

Abstract
Escherichia coli O157:H7, a major Shiga toxin-producing pathogen, has a low infectious dose and causes serious illness in humans. The gastrointestinal tract of cattle is the primary reservoir of E. coli O157:H7, and thus, it is critical to eliminate or reduce E. coli O157:H7 gut colonization. Given that E. coli O157:H7 produces effectors that attenuate inflammatory signaling, we hypothesized that the host inflammatory response acts to perturb E. coli O157:H7 intestinal colonization. Tumor necrosis factor alpha (TNF-α) treatment of HT-29 cells resulted in increased expression of inflammatory cytokine interleukin 1β (IL-1β), IL-8, and TNF-α genes and increased IL-8 protein and resulted in decreased adhesion of E. coli O157:H7. Similarly, E. coli O157:H7 adhesion to cattle colonic explants was reduced by TNF-α treatment. Irrespective of the presence of E. coli O157:H7, TNF-α enhanced activation of p65, the key mediator of NF-κB inflammatory signaling, whereas E. coli O157:H7 infection suppressed this pathway by inhibiting p65 activation in HT-29 cells. To further explore the mechanisms linking the inflammatory response to attenuated E. coli O157:H7 adhesion, mucin 2 (MUC2) expression was analyzed, considering that the intestinal mucus layer is the first defense against enteric pathogens and MUC2 is the major secretory mucin in the intestine. MUC2 expression in HT-29 cells was increased by TNF-α treatment and by E. coli O157:H7 infection. However, reducing mucin expression by blocking mitogen-activated protein kinase (MAPK) extracellular signal-regulated protein kinases 1/2 (ERK1/2) and/or phosphatidylinositol 3-kinase (PI3K)/Akt signaling increased E. coli O157:H7 adherence to HT-29 cells. These data suggest that the inflammatory cytokine response acts to protect host epithelial cells against E. coli O157:H7 colonization, at least in part, by promoting mucin production.
INTRODUCTION
Although the food supply in the United States is one of the safest in the world, outbreaks of food-borne illnesses are not uncommon. Escherichia coli O157:H7 is a major food-borne pathogen that causes hemorrhagic colitis, hemolytic uremic syndrome, and even death (1). Ruminant animals are primary reservoirs for E. coli O157:H7, and fecal shedding of E. coli O157:H7 is a major source for E. coli O157:H7 contamination in food and water. About 10% to 20% of beef cattle shed E. coli O157:H7, and shedding in summer months is higher than in winter months (2). Certain cattle have much higher rates of E. coli O157:H7 shedding (>104 bacteria/g feces) and can maintain a high level of shedding for several weeks or even months (3); such cattle are so-called “supershedders” (4) and are major sources of E. coli O157:H7 contamination in meat, typically via contaminated hides (4, 5). In addition, runoff water from livestock farms can contaminate vegetables, as was shown in a 2006 nationwide spinach recall due to E. coli O157:H7 contamination (6, 7). Eliminating or reducing E. coli O157:H7 gut colonization in cattle could help to solve food safety problems associated with E. coli O157:H7 infection. This effort would be helped by understanding the mechanisms leading to E. coli O157:H7 supershedders.
E. coli O157:H7 interaction with intestinal epithelial cells is the first step in E. coli O157:H7 colonization. Colonization depends on the ability of the bacteria to adhere to the host cells. E. coli O157:H7 possesses intimin and several type III secretion effectors that enable it to intimately attach to intestinal epithelial cells, resulting in the formation of attaching and effacing (A/E) lesions (1, 8, 9). In addition, E. coli O157:H7 possesses numerous fimbrial and nonfimbrial adhesins, which also play important roles in the initial stages of adhesion (10–13). On the other hand, a healthy gastrointestinal (GI) tract contains multiple defensive barriers. The intestinal mucosa is covered by a thick mucus layer, which serves as a major barrier between the epithelium and the lumen of the host GI tract and plays an essential role in innate immune defense (14, 15). In order to maintain homeostasis of the intestinal mucosa, mucus is continuously secreted from the intestinal epithelium. Major components of mucus are the gel-forming mucins (16), highly glycosylated high-molecular-weight proteins that are secreted mainly by goblet cells. Approximately 20 different mucin (MUC1 to MUC20)-encoding genes have been identified. They are broadly grouped as either membrane associated or secretory (17). The prominent component of secretory mucins in the intestine is mucin 2, which is synthesized by goblet cells and stored as mucin granules that expand dramatically upon secretion (16). The rapid secretion and turnover of mucins generates a virtually sterile layer between the gut luminal contents and the epithelial cells, preventing contact with and colonization of the epithelium by pathogens. It has been reported that inflammatory cytokines, including interleukin 4 (IL-4), IL-13, and tumor necrosis factor alpha (TNF-α), promote mucin gene expression in intestinal epithelial cells (18–21). The large amount of mucins and antimicrobial compounds secreted at the site of inflammation is expected to wash away pathogens, preventing pathogen infection and colonization. Therefore, we hypothesized that the inflammatory response acts to protect host epithelial cells against E. coli O157:H7 colonization, at least in part, by promoting mucin production.
MATERIALS AND METHODS
Cell line, media, and bacterial strains.
The human colonic epithelial cell line HT-29 was obtained from the American Type Culture Collection (Manassas, VA). HT-29 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Sigma, St. Louis, MO) supplemented with 10% fetal bovine serum (FBS) (Sigma), 100 units/ml penicillin G, and 100 μg/ml streptomycin (Sigma) at 37°C with 5% CO2. E. coli O157:H7 strain EDL933 was obtained from the Shiga toxin-producing E. coli (STEC) center at Michigan State University and was routinely grown in 5 ml LB broth in 16- by 150-mm culture tubes at 37°C overnight with shaking at 250 rpm.
Adhesion of E. coli O157:H7 to colonic epithelial cells.
HT-29 cells were seeded in 24-well plates, cultured until 80 to 90% confluent, and treated with 0 or 10 ng/ml TNF-α (Cell Signaling Technology, Beverly, MA) for 2 or 12 h. The cells in each well were then washed 3 times with phosphate-buffered saline (PBS) (pH 7.4) and challenged with overnight-diluted E. coli O157:H7 culture (107 CFU/ml) at a multiplicity of infection (MOI) of 10:1. Bacteria and HT-29 cells were cocultured for 3 h at 37°C with 5% CO2. The cell monolayers were then washed 3 times with PBS, and the cells were lysed with 0.2% Triton X-100. Serial dilutions of the lysates were plated on LB agar, and bacterial colonies were counted after 18 h of incubation at 37°C. The percent adhesion was calculated by dividing the number of colonies (recovered E. coli O157:H7) by the total number of E. coli O157:H7 bacteria added and multiplying by 100.
