

Abstract
Pseudomonas aeruginosa is an opportunistic pathogen that causes acute and chronic infections in humans. Pyocins are bacteriocins produced by P. aeruginosa that are usually released through lysis of the producer strains. Expression of pyocin genes is negatively regulated by PrtR, which gets cleaved under SOS response, leading to upregulation of pyocin synthetic genes. Previously, we demonstrated that PrtR is required for the expression of type III secretion system (T3SS), which is an important virulence component of P. aeruginosa. In this study, we demonstrate that mutation in prtR results in reduced bacterial colonization in a mouse acute pneumonia model. Examination of bacterial and host cells in the bronchoalveolar lavage fluids from infected mice revealed that expression of PrtR is induced by reactive oxygen species (ROS) released by neutrophils. We further demonstrate that treatment with hydrogen peroxide or ciprofloxacin, known to induce the SOS response and pyocin production, resulted in an elevated PrtR mRNA level. Overexpression of PrtR by a tac promoter repressed the endogenous prtR promoter activity, and electrophoretic mobility shift assay revealed that PrtR binds to its own promoter, suggesting an autorepressive mechanism of regulation. A high level of PrtR expressed from a plasmid resulted in increased T3SS gene expression during infection and higher resistance against ciprofloxacin. Overall, our results suggest that the autorepression of PrtR contributes to the maintenance of a relatively stable level of PrtR, which is permissive to T3SS gene expression in the presence of ROS while increasing bacterial tolerance to stresses, such as ciprofloxacin, by limiting pyocin production.
INTRODUCTION
To establish colonization and persistence, pathogenic bacteria must counteract the host immune attacks and often the toxicity of antibiotics, as well as competition from other bacterial species. Bacteriocins are antimicrobial compounds produced by almost all bacteria and usually released under environmental stresses (1). The role of bacteriocins is believed to benefit the producers in competition against other closely related strains. Bacteriocins are considered a double-edge sword, as they kill susceptible competitor microbes on one hand, while on the other hand the production of bacteriocins is energetically costly and their release is usually through lysis of the producers (1). Accordingly, production of bacteriocins is under tight regulation (2).
Pseudomonas aeruginosa is a versatile opportunistic pathogen that causes acute and chronic infections in humans (3). The bacteriocins produced by P. aeruginosa are designated pyocins. More than 90% of P. aeruginosa strains are able to produce several types of pyocins (4). Three types of pyocins have been identified based on their structures and mode of actions: R type, F type, and S type (4). Pyocins are believed to play important roles in intraspecies as well as interspecies interactions. Coculturing of a pyocin producer strain with a sensitive strain revealed that high levels of pyocins are produced in an anaerobic biofilm, resulting in dominance of the producer strain (5). Pyocins produced by isolates from cystic fibrosis patients are able to kill Burkholderia cepacia complex (Bcc) strains, isolated either from the same or from different patients, suggesting an active interaction between P. aeruginosa and Bcc within patient lungs (6).
Production of pyocins is under the control of a tight regulatory cascade. PrtR protein, a homologue of λCI, binds to the promoter region of a prtN gene and inhibits its expression (7). PrtN is a transcriptional activator that activates the expression of pyocin biosynthetic genes. Upon activation of the SOS response, RecA promotes PrtR cleavage, leading to the derepression of PrtN and subsequent upregulation of pyocin synthetic genes (7). Hydrogen peroxide (H2O2) and ciprofloxacin have been demonstrated to induce the expression of pyocin genes (8, 9), presumably by causing DNA damage and inhibiting the activity of DNA topoisomerase, respectively leading to the SOS response and cleavage of the PrtR protein. Since production and release of pyocins are metabolically costly and usually lethal to the producer cells, PrtR plays a pivotal role in guarding bacterial survival in response to various environmental stresses. Although the regulation and biological roles of pyocins have been widely studied, the regulatory mechanism of PrtR expression and its role in P. aeruginosa colonization and chronic persistence remain elusive.
A number of observations suggest that PrtR might play a role in P. aeruginosa infections. First, during host infection, P. aeruginosa encounters reactive oxygen species (ROS) generated by neutrophils and usually fluoroquinolones, which are widely used in the treatment of P. aeruginosa infections (10, 11). Therefore, it is likely that PrtR is subjected to cleavage under such stress conditions. Second, we had previously demonstrated that PrtR is required for the expression of P. aeruginosa type III secretion system (T3SS) genes by repressing the expression of PtrB, a specific inhibitor of the T3SS (12). The T3SS plays an important role in the killing of neutrophils and is thus essential for survival and replication of the bacteria within the host environments (13–15).
In this study, we investigated the role of PrtR in a mouse acute pneumonia model and the expression pattern of prtR in vivo. We demonstrate here that PrtR plays a significant role in the bacterial colonization of mice. During infection, expression of PrtR is induced by reactive oxygen species generated by neutrophils. We further demonstrate that the PrtR protein is maintained at a relatively stable level through an autoregulatory mechanism that permits T3SS gene activation in a host environment while conferring resistance against DNA-damaging reagents. Therefore, we have identified a mechanism employed by P. aeruginosa in controlling both virulence and antibiotic resistance.
MATERIALS AND METHODS
Bacterial strains and plasmids.
The bacterial strains and plasmids used in this study are listed in Table 1. Bacteria were grown in L broth (10 g of tryptone, 5 g of yeast extract, and 5 g of NaCl per liter, pH 7.0) or L agar (L broth containing 1.5% [wt/vol] agar) under aerobic conditions at 37°C. Antibiotics were used at the following concentrations: for P. aeruginosa, streptomycin at 200 μg/ml, tetracycline at 100 μg/ml, gentamicin at 150 μg/ml, carbenicillin at 150 μg/ml, neomycin at 100 μg/ml, and spectinomycin at 200 μg/ml; for Escherichia coli, ampicillin at 100 μg/ml, spectinomycin at 50 μg/ml, streptomycin at 25 μg/ml, gentamicin at 10 μg/ml, and tetracycline at 10 μg/ml.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant characteristics or function | Reference or origin |
|---|---|---|
| P. aeruginosa | ||
| PAK | Wild type | David Bradley |
| PAK ΔprtNprtR::Gm | PAK with prtN and prtR disrupted by replacement of Gm cassette; Gmr | 12 |
| PAK ΔprtN::Gm | PAK with prtN disrupted by insertion of Gm cassette; Gmr | 12 |
| PAK ΔprtNprtR/Ptac-exsA | PAK ΔprtNprtR with insertion of a single copy of ExsA driven by tac promoter at attTn7 sites | This study |
| Plasmids | ||
| pUCP19 | Shuttle vector between E. coli and P. aeruginosa | 42 |
| pFlp2 | Containing the recombinase structural gene flp and SacB selection marker; Apr | 43 |
| pUC18T-mini-Tn7T-Gm | For gene insertion in chromosome; Gmr | 44 |
| pTNS3 | Helper plasmid | 45 |
| pE80 | prtR gene of PAK on pUCP19 driven by lac promoter; Apr | 12 |
| pE100 | pMMB67EH-His-SuhB | 46 |
| pE112 | ExsA-FLAG-CTC | 46 |
| pE122 | prtR promoter of PAK fused to promoterless lacZ on pDN19lacZΩ(prtR-1-lacZ); Spr Smr Tcr | This study |
| pE209 | pMMB67EH-His-PrtR | This study |
| pE434 | prtR promoter of PAK fused to promoterless lacZ on pDN19lacZΩ(prtR-2-lacZ); Spr Smr Tcr | This study |
| pE435 | prtR promoter of PAK fused to promoterless lacZ on pDN19lacZΩ(prtR-3-lacZ); Spr Smr Tcr | This study |
| pE707 | exsA gene of PAK on pUC18T-Mini-Tn7T-Gm driven by a tac promoter; Gmr | This study |
| pE727 | prtR gene of PAK on pUC18T-Mini-Tn7T-Gm with full length of the promoter; Gmr | This study |
| pE734 | prtR gene of PAK on pUC18T-Mini-Tn7T-Gm with 91-bp upstream regions of prtR ORF; Gmr | This study |
| pE735 | prtR gene of PAK on pUC18T-Mini-Tn7T-Gm with 58-bp upstream regions of prtR ORF; Gmr | This study |
Plasmid constructions.
