

Abstract
Pseudomallei group Burkholderia species are facultative intracellular parasites that spread efficiently from cell to cell by a mechanism involving the fusion of adjacent cell membranes. Intercellular fusion requires the function of the cluster 5 type VI secretion system (T6SS-5) and its associated valine-glycine repeat protein, VgrG5. Here we show that VgrG5 alleles are conserved and functionally interchangeable between Burkholderia pseudomallei and its relatives B. mallei, B. oklahomensis, and B. thailandensis. We also demonstrate that the integrity of the VgrG5 C-terminal domain is required for fusogenic activity, and we identify sequence motifs, including two hydrophobic segments, that are important for fusion. Mutagenesis and secretion experiments using B. pseudomallei strains engineered to express T6SS-5 in vitro show that the VgrG5 C-terminal domain is dispensable for T6SS-mediated secretion of Hcp5, demonstrating that the ability of VgrG5 to mediate membrane fusion can be uncoupled from its essential role in type VI secretion. We propose a model in which a unique fusogenic activity at the C terminus of VgrG5 facilitates intercellular spread by B. pseudomallei and related species following injection across the plasma membranes of infected cells.
INTRODUCTION
Burkholderia pseudomallei is the causative agent of melioidosis, a serious and often fatal human infection. In Southeast Asia and Australia, where the endemicity of the organism is high, infections are nearly always acquired from the environment (1). Adaptations to avoid predation in the rhizosphere are thought to promote accidental virulence in mammals (2–4). The geographic distribution of B. pseudomallei overlaps with that of the closely related species B. thailandensis (5). Although B. thailandensis is considered relatively nonpathogenic, it has occasionally been associated with human infection, and high inocula can cause disease in mice (6–8). B. mallei, an obligate pathogen of equine hosts that causes glanders, is a clonal descendant of B. pseudomallei that has undergone considerable genome decay, losing the capacity for survival in the environment (3, 9).
Despite their divergent niches and host predilections, B. pseudomallei, B. thailandensis, and B. mallei are facultative intracellular pathogens that exhibit nearly identical intracellular life cycles (10). Following entry into phagocytic or nonphagocytic cells, bacteria escape from vesicles using the Bsa type III secretion system (T3SSBsa), replicate in the cytoplasm, and spread from cell to cell by a mechanism that is facilitated by cytoplasmic motility (11, 12). Movement through the cytoplasm promotes contact with cell membranes and can be provided by BimA-mediated actin polymerization or, for B. thailandensis and Australian isolates of B. pseudomallei, Fla2 flagellar motility (12). Prevailing models for cell-cell spread have invoked protrusion-mediated entry into adjacent cells followed by escape from double membrane vacuoles (2, 3). Direct evidence in support of this mechanism is lacking, however, and the demonstration that cell-cell spread is independent of T3SSBsa following escape from the primary vesicle of the initially infected cell raises doubts about its validity (12). In contrast, abundant evidence shows that Burkholderia species form multinucleated giant cells (MNGCs) during infection (12–15), and we propose that this represents the primary mechanism of cell-cell spread. In B. pseudomallei and B. thailandensis, membrane fusion leading to MNGC formation requires the activity of a type VI secretion system (T6SS) (12, 16).
T6SSs are evolutionarily related to the contractile injection devices of tailed bacteriophages and mediate interactions between Gram-negative bacteria and other prokaryotic or eukaryotic cells (17). T6SSs expressed by pathogenic bacteria are able to translocate proteins or protein domains across the plasma or endosomal membranes of eukaryotic cells (17–20). Although the mechanism of secretion and translocation is still poorly resolved, Basler and colleagues recently demonstrated that a Vibrio cholerae T6SS cycles between assembly, quick contraction, ClpV ATPase-mediated disassembly, and reassembly, supporting a mechanism where contraction of the T6SS sheath provides the energy needed for protein translocation (21). A similar model was described for Pseudomonas aeruginosa, suggesting that this mechanism is conserved (22).
B. pseudomallei genomes encode six T6SS gene clusters, but only cluster 5 (T6SS-5; also known as T6SS-1 [23]) has consistently been shown to be critical for intercellular spread and virulence in animals (16, 24). T6SS-5 is also required for virulence by B. mallei and B. thailandensis (15, 25). Approximately 15 core genes and a variable number of nonconserved accessory elements encode the T6SS “injectisome” (26, 27). Among the core genes are hcp and vgrG, which encode conserved components that play key roles. Hcp forms hexameric rings that polymerize into tubules that are extruded following contraction of the surrounding sheath, facilitating protein translocation across cell surfaces into the extracellular space or across the membranes of target cells (28, 29). Current models suggest that the tip of the apparatus is composed of a VgrG trimer, which functions as part of a puncturing device (19, 30–32). Homology and structural evidence indicate that VgrGs possess a “core motif” that is similar to the neck (gp27) and tail spike (gp5) proteins of bacteriophage T4 (19). In addition, some VgrGs possess extended carboxyl-terminal domains (CTD) that confer effector functions upon injection into eukaryotic cells. The activities of these so-called “evolved” VgrGs include actin cross-linking and the inhibition of phagocytosis by V. cholerae VgrG (19, 20, 33) and ADP ribosylation of actin by Aeromonas hydrophila VgrG1 (34). In this study, we demonstrate that the evolved VgrG5 proteins expressed by Burkholderia species capable of intercellular spread are conserved and functionally interchangeable. We also show that the VgrG5 CTD plays an essential role in membrane fusion that is distinct and separable from the required role of VgrG5 in type VI secretion.
MATERIALS AND METHODS
Bacterial strains and mutant construction.
B. thailandensis E264 (35) and Bp340 [B. pseudomallei 1026b Δ(amrRAB-oprA)] (36) were routinely grown in LB medium without NaCl (LB-NS). Kanamycin or Zeocin was added at a final concentration of 100 μg/ml as needed. In-frame mutations were constructed using allelic exchange with a chlorophenylalanine sensitivity allele, PheS*, as a negative-selection marker on M9 agar containing 0.1% chlorophenylalanine (cPhe) as described previously (37). Gene loci targeted for mutation in B. thailandensis E264 included clpV5 (Bth_II0864), vgrG5 (Bth_II0863), and bimA (Bth_II0875), and vgrG5 (Bp1026b_1596) was mutated in Bp340. Strains constitutively expressing VirA and VirG were constructed by insertion of a mini-Tn7 transposon containing virAG genes from Bp340 (Bp1026b_1587 to -89) downstream from the S12 ribosomal subunit promoter (38) and were selected for by the presence of the Zeocin resistance cassette encoded by the transposon.
Plasmid construction.
Complementation of mutants with partially deleted or full-length VgrG5 was performed using a derivative of the broad-host-range plasmid pBBR1-MCS2 containing the nptII kanamycin resistance gene (39) and a hemagglutinin (HA) tag cloned between the XbaI-SacI sites. The following vgrG5 homologs were amplified from Pseudomallei group Burkholderia species by PCR using the primers listed in Table 1: vgrG5Bp340 (primers 118 and 117), vgrG5HABp340 (primers 118 and 119), HAvgrG5Bp340 (primers 185 and 117), vgrG5Bp-C997 (primers 118 and 153), vgrG5Bp-C850 (primers 118 and 149), vgrG5Bp-C758 (primers 118 and 146), vgrG5Bp-C644 (primers 118 and 147), vgrG5Bp-Δ651-696 (primers 272 and 274 and primers 275 and 273), vgrG5Bp-Δ783-824 (primers 272 and 276 and primers 277 and 273), vgrG5Bp-Δ852-866 (primers 272 and 278 and primers 279 and 273), vgrG-5BtE264 (primers 151 and 117), B. mallei vgrG5 (vgrG5Bm) (primers 150 and 117), B. oklahomensis vgrG5 (vgrG5Bo) (primers 224 and 225), and vgrG5 of the Australian B. pseudomallei strain MSHR668 (vgrG5Bp Aus) (primers 150 and 117). Internal deletions were obtained after amplification, cloning, and ligation of the regions framing the sequence to be eliminated.