E. coli O157:H7 adherence to cattle colonic explants.
Distal colonic tissues (5 to 10 cm from the anus) were aseptically collected from cattle slaughtered in the University of Wyoming Meat Laboratory and transferred to the microbiology laboratory on ice within 5 min. The time lapse between the death of the cattle and processing of explant tissues was less than 30 min. Specimens were washed 5 times with 0.9% (wt/vol) NaCl, and the mucosa was dissected from the underlying tissue with sterile dissecting scissors. The trimmed specimens were reduced to 8-mm round pieces using Miltex disposable biopsy punches (Cardinal Health, OH) and were transferred to individual wells of 24-well plates (without a cover to enhance air circulation). The specimens were cultured in DMEM with or without 10 ng/ml TNF-α for 2 h. E. coli O157:H7 was then added at 107 CFU/well, and culture was continued at 37°C with 5% CO2. Three hours postinfection, E. coli O157:H7-attached explants were washed, serially diluted, plated, and enumerated. The percent adhesion was calculated by dividing the number of colonies (recovered E. coli O157:H7) by the total number of E. coli O157:H7 bacteria added and multiplying by 100. In the inhibitor study, LY294002 (phosphatidylinositol 3-kinase [PI3K]/Akt inhibitors; Cell Signaling Technology) and/or U0126 (mitogen-activated protein kinase [MAPK]/extracellular signal-regulated protein kinases 1/2 [ERK1/2] inhibitor; Cell Signaling Technology) was added to the culture medium (20 μM each) 60 min prior to TNF-α treatment.
Immunofluorescent staining of E. coli O157:H7.
E. coli O157:H7 adhesion was conducted as described above. After interaction, the cells were washed with PBS three times and incubated in DMEM at 37°C for an additional 3 h. The HT-29 monolayer or explants were then fixed in 4% paraformaldehyde. Explant tissues were paraffin embedded, sectioned (5 μm), and rehydrated for immunofluorescent staining. The cells or tissue sections were washed with PBS and then permeabilized with 0.1% Triton X-100 for 10 min. After blocking with 1.5% goat serum for 30 min, the cells or tissue sections were stained with anti-E. coli O157:H7-fluorescein isothiocyanate (FITC) antibody (KPL, Gaithersburg, MD), followed by incubation with 5 μg/ml phalloidin-tetramethyl rhodamine isothiocyanate (TRITC) (Sigma) for 40 min at room temperature, and mounted with Vectashield mounting medium with DAPI (4′,6-diamidino-2-phenylindole) (Vector Laboratory). Attached E. coli O157:H7 was visualized on a Leica DMI6000 B fluorescence microscope equipped with a DFC340 FX digital camera (Leica, Buffalo Grove, IL).
Alcian blue staining of HT-29 cells and cattle gut explants.
HT-29 cell monolayers were seeded and treated or not with 10 ng/ml TNF-α for 12 h as described above and then fixed in 4% paraformaldehyde, and mucins were stained with alcian blue (22). Quantification of mucins produced by HT-29 cells was performed using Image J software (split color channels; NIH, Bethesda, MD). Six randomly selected fields were examined. The results were quantified by dividing the positive (blue) area by the number of cells in each selected area. Cattle gut explants were treated or not with 10 ng/ml TNF-α for 2 h, fixed in 4% (wt/vol) paraformaldehyde, embedded in paraffin, and sectioned (5 μm) as described above. For each colonic explant, 12 5-μm sections evenly spaced over a 450-μm area were obtained. The sections were rehydrated and stained with alcian blue (22). The quantification of mucins in alcian blue-stained explant sections was performed using Image J software as described above. The results were quantified by dividing the positive (blue) area by the total tissue area.
qRT-PCR analyses.
Total RNA was extracted from HT-29 cells using an RNeasy minikit (Qiagen, Valencia, CA) and reverse transcribed using a QuantiTect reverse transcription kit (Qiagen). The resulting cDNA was used as a template for quantitative reverse transcription-PCR (qRT-PCR) analysis of selected genes using a CFX96 Real-Time PCR Detection System (Bio-Rad, CA). SYBR green Master Mix (Bio-Rad, CA) was used for all qRT-PCRs. The primers for qRT-PCR are listed in Table 1. The relative expression of selected genes was normalized to that of the gene encoding glyceraldehyde 3-phosphate dehydrogenase (gapdh). The amplification efficiency was 0.90 to 0.99 (23).
TABLE 1.
Primer sets used for qRT-PCR for HT-29 cells
| Gene name | Accession no. | Product size (bp) | Direction | Sequence (5′→3′) |
|---|---|---|---|---|
| IL-1β | NM_000576 | 126 | Forward | GTGGCAATGAGGATGACTTGT |
| Reverse | TGTAGTGGTGGTCGGAGATTC | |||
| IL-8 | NM_000584 | 88 | Forward | GAGGGTTGTGGAGAAGTTTTTG |
| Reverse | CTGGCATCTTCACTGATTCTTG | |||
| IL-18 | NM_001562 | 100 | Forward | TGAAGATGATGAAAACCTGGAAT |
| Reverse | TTGGTCAATGAAGAGAACTTGGT | |||
| MUC1 | NM_001018017.2 | 108 | Forward | CAGCCAGCGCCTGCCTGAAT |
| Reverse | GCACTGTGAGGAGCAGCAGCA | |||
| MUC2 | M94132.1 | 138 | Forward | CCTGCCGACACCTGCTGCAA |
| Reverse | ACACCAGTAGAAGGGACAGCACCT | |||
| MUC3 | AB038784.1 | 130 | Forward | CTCGTGTTGCCATTGCCTCTCTCG |
| Reverse | CTGCAGGTTGCCTCCAGGTTCAGA | |||
| TNF-α | AF043342 | 100 | Forward | AACCTCCTCTCTGCCATCAA |
| Reverse | GGAAGACCCCTCCCAGATAG | |||
| gapdh | NM_002046 | 94 | Forward | ACAGTCAGCCGCATCTTCTT |
| Reverse | ACGACCAAATCCGTTGACTC |
Immunoblotting.