For DNA manipulations, the standard protocols or the manufacturers' instructions of commercial products were followed. Various lengths of the prtR promoter were amplified from wild-type (WT) P. aeruginosa strain PAK chromosomal DNA (see Table S1 in the supplemental material) and cloned into EcoRI-BamHI sites of pDN19lacZΩ, resulting in pE122, pE434, and pE435. The prtR coding region was isolated as an NdeI-HindIII fragment and cloned into NdeI-HindIII sites of pE100, resulting in pE209, where the prtR gene is under the control of a tac promoter and fused with a His tag on the N terminus. The fragment of Ptac-ExsA was excised from pE112 and subcloned into BamHI-ApaI sites of pUC18T-mini-Tn7T-Gm, resulting in pE707. The prtR open reading frame (ORF) and various lengths of upstream regions were amplified from PAK genomic DNA. To construct C-terminal His-tagged PrtR, a His tag-coding sequence was included in the primer annealing to the C terminus of prtR gene (see Table S1 in the supplemental material). The PCR products were cloned into the SacI-HindIII sites of pUC18T-mini-Tn7T-Gm, resulting in pE727, pE734, and pE735.
Mouse acute pneumonia model.
All animal experiments complied with Nankai University and national guidelines regarding the use of animals in research. Bacteria were grown in L broth overnight and then subcultured into fresh medium and grown at 37°C with aeration to an optical density at 600 nm (OD600) of 1.0. Bacteria were centrifuged and adjusted to 2.5 × 1010 CFU/ml in phosphate-buffered saline (PBS). The exact number of bacteria in each inoculum was determined by serial dilution and plating. Female BALB/c mice (6 to 8 weeks old) were anesthetized with an intraperitoneal injection of 7.5% chloral hydrate (100 μl per mouse). Anesthetized mice were intranasally inoculated with 10 μl of bacterial suspension in each nostril, giving a total infection volume of 20 μl. Bacterial colonization in the lung was determined as previously described (16). Briefly, 16 h postinfection, mice were sacrificed by inhalation of CO2. Lungs were isolated and homogenized in 1% proteose peptone, and bacterial numbers were determined by serial dilution and plating.
CI assay.
The competitive index (CI) assay was performed as previously described (17) with minor modifications. Wild-type PAK and the ptrR::Tn mutant were grown to an OD600 of 1.0. The same number of cells for each bacterial strain were mixed. The bacterial mixture was centrifuged and adjusted to 2.5 × 1010 CFU/ml in PBS. For the in vivo competition assay, 20 μl bacterial suspension was inoculated into each mouse. And for the in vitro competition assay, the bacterial suspension was diluted 50-fold with fresh LB and cultured at 37°C with aeration (200 rpm). Sixteen hours later, the infected mice were sacrificed and lungs were isolated and homogenized in 1% proteose peptone. To determine the numbers of cells of the PAK strain and the ptrR::Tn mutant, the lung homogenates or in vitro-grown bacteria were diluted and plated on LB agar plates in the presence or absence of 100 μg/ml neomycin. Since only the ptrR::Tn mutant can grow in the presence of neomycin, the number of PAK cells can be calculated by subtracting colony numbers on neomycin plates from the colony numbers on plain LB plates. The competitive index (CI) was calculated as follows: CI = (mutant output/WT output)/(mutant input/WT input) (18).
Western blotting.
P. aeruginosa strains were grown in L broth overnight at 37°C and then diluted 20- or 50-fold in fresh LB with or without 5 mM EGTA, respectively, and cultured for 3 h. Supernatant and pellet were separated by centrifugation and mixed with sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer. SDS-PAGE (12% polyacrylamide) gels were used to separate samples from equivalent numbers of bacterial cells. The proteins were transferred onto a polyvinylidene difluoride membrane and probed with rabbit polyclonal antibody against ExoS. For detection of PrtR-His, bacteria grown in LB with or without ciprofloxacin treatment were collected and directly lysed with 2× loading buffer. Samples from equal numbers of bacteria were loaded onto SDS-PAGE. The PrtR-His proteins were detected with anti-His antibody (Tianjin Sungene Biotech). Signals were detected with the ECL-plus kit (Millipore).
RNA extraction and quantitative real-time PCR.
To examine bacterial gene expression levels during infection, mice were sacrificed by CO2 at various time points postinfection. Bronchoalveolar lavage fluid (BALF) was obtained by cannulation of the trachea followed by two instillations of 1 ml sterile PBS with 0.5 mM EDTA (16). Two hundred microliters of the BALF was used for bacterial counting, while the remaining BALF was centrifuged and the pellets were immediately resuspended in 200 μl TRIzol reagent (Invitrogen). Total RNA was isolated as instructed by the manufacturer and further purified with an RNA cleanup kit (Tiangen Biotech). For in vitro-grown bacteria, overnight cultures of bacterial cells were diluted 50-fold into fresh LB medium and grown to an OD600 of 1.0. Total RNA was isolated with an RNeasy Minikit (Tiangen Biotech).
cDNA was synthesized by a PrimeScript Reverse Transcriptase (TaKaRa) with random primers. cDNA was mixed with 5 pmol of forward and reverse primers (see Table S1 in the supplemental material) and iQ SYBR green Supermix (Bio-Rad). Quantitative real-time PCR was conducted using a CFX Connect Real-Time system (Bio-Rad). The 30S ribosomal protein gene rpsL was used as an internal control.
Cell culture and HL-60 cell differentiation.
A549 and Beas-2B cells were maintained in Dulbecco's modified Eagle medium (DMEM), and HL-60 cells were maintained in RPMI 1640. The media were supplemented with 10% (vol/vol) heat-inactivated fetal bovine serum (hiFBS), penicillin G (100 U/ml), and streptomycin (100 μg/ml) at 37°C in 5% (vol/vol) CO2. The HL-60 cell differentiation was performed as previously described (19). Briefly, HL-60 cells were diluted to no more than 4.5 × 105 cells per ml, followed by treatment with 1.3% dimethyl sulfoxide (Sigma) for a period of 6 to 9 days.
Measurement of ROS.
ROS production was measured as previously described (20) with slight modification. A total of 1.5 × 104 A549 cells or 1.3 × 104 Beas-2B cells were seeded in each well of a luminometer plate in DMEM with 10% (vol/vol) FBS. Before the assay, cells were washed once with warm Hank's balanced salt solution (HBSS). Then, 200 μl HBSS containing 100 μM luminol and 5 units of horseradish peroxidase (Sigma) was added to each well. For differentiated neutrophils, 1.5 × 104 cells in 200 μl of HBSS containing 100 μM luminol and 5 units of horseradish peroxidase were added to each well. Cells were incubated for 10 min at 37°C, followed by stimulation with P. aeruginosa strain PAK at a multiplicity of infection (MOI) of 30. Phorbol myristate acetate (PMA) (100 ng/ml) was used as a positive control to stimulate ROS production by differentiated HL-60 cells (19). ROS production was monitored every 2 to 5 min for 2 h with a luminometer (Varioskan Flash, Thermo Scientific).
