

Abstract
Treponema denticola is a predominantly subgingival oral spirochete closely associated with periodontal disease and has been detected in atherosclerosis. This study was designed to evaluate causative links between periodontal disease induced by chronic oral T. denticola infection and atherosclerosis in hyperlipidemic ApoE−/− mice. ApoE−/− mice (n = 24) were orally infected with T. denticola ATCC 35404 and were euthanized after 12 and 24 weeks. T. denticola genomic DNA was detected in oral plaque samples, indicating colonization of the oral cavity. Infection elicited significantly (P = 0.0172) higher IgG antibody levels and enhanced intrabony defects than sham infection. T. denticola-infected mice had higher levels of horizontal alveolar bone resorption than sham-infected mice and an associated significant increase in aortic plaque area (P ≤ 0.05). Increased atherosclerotic plaque correlated with reduced serum nitric oxide (NO) levels and increased serum-oxidized low-density lipoprotein (LDL) levels compared to those of sham-infected mice. T. denticola infection altered the expression of genes known to be involved in atherosclerotic development, including the leukocyte/endothelial cell adhesion gene (Thbs4), the connective tissue growth factor gene (Ctgf), and the selectin-E gene (Sele). Fluorescent in situ hybridization (FISH) revealed T. denticola clusters in both gingival and aortic tissue of infected mice. This is the first study examining the potential causative role of chronic T. denticola periodontal infection and vascular atherosclerosis in vivo in hyperlipidemic ApoE−/− mice. T. denticola is closely associated with periodontal disease and the rapid progression of atheroma in ApoE−/− mice. These studies confirm a causal link for active oral T. denticola infection with both atheroma and periodontal disease.
INTRODUCTION
Atherosclerotic vascular disease (ASVD) is a chronic inflammatory disease of large arteries characterized by the invasion, proliferation, and accumulation of cells from the arterial media (smooth muscle cells) and the circulating blood (monocytes/macrophages and T lymphocytes) in the intimal layer, with deposition of connective tissue and lipids. ASVD is the leading cause of death globally and has very high associated disability and mortality through disabling angina, myocardial infarction (heart attack), arrhythmias, and heart failure as well as cerebrovascular accidents (strokes) and peripheral arterial disease requiring amputations (1). Infectious agents represent a major source of systemic inflammatory response activation with the potential to accelerate plaque growth and instability (2). There is substantial evidence (1–5) demonstrating an association between the induction of inflammatory responses induced by infectious agents and the acceleration of atherosclerosis. Among the various infectious agents, periodontal pathogens are prominent contenders because of the chronic inflammation associated with periodontal disease (PD).
Periodontal diseases are complex multifactorial diseases caused by polymicrobial subgingival biofilm with immune and inflammatory responses. A distinct pathogenic consortium of Porphyromonas gingivalis, Treponema denticola, and Tannerella forsythia is found in subgingival plaque in severe periodontitis. T. denticola is the predominant spirochete in human subgingival plaque and is associated with chronic periodontitis, acute necrotizing ulcerative gingivitis, endodontic infections, and acute dental abscesses (6–8). T. denticola possesses several virulence factors, such as the major surface protein (MSP), cell-associated lipooligosaccharide, chymotrypsin-like protease (dentilisin), peptidoglycan, cystalysin, several peptidases, and a phosphatase which causes host immune cells to express molecular mediators that destroy periodontal connective tissue (7–9).
We have reported previously that oral infections with T. denticola resulted in colonization of the rat oral cavity, induction of gingival inflammation, a specific immune response, and significant alveolar bone loss (10, 11). In a murine calvarial model of inflammation, bone resorption was characterized by distinct host transcriptional profiles (12) (inflammatory mediators, cell adhesion, extracellular matrix [ECM] interactions, and cell cycle components) that demonstrate the acute pathogenicity of T. denticola. These features of aggressive oral pathology have been reported to correlate with the detection of T. denticola aggregated antigenic particles in human atherosclerotic lesions (13) in carotid arterial specimens and atheromatous plaques by fluorescent in situ hybridization (FISH) (14); however, a direct causative relation has not been proven. Additionally, seven oral spirochetes, including T. denticola, have been detected in Alzheimer patients' brains (15). Recently, Okuda et al. (16) demonstrated that T. denticola activates human endothelial cells by inducing interleukin-8 (IL-8) and macrophage chemoattractant protein-1 expression.