Immunoblotting analysis was conducted according to procedures previously described (23). Antibodies against protein kinase B (Akt) (catalog number 9272), phospho-Akt (catalog number 9271), ERK1/2 (catalog number 9102), phospho-ERK1/2 (catalog number 9101), NF-κB p65 (catalog number 4764), and phospho-p65 (Ser 536) (catalog number 3033) were purchased from Cell Signaling Technology (Beverly, MA). Anti-GAPDH antibody (catalog number MA116757) was purchased from Affinity BioReagents (Golden, CO). Binding of antibodies was detected using horseradish peroxidase (HRP)-coupled anti-rabbit or anti-mouse immunoglobulin and visualized by chemiluminescence. The density of bands was quantified using an Imager Scanner II (Amersham Bioscience) and ImageQuant TL software (Amersham Bioscience) and normalized to the GAPDH content.
IL-8 and IL-18 analysis by ELISA.
After E. coli O157:H7 infection, spent HT-29 cell medium was collected and analyzed for IL-8 using a human IL-8 enzyme-linked immunosorbent assay (ELISA) kit (Thermo Scientific, IL) according to the manufacturer's directions. The intra-assay coefficient of variation (CV) and the interassay CV were <10%. IL-18 levels were analyzed using a human IL-18 ELISA kit (eBioscience, San Diego, CA) according to the manufacturer's directions. The intra-assay CV and the interassay CV were 6.5% and 8.1%, respectively.
Statistical analyses.
Statistical analyses were conducted as previously described (23). Data were analyzed as a complete randomized design using GLM (General Linear Model of Statistical Analysis System; SAS, 2000). Means ± standard errors of the mean (SEM) are reported. A P value of <0.05 was considered significant.
RESULTS
TNF-α treatment reduced E. coli O157:H7 adhesion.
To test the effect of the inflammatory response on E. coli O157:H7 adhesion to epithelial cells, we treated HT-29 cells with TNF-α, which is known to induce inflammatory cytokine expression (24). As indicated in Fig. 1, TNF-α pretreatment resulted in increased mRNA expression of the proinflammatory cytokines IL-1β, IL-8, and TNF-α in HT-29 cells (Fig. 1A). Furthermore, TNF-α treatment resulted in increased production of IL-8 protein (Fig. 1B), which was associated with increased phosphorylation of NF-κB p65 (Fig. 1C). The NF-κB signaling pathway is a major regulator of inflammation, and its activation is associated with phosphorylation of p65 (25). Longer (12-h) TNF-α exposure resulted in enhanced p65 activation in HT-29 cells compared with shorter (2-h) exposure (Fig. 1C). Given that a longer duration of TNF-α exposure is more similar to chronic inflammation in the gut in vivo, we chose 12-h TNF-α pretreatment for further experiments using HT-29 cells.
FIG 1.

TNF-α treatment induces inflammatory response in HT-29 cells. (A) Relative mRNA expression of proinflammatory cytokines in HT-29 cells untreated (Con) or treated with 10 ng/ml TNF-α (TNF-α) for 12 h. (B) IL-8 production in HT-29 cells 2 h or 12 h after TNF-α treatment. (C) NF-κB p65 signaling in HT-29 cells at 2 h or 12 h after TNF-α treatment. (Top) Representative Western blotting bands. (Bottom) Statistical data. Shown are means and SEM; n = 4. **, P < 0.01; *, P < 0.05.
Accompanying the increased inflammatory cytokine response, the adhesion of E. coli O157:H7 to HT-29 cells treated with TNF-α was reduced compared with that of untreated controls (Fig. 2A). We evaluated the significance of this observation using colonic explant tissues separated from the rectoanal region of beef cattle, considering that the terminal rectoanal junction (RAJ) is the principal colonization site of E. coli O157:H7 (26, 27). We note that this claim has recently been disputed, and it has been proposed that E. coli O157:H7 colonizes other sites in the GI tract, including the small intestine, in addition to the RAJ (28). In agreement with our findings using HT-29 cells, TNF-α treatment also reduced adhesion of E. coli O157:H7 in colonic explant tissues (Fig. 2B). Since bacteria can also attach to the exposed basal lamina and other tissues and cells of explant tissues, in addition to epithelial cells, the bacterial counts might be artificially increased, which might partially mask the treatment effect. Immunofluorescent staining indicated that fewer E. coli O157:H7 bacteria (green fluorescent signal) were associated with HT-29 cells (Fig. 2C) or colonic explant tissues treated with TNF-α (Fig. 2D), showing once again that TNF-α treatment reduced E. coli O157:H7 adherence to epithelial cells. Examination of paraffin-embedded post-E. coli O157:H7-infected cattle explant sections imaged under a bright-field microscope (Fig. 2E) demonstrated that the overall integrity of epithelial cells after E. coli O157:H7 infection was preserved.
FIG 2.

TNF-α treatment reduces adhesion of E. coli O157:H7 to HT-29 cells and cattle colonic explant tissues. (A) Adhesion of E. coli O157:H7 to HT-29 cells untreated (Con) or treated with 10 ng/ml TNF-α (TNF-α) for 2 h or 12 h. **, P < 0.01. (B) Adhesion of E. coli O157:H7 to cattle colonic epithelial cells untreated or treated with 10 ng/ml TNF-α for 2 h. *, P < 0.05. Shown are means and SEM; n = 8 for each treatment of HT-29 cells and explant. (C and D) Representative images of immunofluorescent staining of E. coli O157:H7 adherence to HT-29 cells (C) and cattle colonic explant tissues (D) treated or not with 10 ng/ml TNF-α. Blue, DAPI-stained nucleus; green, FITC-labeled E. coli O157:H7; red, phalloidin-TRITC to indicate cell skeletal elements. (E) H&E staining of postinfection cattle colonic explant tissues untreated (Con) or treated with 10 ng/ml TNF-α (TNF-α) for 2 h.