Overexpression and purification of PrtR.
Plasmid pE209 with His-PrtR driven by a tac promoter was transformed into wild-type PAK. Expression of the His-PrtR fusion protein was induced by 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 4 h, and 200 ml of the bacterial culture was centrifuged at 5,000 × g for 10 min. The bacterial pellet was resuspended in 5 ml lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole [pH 8.0]) and subjected to sonication on ice. After centrifugation at 13,000 × g for 10 min, the pellet was washed twice with inclusion body washing buffer (50 mM Tris-HCl [pH 8.0], 0.5 M NaCl, 2 M urea) and then dissolved in inclusion body dissolving buffer (20 mM Tris-HCl [pH 8.0], 0.5 M NaCl, 8 M urea) for 8 h. The denatured fusion protein was purified with a metal affinity chromatography column (Qiagen). To refold PrtR, 4 ml purified protein in a dialysis bag (molecular weight cutoff [MWCO], 10,000) was immersed in 200 ml PBS at 4°C. The solution was replaced with fresh PBS every 8 or 12 h, for a total of 6 times.
EMSA.
The electrophoretic mobility shift assays (EMSA) were performed as previously described (21). Briefly, the 40-bp complementary single-stranded oligonucleotides Binding 1 and Binding 2 or Nbinding 1 and Nbinding 2 (see Table S1 in the supplemental material), respectively, were annealed together. PrtR was added to the DNA fragments at the indicated amounts (0.5 pmol DNA fragments, 1 pmol PrtR) and incubated for 10 min at room temperature in a complex buffer (50 mM Tris-HCl [pH 7.5], 20 mM NaCl, 1 mM EDTA, and 1 mM dithiothreitol) in a total volume of 10 μl. Reaction mixtures were subjected to electrophoresis on a 10% polyacrylamide gel in Tris-borate-EDTA (TBE) buffer. Motility shift was observed by ethidium bromide staining of the DNA.
Biofilm resistance against ciprofloxacin.
Bacteria were cultivated in 96-well culture plates for biofilm formation as previously described (22). The resistance of biofilms against ciprofloxacin was measured as previously described with minor modification (23, 24). Briefly, overnight cultures were diluted to an OD600 of 0.025 with fresh LB medium. The diluted cultures (150 μl) were transferred to each well of 96-well plates and incubated for 24 h at 37°C. After removing planktonic bacteria, biofilms were treated for 60 min with 150 μl LB medium containing 0.625 μg/ml ciprofloxacin. Wells were washed twice with double-distilled water (ddH2O), and biofilms were stained with 0.1% crystal violet for 10 min, followed by two washes with ddH2O. For quantification, the biofilm-associated dye was dissolved in 70% ethanol and OD590 was measured. Bacterial cell numbers in the biofilm were determined as previously described with minor modifications (25). Briefly, after the biofilm was treated with ciprofloxacin, the medium in each well was removed. Two hundred microliters of fresh LB medium was added to each well, and the 96-well plate was subjected to sonication at a frequency of 40 kHz, with a power output of 300 W, at 37°C for 5 min. After sonication, the exact number of surviving bacteria in the biofilm of each well was determined by serial dilution and plating.
RESULTS
Role of PrtR in P. aeruginosa colonization.
To evaluate the role of PrtR in pathogenesis, we utilized a mouse acute pneumonia model. Since the growth rate of the prtR::Tn5 mutant in our previous study (12) was much lower than that of the wild-type strain (presumably due to the hyperproduction of pyocins), in our experiments we initially used the ΔprtNprtR::Gm and ΔprtN::Gm mutant strains, whose growth rates were comparable to that of the wild-type strain. Six- to 8-week-old female BALB/c mice were infected with 5 × 108 bacteria intranasally. Sixteen hours postinfection (hpi), lungs were isolated and homogenized. Bacterial loads were determined by serial dilution and plate counting. Compared to the wild-type strain, the population of the ΔprtNprtR::Gm strain was significantly lower, whereas mutation in prtN alone did not affect the colonization (Fig. 1A). These results suggested that PrtR but not PrtN is involved in the colonization of P. aeruginosa. To further confirm the role of PrtR in P. aeruginosa pathogenesis, we compared the competitive index (CI) of the prtR::Tn mutant with that of wild-type PAK under in vivo infection and in vitro culture conditions. When cultured in vitro, the CI of the prtR::Tn mutant against PAK was approximately 0.5, whereas it dropped to 0.16 in mouse lung infection (Fig. 1B), suggesting a defect of the prtR::Tn mutant in lung colonization.
FIG 1.

Role of PrtR in a mouse acute pneumonia model. (A) Role of PrtR in the colonization of P. aeruginosa. Female BALB/c mice (6 to 8 weeks old) were intranasally inoculated with 5 × 108 bacterial cells of wild-type PAK or its isogenic ΔprtN::Gm, ΔprtNprtR::Gm, or ΔprtNprtR::Gm mutant containing exsA driven by a tac promoter (ΔprtNR::Gm/Ptac-exsA). Mice were sacrificed 16 h postinfection (hpi). Lungs were isolated and homogenized. Bacterial loads were determined by serial dilution and plating. Bars represent medians, and error bars represent standard errors of the means (SEM). Significance by the Mann-Whitney test: *, P < 0.05; **, P < 0.01. (B) In vivo and in vitro competitive index (CI) between the prtR::Tn mutant and wild-type PAK. The two strains were mixed 1:1 and used for infection in a mouse acute pneumonia model (eight mice) or coculture in vitro. The competitive index was calculated as follows: CI = (mutant output/WT output)/(mutant input/WT input). **, P < 0.01, by Student's t test. (C) Cellular and secreted ExoS in PAK or ΔprtN::Gm, ΔprtNprtR::Gm, and ΔprtNR::Gm/Ptac-ExsA mutant strains. Overnight bacterial cultures were diluted to 2% in LB or 5% in LB plus 5 mM EGTA and grown at 37°C for 3 h. Supernatants and pellets from equivalent bacterial cell numbers were loaded, separated by SDS-PAGE gels, and immunoblotted with anti-ExoS antibody. ExoS is indicated by arrows. ΔprtNR::Gm represents ΔprtNprtR::Gm.
Since PrtR is required for the expression of T3SS genes (12), we examined whether the attenuated virulence in the ΔprtNprtR::Gm mutant was due to a defect in the T3SS. In the acute pneumonia model, bronchoalveolar lavage fluids (BALFs) were collected from mice infected with wild-type strain PAK at various time points. Total RNAs from the BALFs were isolated, and the mRNA levels of T3SS genes exoS, exsC, pcrV, and pscC as well as the T3SS repressor gene ptrB were determined by real-time PCR. The 30S ribosomal protein gene rpsL was used as an internal standard, and all reported changes were normalized to the levels of RpsL RNA. Compared to the wild-type strain, expression of these T3SS genes was lower in the ΔprtNprtR::Gm strain, whereas the prtB level was higher at indicated time points (Table 2). Overexpression of ExsA in the ΔprtNprtR::Gm strain significantly increased the expression and secretion of ExoS in vitro (Fig. 1C). Furthermore, colonization of the ΔprtNprtR::Gm strain was restored by overexpression of the exsA gene (Fig. 1A). These results suggested that a defect in T3SS of the ΔprtNprtR::Gm mutant plays a major role in the attenuation of virulence. As for the prtR::Tn mutant, a combination of slow growth (presumably due to overproduction of pyocins) and defect of T3SS (12) might contribute to the attenuation in lung colonization.