After successful colonization of the oral cavity, these bacteria can penetrate gingival tissues and become disseminated through blood vessels, with the potential to seed the heart and the cardiovascular endothelium in medium to large arteries, such as the aorta, coronary, and carotid arteries. These infectious spirochetes can also stimulate inflammatory cytokines either through direct invasion or through increased damage due to the activation of inflammatory cell responses (17). This response is in part because of the spirochete's inherent ability to release highly proteolytic vesicles, which degrade cellular tight junction proteins and the intracellular matrix (18). Although T. denticola is both an oral pathogen and associated with areas of atheroma formation, a direct causative association between T. denticola oral infection and arterial plaque growth has not yet been demonstrated in humans. We assess here the impact of active T. denticola chronic oral infection and disease on atherosclerotic plaque growth in a hyperlipidemic ApoE−/− mouse model with concomitant analysis of arterial infection, endothelial dysfunction and active inflammatory responses, modified lipid profiles, and modified gene expression.
MATERIALS AND METHODS
Bacterial strain and growth conditions.
T. denticola ATCC 35404 was grown under anaerobic conditions (85% N2, 10% H2, and 5% CO2) at 37°C in a Coy anaerobic chamber as described previously (19). For detailed methods, see the supplemental material.
Mouse strain and infection.
Twenty-four 10-week-old male ApoE−/− mice (20) (strain B6.129P2-ApoEtm1Unc/J) were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). A total of 109 T. denticola cells were administered orally to mice every third week for four consecutive days until euthanized at 12 and 24 weeks (Fig. 1A). Sham-infected mice received a 1:1 mixture of reduced transport medium (RTF) and 4% carboxymethyl cellulose (CMC). For detailed methods, see the supplemental material. Blood was collected during euthanasia at 12 and 24 weeks after initial infection, and sera were stored at −20°C for immunoglobulin G (IgG) and IgM antibody analysis (21). All animal procedures were approved by the University of Florida Institutional Animal Care and Use Committee (IACUC), protocol number 201004539.
FIG 1.
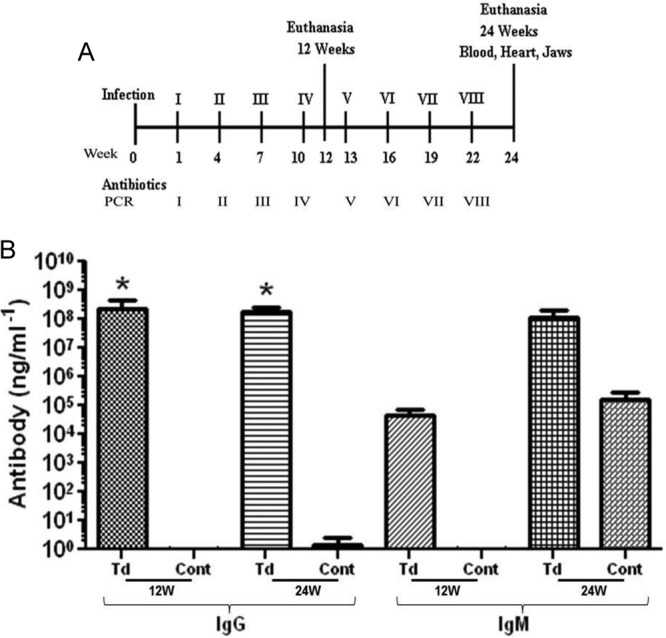
T. denticola infection schedule and spirochete-specific serum antibody levels in ApoE−/− mice. (A) Schematic diagram of the experimental design illustrating T. denticola infections, oral plaque sampling, euthanasia, and collection of blood and tissue specimens. (B) Serum IgG and IgM antibody levels in T. denticola-infected mice (Td) and sham-infected mice (Cont). Each bar represents the group mean (n = 12 mice) antibody level. T. denticola-infected mice at both 12 and 24 weeks had statistically significant higher levels of IgG than the sham-infected controls (*, P ≤ 0.05). All the tests were performed in triplicates. Data points and error bars represent means and standard deviations from three independent experiments.