In line with its proinflammatory function, TNF-α increased the phosphorylation of p65, the key mediator of NF-κB inflammatory signaling, while E. coli O157:H7 infection resulted in suppression of phosphorylated p65 and therefore NF-κB activation (Fig. 3A). The ability of E. coli O157:H7 to reduce NF-κB activation, thereby diminishing the host inflammatory response, correlated with increased E. coli O157:H7 colonization. As indicated in Fig. 3B, the presence of E. coli O157:H7 resulted in enhanced expression of IL-1β and TNF-α mRNAs in HT-29 cells, and TNF-α incubation further enhanced TNF-α mRNA (Fig. 3B). IL-8, a chemotactic cytokine for T lymphocytes and neutrophils, is induced under inflammatory conditions (29). E. coli O157:H7 infection resulted in exaggerated IL-8 mRNA expression and protein production, which was further increased by TNF-α treatment (Fig. 3C). IL-18 is another important proinflammatory cytokine. Consistent with its effects on NF-κB p65 signaling pathways, TNF-α treatment stimulated the activation of IL-18, as indicated by increased mRNA expression and secretion, which were blocked after E. coli O157:H7 infection (Fig. 3C).
FIG 3.

Inflammatory responses in HT-29 cells untreated (Con) or treated with 10 ng/ml TNF-α for 12 h (TNF-α) and infected (+) or not (−) with E. coli O157:H7. (A) NF-κB p65 signaling analyzed by Western blotting. (Top) Representative bands. (Bottom) Statistical data. (B) IL-1β and TNF-α mRNA expression. (C) IL-8 and IL-18 mRNA expression and protein production. **, P < 0.01; *, P < 0.05. Shown are means and SEM; n = 4.
TNF-α treatment enhanced mucin production, which possibly contributed to the reduced E. coli O157:H7 epithelial adhesion.
The mucus layer functions as a critical protective barrier against bacterial adhesion to and invasion of the epithelial layer; therefore, we analyzed mucin mRNA expression in HT-29 cells in the presence of E. coli O157:H7 and/or TNF-α. In the presence or absence of E. coli O157:H7 infection, for both 2 h (Fig. 4A) and 12 h (Fig. 4B), TNF-α treatments resulted in enhanced MUC1, MUC2, and MUC3 mRNA expression. Because mucin proteins are highly glycosylated and have high and variable molecular weights, we were not able to analyze the mucin protein content by Western blotting. To determine whether there was a change in mucin protein levels in response to TNF-α treatment, we used alcian blue staining on HT-29 cells (Fig. 4C) and on cattle colonic explants (Fig. 4D), each of which indicated increased mucin excretion/production in response to TNF-α treatment. Of note, the detected mucin level in explant tissues might be an underestimate caused by possible loss of crypt epithelium due to postmortem autolysis, processing, and culturing. These results are consistent with our hypothesis that TNF-α reduces E. coli O157:H7 attachment by enhancing mucin secretion and synthesis.
FIG 4.

Mucin expression and production in response to TNF-α treatment and E. coli O157:H7 infection. (A and B) mRNA expression of selected mucin genes in HT-29 cells untreated (Con) or treated with 10 ng/ml TNF-α (TNF-α) for 2 h (A) and 12 h (B) and infected (+) or not (−) with E. coli O157:H7. (C) Alcian blue staining of HT-29 cells not pretreated (Con) or pretreated with 10 ng/ml TNF-α (TNF-α) for 12 h. (Left) Representative images. (Right) Statistical data. (D) Alcian blue mucin staining in cattle colonic explant tissues not pretreated or pretreated with 10 ng/ml TNF-α for 2 h. (Left) Representative images. (Right) Statistical data. **, P < 0.01; *, P < 0.05. Shown are means and SEM; n = 4 for mRNA expression; n = 8 for alcian blue staining.
Blocking mucin production by inhibition of PI3K/Akt and MAPK/ERK1/2 signaling.
To explore possible mechanisms by which TNF-α enhances mucin production, we analyzed PI3K/Akt and MAPK/ERK1/2 signaling pathways. Both E. coli O157:H7 infection and TNF-α treatment enhanced Akt signaling (Fig. 5A), a pathway-regulating protein synthesis, including the synthesis of mucins, as indicated by increased phosphorylation of Akt and an increased ratio of phospho-Akt to total Akt. Similarly, TNF-α treatment activated ERK1/2 signaling in HT-29 cells; however, upon E. coli O157:H7 infection, ERK1/2 signaling was downregulated (Fig. 5B).
FIG 5.

Akt and mitogen-activated protein kinase/ERK1/2 signaling in HT-29 cells untreated (Con) or treated with 10 ng/ml TNF-α (TNF-α) and uninfected (−) or infected with E. coli O157:H7 (+). (A) Akt signaling. (Top) Representative Western blotting bands. (Bottom) Statistical data. (B) ERK1/2 signaling. (Top) Representative Western blotting bands. (Bottom) Statistical data. HT-29 cells at 80 to 90% confluence were incubated without or with 10 ng/ml TNF-α for 12 h, and then the HT-29 cells were subjected to E. coli O157:H7 infection for 3 h and washed, lysed, and collected. The cell lysates were subjected to 10% SDS-PAGE, and total and phosphorylated Akt (A) and ERK1/2 (B) were detected by probing with their respective antibodies. The ratios of phosphorylated Akt to total Akt and phosphorylated ERK1/2 to total ERK1/2 were used to indicate activation/inhibition of Akt and ERK1/2 signaling, respectively. **, P < 0.01; *, P < 0.05. Shown are means and SEM; n = 4.
To elucidate the role of PI3K/Akt and/or MAPK/ERK1/2 signaling in mucin production and E. coli O157:H7 epithelial cell adherence, LY294002, a specific inhibitor of the PI3K/Akt pathway, and U0126, a specific inhibitor of MAPK/ERK1/2 signaling, were used to block PI3K/Akt and/or MAPK/ERK1/2 signaling. Expression of MUC2 mRNA was inhibited in HT-29 cells treated or not with TNF-α following addition of LY294002 and/or U0126. The effect of U0126 was far more dramatic than that of LY294002 (Fig. 6A). The ability of TNF-α to reduce adhesion of E. coli O157:H7 to epithelial cells was negated by treatment with LY294002 and/or U0126. These data indicate that mucin expression and adhesion of E. coli O157:H7 to gut epithelial cells following TNF-α treatment are at least in part regulated by the PI3K/Akt and MAPK/ERK1/2 pathways. These data also provide further correlative evidence that blocking mucin expression enhances E. coli O157:H7 adhesion to epithelial cells.