TABLE 2.
Expression of T3SS genes and ptrB during infection
| Gene ID | Gene name | Fold change (±SD) in PAK ΔprtNprtR::Gma |
|||
|---|---|---|---|---|---|
| 3 hpi | 6 hpi | 9 hpi | 13 hpi | ||
| PA0612 | ptrB | 235.7 ± 11.55 | 163.14 ± 19.15 | 110.28 ± 1.08 | 113 ± 2.77 |
| PA1706 | pcrV | −6.57 ± 1.09 | −2.85 ± 0.27 | −1.08 ± 0.03 | −1.34 ± 0.03 |
| PA1710 | exsC | −1.75 ± 0.008 | −2.2 ± 0.15 | −1.27 ± 0.1 | −1.33 ± 0.06 |
| PA1716 | pscC | −2.75 ± 0.013 | −1.5 ± 0.04 | −1.34 ± 0.18 | −1.27 ± 0.006 |
| PA3841 | exoS | −10.45 ± 0.46 | −3.8 ± 0.13 | −2.19 ± 0.09 | −1.75 ± 0.08 |
Mice infected with wild-type PAK or the ΔprtNprtR::Gm mutant were sacrificed at the indicated time points. BALFs were collected, and total RNAs were extracted. mRNA levels were determined by real-time PCR. The values are relative mRNA levels compared to those in wild-type PAK at the same time point. All reported changes are normalized to the levels of RpsL RNA.
Inducing signals for prtR expression.
To further understand the role of PrtR in pathogenesis, we examined the expression levels of prtR during infection. BALFs from wild-type-PAK-infected mice were used to isolate total RNA, and the mRNA levels of prtR were examined by real-time PCR. We observed a slight increase of the prtR mRNA level at 3 hpi, followed by a drastic increase at 6 hpi (Fig. 2). At 9 and 13 hpi, the prtR mRNA level dropped to a level similar to that in the BALFs isolated immediately after bacterial inoculation (Fig. 2). These results suggest that at 6 hpi the host environment provided a strong inducing signal for the expression of PrtR.
FIG 2.
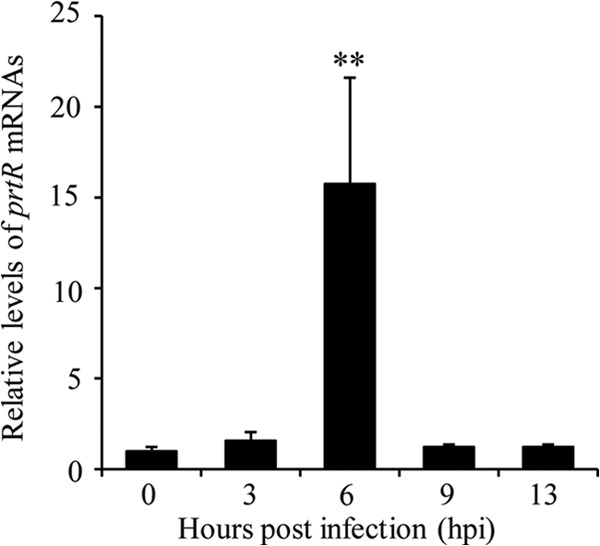
Expression of PrtR during mouse lung infection. The BALFs from three wild-type-PAK-infected mice were collected at the indicated time points. The BALF at 0 hpi was collected immediately after bacterial inoculation. Total RNA was isolated from the BALF, and the expression levels of prtR were determined by real-time PCR. The 30S ribosomal protein gene rpsL was used as an internal standard, and all reported changes are normalized to the levels of RpsL RNA. **, P < 0.01 compared to the other time points by Student's t test.
To identify the host signals that induce the expression of PrtR, we analyzed the host cell in the BALFs. At 3 and 6 hpi, increasing numbers of cells were observed in the BALFs, whereas fewer cells were found in the BALFs immediately after bacterial infection (Fig. 3A). Wright staining revealed that the majority of the cells in the BALFs from infected mice were neutrophils (Fig. 3B), which is consistent with previous observations (26–29). Compared to what was seen at 6 hpi, there were much-more-severe hemorrhages in the lungs at 9 and 13 hpi. Although similar numbers of neutrophils were observed in the BALFs isolated at 6, 9, and 13 hpi (Fig. 3A), the ratio of red blood cells to neutrophils increased from 10:1 at 6 hpi to 1,000:1 at 9 and 13 hpi (data not shown), which is similar to the ratio in the circulating blood (30). In combination, it is likely that the increasing neutrophils might play a role in the induction of PrtR expression, whereas the influx of red blood cells or other environmental changes at later time points might lead to decreased PrtR expression.
FIG 3.

Signals that induce PrtR expression. (A) Numbers of host cells (means ± SEM) in the BALFs at indicated time points. **, P < 0.01 by the Mann-Whitney test. (B) Cells in the BALFs stained with Wright's stain. The neutrophil percentage is 76.6% ± 9.91% (average ± standard deviation), as counted based on cell morphology of samples from 3 infected mice. (C) A549 cells, Beas-2B cells, HL-60 cells, and differentiated HL-60 (dHL-60) cells were infected with PAK (MOI = 30). After 1 h of incubation, total RNA was extracted. The mRNA levels of PrtR were determined by real-time PCR. All reported changes are normalized to the levels of RpsL RNA. White bars (0 h) indicate RNA that was extracted immediately after bacteria were mixed with cells. (D) Luminometry of ROS production by A549, Beas-2B, HL-60, differentiated HL-60 cells infected with PAK (MOI = 30), and differentiated HL-60 cells treated with PMA. RU, relative units.
To test whether neutrophils play a role in the induction of PrtR expression, we incubated wild-type PAK cells with neutrophils (differentiated from HL-60 cells) and human alveolar basal epithelial cells (A549) as well as bronchial epithelial cells (Beas-2B), which are the cell types that bacteria are most likely to encounter during lung infection. In contrast to epithelial cells, neutrophils induced upregulation of PrtR (Fig. 3C), suggesting that neutrophils provide a major inducing signal for the PrtR expression.
Neutrophils are immune cells that generate a large amount of reactive oxygen species (ROS) to kill invading microorganisms (27). Compared to the epithelial cells, differentiated HL-60 cells generated drastically larger amounts of ROS upon P. aeruginosa infection (Fig. 3D). Undifferentiated HL-60 cells, which produced minimal levels of ROS (Fig. 3D), did not induce PrtR expression (Fig. 3C). In addition, treatment of wild-type PAK cells with H2O2 resulted in upregulation of PrtR (Table 3). These results suggested that ROS generated by neutrophils during infection play an important role in induction of PrtR expression.
TABLE 3.
Comparison of expression changes for various pyocin-related genes as analyzed by relative real-time PCR
| Gene ID | Gene name | Fold change in PAK strain |
|
|---|---|---|---|
| H2O2 | CIPa | ||
| PA0610 | prtN | 156 | 81.9 |
| PA0611 | prtR | 25.5 | 28.0 |
| PA0613 | 117.4 | 673.3 | |
| PA0614 | 77.8 | 541.2 | |
| PA1769b | 1.0 | 1.1 | |
CIP, ciprofloxacin.
PA1769 was used as a negative control.
Autoregulation of PrtR.
