 201:855–865. 10.1086/651019 [DOI] [PubMed] [Google Scholar]
- 14.Li J, Zhu H, Feng W, Liu M, Song Y, Zhang X, Zhou Y, Bei W, Lei B. 2013. Regulation of inhibition of neutrophil infiltration by the two-component regulatory system CovRS in subcutaneous murine infection with group A streptococcus. Infect. Immun. 81:974–983. 10.1128/IAI.01218-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ikebe T, Ato M, Matsumura T, Hasegawa H, Sata T, Kobayashi K, Watanabe H. 2010. Highly frequent mutations in negative regulators of multiple virulence genes in group A streptococcal toxic shock syndrome isolates. PLoS Pathog. 6:e1000832. 10.1371/journal.ppat.1000832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shea PR, Beres SB, Flores AR, Ewbank AL, Gonzalez-Lugo JH, Martagon-Rosado AJ, Martinez-Gutierrez JC, Rehman HA, Serrano-Gonzalez M, Fittipaldi N, Ayers SD, Webb P, Willey BM, Low DE, Musser JM. 2011. Distinct signatures of diversifying selection revealed by genome analysis of respiratory tract and invasive bacterial populations. Proc. Natl. Acad. Sci. U. S. A. 108:5039–5044. 10.1073/pnas.1016282108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levin JC, Wessels MR. 1998. Identification of csrR/csrS, a genetic locus that regulates hyaluronic acid capsule synthesis in group A Streptococcus. Mol. Microbiol. 30:209–219. 10.1046/j.1365-2958.1998.01057.x [DOI] [PubMed] [Google Scholar]
- 18.Heath A, DiRita VJ, Barg NL, Engleberg NC. 1999. A two-component regulatory system, CsrR-CsrS, represses expression of three Streptococcus pyogenes virulence factors, hyaluronic acid capsule, streptolysin S, and pyrogenic exotoxin B. Infect. Immun. 67:5298–5305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Federle MJ, McIver KS, Scott JR. 1999. A response regulator that represses transcription of several virulence operons in the group A Streptococcus. J. Bacteriol. 181:3649–3657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Treviño J, Perez N, Ramirez-Peña E, Liu Z, Shelburne SA, Musser JM, Sumby P. 2009. CovS simultaneously activates and inhibits the CovR-mediated repression of distinct subsets of group A Streptococcus virulence factor-encoding genes. Infect. Immun. 77:3141–3149. 10.1128/IAI.01560-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aziz RK, Pabst MJ, Jeng A, Kansal R, Low DE, Nizet V, Kotb M. 2004. Invasive M1T1 group A Streptococcus undergoes a phase-shift in vivo to prevent proteolytic degradation of multiple virulence factors by SpeB. Mol. Microbiol. 51:123–134. 10.1046/j.1365-2958.2003.03797.x [DOI] [PubMed] [Google Scholar]
- 22.Engleberg NC, Heath A, Vardaman K, DiRita VJ. 2004. Contribution of CsrR-regulated virulence factors to the progress and outcome of murine skin infections by Streptococcus pyogenes. Infect. Immun. 72:623–628. 10.1128/IAI.72.2.623-628.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cole JN, McArthur JD, McKay FC, Sanderson-Smith ML, Cork AJ, Ranson M, Rohde M, Itzek A, Sun H, Ginsburg D, Kotb M, Nizet V, Chhatwal GS, Walker MJ. 2006. Trigger for group A streptococcal M1T1 invasive disease. FASEB J. 20:1745–1747. 10.1096/fj.06-5804fje [DOI] [PubMed] [Google Scholar]
- 24.Zhu H, Liu M, Sumby P, Lei B. 2009. The secreted esterase of group A streptococcus is important for invasive skin infection and dissemination in mice. Infect. Immun. 77:5225–5232. 10.1128/IAI.00636-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu M, Zhu H, Li J, Garcia CC, Feng W, Kirpotina LN, Hilmer J, Tavares LP, Layton AW, Quinn MT, Bothner B, Teixeira MM, Lei B. 2012. Group A Streptococcus secreted esterase hydrolyzes platelet-activating factor to impede neutrophil recruitment and facilitate innate immune evasion. PLoS Pathog. 8:e1002624. 10.1371/journal.ppat.1002624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Engleberg NC, Heath A, Miller A, Rivera C, DiRita VJ. 2001. Spontaneous mutations in the CsrRS two-component regulatory system of Streptococcus pyogenes result in enhanced virulence in a murine model of skin and soft tissue infection. J. Infect. Dis. 183:1043–1054. 10.1086/319291 [DOI] [PubMed] [Google Scholar]