FIG 6.
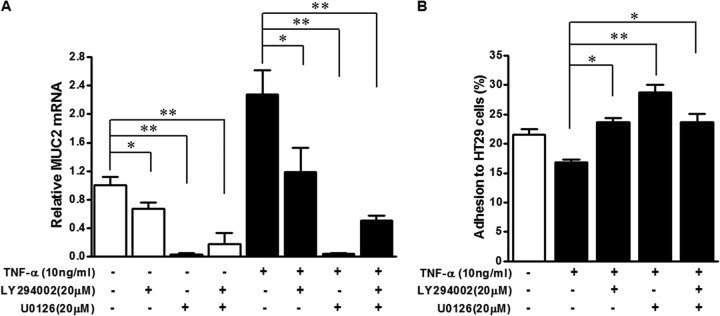
Akt and ERK1/2 inhibition blunt MUC2 expression and increase E. coli O157:H7 adhesion to HT-29 cells. (A) MUC2 mRNA expression in HT-29 cells untreated (−) or treated with 10 ng/ml TNF-α (+) and untreated (−) or treated with PI3K/Akt inhibitor (LY294002) or MAPK/ERK1/2 inhibitor (U0126) (+). (B) Adhesion of E. coli O157:H7 to HT-29 cells untreated (−) or treated with 10 ng/ml TNF-α (+) and untreated (−) or treated with LY294002 or U0126 (+). **, P < 0.01; *, P < 0.05. Shown are means and SEM; n = 4 for mRNA expression and n = 8 for adhesion.
DISCUSSION
Mucins produced by goblet cells form a mucus layer that provides a defensive barrier between the epithelial cells and invading pathogens, including E. coli O157:H7. In order to colonize epithelial cells, E. coli O157:H7 must penetrate the mucus layer. The precise role that mucins play in E. coli O157:H7 epithelial colonization has not been fully elucidated. Mucin2 is the major secreted mucin, whereas mucin2 and mucin3 are membrane-bound mucins in the intestine. HT-29 cells express MUC2 (18, 30). Mucin expression is upregulated by inflammatory cytokines (18–21), which are induced by NF-κB signaling (31), as well as STAT signaling (32). Consistent with these reports, we observed that TNF-α treatment enhanced mucin mRNA expression in HT-29 cells and enhanced mucin staining in both HT-29 cells and cattle colonic explant tissues. These data are consistent with previous reports showing that upon E. coli O157:H7 infection, MUC2 mRNA expression increased, which is in line with an innate defense role. Consistent with our observations, it has been reported that mucin secretion is increased in response to intestinal infection with Citrobacter rodentium (33) and Shigella dysenteriae (34).
The inflammatory response is an innate defense mechanism. However, some pathogens can disrupt host inflammatory responses to avoid recognition by the innate immune system and may even exploit it to enhance their colonization. It has been previously reported that enterohemorrhagic E. coli (EHEC) infection suppresses TNF-α-induced activation of the NF-κB signal transduction cascade in host cells (35–37). Our results indicated that E. coli O157:H7 attenuated both basal and TNF-α-induced activation of NF-κB p65 signaling in HT-29 cells 3 h postinfection. In contrast to our results, a previous study indicated that EHEC infection enhanced NF-κB signaling in T84 cells (24). IL-18 is an important inflammatory cytokine involved in both innate and adaptive immunity. IL-18 mRNA expression in cardiomyocytes was upregulated by TNF-α through activation of NF-κB (38). Consistently, we found that TNF-α enhanced IL-18 expression and secretion in HT-29 cells, while E. coli O157:H7 infection suppressed IL-18 production, which is consistent with data indicating that E. coli O157:H7 infection downregulates NF-κB signaling (35–37).
Despite the suppression of NF-κB signaling and IL-18 secretion, E. coli O157:H7 upregulated IL-8 production in control and TNF-α-treated HT-29 cells upon infection, indicating that E. coli O157:H7 infection might stimulate IL-8 expression/production independently of the NF-κB signaling pathway. This is in agreement with previous studies showing that E. coli O157:H7 infection elevated IL-8 mRNA expression in T84 cells (24, 39) and Caco-2 cells (40). In addition, Bellmeyer et al. (24) reported that EHEC infection reduced IL-8 production in HT-29 cells in response to TNF-α challenge. MAPK/ERK1/2 signaling was reported to be involved in upregulation of IL-8 mRNA expression by epithelial cells in response to E. coli O157:H7 infection (39, 40). Phosphorylation of ERK 1/2 was increased in Caco-2 cells 1 to 2 h post-EHEC infection; however, the extent of phosphorylation of ERK was dramatically decreased by 3 h postinfection (40). We found that E. coli O157:H7 inhibited ERK 1/2 activation in HT-29 cells 3 h postinfection. Similar results were found in Caco-2 cells (40), but not in T84 cells (39). A possible reason for these discrepant results could be differences among these cell lines in expression of globotriaosylceramide-3 (Gb3), a receptor for Shiga toxins. Only about 5% of T84 cells are Gb3 positive, while 50% of Caco-2 cells and 90% of HT-29 cells are Gb3 positive (41). Thus, in Caco-2 and HT-29 cells, Shiga toxins might be involved in inhibiting the host immune response via a largely Gb3-mediated mechanism, which would have minor influence in T84 cells (42, 43). Inhibition of both NF-κB and ERK 1/2 signaling may be strategies by which E. coli O157:H7 evades the host response, facilitating pathogen survival and further colonization of the GI tract.