It had been reported that H2O2 treatment upregulates pyocin synthesis genes, suggesting a role of H2O2 in inducing the SOS response and subsequent cleavage of the PrtR (9). Consistent with this, we observed upregulation of prtN and of PA0614 (a pyocin gene) as well as PA0613 (directly regulated by PrtR [12]) in H2O2-treated wild-type PAK cells (Table 3). Treatment with ciprofloxacin, which inhibits DNA synthesis and elicits the SOS response, also induced the expression of prtN, PA0614, and PA0613 as well as prtR (Table 3), again consistent with a previous report (8). These results suggest that cleavage of the PrtR might lead to the upregulation of prtR itself, thus constituting an autoregulatory mechanism.
Since PrtR functions as a repressor, it is possible that PrtR directly represses its own expression or that the transcription of PrtR is activated by PrtN, whose expression is controlled by PrtR. To test these possibilities, we constructed a prtR-1-lacZ transcriptional fusion containing a 505-bp fragment upstream of the prtR coding region. In the ΔprtNR::Gm mutant, the expression of prtR-1-lacZ was higher than that in the wild-type strain or the ΔprtN::Gm mutant (Fig. 4A). Furthermore, overexpression of PrtR in these strains drastically inhibited the expression of prtR-1-lacZ (Fig. 4A). These results suggest that PrtR is likely to directly repress its own expression.
FIG 4.

Autoregulation of PrtR. (A) Expression of prtR-1-lacZ in PAK and in ΔprtNprtR::Gm and ΔprtN::Gm mutants containing empty vector or PrtR-overexpressing plasmid. **, P < 0.01, compared to PAK or ΔprtN::Gm mutant by one-way analysis of variance (ANOVA) test. (B) Fragments of the prtR promoter region fused with promoterless lacZ gene or prtR-His fusion. The sequence shown is the putative PrtR binding site. Italic nucleotides represent conserved sequences in the putative PrtR-binding sites in prtR and prtN promoters. (C) Expression of prtR-1-lacZ, prtR-2-lacZ, and prtR-3-lacZ in PAK containing empty vector or prtR overexpression plasmid. *, P < 0.05 compared with prtR-1-lacZ or prtR-2-lacZ in PAK containing empty vector, by one-way ANOVA test; **, P < 0.01, compared with prtR-1-lacZ or prtR-2-lacZ in PAK containing prtR overexpression plasmid, by one-way ANOVA test. (D) Expression of PrtR-His with different lengths of promoter in PAK containing empty vector or prtR overexpression plasmid. (E) EMSA displaying binding of PrtR with the potential PrtR binding site in the promoter of prtR. A 0.5-pmol volume of double-stranded DNA fragment (containing the consensus motif or an altered conserved sequence) was electrophoresed alone (lanes 1 and 3) or after incubation with 1 pmol of purified PrtR protein (lanes 2 and 4).
Previously, we identified a putative PrtR binding sequence between the coding regions of prtR and ptrB genes (12) (Fig. 4B). Since the putative PrtR binding site is adjacent to the prtR coding region, it is likely that PrtR controls its own expression through binding of this site. To test this possibility, we constructed two additional prtR-lacZ transcriptional fusions. In prtR-2-lacZ, a 91-bp fragment upstream of the prtR coding region including the putative PrtR binding site, was fused with the promoterless lacZ gene, whereas in prtR-3-lacZ, the PrtR binding site was excluded (Fig. 4B). In wild-type PAK, prtR-1-lacZ and prtR-2-lacZ had similar levels of lacZ expression. However, removal of the PrtR binding site resulted in significantly higher lacZ expression, as displayed by PAK harboring the prtR-3-lacZ reporter plasmid (Fig. 4C). In addition, overexpression of PrtR significantly reduced the expression of prtR-1-lacZ and prtR-2-lacZ but not that of prtR-3-lacZ (Fig. 4C). Next, we constructed C-terminal His-tagged full-length PrtR fusion proteins (PrtR-His) with various prtR upstream regions as in the lacZ transcriptional fusions (Fig. 4B). Consistent with the lacZ reporter results, exclusion of the PrtR binding site resulted in drastic increase of the PrtR-His protein level (Fig. 4D). Overexpression of PrtR diminished the expression of PrtR-His with promoters containing the PrtR binding site (PprtR-1 and PprtR-2, as in Fig. 4B), but not the construct without the binding site (PprtR-3, as in Fig. 4B) (Fig. 4D). These results suggest that PrtR represses its own expression through the putative PrtR binding site.
To further confirm the autoregulation of PrtR, we expressed and purified the His-PrtR fusion protein from P. aeruginosa and performed an EMSA to assess the binding of PrtR to the putative PrtR binding promoter sequence. As shown in Fig. 4E, in the presence of PrtR protein, the mobility of the DNA fragment with the putative PrtR binding sequence was reduced. Alteration of the conserved sequence shared by PrtR binding sites in prtR and prtN promoters abolished the delay in mobility (Fig. 4E). Overall, these results suggested that PrtR represses its own expression through direct binding to its promoter.
Expression of PrtR under SOS response-inducing environmental stress.
So far, our results indicate that the autoregulation of PrtR contributes to the maintenance of a relatively stable level of PrtR in response to DNA damage. To test the protein levels of PrtR during the SOS response, we utilized wild-type PAK containing the prtR-His fusion protein driven by the native promoter with or without the PrtR binding site (PprtR-1 and PprtR-3, as in Fig. 4B). Since ROS causes damage to proteins and lipids in addition to DNA, we used ciprofloxacin, which specifically targets DNA topoisomerase, to induce the SOS response. When driven by PprtR-1 (containing the PrtR binding site), the PrtR-His level was increased by a small amount of ciprofloxacin (0.5 μg/ml) (Fig. 5A), suggesting a derepression of the promoter. Larger amounts of ciprofloxacin decreased the PrtR-His level, indicating that the synthesis of PrtR was outcompeted by accelerated cleavage of PrtR as the SOS response intensified. However, exclusion of the PrtR binding site (PprtR-3) abolished the induction of PrtR expression. As shown in Fig. 5B, treatment with increasing amounts of ciprofloxacin resulted in a gradual reduction of PrtR-His levels. These results revealed that the autoregulation of PrtR prevents quick depletion of PrtR under the SOS response.
FIG 5.

Protein levels of PrtR-His under ciprofloxacin treatment. Wild-type PAK cells containing PrtR-His driven by the prtR promoter with the PrtR binding site (PprtR-1) (A) or without the binding site (PprtR-3) (B) were treated with indicated concentrations of ciprofloxacin for 1 h. Samples from equal numbers of bacteria were loaded onto SDS-PAGE gels, and levels of PrtR-His were detected with anti-His antibody. Note that due to the low expression level of prtR-His driven by PprtR-1, the exposure time in panel A was much longer than that in panel B.
Overexpression of PrtR increases T3SS gene expression during infection.
Our results above suggested that when P. aeruginosa is exposed to ROS released from neutrophils, the autoregulation of PrtR sustains a stable level of PrtR, thus permitting the expression of T3SS. If this is true, a high level of PrtR should result in increased T3SS expression during infection. To test this hypothesis, we infected mice with wild-type PAK containing empty vector or PrtR overexpressing plasmid pE80. At 6 hpi, expression levels of PrtB and T3SS genes were determined by real-time PCR. Consistent with our prediction, overexpression of PrtR resulted in a reduced ptrB mRNA level and increased T3SS gene expression (Fig. 6). Overall, these results suggest that during infection, the initial degradation of PrtR triggered by the SOS response is replenished by newly synthesized PrtR through the autoregulatory circuit, which contributes to the repression of ptrB, thus allowing the T3SS to respond to the in vivo inducing signals.
FIG 6.