TABLE 1.
Oligonucleotide primers used in this study
| Primer | Sequence (5′–3′)a |
|---|---|
| 117 | GTAATCTCTAGATCAGCCGAGCTGGATCAGTTGGCCGTC |
| 118 | GGAGTTCCATATGTCTTCGTCCCCCCGCC |
| 119 | ATGCGCTCTAGAGCCGAGCTGGATCAGTTGGC |
| 145 | GACTGATCTAGATCATGCGCTGCCGCAGCCGGCCGCGATC |
| 146 | GTACTATCTAGATCACGCGTGCGTCGACCACGAGACGC |
| 147 | GATACATCTAGATCAAGCTTGTCGAGCTGCTTTTGCAG |
| 149 | GTTCAGTCTAGATCACGTGGGGATGAGCGCGCTGAC |
| 150 | GGAGTTCCATATGCCTTCGTCCCCCCGCCACG |
| 151 | GGAGTTCCATATGTCTTCGTCCCATCGACACTACG |
| 153 | GACTGATCTAGATCACAGCGACGCGCTGCCGTTCAGC |
| 185 | GGAGTTCCATATGGCGTACCCGTACGACGTGCCGGACTACGCGTCTTCGTCCCCCCGCC |
| 224 | GGAGTTCCATATGTCTTCGTCCCACCCACAG |
| 225 | GTAATCTCTAGATCAGCCGAGCTGGATCAGTTGG |
| 272 | GAATCTCATATGTCTTCGTCCCCCCGCCACG |
| 273 | GTAATCTCTAGATCAGCCGAGCTGGATCAGTTGGCCG |
| 274 | CTAGCAGCTAGCCTTGCGCACCGTGCCCTTGAGC |
| 275 | CTAGTCGCTAGCGTCAGCACGGCGCTGATGGCGAC |
| 276 | CTAGCTGCTAGCGTGCTTCGCCGCTTCGACGCCC |
| 277 | ATCGTTGCTAGCCTCGAAGCCGACATTTCGGAAAAC |
| 278 | TACGAAGCTAGCCGGCGTGGGGATGAGCGCGCTGAC |
| 279 | TACGTTGCTAGCCTCGTCGAGCTGAAGGCCAAGCAGA |
Stop/start codons are italicized; restriction sites are underlined; the HA tag sequence is shown in boldface.
Assays for the formation of multinucleated giant cells.
HEK293 cell lines that stably express enhanced green fluorescent protein (eGFP) or a monomeric strawberry red fluorescent protein (msRFP) were infected with B. pseudomallei or B. thailandensis derivatives as described previously (12). Briefly, GFP- and RFP expressing cells were mixed at a 1:1 ratio and were seeded at a final concentration of 1.8 × 106 per well precoated with a 1/40 dilution of liquid Matrigel (Becton, Dickinson). Cells were infected at a multiplicity of infection (MOI) of 1 × 10−3, and 1 h postinfection, cells were washed, and remaining extracellular bacteria were killed by adding gentamicin (100 μg/ml). After 18 h, cells were fixed with phosphate-buffered saline (PBS) plus 10% formalin, and MNGCs were observed by fluorescence microscopy. The number of MNGCs formed per bacterial CFU was determined, and values are reported as the means ± standard deviations (SD) for a minimum of 3 independent experiments.
Actin staining.
HEK293 cells were grown on coverslips in 12-well plates at 2 × 105 cells/well and were infected with B. thailandensis E264 or its ΔvgrG5 or ΔbimA mutant at an MOI of 10. After 1 h of infection, extracellular bacteria were killed by the addition of gentamicin (100 μg/ml), and cells were prepared for microscopy as described previously (12). Antibodies were used at the following dilutions: rabbit anti-B. thailandensis antiserum, 1:1,000; Alexa Fluor 488-labeled phalloidin and Alexa Fluor 633-labeled goat anti-rabbit antiserum (Molecular Probes), 1:200. Permanent mounts of specimens were created using ProLong Gold with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen). Fluorescence imaging was performed using a Leica SP5-II-AOBS confocal microscope setup. Images were processed with the LAS-AF software suite (Leica) or with Adobe Photoshop CS5.
Intracellular replication assays.
Cells growing in 6- or 12-well plates were infected with Bp340 or its derivatives at an MOI of 10:1. One hour after infection, cells were washed with Hanks' balanced salts, and the remaining extracellular bacteria were killed by the addition of 100 μg/ml gentamicin. At the times postinfection indicated in Fig. 4A, cells were washed with Hanks' balanced salts, harvested by trypsinization, and lysed with 0.2% Triton X-100 plus 20 mM MgSO4 and 50 μg/ml DNase I (to reduce lysate viscosity). The numbers of CFU per well were determined for serial dilutions of the lysate and were normalized with respect to the growth surface area (CFU/cm2). The results are expressed as means ± SD for 3 independent experiments.
FIG 4.
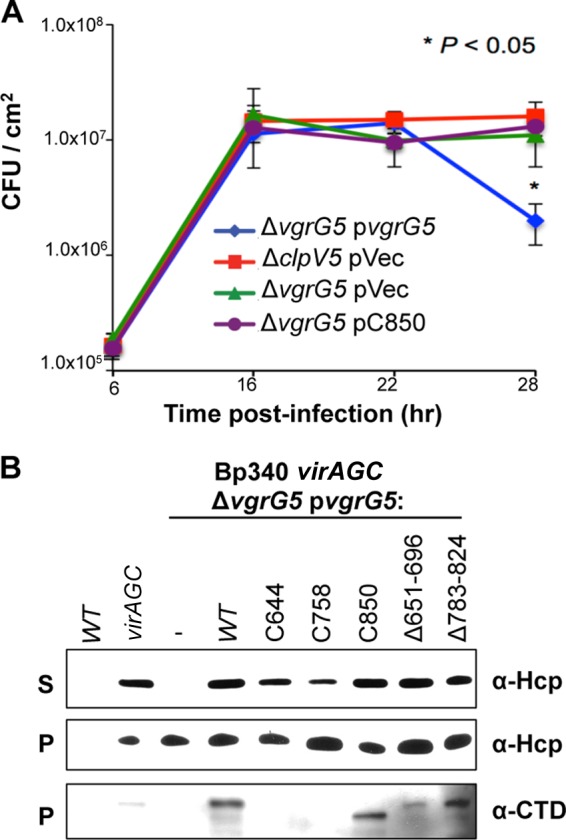
Differential roles of the VgrG5 CTD in T6SS-associated membrane damage and T6SS-5 activity. (A) Assays of intracellular replication of Bp340 ΔclpV5 or Bp340 ΔvgrG5 complemented with an empty plasmid vector (pVec) or with plasmid-expressed vgrG5 or vgrG5-C850. Values are means ± SD for 3 independent experiments (*, P < 0.05). (B) Western blot analysis of bacterial supernatant (S) or pellet (P) fractions from Bp340, Bp340 virAG(Con), Bp340 virAG(Con) ΔvgrG5, or Bp340 virAG(Con) ΔvgrG5 expressing WT or mutant vgrG5 from a replicating plasmid. Blots were probed with an antibody against Hcp5 (α-Hcp) or against the VgrG5 CTD (α-CTD). virAG(Con), virAG constitutively expressed. Although T6SS apparatuses contain numerous Hcp monomers, which are released upon disengagement, they are predicted to include only one VgrG trimer, at the tip (30). VgrG5 derivatives that retain the α-CTD antibody epitope (Fig. 3A) can be detected in cell pellets but are not exported in sufficient quantities to be reproducibly detected in the supernatant fractions of broth-grown cells.