As a key inflammatory cytokine, the role of TNF-α in inflammatory signaling has been well defined; however, its role in pathogen colonization of host cells is less defined. TNF-α exerts its biological effects mainly through TNF-α receptor 1 (TNFR1) and TNFR2 (44). Very recently, it was demonstrated that these two receptors have different functions in mucin expression and secretion. TNFR1 is needed for goblet cells to secrete mucus, while TNFR2 is needed for upregulation of mucin expression and synthesis (21). Both TNF-α receptors activate inflammatory NF-κB signaling. In addition, these receptors also activate PI3K/Akt and ERK1/2 signaling (45, 46). In line with previous observations (19), we observed that TNF-α treatment augmented both NF-κB and PI3K/Akt signaling in HT-29 cells. In addition, HT-29 cells exhibited enhanced Akt activation in response to E. coli O157:H7 infection. To further assess the roles of PI3K/Akt and MAPK/ERK1/2 signaling in mucin production and in E. coli O157:H7 epithelial adherence in response to TNF-α treatment, we used the specific inhibitors LY294002 and U0126 to inhibit PI3K/Akt and MAPK/ERK1/2 signaling, respectively (47, 48). Inhibition of PI3K/Akt and/or MAPK/ERK1/2 signaling reduced MUC2 expression and enhanced E. coli O157:H7 adherence, consistent with previous studies showing that Akt signaling (19) and ERK1/2 signaling (18) were involved in TNF-α-augmented MUC2 mRNA expression. Of note, in our study, U0126 was more effective than LY294002 in inhibiting MUC2 expression and promoting E. coli O157:H7 adhesion. This effect may be due to the dose and efficacy of the inhibitors and may not necessarily indicate that ERK1/2 is more critical for MUC2 expression than Akt.
Our data indicate that the inflammatory response induced by TNF-α reduces E. coli O157:H7 adhesion to gut epithelial cells and explants. The decreased adhesion was partially mediated by activating Akt and ERK1/2 signaling, which also enhanced the production and secretion of mucins (Fig. 7). These data suggest that the inflammatory cytokine response has a critical role in preventing E. coli O157:H7 epithelial colonization. Mucins, previously underappreciated factors in E. coli O157:H7 colonization, may have key roles in mediating E. coli O157:H7 gut colonization; thus, stimulation of gut epithelium inflammatory responses and associated mucin secretion might be a useful strategy to reduce E. coli O157:H7 gut colonization in beef cattle.
FIG 7.
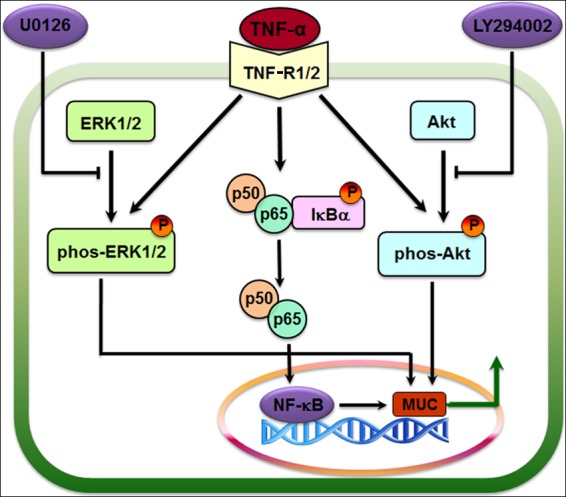
Proposed signaling pathways regulating MUC synthesis and secretion. TNF-α induces an inflammatory response through the NF-κB signaling pathway mediated by TNFR, with concomitant activation of MAPK/ERK1/2 and PI3K/Akt pathways. These pathways synergistically regulate MUC expression and synthesis. LY294002 and U0126 are specific inhibitors of the PI3K and ERK1/2 signaling pathways, respectively. The arrows indicate demonstrated effects. Steps blocked by inhibitors are indicated.
ACKNOWLEDGMENTS
This work was supported by USDA-AFRI 2010-65201-20599, the Agricultural Experiment Station at the University of Wyoming, and the Agricultural Research Center at Washington State University.
Footnotes
Published ahead of print 24 February 2014
REFERENCES
- 1.Melton-Celsa A, Mohawk K, Teel L, O'Brien A. 2012. Pathogenesis of Shiga-toxin producing Escherichia coli. Curr. Top. Microbiol. Immunol. 357:67–103. 10.1007/82_2011_176 [DOI] [PubMed] [Google Scholar]
- 2.Alam MJ, Zurek L. 2006. Seasonal prevalence of Escherichia coli O157:H7 in beef cattle feces. J. Food Prot. 69:3018–3020 [DOI] [PubMed] [Google Scholar]
- 3.Lim JY, Li J, Sheng H, Besser TE, Potter K, Hovde CJ. 2007. Escherichia coli O157:H7 colonization at the rectoanal junction of long-duration culture-positive cattle. Appl. Environ. Microbiol. 73:1380–1382. 10.1128/AEM.02242-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chase-Topping M, Gally D, Low C, Matthews L, Woolhouse M. 2008. Super-shedding and the link between human infection and livestock carriage of Escherichia coli O157. Nat. Rev. Microbiol. 6:904–912. 10.1038/nrmicro2029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arthur TM, Keen JE, Bosilevac JM, Brichta-Harhay DM, Kalchayanand N, Shackelford SD, Wheeler TL, Nou X, Koohmaraie M. 2009. Longitudinal study of Escherichia coli O157:H7 in a beef cattle feedlot and role of high-level shedders in hide contamination. Appl. Environ. Microbiol. 75:6515–6523. 10.1128/AEM.00081-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grant J, Wendelboe AM, Wendel A, Jepson B, Torres P, Smelser C, Rolfs RT. 2008. Spinach-associated Escherichia coli O157:H7 outbreak, Utah and New Mexico, 2006. Emerg. Infect. Dis. 14:1633–1636. 10.3201/eid1410.071341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.CDC. 2006. Ongoing multistate outbreak of Escherichia coli serotype O157:H7 infections associated with consumption of fresh spinach—United States, September 2006. MMWR Morb. Mortal. Wkly. Rep. 55:1045–1046 [PubMed] [Google Scholar]
- 8.Xicohtencatl-Cortes J, Monteiro-Neto V, Saldana Z, Ledesma MA, Puente JL, Giron JA. 2009. The type 4 pili of enterohemorrhagic Escherichia coli O157:H7 are multipurpose structures with pathogenic attributes. J. Bacteriol. 191:411–421. 10.1128/JB.01306-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frankel G, Phillips AD. 2008. Attaching effacing Escherichia coli and paradigms of Tir-triggered actin polymerization: getting off the pedestal. Cell Microbiol. 10:549–556. 10.1111/j.1462-5822.2007.01103.x [DOI] [PubMed] [Google Scholar]