- 27.Walker MJ, Hollands A, Sanderson-Smith ML, Cole JN, Kirk JK, Henningham A, McArthur JD, Dinkla K, Aziz RK, Kansal RG, Simpson AJ, Buchanan JT, Chhatwal GS, Kotb M, Nizet V. 2007. DNase Sda1 provides selection pressure for a switch to invasive group A streptococcal infection. Nat. Med. 13:981–985. 10.1038/nm1612 [DOI] [PubMed] [Google Scholar]
- 28.Cole JN, Pence MA, von Köckritz-Blickwede M, Hollands A, Gallo RL, Walker MJ, Nizet V. 2010. M protein and hyaluronic acid capsule are essential for in vivo selection of covRS mutations characteristic of invasive serotype M1T1 group A Streptococcus. mBio 1(4):e00191-10. 10.1128/mBio.00191-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Venturini C, Ong CL, Gillen CM, Ben-Zakour NL, Maamary PG, Nizet V, Beatson SA, Walker MJ. 2013. Acquisition of the Sda1-encoding bacteriophage does not enhance virulence of the M1 Streptococcus pyogenes strain SF370. Infect. Immun. 81:2062–2069. 10.1128/IAI.00192-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsatsaronis JA, Hollands A, Cole JN, Maamary PG, Gillen CM, Zakour NL, Kotb M, Nizet V, Beatson SA, Walker MJ, Sanderson-Smith ML. 2013. Streptococcal collagen-like protein A and general stress protein 24 are immunomodulating virulence factors of group A Streptococcus. FASEB J. 27:2633–2643. 10.1096/fj.12-226662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed. National Academies Press, Washington, DC [Google Scholar]
- 32.Ferretti JJ, McShan WM, Ajdic D, Savic DJ, Savic G, Lyon K, Primeaux C, Sezate S, Suvorov AN, Kenton S, Lai HS, Lin SP, Qian Y, Jia HG, Najar FZ, Ren Q, Zhu H, Song L, White J, Yuan X, Clifton SW, Roe BA, McLaughlin R. 2001. Complete genome sequence of an M1 strain of Streptococcus pyogenes. Proc. Natl. Acad. Sci. U. S. A. 98:4658–4663. 10.1073/pnas.071559398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cacalano G, Lee J, Kikly K, Ryan AM, Pitts-Meek S, Hultgren B, Wood WI, Moore MW. 1994. Neutrophil and B cell expansion in mice that lack the murine IL-8 receptor homolog. Science 265:682–684. 10.1126/science.8036519 [DOI] [PubMed] [Google Scholar]
- 34.Ma Y, Bryant AE, Salmi DB, Hayes-Schroer SM, McIndoo E, Aldape MJ, Stevens DL. 2006. Identification and characterization of bicistronic speB and prsA gene expression in the group A Streptococcus. J. Bacteriol. 188:7626–7634. 10.1128/JB.01059-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bradley PP, Priebat DA, Christensen RD, Rothstein G. 1982. Measurement of cutaneous inflammation: estimation of neutrophil content with an enzyme marker. J. Investig. Dermatol. 78:206–209. 10.1111/1523-1747.ep12506462 [DOI] [PubMed] [Google Scholar]
- 36.Liu G, Liu M, Xie G, Lei B. 2013. Characterization of streptococcal platelet-activating factor acetylhydrolase variants that are involved in innate immune evasion. Infect. Immun. 81:3128–3138. 10.1128/IAI.00398-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu M, Hanks TS, Zhang J, McClure MJ, Siemsen DW, Elser JL, Quinn MT, Lei B. 2006. Defects in ex vivo and in vivo growth and sensitivity to osmotic stress of group A Streptococcus caused by interruption of response regulator gene vicR. Microbiology 152:967–978. 10.1099/mic.0.28706-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. 2008. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J. Leukoc. Biol. 83:64–70. 10.1189/jlb.0407247 [DOI] [PubMed] [Google Scholar]