In vitro type VI secretion assays.
Overnight cultures of bacteria were diluted 200-fold in LB-NS containing antibiotics when necessary. At an optical density at 600 nm (OD600) of 0.5 to 0.7, 1-ml aliquots were centrifuged, washed, and resuspended in 400 μl of Laemmli buffer (pellet fractions). The culture supernatants were again centrifuged at 4,750 × g for 15 min, and the supernatants were filtered through a 0.2-μm syringe-tip filter and were precipitated with 10% trichloroacetic acid (TCA). After overnight incubation at 4°C, the supernatants were centrifuged at 18,900 × g for 15 min at 4°C, and the pellets were washed with 1 ml acetone and were resuspended in Laemmli buffer (supernatant fractions). The pellet and supernatant fractions were normalized according to the OD600 of the bacterial culture at the time of harvest. Pellet or supernatant samples were loaded onto a 4-to-15% TGX Tris-glycine protein gel (Bio-Rad), subjected to SDS-PAGE, and transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were blocked with PBS–5% skim milk for 1 h, incubated with a primary antibody (either rat anti-Bp Hcp5 or rabbit anti-VgrG5 CTD) at 1:3,000 for 1 h, and washed 3 times with PBS–1% skim milk for 10 min. Membranes were incubated with a secondary antibody (horseradish peroxidase-labeled IgG conjugate; Amersham) diluted 1:2,000 in PBS–1% skim milk, which was added for 30 min, and were then washed 3 times in PBS for 10 min each time.
RESULTS
VgrG5 is required for cell fusion and intercellular spread.
We have reported recently that mutation of the clpV5 ATPase allele located in T6SS gene cluster 5 significantly reduces the efficiency of cell fusion and the formation of MNGCs following invasion by B. pseudomallei or B. thailandensis, or following the direct injection of B. thailandensis into the cytoplasm using a photothermal nanoblade (12). To investigate whether vgrG5 is similarly required for cell fusion, we constructed in-frame deletions of the vgrG5 coding sequences in B. pseudomallei Bp340 and B. thailandensis E264. MNGC formation assays were conducted by infecting 1:1 mixtures of HEK293 cells transduced with lentiviral vectors expressing msRFP or eGFP, where regions of fused cells appear yellow on image overlays. As shown in Fig. 1A and B, Bp340 and B. thailandensis E264 ΔvgrG5 mutants were unable to induce MNGCs at 18 h postinfection, in contrast to the wild-type (WT) parental strains. This phenotype was fully reversible by complementation with cognate vgrG5 alleles expressed from a replicating plasmid in trans. These effects were quantified by measuring MNGC formation per CFU. This is equivalent to PFU per CFU, since we have shown previously that Burkholderia plaques on cell monolayers bathed in antibiotics result from the expansion and lysis of MNGCs (12). As expected, ΔvgrG5 mutants escape from endosomes and polymerize actin in a BimA-dependent manner (Fig. 1C). Together, these results show that VgrG5 is required for cell fusion and subsequent plaque formation but is dispensable for endosome escape and actin polymerization.
FIG 1.
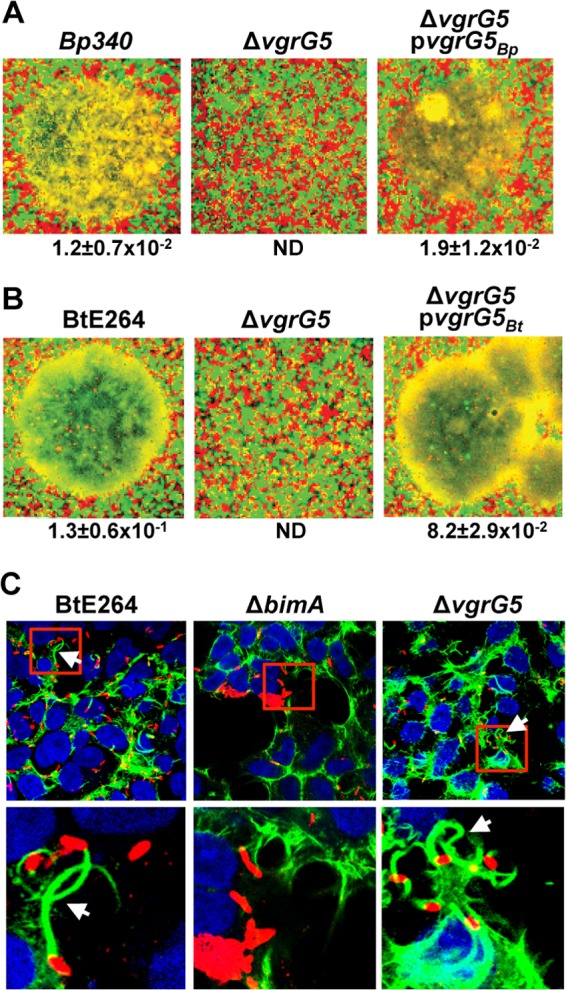
VgrG5 is required for membrane fusion. (A and B) MNGCs were visualized 18 h after infection of HEK293-eGFP or -msRFP cells mixed in 1:1 ratios with WT, ΔvgrG5 mutant, or complemented ΔvgrG5 mutant (carrying a cognate vgrG5 allele expressed in trans on a replicating plasmid) Bp340 (A) or B. thailandensis E264 (B). Green and red, individual cells; yellow, fused MNGCs. Representative MNGCs are shown for WT and complemented strains. No MNGCs were observed in multiple fields for ΔvgrG5 mutants. The numbers of MNGCs per bacterial CFU are reported below the images as the means ± SD for a minimum of 3 independent experiments. ND, not detected (<1 × 10−4 MNGC/CFU). (C) Immunofluorescence microscopy showing actin tails (arrows) formed by B. thailandensis E264 or its ΔbimA or ΔvgrG5 derivative. Blue, DAPI-stained nuclei;, red, immunostained bacteria; green, polymerized actin stained with Alexa Fluor 488-labeled phalloidin. Images in the lower panel are close-ups of areas boxed in red in the upper panel.
VgrG5 is conserved and functionally interchangeable between Burkholderia pseudomallei and near-neighbor species.
Sequence comparisons with VgrG5 encoded by the B. pseudomallei reference genome from strain 1026b (40), the parent of Bp340 (36), indicates high levels of conservation with orthologs in other Pseudomallei group Burkholderia species and strains (Fig. 2A). VgrG5 sequences from Australian (MSHR668) and Southeast Asian (1026b) strains, which represent the two major clades of the species B. pseudomallei (41), and from B. mallei are nearly identical (≥99% amino acid [aa] identity) but diverge from those of B. thailandensis and B. oklahomensis, which share 90% and 82% identity, respectively, with the 1026b sequence. Although little is known regarding virulence mechanisms, B. oklahomensis has been isolated from soil and human wound infections and is minimally pathogenic in animal models (42–44). The next closest VgrG5 relative within sequenced members of the Burkholderia genus is from Burkholderia ambifaria, a member of the Burkholderia cepacia complex (45), with 31% overall sequence identity and 46% similarity. The phylogenetic tree in Fig. 2B supports the close evolutionary relationships between VgrG5 proteins encoded by Pseudomallei group Burkholderia species and B. oklahomensis. The 640-aa N-terminal domain (NTD) of VgrG5 homologs contains the VgrG “core elements,” which resemble the gp27 and gp5 tail spike proteins of bacteriophage T4. In contrast, the 367-aa CTD (aa 641 to 1007) is uniquely present in VgrG5 homologs (see below).