- 10.Low AS, Holden N, Rosser T, Roe AJ, Constantinidou C, Hobman JL, Smith DG, Low JC, Gally DL. 2006. Analysis of fimbrial gene clusters and their expression in enterohaemorrhagic Escherichia coli O157:H7. Environ. Microbiol. 8:1033–1047. 10.1111/j.1462-2920.2006.00995.x [DOI] [PubMed] [Google Scholar]
- 11.Krogfelt KA. 1991. Bacterial adhesion: genetics, biogenesis, and role in pathogenesis of fimbrial adhesins of Escherichia coli. Rev. Infect. Dis. 13:721–735. 10.1093/clinids/13.4.721 [DOI] [PubMed] [Google Scholar]
- 12.Jordan DM, Cornick N, Torres AG, Dean-Nystrom EA, Kaper JB, Moon HW. 2004. Long polar fimbriae contribute to colonization by Escherichia coli O157:H7 in vivo. Infect. Immun. 72:6168–6171. 10.1128/IAI.72.10.6168-6171.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rendon MA, Saldana Z, Erdem AL, Monteiro-Neto V, Vazquez A, Kaper JB, Puente JL, Giron JA. 2007. Commensal and pathogenic Escherichia coli use a common pilus adherence factor for epithelial cell colonization. Proc. Natl. Acad. Sci. U. S. A. 104:10637–10642. 10.1073/pnas.0704104104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lievin-Le Moal V, Servin AL. 2006. The front line of enteric host defense against unwelcome intrusion of harmful microorganisms: mucins, antimicrobial peptides, and microbiota. Clin. Microbiol. Rev. 19:315–337. 10.1128/CMR.19.2.315-337.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maldonado-Contreras AL, McCormick BA. 2011. Intestinal epithelial cells and their role in innate mucosal immunity. Cell Tissue Res. 343:5–12. 10.1007/s00441-010-1082-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johansson ME, Larsson JM, Hansson GC. 2011. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc. Natl. Acad. Sci. U. S. A. 108(Suppl 1):4659–4665. 10.1073/pnas.1006451107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim YS, Ho SB. 2010. Intestinal goblet cells and mucins in health and disease: recent insights and progress. Curr. Gastroenterol. Rep. 12:319–330. 10.1007/s11894-010-0131-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iwashita J, Sato Y, Sugaya H, Takahashi N, Sasaki H, Abe T. 2003. mRNA of MUC2 is stimulated by IL-4, IL-13 or TNF-alpha through a mitogen-activated protein kinase pathway in human colon cancer cells. Immunol. Cell Biol. 81:275–282. 10.1046/j.1440-1711.2003.t01-1-01163.x [DOI] [PubMed] [Google Scholar]
- 19.Ahn DH, Crawley SC, Hokari R, Kato S, Yang SC, Li JD, Kim YS. 2005. TNF-alpha activates MUC2 transcription via NF-kappaB but inhibits via JNK activation. Cell Physiol. Biochem. 15:29–40. 10.1159/000083636 [DOI] [PubMed] [Google Scholar]
- 20.Enss ML, Cornberg M, Wagner S, Gebert A, Henrichs M, Eisenblatter R, Beil W, Kownatzki R, Hedrich HJ. 2000. Proinflammatory cytokines trigger MUC gene expression and mucin release in the intestinal cancer cell line LS180. Inflamm. Res. 49:162–169. 10.1007/s000110050576 [DOI] [PubMed] [Google Scholar]
- 21.McElroy SJ, Prince LS, Weitkamp JH, Reese J, Slaughter JC, Polk DB. 2011. Tumor necrosis factor receptor 1-dependent depletion of mucus in immature small intestine: a potential role in neonatal necrotizing enterocolitis. Am. J. Physiol. Gastrointest. Liver Physiol. 301:G656–G666. 10.1152/ajpgi.00550.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pellegrinet L, Rodilla V, Liu Z, Chen S, Koch U, Espinosa L, Kaestner KH, Kopan R, Lewis J, Radtke F. 2011. dll1- and dll4-mediated notch signaling are required for homeostasis of intestinal stem cells. Gastroenterology 140:1230–1240 e1231–e1237. 10.1053/j.gastro.2011.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu MJ, Du M, Nathanielsz PW, Ford SP. 2010. Maternal obesity up-regulates inflammatory signaling pathways and enhances cytokine expression in the mid-gestation sheep placenta. Placenta 31:387–391. 10.1016/j.placenta.2010.02.002 [DOI] [PubMed] [Google Scholar]
- 24.Bellmeyer A, Cotton C, Kanteti R, Koutsouris A, Viswanathan VK, Hecht G. 2009. Enterohemorrhagic Escherichia coli suppresses inflammatory response to cytokines and its own toxin. Am. J. Physiol. Gastrointest. Liver Physiol. 297:G576–G581. 10.1152/ajpgi.00050.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dijkstra G, Moshage H, Jansen PL. 2002. Blockade of NF-kappaB activation and donation of nitric oxide: new treatment options in inflammatory bowel disease? Scand. J. Gastroenterol. Suppl. 236:37–41 [DOI] [PubMed] [Google Scholar]
- 26.Naylor SW, Low JC, Besser TE, Mahajan A, Gunn GJ, Pearce MC, McKendrick IJ, Smith DG, Gally DL. 2003. Lymphoid follicle-dense mucosa at the terminal rectum is the principal site of colonization of enterohemorrhagic Escherichia coli O157:H7 in the bovine host. Infect. Immun. 71:1505–1512. 10.1128/IAI.71.3.1505-1512.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Low JC, McKendrick IJ, McKechnie C, Fenlon D, Naylor SW, Currie C, Smith DG, Allison L, Gally DL. 2005. Rectal carriage of enterohemorrhagic Escherichia coli O157 in slaughtered cattle. Appl. Environ. Microbiol. 71:93–97. 10.1128/AEM.71.1.93-97.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keen JE, Laegreid WW, Chitko-McKown CG, Durso LM, Bono JL. 2010. Distribution of Shiga-toxigenic Escherichia coli O157 in the gastrointestinal tract of naturally O157-shedding cattle at necropsy. Appl. Environ. Microbiol. 76:5278–5281. 10.1128/AEM.00400-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kunsch C, Rosen CA. 1993. NF-kappa B subunit-specific regulation of the interleukin-8 promoter. Mol. Cell. Biol. 13:6137–6146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mack DR, Michail S, Wei S, McDougall L, Hollingsworth MA. 1999. Probiotics inhibit enteropathogenic E. coli adherence in vitro by inducing intestinal mucin gene expression. Am. J. Physiol. 276:G941–G950 [DOI] [PubMed] [Google Scholar]