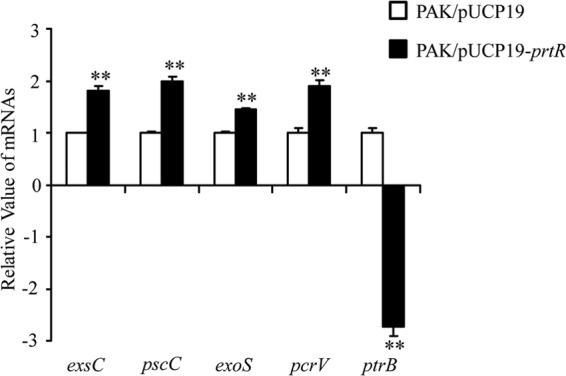
Effect of PrtR overexpression on T3SS genes and ptrB expression during infection. Mice were infected with PAK containing empty vector or PrtR overexpression plasmid. BALFs were isolated at 6 hpi, and RNAs were purified. mRNA levels of T3SS genes and ptrB were quantified by real-time PCR. All reported changes are normalized to the levels of RpsL RNA. **, P < 0.01 compared to PAK containing empty vector by Student's t test.
Role of PrtR in biofilm resistance against ciprofloxacin.
Besides ptrB, PrtR also represses the expression of prtN and the production of pyocins upon DNA damage. Ciprofloxacin, which inhibits DNA synthesis, strongly induces pyocin production (Table 3) (8), thus imposing an additional detrimental effect on the survival of P. aeruginosa. Therefore, it is likely that the autoregulation of prtR prevents a quick depletion of PrtR and subsequent surge of pyocin production during the SOS response, contributing to bacterial survival. To test this possibility, we overexpressed PrtR in the wild-type PAK cells and examined the effect on bacterial survival in the presence of ciprofloxacin. When cells were cultured in LB medium with agitation, we did not observe an obvious increase in the resistance by overexpressing PrtR (data not shown), presumably due to the overwhelming DNA replication stalling caused by ciprofloxacin. Next, we examined whether overexpression of PrtR could increase the resistance of biofilm, inside which sessile bacteria are embedded in an extracellular matrix and the concentration of ciprofloxacin gradually drops. Compared to the wild-type strain carrying empty vector, the PrtR-overexpressing strain formed a similar level of biofilm (Fig. 7A). To examine the resistance of biofilm, planktonic bacteria were removed and the sessile bacteria were treated with ciprofloxacin at a concentration of 4× MIC for 60 min. Biofilms formed by the wild-type strain carrying empty vector were diminished after the treatment, whereas overexpression of PrtR protected the biofilm, as quantified by crystal violet staining (Fig. 7A and B) as well as CFU determination (Fig. 7C). Expression of pyocin synthetic genes in the PrtR-overexpressing strain was lower than that in the wild-type strain upon ciprofloxacin treatment (Fig. 7D). Since PrtR represses pyocin genes through directly inhibiting PrtN expression, overexpression of PrtR had no effect on the resistance of the ΔprtN mutant (Fig. 7A, lower two panels, and Fig. 7B and C). In addition, mutation of prtN or of prtN and prtR increased the bacterial resistance against ciprofloxacin (Fig. 7E), confirming that reduction of pyocin production can result in higher resistance against ciprofloxacin. Overall, these results suggest that the autoregulation of PrtR contributes to the bacterial resistance against ciprofloxacin by repressing pyocin production.
FIG 7.

Role of PrtR in biofilm resistance against ciprofloxacin. PAK cells containing empty vector or PrtR overexpression plasmid were inoculated in wells of a 96-well plate. (A) Crystal violet staining of biofilms. Upper panel, biofilms without ciprofloxacin treatment; lower panel, biofilms treated with ciprofloxacin at a concentration of 4× MIC (0.625 μg/ml) for 60 min. (B) The biofilm-associated dye was quantified by optical density measurement. (C) The biofilms were dissociated from the wells by gentle sonication, and bacterial numbers were quantified by plating. The bacterial survival rates after ciprofloxacin treatment were calculated based on live bacterial numbers in biofilms with or without ciprofloxacin treatment. **, P < 0.01, *, P < 0.05, compared to the values of wild-type PAK containing empty vector by one-way ANOVA test. (D) mRNA levels of indicated genes in biofilms formed by PAK containing empty vector or PrtR overexpression plasmid after ciprofloxacin treatment. All reported changes are normalized to the levels of RpsL RNA. **, P < 0.01 compared to PAK containing empty vector by Student's t test. (E) Survival rates of the biofilms formed by PAK and by ΔprtNprtR::Gm and ΔprtN::Gm mutants after ciprofloxacin treatment. *, P < 0.05 compared to PAK by one-way ANOVA test. CIP, ciprofloxacin.
DISCUSSION
In P. aeruginosa, PrtR represses the production of pyocins by inhibiting the expression of PrtN, an activator of pyocin synthetic genes. In addition, PrtR is required for T3SS gene expression through its repressive role on PtrB. In this study, we investigated the role of PrtR in the pathogenesis of P. aeruginosa in a mouse acute pneumonia model. Mutation in prtN did not affect the colonization, suggesting that pyocins are not essential for pathogenesis in this animal model. However, double mutation of prtN and prtR significantly reduced the bacterial load. And the competitive index between the prtR::Tn mutant and wild-type PAK was significantly lower in lung infection than that in the in vitro growth, suggesting a role of PrtR in colonization. We further demonstrated that the expression of PrtR is induced by ROS released by neutrophils during the early infection. At later time points, the PrtR expression level decreased, accompanied by severe hemorrhage in the lung and a large amount of red blood cells in the BALFs. Previous reports revealed that red blood cells can scavenge H2O2 produced by neutrophils (31) and protect against H2O2-mediated cell or tissue damage (32–35). Thus, it is likely that large amounts of red blood cells might suppress PrtR expression by reducing ROS in the lung environment.
Hydrogen peroxide and ciprofloxacin, known to induce DNA damage and subsequent pyocin gene expression, also upregulated the expression of PrtR. The simultaneous degradation of PrtR protein and upregulation of PrtR during infection suggest an autorepressive mechanism. Overexpression of PrtR indeed reduced the transcription of prtR, further supporting the autorepression of PrtR. In addition, a low level of ciprofloxacin induced upregulation of PrtR driven by its endogenous promoter, whereas exclusion of the PrtR binding site abolished the induction.
Based on these observations, we postulate that shortly after entering the host environment, P. aeruginosa encounters neutrophils that release ROS to kill invading microorganisms. The ROS triggers the bacterial SOS response through DNA damage and eventually results in cleavage of the PrtR. However, this leads to elevated PrtR protein synthesis due to the autorepressive mechanism. Through such a regulatory circuit, PrtR is maintained at a relatively stable level, keeping the expression of PrtB low and thus allowing T3SS genes to respond to in vivo inducing signals. Consistent with this, when PrtR was overexpressed by an exogenous tac promoter, higher T3SS gene expression was observed during infection, suggesting a correlation between PrtR level and T3SS expression. Therefore, the autorepression of PrtR ensures sufficient expression of T3SS when the bacteria are under limited attack by ROS released by neutrophils, so that the bacteria can kill neutrophils to establish colonization successfully.