- 39.Bos A, Wever R, Roos D. 1978. Characterization and quantification of the peroxidase in human monocytes. Biochim. Biophys. Acta 525:37–44. 10.1016/0005-2744(78)90197-3 [DOI] [PubMed] [Google Scholar]
- 40.Tatsuno I, Okada R, Zhang Y, Isaka M, Hasegawa T. 2013. Partial loss of CovS function in Streptococcus pyogenes causes severe invasive disease. BMC Res. Notes 6:126. 10.1186/1756-0500-6-126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miyoshi-Akiyama T, Ikebe T, Watanabe H, Uchiyama T, Kirikae T, Kawamura Y. 2006. Use of DNA arrays to identify a mutation in the negative regulator, csrR, responsible for the high virulence of a naturally occurring type M3 group A streptococcus clinical isolate. J. Infect. Dis. 193:1677–1684. 10.1086/504263 [DOI] [PubMed] [Google Scholar]
- 42.Horstmann N, Sahasrabhojane P, Suber B, Kumaraswami M, Olsen RJ, Flores A, Musser JM, Brennan RG, Shelburne SA., III 2011. Distinct single amino acid replacements in the control of virulence regulator protein differentially impact streptococcal pathogenesis. PLoS Pathog. 7:e1002311. 10.1371/journal.ppat.1002311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mishalian I, Ordan M, Peled A, Maly A, Eichenbaum MB, Ravins M, Aychek T, Jung S, Hanski E. 2011. Recruited macrophages control dissemination of group A Streptococcus from infected soft tissues. J. Immunol. 187:6022–6031. 10.4049/jimmunol.1101385 [DOI] [PubMed] [Google Scholar]
- 44.Edwards RJ, Taylor GW, Ferguson M, Murray S, Rendell N, Wrigley A, Bai Z, Boyle J, Finney SJ, Jones A, Russell HH, Turner C, Cohen J, Faulkner L, Sriskandan S. 2005. Specific C-terminal cleavage and inactivation of interleukin-8 by invasive disease isolates of Streptococcus pyogenes. J. Infect. Dis. 192:783–790. 10.1086/432485 [DOI] [PubMed] [Google Scholar]
- 45.Zinkernagel AS, Timmer AM, Pence MA, Locke JB, Buchanan JT, Turner CE, Mishalian I, Sriskandan S, Hanski E, Nizet V. 2008. The IL-8 protease SpyCEP/ScpC of group A Streptococcus promotes resistance to neutrophil killing. Cell Host Microbe 4:170–178. 10.1016/j.chom.2008.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sumby P, Zhang S, Whitney AR, Falugi F, Grandi G, Graviss EA, Deleo FR, Musser JM. 2008. A chemokine-degrading extracellular protease made by group A Streptococcus alters pathogenesis by enhancing evasion of the innate immune response. Infect. Immun. 76:978–985. 10.1128/IAI.01354-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wexler DE, Chenoweth DE, Cleary PP. 1985. Mechanism of action of the group A streptococcal C5a inactivator. Proc. Natl. Acad. Sci. U. S. A. 82:8144–8148. 10.1073/pnas.82.23.8144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perez-Casal J, Caparon MG, Scott JR. 1992. Introduction of the emm6 gene into an emm-deleted strain of Streptococcus pyogenes restores its ability to resist phagocytosis. Res. Microbiol. 143:549–558. 10.1016/0923-2508(92)90112-2 [DOI] [PubMed] [Google Scholar]
- 49.Ashbaugh CD, Moser TJ, Shearer MH, White GL, Kennedy RC, Wessels MR. 2000. Bacterial determinants of persistent throat colonization and the associated immune response in a primate model of human group A streptococcal pharyngeal infection. Cell. Microbiol. 2:283–292. 10.1046/j.1462-5822.2000.00050.x [DOI] [PubMed] [Google Scholar]
- 50.Miyoshi-Akiyama T, Takamatsu D, Koyanagi M, Zhao J, Imanishi K, Uchiyama T. 2005. Cytocidal effect of Streptococcus pyogenes on mouse neutrophils in vivo and the critical role of streptolysin S. J. Infect. Dis. 192:107–116. 10.1086/430617 [DOI] [PubMed] [Google Scholar]