FIG 2.

vgrG5 alleles from Pseudomallei group Burkholderia species are functionally interchangeable. (A) Alignment of VgrG proteins by BLAST. Gray bars, amino-terminal VgrG “core” region with homology to the gp27/gp5 T4 bacteriophage tail spike complex proteins; red bars, conserved CTD; striped bars, protein regions with no similarities. The boundary of the CTD was assigned based on the alignment of the Burkholderia VgrG5 sequence to the sequences of VgrG proteins in other species. The percentage of identity with the B. pseudomallei 1026b VgrG5 coding sequence is given for each sequence diagramed here. Bp, B. pseudomallei; Bm, B. mallei; Bt, B. thailandensis; Bo, B. oklahomensis. (B) Phylogenetic tree based on comparisons of sequences of VgrG5 proteins from an expanded set of Burkholderia isolates and of VgrG and VgrG1 proteins from V. cholerae and P. aeruginosa, respectively. Analysis was based on the JTT matrix-based model (62) and was conducted in MEGA5 (63). (C) MNGC assays. HEK293 cells were infected with a Bp340 ΔvgrG5 mutant complemented with a plasmid expressing vgrG5 from the Australian B. pseudomallei strain MSHR668 (Bp Aus), B. mallei ATCC 23344, B. thailandensis E264, or B. oklahomensis C6786. (D and E) Numbers of MNGCs per CFU 18 h after infection of HEK293 cells with B. pseudomallei Bp340 (D) or B. thailandensis E264 (E) containing ΔvgrG5 deletions and complementing plasmids as indicated. No fusion activity was observed in ΔvgrG5 strains containing the empty vector. Values are means ± SD for 3 independent experiments (*, P < 0.05).
In light of their high degree of conservation, we investigated whether vgrG5 alleles from different Burkholderia species could cross-complement the cell-cell spread defect of a B. pseudomallei vgrG5 mutant. vgrG5 loci from B. mallei ATCC 23344, B. pseudomallei MSHR668, B. oklahomensis C6786, or B. thailandensis E264 were expressed on a plasmid vector in Bp340 ΔvgrG5. All vgrG5-encoded orthologs were able to restore cell fusion capability and MNGC formation (Fig. 2C and D). Complementation of the Bp340 ΔvgrG5 mutation with homologous loci from other species was remarkably efficient, with no statistically significant difference in the efficiency of MNGC formation between these loci and the cognate Bp340 vgrG5 allele. Analogous results were observed in complementation experiments with a B. thailandensis E264 ΔvgrG5 strain (Fig. 2E). These results demonstrate that vgrG5 alleles are functionally equivalent among Burkholderia strains that share the ability to form MNGCs, supporting a conserved role for VgrG5 proteins in cell-cell fusion. This is intriguing considering the highly divergent ecological niches occupied by these species, which range from the rhizosphere to obligate mammalian hosts. Our results also suggest that B. thailandensis, a biosafety level 2 (BSL2) organism, and B. mallei or B. pseudomallei, which are tier 1 BSL3 pathogens, can be used interchangeably to study VgrG5-mediated membrane fusion.
The VgrG5 C terminus is critical for cell fusion.
In experiments focused on examining the expression of VgrG5, we noticed that strains with epitope tags at the N terminus retained function while derivatives tagged at the C terminus were incapable of MNGC formation. Considering the unique nature of the VgrG5 CTD and its conservation between Burkholderia species, we tested the hypothesis that it plays a role in membrane fusion. In the experiment presented in Fig. 3, a series of truncated vgrG5 derivatives were expressed in Bp340 ΔvgrG5 and were tested in complementation assays. None of the truncation mutants were able to restore MNGC formation, indicating the loss of intercellular spread (Fig. 3A and B). The distal C terminus of VgrG5 appears to be particularly sensitive to perturbation, since the removal of only 10 aa, or the addition of a triple HA tag (50 aa), abolishes function.
FIG 3.

Mutational analysis of the VgrG5 CTD. (A) Bp340 vgrG5 and derivatives with truncation and deletion mutations. “HA” stands for a C-terminal HA epitope tag. The abilities of plasmids carrying wild-type or mutant vgrG5 alleles to complement MNGC formation by Bp340 ΔvgrG5 are shown on the right. TM, transmembrane domain; yellow box, sequence similarity to the enzymatic domain of E. coli hydroxymyristoyl glucosamine-N-transferase; Ab, epitope for antibody against the VgrG5 CTD. (B) Numbers of MNGCs per CFU obtained 18 h after infection of HEK293 cells with Bp340, Bp340 ΔvgrG5, or Bp340 ΔvgrG5 mutants complemented with a wild-type or mutant vgrG5 allele expressed on a replicating plasmid in trans. No fusion activity was observed in ΔvgrG5 strains containing the empty vector. Values are means ± SD for 3 independent experiments. *, P < 0.05. (C) Alignment of VgrG5 C-terminal domains of B. mallei ATCC 23344 (Bm), B. thailandensis E264 (Bt), and B. oklahomensis C6786 (Bo) to that of B. pseudomallei 1026b (Bp) and bioinformatic predictions of internal features. Cons., consensus. Red letters indicate amino acid identity; residues shown in blue/black are nonidentical but generally conserved. Potential regions likely to exhibit TM topology are highlighted in blue. The TM1 and TM2 regions were highly predicted using the TMHMM or TMpred algorithm (92% or 88% confidence, respectively). The TM3 region was identified only by TMpred, with a lower probability (24%). A domain with moderate similarity (30% confidence; 15% aa identity) to E. coli hydroxymyristoyl glucosamine-N-transferase (Enz.) (yellow highlight) was identified using the Phyre2 homology recognition engine. The epitope used to raise polyclonal antisera to the VgrG5 C-terminal domain is highlighted in pink.
In contrast to the N-terminal sequences of VgrG5, where conserved elements are present, the CTD is relatively devoid of obvious similarities with characterized functional domains in other bacterial, archeal, or eukaryotic proteins, including membrane fusogens. Secondary structure and fold recognition algorithms were used to identify potential structural motifs in the VgrG5 CTD. Since fusogenic proteins often include transmembrane (TM) helices, we used the transmembrane helix prediction platforms TMpred (46), TMHMM (47), and MEMSAT (48) to identify three TM regions at positions 651 to 671 (TM1), 685 to 703 (TM2), and 850 to 867 (TM3) (Fig. 3A and C). TM1 and TM2 are separated by a short (13-aa) hydrophilic linker, and both are highly predicted TM helices. TM3, which includes several prolines, was predicted with lower confidence. Using the Phyre2 fold recognition algorithm (49), sequences from position 785 to 833 were found to exhibit moderate similarity to the enzymatic domain of Escherichia coli hydroxymyristoyl glucosamine-N-transferase. Since the putative TM and transferase sequences are highly conserved among Pseudomallei group species (Fig. 3C), mutant vgrG5 alleles lacking these regions were constructed and were tested for their abilities to complement MNGC formation in Bp340 ΔvgrG5. As shown in Fig. 3A and B, deletion of the TM1-2 region (aa 651 to 696) or the predicted enzymatic domain completely or partially eliminated MNGC formation, respectively, whereas deletion of TM3 (aa 851 to 866) had no effect. This finding indicates that, in addition to the extreme C-terminal end of VgrG5, internal sequences that include potential TM regions are required for cell fusion.
The VgrG5 C-terminal domain is required for T6SS-mediated cell death.