As an opportunistic pathogen, P. aeruginosa causes infections in immunocompromised patients. It is a major cause of bacteremic pneumonia in neutropenic cancer patients (36). In neutropenic patients, other immune cells can contribute to pathogen clearance. A recent study demonstrated that passive immunization with an O-antigen monoclonal antibody protected neutropenic mice from P. aeruginosa infection (37), suggesting that other cells are involved in antibody-mediated clearance of bacteria. Another study demonstrated that monocytes and macrophages are recruited to the infection site and play a major role in clearance of P. aeruginosa in neutropenic mice (38). Therefore, P. aeruginosa is very likely to encounter monocytes and macrophages in neutropenic mice. Since monocytes and macrophages are able to generate ROS (39), it is likely that PrtR might also play a role in P. aeruginosa infection in neutropenic mice.
Since ciprofloxacin induces PrtR cleavage and subsequent upregulation of pyocin genes (8), the autorepression mechanism of PrtR reduces the detrimental production and release of pyocins, thus ensuring the bacterial survival. In this aspect, the autorepression of PrtR can be regarded as an additional resistance mechanism against antibiotics that induce the SOS response, such as ciprofloxacin.
In this study, we further demonstrated that PrtR achieves autorepression by directly binding to its own promoter region. Interestingly, two PrtR binding sequences have been identified in the prtN promoter region (7, 12), which might function as a double lock, subjecting to much tighter regulation. We suspect that under the SOS response, when PrtR is cleaved, the repression of prtR transcription is relieved first, before the prtN promoter gets derepressed. This way, the bacteria can maintain a stable PrtR level to keep PrtN repressed, blocking or at least delaying pyocin production until bacterial suicide becomes absolutely necessary.
PrtR is a homologue of λ CI, which is a DNA binding protein. On the λ phage genome, there are 6 CI binding sites with three on each side of the cI gene. At high concentration, the CI protein forms tetramers, which bind DNA and shut off the transcription of the cI gene (40, 41). Whether PrtR forms polymers to bind DNA and shuts off target gene transcription is not known at present.
In conclusion, our data suggest that the autoregulation of PrtR contributes to a relative stable level of PrtR under mild stress environments, which plays a dual role in P. aeruginosa infection. First, it sustains the activation of T3SS, which is essential for the bacterial colonization. Second, it limits pyocin production, which increases bacterial survival under stress conditions, such as in the presence of ROS or ciprofloxacin.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Science Foundation of China (31170128 and 31370168), National Basic Research Program of China (973 Program, 2012CB518700), Science and Technology Committee of Tianjin (13JCYBJC36700), Ph.D. Programs Foundation of Ministry of Education of China (20120031120019), and a startup fund from Nankai University.
Footnotes
Published ahead of print 3 February 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01388-13.
REFERENCES
- 1.Riley MA, Wertz JE. 2002. Bacteriocins: evolution, ecology, and application. Annu. Rev. Microbiol. 56:117–137. 10.1146/annurev.micro.56.012302.161024 [DOI] [PubMed] [Google Scholar]
- 2.Zgur-Bertok D. 2012. Regulating colicin synthesis to cope with stress and lethality of colicin production. Biochem. Soc. Trans. 40:1507–1511. 10.1042/BST20120184 [DOI] [PubMed] [Google Scholar]
- 3.Driscoll JA, Brody SL, Kollef MH. 2007. The epidemiology, pathogenesis and treatment of Pseudomonas aeruginosa infections. Drugs 67:351–368. 10.2165/00003495-200767030-00003 [DOI] [PubMed] [Google Scholar]
- 4.Michel-Briand Y, Baysse C. 2002. The pyocins of Pseudomonas aeruginosa. Biochimie 84:499–510. 10.1016/S0300-9084(02)01422-0 [DOI] [PubMed] [Google Scholar]
- 5.Waite RD, Curtis MA. 2009. Pseudomonas aeruginosa PAO1 pyocin production affects population dynamics within mixed-culture biofilms. J. Bacteriol. 191:1349–1354. 10.1128/JB.01458-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bakkal S, Robinson SM, Ordonez CL, Waltz DA, Riley MA. 2010. Role of bacteriocins in mediating interactions of bacterial isolates taken from cystic fibrosis patients. Microbiology 156:2058–2067. 10.1099/mic.0.036848-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matsui H, Sano Y, Ishihara H, Shinomiya T. 1993. Regulation of pyocin genes in Pseudomonas aeruginosa by positive (prtN) and negative (prtR) regulatory genes. J. Bacteriol. 175:1257–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brazas MD, Hancock RE. 2005. Ciprofloxacin induction of a susceptibility determinant in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 49:3222–3227. 10.1128/AAC.49.8.3222-3227.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang W, Small DA, Toghrol F, Bentley WE. 2005. Microarray analysis of Pseudomonas aeruginosa reveals induction of pyocin genes in response to hydrogen peroxide. BMC Genomics 6:115. 10.1186/1471-2164-6-115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kolpen M, Hansen CR, Bjarnsholt T, Moser C, Christensen LD, van Gennip M, Ciofu O, Mandsberg L, Kharazmi A, Doring G, Givskov M, Hoiby N, Jensen PO. 2010. Polymorphonuclear leucocytes consume oxygen in sputum from chronic Pseudomonas aeruginosa pneumonia in cystic fibrosis. Thorax 65:57–62. 10.1136/thx.2009.114512 [DOI] [PubMed] [Google Scholar]
- 11.Tamma PD, Cosgrove SE, Maragakis LL. 2012. Combination therapy for treatment of infections with gram-negative bacteria. Clin. Microbiol. Rev. 25:450–470. 10.1128/CMR.05041-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu W, Jin S. 2005. PtrB of Pseudomonas aeruginosa suppresses the type III secretion system under the stress of DNA damage. J. Bacteriol. 187:6058–6068. 10.1128/JB.187.17.6058-6068.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diaz MH, Shaver CM, King JD, Musunuri S, Kazzaz JA, Hauser AR. 2008. Pseudomonas aeruginosa induces localized immunosuppression during pneumonia. Infect. Immun. 76:4414–4421. 10.1128/IAI.00012-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diaz MH, Hauser AR. 2010. Pseudomonas aeruginosa cytotoxin ExoU is injected into phagocytic cells during acute pneumonia. Infect. Immun. 78:1447–1456. 10.1128/IAI.01134-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Howell HA, Logan LK, Hauser AR. 2013. Type III secretion of ExoU is critical during early Pseudomonas aeruginosa pneumonia. mBio 4:e00032–13. 10.1128/mBio.00032-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu W, Huang J, Duan B, Traficante DC, Hong H, Risech M, Lory S, Priebe GP. 2012. Th17-stimulating protein vaccines confer protection against Pseudomonas aeruginosa pneumonia. Am. J. Respir. Crit. Care Med. 186:420–427. 10.1164/rccm.201202-0182OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lane MC, Lockatell V, Monterosso G, Lamphier D, Weinert J, Hebel JR, Johnson DE, Mobley HL. 2005. Role of motility in the colonization of uropathogenic Escherichia coli in the urinary tract. Infect. Immun. 73:7644–7656. 10.1128/IAI.73.11.7644-7656.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Champion OL, Cooper IA, James SL, Ford D, Karlyshev A, Wren BW, Duffield M, Oyston PC, Titball RW. 2009. Galleria mellonella as an alternative infection model for Yersinia pseudotuberculosis. Microbiology 155:1516–1522. 10.1099/mic.0.026823-0 [DOI] [PubMed] [Google Scholar]