- 51.Ato M, Ikebe T, Kawabata H, Takemori T, Watanabe H. 2008. Incompetence of neutrophils to invasive group A Streptococcus is attributed to induction of plural virulence factors by dysfunction of a regulator. PLoS One 3:e3455. 10.1371/journal.pone.0003455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Timmer AM, Timmer JC, Pence MA, Hsu LC, Ghochani M, Frey TG, Karin M, Salvesen GS, Nizet V. 2009. Streptolysin O promotes group A Streptococcus immune evasion by accelerated macrophage apoptosis. J. Biol. Chem. 284:862–871. 10.1074/jbc.M804632200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lei B, DeLeo FR, Hoe NP, Graham MR, Mackie SM, Cole RL, Liu M, Hill HR, Low DE, Federle MJ, Scott JR, Musser JM. 2001. Evasion of human innate and acquired immunity by a bacterial homolog of CD11b that inhibits opsonophagocytosis. Nat. Med. 7:1298–1305. 10.1038/nm1201-1298 [DOI] [PubMed] [Google Scholar]
- 54.Sumby P, Barbian KD, Gardner DJ, Whitney AR, Welty DM, Long RD, Bailey JR, Parnell MJ, Hoe NP, Adams GG, Deleo FR, Musser JM. 2005. Extracellular deoxyribonuclease made by group A Streptococcus assists pathogenesis by enhancing evasion of the innate immune response. Proc. Natl. Acad. Sci. U. S. A. 102:1679–1684. 10.1073/pnas.0406641102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schnitzler N, Haase G, Podbielski A, Lütticken R, Schweizer KG. 1999. A co-stimulatory signal through ICAM-beta2 integrin-binding potentiates neutrophil phagocytosis. Nat. Med. 5:231–235. 10.1038/5597 [DOI] [PubMed] [Google Scholar]
- 56.Nizet V, Ohtake T, Lauth X, Trowbridge J, Rudisill J, Dorschner RA, Pestonjamasp V, Piraino J, Huttner K, Gallo RL. 2001. Innate antimicrobial peptide protects the skin from invasive bacterial infection. Nature 414:454–457. 10.1038/35106587 [DOI] [PubMed] [Google Scholar]
- 57.DeMaster E, Schnitzler N, Cheng Q, Cleary P. 2002. M(+) group A streptococci are phagocytized and killed in whole blood by C5a-activated polymorphonuclear leukocytes. Infect. Immun. 70:350–359. 10.1128/IAI.70.1.350-359.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Matsumura T, Ato M, Ikebe T, Ohnishi M, Watanabe H, Kobayashi K. 2012. Interferon-γ-producing immature myeloid cells confer protection against severe invasive group A Streptococcus infections. Nat. Commun. 3:678. 10.1038/ncomms1677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Navarini AA, Lang KS, Verschoor A, Recher M, Zinkernagel AS, Nizet V, Odermatt B, Hengartner H, Zinkernagel RM. 2009. Innate immune-induced depletion of bone marrow neutrophils aggravates systemic bacterial infections. Proc. Natl. Acad. Sci. U. S. A. 106:7107–7112. 10.1073/pnas.0901162106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aziz RK, Kansal R, Aronow BJ, Taylor WL, Rowe SL, Kubal M, Chhatwal GS, Walker MJ, Kotb M. 2010. Microevolution of group A streptococci in vivo: capturing regulatory networks engaged in sociomicrobiology, niche adaptation, and hypervirulence. PLoS One 5:e9798. 10.1371/journal.pone.0009798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Turner CE, Kurupati P, Jones MD, Edwards RJ, Sriskandan S. 2009. Emerging role of the interleukin-8 cleaving enzyme SpyCEP in clinical Streptococcus pyogenes infection. J. Infect. Dis. 200:555–563. 10.1086/603541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Beres SB, Sylva GL, Barbian KD, Lei B, Hoff JS, Mammarella ND, Liu MY, Smoot JC, Porcella SF, Parkins LD, Campbell DS, Smith TM, McCormick JK, Leung DY, Schlievert PM, Musser JM. 2002. Genome sequence of a serotype M3 strain of group A Streptococcus: phage-encoded toxins, the high-virulence phenotype, and clone emergence. Proc. Natl. Acad. Sci. U. S. A. 99:10078–10083. 10.1073/pnas.152298499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Green NM, Zhang S, Porcella SF, Nagiec MJ, Barbian KD, Beres SB, LeFebvre RB, Musser JM. 2005. Genome sequence of a serotype M28 strain of group A streptococcus: potential new insights into puerperal sepsis and bacterial disease specificity. J. Infect. Dis. 192:760–770. 10.1086/430618 [DOI] [PubMed] [Google Scholar]