Using intracellular growth assays with cell monolayers bathed in antibiotics, we have shown previously that B. thailandensis ΔclpV5 mutants replicate to higher numbers than the WT parental strain (12). We interpreted these results as suggesting that T6SS-5 activity is associated with membrane damage and leakage of antibiotics into the cytosol, possibly due to abortive attempts at membrane fusion. To further address this hypothesis, we examined the roles of VgrG5 and its CTD in this phenotype by using B. pseudomallei.
Replication assays were performed using Bp340 ΔvgrG5 derivatives complemented with either full-length vgrG5, a mutant containing a 157-aa C-terminal truncation that eliminates fusogenic activity (vgrG5-C850 [Fig. 3A]), or the empty plasmid vector. HEK293 cells were first infected and then overlaid with gentamicin, and the number of intracellular bacteria was determined over a 28-h time course. As shown in Fig. 4A, all strains behaved identically up to 22 h, after which levels of the ΔvgrG5 strain complemented with WT vgrG5 begin to decrease due to exposure to extracellular antibiotics. The growth kinetics of the ΔclpV5 and ΔvgrG5 strains were indistinguishable, and by 28 h, these T6SS-5 mutant strains reached intracellular numbers that were nearly an order of magnitude higher that those of the vgrG5-complemented strain. Importantly, the replication levels of the ΔvgrG5 strain complemented with the vgrG5-C850 allele were indistinguishable from those of the empty-vector control. Although the differential susceptibilities of WT and mutant VgrG5-complemented bacteria to antibiotics at later time points are consistent with “collateral damage” to cell membranes mediated by VgrG5, it might also be explained by an inherent instability of MNGCs. Regardless, these results demonstrate that the ability to fuse cell membranes correlates with the accelerated decline in bacterial numbers observed in intracellular growth assays, which, in turn, correlates with the presence of an intact VgrG5 CTD.
The role of the VgrG5 CTD in membrane fusion can be uncoupled from the role of VgrG5 in T6SS activity.
VgrG proteins are required for the proper function of T6SS injection machines (30, 50, 51). Accordingly, our results thus far are consistent with the idea that the requirement for the VgrG5 CTD in membrane fusion is solely a reflection of the essential role of VgrG5 in T6SS activity. In this scenario, candidate fusogenic factors might include unidentified effectors or VgrG5-associated PAAR proteins (32), and VgrG5 CTD mutations that eliminate fusion should also abrogate secretion. An alternative hypothesis is that the CTD of VgrG5 plays a specific role in membrane fusion, independent of the requirement for the rest of the protein in secretion. Previous observations by Burtnick and colleagues showed that an N-terminal segment of VgrG5, consisting of only the first 188 aa, is sufficient for Hcp5 secretion (16). Extrapolating from their results, we predicted that the intercellular spread phenotype associated with the VgrG5 CTD could be “uncoupled” from the role of N-terminal sequences in T6SS-5 activity, suggesting a direct role for the CTD in membrane fusion.
Although T6SS-5 is poorly expressed and minimally active under standard laboratory growth conditions, it can be induced by overexpression of the two-component regulatory system VirAG (16). We constructed a constitutive virAG [virAG(Con)] derivative of Bp340 utilizing a single-copy Tn7 integration vector that expresses virAG from the S12 ribosomal subunit promoter (4). As shown in Fig. 4B, Hcp5 was detected by Western blotting with anti-Hcp5 antisera in both the pellet and supernatant fractions of Bp340 virAG(Con), but not in those of WT Bp340. Deletion of vgrG5 in the virAG(Con) background greatly reduces the ability to detect Hcp5 in supernatant fractions, but not in pellet fractions, verifying that Hcp5 secretion is dependent on VgrG5 (16). Hcp5 secretion was restored by complementation of the virAG(Con) ΔvgrG5 strain with full-length vgrG5 expressed in trans. Most importantly, it was also restored by vgrG5 alleles carrying truncations or internal deletions in the CTD. Since the CTD mutations analyzed in Fig. 4B nearly (CΔ783-824) or completely (C644, C758, C850, CΔ651-696) abolished cell-cell spread, we conclude that the function of the CTD in membrane fusion is distinct and independent from the required role of VgrG5 in type VI secretion.
DISCUSSION
Functional conservation of VgrG5.
B. pseudomallei is a diverse species separated into Australian and Southeast Asian clades, having diverged from a B. thailandensis-like ancestor on the Australian subcontinent an estimated 0.3 to 4.2 million years ago (52). B. mallei, a clonal species with limited diversity, was established as a host-restricted derivative of B. pseudomallei prior to its introduction into Southeast Asia. The origins of B. oklahomensis have not been studied in detail, although it is clearly a Pseudomallei group species that shares a common ancestor with B. thailandensis and B. pseudomallei (43). In agreement with this scenario, although VgrG5 orthologs are highly similar, the B. oklahomensis and B. thailandensis proteins are more closely related to each other than they are to B. pseudomallei or B. mallei orthologs. In this study, we show that VgrG5 orthologs from all of these species are functionally interchangeable with respect to their abilities to mediate cell fusion, suggesting that similar mechanisms are involved.
The requirement for T6SS-5 in virulence and cell-cell spread by B. pseudomallei, B. mallei, and B. thailandensis is well established (12, 15, 16, 23, 25). The conservation of a functionally equivalent, albeit divergent, vgrG5 allele in B. oklahomensis was surprising, however, since isolates tested to date appear to be incapable of MNGC formation in mammalian cells in vitro (44, 53). In lieu of an explanation for the lack of cell-cell spread by B. oklahomensis, the divergence of its VgrG5 ortholog (Fig. 2A and B) provides an indication of the degree to which the protein can tolerate variability without losing fusogenic activity. Perhaps most importantly, B. oklahomensis, B. thailandensis, and B. pseudomallei are primarily soil organisms, and virulence determinants such as VgrG5 and the ability to fuse cells have likely evolved in response to competition and predation in the rhizosphere (54).
VgrG5 structural features.
Comparison of the Bp340 VgrG5 protein sequence to the NCBI RefSeq protein database (55) reveals regions similar to the hallmark domains of Gram-negative VgrG proteins, which include sequences homologous to the gp25 and gp27 components of contractile phage tail fibers. Although the CTD is clearly required for fusogenic activity, BLAST-P and iterative PSI-BLAST analysis did not reveal significant similarities with known fusion proteins. Since widely varying sequences, structures, and mechanisms can mediate membrane fusion (42), a lack of primary sequence similarity with known fusogens does not preclude the possibility that a protein or protein domain participates in membrane fusion.
Using bioinformatic strategies, we were able to identify three potential transmembrane helices that are common components of fusion proteins such as gp41 of HIV-1 (56). A region containing tandem TM sequences (TM1-2) separated by a short hydrophilic linker, which received the highest predictive scores, is essential for cell fusion, whereas the putative TM3 helix is dispensable. In addition, the Phyre2 fold recognition algorithm indicated a weak match to enzymatic sequences with hydroxymyristoyl transferase/hydrolase activity. Although the relevance of this similarity is unclear, deletion of this region caused a partial but significant loss of function. More importantly, mutations in the CTD that reduce or eliminate cell-cell spread have little, if any, effect on Hcp5 secretion, demonstrating two separable functions for VgrG5. Furthermore, the CTD plays a clear role in a murine model of B. thailandensis infection. As shown in the accompanying article by Schwarz and colleagues, C57BL/6 mice do not succumb to infection following aerosol challenge with a B. thailandensis VgrG-5 CTD deletion mutant, whereas the WT parental strain causes complete lethality by 2 to 4 days postinfection (57). It will be important to determine if similar results are obtained with highly pathogenic Burkholderia species.
Membrane fusion.