- 19.Chen A, Seifert HS. 2011. Neisseria gonorrhoeae-mediated inhibition of apoptotic signalling in polymorphonuclear leukocytes. Infect. Immun. 79:4447–4458. 10.1128/IAI.01267-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu W, Hsu YM, Bi L, Songyang Z, Lin X. 2009. CARD9 facilitates microbe-elicited production of reactive oxygen species by regulating the LyGDI-Rac1 complex. Nat. Immunol. 10:1208–1214. 10.1038/ni.1788 [DOI] [PubMed] [Google Scholar]
- 21.Bertram R, Rigali S, Wood N, Lulko AT, Kuipers OP, Titgemeyer F. 2011. Regulon of the N-acetylglucosamine utilization regulator NagR in Bacillus subtilis. J. Bacteriol. 193:3525–3536. 10.1128/JB.00264-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brencic A, McFarland KA, McManus HR, Castang S, Mogno I, Dove SL, Lory S. 2009. The GacS/GacA signal transduction system of Pseudomonas aeruginosa acts exclusively through its control over the transcription of the RsmY and RsmZ regulatory small RNAs. Mol. Microbiol. 73:434–445. 10.1111/j.1365-2958.2009.06782.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liao J, Schurr MJ, Sauer K. 2013. The MerR-like regulator BrlR confers biofilm tolerance by activating multidrug efflux pumps in Pseudomonas aeruginosa biofilms. J. Bacteriol. 195:3352–3363. 10.1128/JB.00318-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Billings N, Millan M, Caldara M, Rusconi R, Tarasova Y, Stocker R, Ribbeck K. 2013. The extracellular matrix component Psl provides fast-acting antibiotic defense in Pseudomonas aeruginosa biofilms. PLoS Pathog. 9:e1003526. 10.1371/journal.ppat.1003526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bjerkan G, Witso E, Bergh K. 2009. Sonication is superior to scraping for retrieval of bacteria in biofilm on titanium and steel surfaces in vitro. Acta Orthop. 80:245–250. 10.3109/17453670902947457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Priebe GP, Walsh RL, Cederroth TA, Kamei A, Coutinho-Sledge YS, Goldberg JB, Pier GB. 2008. IL-17 is a critical component of vaccine-induced protection against lung infection by lipopolysaccharide-heterologous strains of Pseudomonas aeruginosa. J. Immunol. 181:4965–4975 http://www.jimmunol.org/content/181/7/4965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Craig A, Mai J, Cai S, Jeyaseelan S. 2009. Neutrophil recruitment to the lungs during bacterial pneumonia. Infect. Immun. 77:568–575. 10.1128/IAI.00832-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dubin PJ, Martz A, Eisenstatt JR, Fox MD, Logar A, Kolls JK. 2012. Interleukin-23-mediated inflammation in Pseudomonas aeruginosa pulmonary infection. Infect. Immun. 80:398–409. 10.1128/IAI.05821-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Power MR, Peng Y, Maydanski E, Marshall JS, Lin TJ. 2004. The development of early host response to Pseudomonas aeruginosa lung infection is critically dependent on myeloid differentiation factor 88 in mice. J. Biol. Chem. 279:49315–49322. 10.1074/jbc.M402111200 [DOI] [PubMed] [Google Scholar]
- 30.Aoshiba K, Nakajima Y, Yasui S, Tamaoki J, Nagai A. 1999. Red blood cells inhibit apoptosis of human neutrophils. Blood 93:4006–4010 [PubMed] [Google Scholar]
- 31.Winterbourn CC, Stern A. 1987. Human red cells scavenge extracellular hydrogen peroxide and inhibit formation of hypochlorous acid and hydroxyl radical. J. Clin. Invest. 80:1486–1491. 10.1172/JCI113230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Asbeck BS, Hoidal J, Vercellotti GM, Schwartz BA, Moldow CF, Jacob HS. 1985. Protection against lethal hyperoxia by tracheal insufflation of erythrocytes: role of red cell glutathione. Science 227:756–759. 10.1126/science.2982213 [DOI] [PubMed] [Google Scholar]
- 33.Toth KM, Clifford DP, Berger EM, White CW, Repine JE. 1984. Intact human erythrocytes prevent hydrogen peroxide-mediated damage to isolated perfused rat lungs and cultured bovine pulmonary artery endothelial cells. J. Clin. Invest. 74:292–295. 10.1172/JCI111414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Agar NS, Sadrzadeh SM, Hallaway PE, Eaton JW. 1986. Erythrocyte catalase. A somatic oxidant defense? J. Clin. Invest. 77:319–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown JM, Grosso MA, Terada LS, Beehler CJ, Toth KM, Whitman GJ, Harken AH, Repine JE. 1989. Erythrocytes decrease myocardial hydrogen peroxide levels and reperfusion injury. Am. J. Physiol. 256:H584–588 [DOI] [PubMed] [Google Scholar]
- 36.Carratala J, Roson B, Fernandez-Sevilla A, Alcaide F, Gudiol F. 1998. Bacteremic pneumonia in neutropenic patients with cancer: causes, empirical antibiotic therapy, and outcome. Arch. Intern. Med. 158:868–872. 10.1001/archinte.158.8.868 [DOI] [PubMed] [Google Scholar]
- 37.Secher T, Fas S, Fauconnier L, Mathieu M, Rutschi O, Ryffel B, Rudolf M. 2013. The anti-Pseudomonas aeruginosa antibody Panobacumab is efficacious on acute pneumonia in neutropenic mice and has additive effects with meropenem. PLoS One 8:e73396. 10.1371/journal.pone.0073396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kamei A, Wu W, Traficante DC, Koh AY, Van Rooijen N, Pier GB, Priebe GP. 2013. Collaboration between macrophages and vaccine-induced CD4+ T cells confers protection against lethal Pseudomonas aeruginosa pneumonia during neutropenia. J. Infect. Dis. 207:39–49. 10.1093/infdis/jis657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wink DA, Hines HB, Cheng RY, Switzer CH, Flores-Santana W, Vitek MP, Ridnour LA, Colton CA. 2011. Nitric oxide and redox mechanisms in the immune response. J. Leukoc. Biol. 89:873–891. 10.1189/jlb.1010550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stayrook S, Jaru-Ampornpan P, Ni J, Hochschild A, Lewis M. 2008. Crystal structure of the lambda repressor and a model for pairwise cooperative operator binding. Nature 452:1022–1025. 10.1038/nature06831 [DOI] [PubMed] [Google Scholar]
- 41.Hochschild A, Lewis M. 2009. The bacteriophage lambda CI protein finds an asymmetric solution. Curr. Opin. Struct. Biol. 19:79–86. 10.1016/j.sbi.2008.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olsen RH, DeBusscher G, McCombie WR. 1982. Development of broad-host-range vectors and gene banks: self-cloning of the Pseudomonas aeruginosa PAO chromosome. J. Bacteriol. 150:60–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86. 10.1016/S0378-1119(98)00130-9 [DOI] [PubMed] [Google Scholar]
- 44.Choi KH, Schweizer HP. 2006. mini-Tn7 insertion in bacteria with single attTn7 sites: example Pseudomonas aeruginosa. Nat. Protoc. 1:153–161. 10.1038/nprot.2006.24 [DOI] [PubMed] [Google Scholar]
- 45.Choi KH, Gaynor JB, White KG, Lopez C, Bosio CM, Karkhoff-Schweizer RR, Schweizer HP. 2005. A Tn7-based broad-range bacterial cloning and expression system. Nat. Methods 2:443–448. 10.1038/nmeth765 [DOI] [PubMed] [Google Scholar]
- 46.Li K, Xu C, Jin Y, Sun Z, Liu C, Shi J, Chen G, Chen R, Jin S, Wu W. 2013. SuhB is a regulator of multiple virulence genes and essential for pathogenesis of Pseudomonas aeruginosa. mBio 4:e00419–13. 10.1128/mBio.00419-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.