- 31.Wu J, Gong J, Geng J, Song Y. 2008. Deoxycholic acid induces the overexpression of intestinal mucin, MUC2, via NF-kB signaling pathway in human esophageal adenocarcinoma cells. BMC Cancer 8:333. 10.1186/1471-2407-8-333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mejias-Luque R, Linden SK, Garrido M, Tye H, Najdovska M, Jenkins BJ, Iglesias M, Ernst M, de Bolos C. 2010. Inflammation modulates the expression of the intestinal mucins MUC2 and MUC4 in gastric tumors. Oncogene 29:1753–1762. 10.1038/onc.2009.467 [DOI] [PubMed] [Google Scholar]
- 33.Linden SK, Florin TH, McGuckin MA. 2008. Mucin dynamics in intestinal bacterial infection. PLoS One 3:e3952. 10.1371/journal.pone.0003952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prakash R, Bharathi Raja S, Devaraj H, Devaraj SN. 2011. Up-regulation of MUC2 and IL-1beta expression in human colonic epithelial cells by Shigella and its interaction with mucins. PLoS One 6:e27046. 10.1371/journal.pone.0027046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao X, Wan F, Mateo K, Callegari E, Wang D, Deng W, Puente J, Li F, Chaussee MS, Finlay BB, Lenardo MJ, Hardwidge PR. 2009. Bacterial effector binding to ribosomal protein s3 subverts NF-kappaB function. PLoS Pathog. 5:e1000708. 10.1371/journal.ppat.1000708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gareau MG, Ho NK, Brenner D, Sousa AJ, Lebourhis L, Mak TW, Girardin SE, Philpott DJ, Sherman PM. 2011. Enterohaemorrhagic, but not enteropathogenic, Escherichia coli infection of epithelial cells disrupts signalling responses to tumour necrosis factor-alpha. Microbiology 157:2963–2973. 10.1099/mic.0.051094-0 [DOI] [PubMed] [Google Scholar]
- 37.Hauf N, Chakraborty T. 2003. Suppression of NF-kappa B activation and proinflammatory cytokine expression by Shiga toxin-producing Escherichia coli. J. Immunol. 170:2074–2082 [DOI] [PubMed] [Google Scholar]
- 38.Chandrasekar B, Colston JT, de la Rosa SD, Rao PP, Freeman GL. 2003. TNF-alpha and H2O2 induce IL-18 and IL-18R beta expression in cardiomyocytes via NF-kappa B activation. Biochem. Biophys. Res. Commun. 303:1152–1158. 10.1016/S0006-291X(03)00496-0 [DOI] [PubMed] [Google Scholar]
- 39.Dahan S, Busuttil V, Imbert V, Peyron JF, Rampal P, Czerucka D. 2002. Enterohemorrhagic Escherichia coli infection induces interleukin-8 production via activation of mitogen-activated protein kinases and the transcription factors NF-kappaB and AP-1 in T84 cells. Infect. Immun. 70:2304–2310. 10.1128/IAI.70.5.2304-2310.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berin MC, Darfeuille-Michaud A, Egan LJ, Miyamoto Y, Kagnoff MF. 2002. Role of EHEC O157:H7 virulence factors in the activation of intestinal epithelial cell NF-kappaB and MAP kinase pathways and the upregulated expression of interleukin 8. Cell Microbiol. 4:635–648. 10.1046/j.1462-5822.2002.00218.x [DOI] [PubMed] [Google Scholar]
- 41.Kovbasnjuk O, Mourtazina R, Baibakov B, Wang T, Elowsky C, Choti MA, Kane A, Donowitz M. 2005. The glycosphingolipid globotriaosylceramide in the metastatic transformation of colon cancer. Proc. Natl. Acad. Sci. U. S. A. 102:19087–19092. 10.1073/pnas.0506474102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gobert AP, Vareille M, Glasser AL, Hindre T, de Sablet T, Martin C. 2007. Shiga toxin produced by enterohemorrhagic Escherichia coli inhibits PI3K/NF-kappaB signaling pathway in globotriaosylceramide-3-negative human intestinal epithelial cells. J. Immunol. 178:8168–8174 [DOI] [PubMed] [Google Scholar]
- 43.Ho NK, Ossa JC, Silphaduang U, Johnson R, Johnson-Henry KC, Sherman PM. 2012. Enterohemorrhagic Escherichia coli O157:H7 Shiga toxins inhibit gamma interferon-mediated cellular activation. Infect. Immun. 80:2307–2315. 10.1128/IAI.00255-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Naude PJ, den Boer JA, Luiten PG, Eisel UL. 2011. Tumor necrosis factor receptor cross-talk. FEBS J. 278:888–898. 10.1111/j.1742-4658.2011.08017.x [DOI] [PubMed] [Google Scholar]
- 45.Hendarmin L, Sandra F, Nakao Y, Ohishi M, Nakamura N. 2005. TNFalpha played a role in induction of Akt and MAPK signals in ameloblastoma. Oral Oncol. 41:375–382. 10.1016/j.oraloncology.2004.09.014 [DOI] [PubMed] [Google Scholar]
- 46.Rivas MA, Carnevale RP, Proietti CJ, Rosemblit C, Beguelin W, Salatino M, Charreau EH, Frahm I, Sapia S, Brouckaert P, Elizalde PV, Schillaci R. 2008. TNF alpha acting on TNFR1 promotes breast cancer growth via p42/P44 MAPK, JNK, Akt and NF-kappa B-dependent pathways. Exp. Cell Res. 314:509–529. 10.1016/j.yexcr.2007.10.005 [DOI] [PubMed] [Google Scholar]
- 47.Takenouchi-Ohkubo N, Moro I, Mukae S, Kaneko Y, Komiyama K. 2008. Tumour necrosis factor-alpha-mediated human polymeric immunoglobulin receptor expression is regulated by both mitogen-activated protein kinase and phosphatidylinositol-3-kinase in HT-29 cell line. Immunology 123:500–507. 10.1111/j.1365-2567.2007.02716.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lea MA, Ibeh C, Shah N, Moyer MP. 2007. Induction of differentiation of colon cancer cells by combined inhibition of kinases and histone deacetylase. Anticancer Res. 27:741–748 [PubMed] [Google Scholar]