The fusion of two stable cellular membranes is a complex event that requires close membrane apposition and energy to disorder lipid bilayers (58, 59). We have shown recently that intercellular spread is facilitated by bacterial motility in the cytosol, which can be provided by BimA-mediated actin polymerization or lateral flagella in Burkholderia strains carrying the fla2 locus (12). In the hypothesis shown in Fig. 5, we propose that the force generated by bacterial movement is responsible for bringing the membranes of infected cells into close contact with neighboring cells, in a manner analogous to the roles of ligand-receptor interactions in bridging the fusion machinery of enveloped viruses with target cells (56, 60). In a second step, membrane contact or some other cue triggers contraction of the T6SS-5 sheath, and the resulting energy drives the VgrG5 CTD across one or both cell surfaces, creating a disordered “hemifusion zone” (59) that can resolve to merge both membranes.
FIG 5.
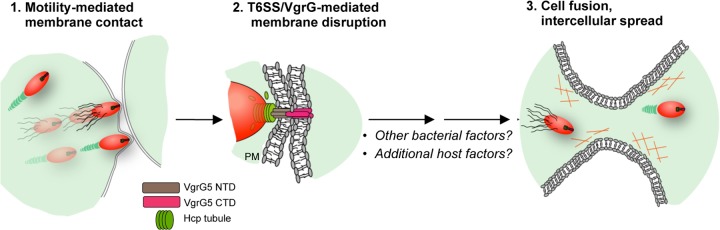
VgrG5-induced membrane fusion during Burkholderia infection: a hypothesis. Lateral flagella or actin polymerization provides motility, facilitating contact between bacteria and the plasma membrane (PM), bringing adjacent membranes into close apposition (stage 1). After contact with the membrane, T6SS-5 is deployed, and VgrG5 is inserted across the infected and adjacent cell surfaces, inducing a region of localized disturbance of lipid bilayers (stage 2) that ultimately leads to membrane fusion (stage 3). See the text for details.
The VgrG5 CTD is clearly required for cell fusion, but it seems unlikely to be sufficient. As suggested in Fig. 5, additional bacterial factors, such as surface proteins and secreted effectors, could be required. Host factors such as intercellular adhesin proteins may also play a role, and one can imagine numerous different mechanisms that could be involved. A recent study by Suparak and colleagues demonstrated that monoclonal antibodies against macrophage surface and adhesion proteins were able to block fusion in a concentration-dependent manner, supporting the possible involvement of host factors (61).
We reported previously that direct injection of B. thailandensis into a single mammalian cell is sufficient for MNGC formation (12). This is consistent with the idea that the fusion process is asymmetric in that it requires fusogenic activity only on one of two interacting membranes, as with enveloped viruses. Although the exact mechanisms at play are unknown, the ecological context of the problem offers some important clues. Pseudomallei group Burkholderia species are able to fuse a diverse array of mammalian cell types in vitro, including epithelial and phagocytic cells from different mammals (12, 16, 25), using a mechanism that is highly conserved. For the soil-dwelling species, however, the most likely natural targets are predators in the rhizosphere, such as amoebae and nematodes. Accordingly, it seems reasonable to assume that any required features of target cells are conserved from humans to amoebae. An attractive feature of the model in Fig. 5 is that it relies on bacterial factors and shared, fundamental properties of eukaryotic plasma membranes.
ACKNOWLEDGMENTS
We declare no conflicts of interest.
We thank Joseph Mougous and Sandra Schwarz (Department of Microbiology, University of Washington, Seattle, WA, USA) for providing the antibody against the VgrG CTD, Mary Burtnick (Department of Microbiology and Immunology, University of South Alabama, Mobile, AL, USA) for the antibody against B. pseudomallei Hcp5, and Bart Currie (Menzies School of Health Research, Darwin, Australia) for strain MSHR668.
This work was supported by grants from the Defense Threat Reduction Agency of the Department of Defense (HDTRA1-11-1-0003) and the Pacific Southwest Regional Center of Excellence in Biodefense and Emerging Infectious Diseases (NIH U54 A1065359) to J.F.M.
Footnotes
Published ahead of print 13 January 2014
REFERENCES
- 1.Wiersinga WJ, Currie BJ, Peacock SJ. 2012. Melioidosis. N. Engl. J. Med. 367:1035–1044. 10.1056/NEJMra1204699 [DOI] [PubMed] [Google Scholar]
- 2.Wiersinga WJ, Van Der Poll T, White NJ, Day NP, Peacock SJ. 2006. Melioidosis: insights into the pathogenicity of Burkholderia pseudomallei. Nat. Rev. Microbiol. 4:272–282. 10.1038/nrmicro1385 [DOI] [PubMed] [Google Scholar]
- 3.Galyov EE, Brett PJ, DeShazer D. 2010. Molecular insights into Burkholderia pseudomallei and Burkholderia mallei pathogenesis. Annu. Rev. Microbiol. 64:495–517. 10.1146/annurev.micro.112408.134030 [DOI] [PubMed] [Google Scholar]
- 4.Nandi T, Ong C, Singh AP, Boddey J, Atkins T, Sarkar-Tyson M, Essex-Lopresti AE, Chua HH, Pearson T, Kreisberg JF, Nilsson C, Ariyaratne P, Ronning C, Losada L, Ruan Y, Sung W-K, Woods D, Titball RW, Beacham I, Peak I, Keim P, Nierman WC, Tan P. 2010. A genomic survey of positive selection in Burkholderia pseudomallei provides insights into the evolution of accidental virulence. PLoS Pathog. 6:e1000845. 10.1371/journal.ppat.1000845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim HS, Schell MA, Yu Y, Ulrich RL, Sarria SH, Nierman WC, DeShazer D. 2005. Bacterial genome adaptation to niches: divergence of the potential virulence genes in three Burkholderia species of different survival strategies. BMC Genomics 6:174. 10.1186/1471-2164-6-174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dharakul T, Tassaneetrithep B, Trakulsomboon S, Songsivilai S. 1999. Phylogenetic analysis of Ara+ and Ara− Burkholderia pseudomallei isolates and development of a multiplex PCR procedure for rapid discrimination between the two biotypes. J. Clin. Microbiol. 37:1906–1912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lertpatanasuwan N, Sermsri K, Petkaseam A, Trakulsomboon S, Thamlikitkul V, Suputtamongkol Y. 1999. Arabinose-positive Burkholderia pseudomallei infection in humans: case report. Clin. Infect. Dis. 28:927–928. 10.1086/517252 [DOI] [PubMed] [Google Scholar]
- 8.Morici LA, Heang J, Tate T, Didier PJ, Roy CJ. 2010. Differential susceptibility of inbred mouse strains to Burkholderia thailandensis aerosol infection. Microb. Pathog. 48:9–17. 10.1016/j.micpath.2009.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Losada L, Ronning CM, DeShazer D, Woods D, Fedorova N, Kim HS, Shabalina SA, Pearson TR, Brinkac L, Tan P, Nandi T, Crabtree J, Badger J, Beckstrom-Sternberg S, Saqib M, Schutzer SE, Keim P, Nierman WC. 2010. Continuing evolution of Burkholderia mallei through genome reduction and large-scale rearrangements. Genome Biol. Evol. 2:102–116. 10.1093/gbe/evq003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Allwood EM, Devenish RJ, Prescott M, Adler B, Boyce JD. 2011. Strategies for intracellular survival of Burkholderia pseudomallei. Front. Microbiol. 2:170. 10.3389/fmicb.2011.00170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stevens JM, Galyov EE, Stevens MP. 2006. Actin-dependent movement of bacterial pathogens. Nat. Rev. Microbiol. 4:91–101. 10.1038/nrmicro1320 [DOI] [PubMed] [Google Scholar]
- 12.French CT, Toesca IJ, Wu TH, Teslaa T, Beaty SM, Wong W, Liu M, Schröder I, Chiou P-Y, Teitell MA, Miller JF. 2011. Dissection of the Burkholderia intracellular life cycle using a photothermal nanoblade. Proc. Natl. Acad. Sci. U. S. A. 108:12095–12100. 10.1073/pnas.1107183108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kespichayawattana W, Rattanachetkul S, Wanun T, Utaisincharoen P, Sirisinha S. 2000. Burkholderia pseudomallei induces cell fusion and actin-associated membrane protrusion: a possible mechanism for cell-to-cell spreading. Infect. Immun. 68:5377–5384. 10.1128/IAI.68.9.5377-5384.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ray K, Marteyn B, Sansonetti PJ, Tang CM. 2009. Life on the inside: the intracellular lifestyle of cytosolic bacteria. Nat. Rev. Microbiol. 7:333–340. 10.1038/nrmicro2112 [DOI] [PubMed] [Google Scholar]
- 15.Schwarz S, West TE, Boyer F, Chiang WC, Carl MA, Hood RD, Rohmer L, Tolker-Nielsen T, Skerrett SJ, Mougous JD. 2010. Burkholderia type VI secretion systems have distinct roles in eukaryotic and bacterial cell interactions. PLoS Pathog. 6:e1001068. 10.1371/journal.ppat.1001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burtnick MN, Brett PJ, Harding SV, Ngugi SA, Ribot WJ, Chantratita N, Scorpio A, Milne TS, Dean RE, Fritz DL, Peacock SJ, Prior JL, Atkins TP, Deshazer D. 2011. The cluster 1 type VI secretion system is a major virulence determinant in Burkholderia pseudomallei. Infect. Immun. 79:1512–1525. 10.1128/IAI.01218-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Veesler D, Cambillau C. 2011. A common evolutionary origin for tailed-bacteriophage functional modules and bacterial machineries. Microbiol. Mol. Biol. Rev. 75:423–433. 10.1128/MMBR.00014-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Russell AB, Hood RD, Bui NK, Leroux M, Vollmer W, Mougous JD. 2011. Type VI secretion delivers bacteriolytic effectors to target cells. Nature 475:343–347. 10.1038/nature10244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pukatzki S, Ma AT, Revel AT, Sturtevant D, Mekalanos JJ. 2007. Type VI secretion system translocates a phage tail spike-like protein into target cells where it cross-links actin. Proc. Natl. Acad. Sci. U. S. A. 104:15508–15513. 10.1073/pnas.0706532104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma AT, McAuley S, Pukatzki S, Mekalanos JJ. 2009. Translocation of a Vibrio cholerae type VI secretion effector requires bacterial endocytosis by host cells. Cell Host Microbe 5:234–243. 10.1016/j.chom.2009.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Basler M, Pilhofer M, Henderson GP, Jensen GJ, Mekalanos JJ. 2012. Type VI secretion requires a dynamic contractile phage tail-like structure. Nature 483:182–186. 10.1038/nature10846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lossi NS, Manoli E, Forster A, Dajani R, Pape T, Freemont P, Filloux A. 2013. The HsiB1C1 (TssB-TssC) complex of the Pseudomonas aeruginosa type VI secretion system forms a bacteriophage tail sheathlike structure. J. Biol. Chem. 288:7536–7548. 10.1074/jbc.M112.439273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schell MA, Ulrich RL, Ribot WJ, Brueggemann EE, Hines HB, Chen D, Lipscomb L, Kim HS, Mrazek J, Nierman WC, Deshazer D. 2007. Type VI secretion is a major virulence determinant in Burkholderia mallei. Mol. Microbiol. 64:1466–1485. 10.1111/j.1365-2958.2007.05734.x [DOI] [PubMed] [Google Scholar]
- 24.Shalom G, Shaw JG, Thomas MS. 2007. In vivo expression technology identifies a type VI secretion system locus in Burkholderia pseudomallei that is induced upon invasion of macrophages. Microbiology 153:2689–2699. 10.1099/mic.0.2007/006585-0 [DOI] [PubMed] [Google Scholar]
- 25.Burtnick MN, DeShazer D, Nair V, Gherardini FC, Brett PJ. 2010. Burkholderia mallei cluster 1 type VI secretion mutants exhibit growth and actin polymerization defects in RAW 264.7 murine macrophages. Infect. Immun. 78:88–99. 10.1128/IAI.00985-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bingle LE, Bailey CM, Pallen MJ. 2008. Type VI secretion: a beginner's guide. Curr. Opin. Microbiol. 11:3–8. 10.1016/j.mib.2008.01.006 [DOI] [PubMed] [Google Scholar]
- 27.Cascales E. 2008. The type VI secretion toolkit. EMBO Rep. 9:735–741. 10.1038/embor.2008.131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mougous JD, Cuff ME, Raunser S, Shen A, Zhou M, Gifford CA, Goodman AL, Joachimiak G, Ordonez CL, Lory S, Walz T, Joachimiak A, Mekalanos JJ. 2006. A virulence locus of Pseudomonas aeruginosa encodes a protein secretion apparatus. Science 312:1526–1530. 10.1126/science.1128393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ballister ER, Lai AH, Zuckermann RN, Cheng Y, Mougous JD. 2008. In vitro self-assembly of tailorable nanotubes from a simple protein building block. Proc. Natl. Acad. Sci. U. S. A. 105:3733–3738. 10.1073/pnas.0712247105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cascales E, Cambillau C. 2012. Structural biology of type VI secretion systems. Philos. Trans. R. Soc. Lond. B Biol. Sci. 367:1102–1111. 10.1098/rstb.2011.0209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pukatzki S, Ma AT, Sturtevant D, Krastins B, Sarracino D, Nelson WC, Heidelberg JF, Mekalanos JJ. 2006. Identification of a conserved bacterial protein secretion system in Vibrio cholerae using the Dictyostelium host model system. Proc. Natl. Acad. Sci. U. S. A. 103:1528–1533. 10.1073/pnas.0510322103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shneider MM, Buth SA, Ho BT, Basler M, Mekalanos JJ, Leiman PG. 2013. PAAR-repeat proteins sharpen and diversify the type VI secretion system spike. Nature 500:350–353. 10.1038/nature12453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sheahan KL, Cordero CL, Satchell KJ. 2004. Identification of a domain within the multifunctional Vibrio cholerae RTX toxin that covalently cross-links actin. Proc. Natl. Acad. Sci. U. S. A. 101:9798–9803. 10.1073/pnas.0401104101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suarez G, Sierra JC, Erova TE, Sha J, Horneman AJ, Chopra AK. 2010. A type VI secretion system effector protein, VgrG1, from Aeromonas hydrophila that induces host cell toxicity by ADP ribosylation of actin. J. Bacteriol. 192:155–168. 10.1128/JB.01260-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brett PJ, DeShazer D, Woods DE. 1998. Burkholderia thailandensis sp. nov., a Burkholderia pseudomallei-like species. Int. J. Syst. Bacteriol. 48(Part 1):317–320. 10.1099/00207713-48-1-317 [DOI] [PubMed] [Google Scholar]
- 36.Mima T, Schweizer HP. 2010. The BpeAB-OprB efflux pump of Burkholderia pseudomallei 1026b does not play a role in quorum sensing, virulence factor production, or extrusion of aminoglycosides but is a broad-spectrum drug efflux system. Antimicrob. Agents Chemother. 54:3113–3120. 10.1128/AAC.01803-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barrett AR, Kang Y, Inamasu KS, Son MS, Vukovich JM, Hoang TT. 2008. Genetic tools for allelic replacement in Burkholderia species. Appl. Environ. Microbiol. 74:4498–4508. 10.1128/AEM.00531-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kumar A, Dalton C, Cortez-Cordova J, Schweizer HP. 2010. Mini-Tn7 vectors as genetic tools for single copy gene cloning in Acinetobacter baumannii. J. Microbiol. Methods 82:296–300. 10.1016/j.mimet.2010.07.002 [DOI] [PubMed] [Google Scholar]
- 39.Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop RM, II, Peterson KM. 1995. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166:175–176. 10.1016/0378-1119(95)00584-1 [DOI] [PubMed] [Google Scholar]
- 40.Hayden HS, Lim R, Brittnacher MJ, Sims EH, Ramage ER, Fong C, Wu Z, Crist E, Chang J, Zhou Y, Radey M, Rohmer L, Haugen E, Gillett W, Wuthiekanun V, Peacock SJ, Kaul R, Miller SI, Manoil C, Jacobs MA. 2012. Evolution of Burkholderia pseudomallei in recurrent melioidosis. PLoS One 7:e36507. 10.1371/journal.pone.0036507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.U'Ren JM, Hornstra H, Pearson T, Schupp JM, Leadem B, Georgia S, Sermswan RW, Keim P. 2007. Fine-scale genetic diversity among Burkholderia pseudomallei soil isolates in northeast Thailand. Appl. Environ. Microbiol. 73:6678–6681. 10.1128/AEM.00986-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCormick JB, Weaver RE, Hayes PS, Boyce JM, Feldman RA. 1977. Wound infection by an indigenous Pseudomonas pseudomallei-like organism isolated from the soil: case report and epidemiologic study. J. Infect. Dis. 135:103–107. 10.1093/infdis/135.1.103 [DOI] [PubMed] [Google Scholar]
- 43.Glass MB, Steigerwalt AG, Jordan JG, Wilkins PP, Gee JE. 2006. Burkholderia oklahomensis sp. nov., a Burkholderia pseudomallei-like species formerly known as the Oklahoma strain of Pseudomonas pseudomallei. Int. J. Syst. Evol. Microbiol. 56:2171–2176. 10.1099/ijs.0.63991-0 [DOI] [PubMed] [Google Scholar]
- 44.Deshazer D. 2007. Virulence of clinical and environmental isolates of Burkholderia oklahomensis and Burkholderia thailandensis in hamsters and mice. FEMS Microbiol. Lett. 277:64–69. 10.1111/j.1574-6968.2007.00946.x [DOI] [PubMed] [Google Scholar]
- 45.Carvalho AP, Ventura GM, Pereira CB, Leao RS, Folescu TW, Higa L, Teixeira LM, Plotkowski MC, Merquior VL, Albano RM, Marques EA. 2007. Burkholderia cenocepacia, B. multivorans, B. ambifaria and B. vietnamiensis isolates from cystic fibrosis patients have different profiles of exoenzyme production. APMIS 115:311–318. 10.1111/j.1600-0463.2007.apm_603.x [DOI] [PubMed] [Google Scholar]
- 46.Hofmann K, Stoffel W. 1993. TMBASE—a database of membrane spanning protein segments. Biol. Chem. Hoppe-Seyler 374:166 [Google Scholar]
- 47.Möller S, Croning MD, Apweiler R. 2001. Evaluation of methods for the prediction of membrane spanning regions. Bioinformatics 17:646–653. 10.1093/bioinformatics/17.7.646 [DOI] [PubMed] [Google Scholar]
- 48.Jones DT, Taylor WR, Thornton JM. 1994. A model recognition approach to the prediction of all-helical membrane protein structure and topology. Biochemistry 33:3038–3049. 10.1021/bi00176a037 [DOI] [PubMed] [Google Scholar]
- 49.Söding J. 2005. Protein homology detection by HMM-HMM comparison. Bioinformatics 21:951–960. 10.1093/bioinformatics/bti125 [DOI] [PubMed] [Google Scholar]
- 50.Zheng J, Ho B, Mekalanos JJ. 2011. Genetic analysis of anti-amoebae and anti-bacterial activities of the type VI secretion system in Vibrio cholerae. PLoS One 6:e23876. 10.1371/journal.pone.0023876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zheng J, Leung KY. 2007. Dissection of a type VI secretion system in Edwardsiella tarda. Mol. Microbiol. 66:1192–1206. 10.1111/j.1365-2958.2007.05993.x [DOI] [PubMed] [Google Scholar]
- 52.Pearson T, Giffard P, Beckstrom-Sternberg S, Auerbach R, Hornstra H, Tuanyok A, Price EP, Glass MB, Leadem B, Beckstrom-Sternberg JS, Allan GJ, Foster JT, Wagner DM, Okinaka RT, Sim SH, Pearson O, Wu Z, Chang J, Kaul R, Hoffmaster AR, Brettin TS, Robison RA, Mayo M, Gee JE, Tan P, Currie BJ, Keim P. 2009. Phylogeographic reconstruction of a bacterial species with high levels of lateral gene transfer. BMC Biol. 7:78. 10.1186/1741-7007-7-78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wand ME, Muller CM, Titball RW, Michell SL. 2011. Macrophage and Galleria mellonella infection models reflect the virulence of naturally occurring isolates of B. pseudomallei, B. thailandensis and B. oklahomensis. BMC Microbiol. 11:11. 10.1186/1471-2180-11-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ooi WF, Ong C, Nandi T, Kreisberg JF, Chua HH, Sun G, Chen Y, Mueller C, Conejero L, Eshaghi M, Ang RM, Liu J, Sobral BW, Korbsrisate S, Gan YH, Titball RW, Bancroft GJ, Valade E, Tan P. 2013. The condition-dependent transcriptional landscape of Burkholderia pseudomallei. PLoS Genet. 9:e1003795. 10.1371/journal.pgen.1003795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402. 10.1093/nar/25.17.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ashkenazi A, Shai Y. 2011. Insights into the mechanism of HIV-1 envelope induced membrane fusion as revealed by its inhibitory peptides. Eur. Biophys. J. 40:349–357. 10.1007/s00249-010-0666-z [DOI] [PubMed] [Google Scholar]
- 57.Schwarz S, Singh P, Robertson JD, Le Roux M, Skerrett SJ, Goodlett DR, West TE, Mougous JD. 2014. VgrG-5 is a Burkholderia thailandensis type VI secretion system-exported protein required for multinucleated giant cell formation and virulence. Infect. Immun. 82:1445–1452. 10.1128/IAI.01368-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marsden HR, Tomatsu I, Kros A. 2011. Model systems for membrane fusion. Chem. Soc. Rev. 40:1572–1585. 10.1039/c0cs00115e [DOI] [PubMed] [Google Scholar]
- 59.Martens S, McMahon HT. 2008. Mechanisms of membrane fusion: disparate players and common principles. Nat. Rev. Mol. Cell Biol. 9:543–556. 10.1038/nrm2417 [DOI] [PubMed] [Google Scholar]
- 60.Backovic M, Jardetzky TS. 2011. Class III viral membrane fusion proteins. Adv. Exp. Med. Biol. 714:91–101. 10.1007/978-94-007-0782-5_3 [DOI] [PubMed] [Google Scholar]
- 61.Suparak S, Muangsombut V, Riyapa D, Stevens JM, Stevens MP, Lertmemongkolchai G, Korbsrisate S. 2011. Burkholderia pseudomallei-induced cell fusion in U937 macrophages can be inhibited by monoclonal antibodies against host cell surface molecules. Microbes Infect. 13:1006–1011. 10.1016/j.micinf.2011.06.007 [DOI] [PubMed] [Google Scholar]
- 62.Jones DT, Taylor WR, Thornton JM. 1992. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 8:275–282 [DOI] [PubMed] [Google Scholar]
- 63.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739. 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]