Oral colonization/infection with T. denticola.
Oral microbial samples from mice were collected using sterile cotton swabs at pre- and postinfection times. Mouse oral cavities were swabbed 72 h after infection, and microbial samples were collected from all the infected ApoE−/− mice (Fig. 1A). Subsequently, PCR was performed using 16S rRNA gene species-specific PCR oligonucleotide primers 5′-TAATACCGAATGTGCTCATTTACAT-3′ and 5′-CTGCCATATCTCTATGTCATTGCTCTT-3′ (10), by using a Bio-Rad thermal cycler as described previously (21).
Serum antibody analysis.
Blood was collected from the cardiac puncture of each mouse at the time of euthanasia and was used to determine IgG and IgM antibody concentrations, using a standard enzyme-linked immunosorbent assay (ELISA) protocol (19, 21). For detailed methods, see the supplemental material.
Morphometric analysis of horizontal alveolar bone resorption.
The pattern of alveolar bone resorption induced by T. denticola was measured by the morphometric method (19, 21). For detailed methods, see the supplemental material.
Histomorphometric analysis of atherosclerotic plaque.
Hearts (n = 6) and aortas (n = 6) were fixed in 10% neutral buffered formalin and embedded in paraffin. For detailed methods, see the supplemental material. Mean plaque area, lumen and internal elastic lamina (IEL) areas, and intimal and medial thickness ratios were measured by using the Image Pro system MC 6.0 software program (Olympus America, Center Valley, PA, USA).
FISH.
Fluorescence in situ hybridization (FISH) was performed on formalin-fixed paraffin-embedded jaw and aortic tissue sections using a 16S rRNA T. denticola-specific oligonucleotide probe, 5′-CATGACTACCGTCATCAAAGAAGC-3′, labeled with Alexa Fluor 568 (22). For detailed methods, see the supplemental material.
Determination of serum lipoprotein and SAA in ApoE−/− mice.
Serum contents of lipoproteins (chylomicrons, very low-density lipoprotein [VLDL], low-density lipoprotein [LDL], and high-density lipoprotein [HDL]), total cholesterol, and triglycerides were determined by the high-pressure liquid chromatography principle using the method of LipoSearch (Skylight Biotech Inc., Akita, Japan). Acute-phase reactant serum amyloid A (SAA) concentrations were determined using the mouse serum amyloid A ELISA kit (Kamiya Biomedical Company, Seattle, WA). For detailed methods, see the supplemental material.
Determination of serum nitric oxide (NO) and oxidized LDL levels in ApoE−/− mice.
Sera obtained from the blood collected from the cardiac puncture of mice at the time of euthanasia from T. denticola-infected and sham-infected mice (n = 6) at 24 weeks were used to detect serum NO (μM) concentrations using a nitric oxide fluorometric assay kit (Bio Vision Inc., Milpitas, CA) and oxidized LDL levels using the mouse-oxidized low-density lipoprotein antibody (OLAb) ELISA kit (TSZ ELISA, Waltham, MA, USA).
Mouse atherosclerosis RT2 profiler PCR array.
Expression levels of 84 preselected atherosclerosis-related genes were examined in aortas of infected (n = 3) and control (n = 3) mice by quantitative reverse transcription-PCR (qRT-PCR) with the RT2 profiler mouse atherosclerosis PCR array (SABiosciences, Valencia, CA) (23). For detailed methods, see the supplemental material.
Evaluation of inflammatory cytokine array in mouse serum.
Mouse sera from infected (n = 10, both at 12 and 24 weeks) and control (n = 10, both time points) mice were pooled and used to detect 40 inflammatory cytokines on the RayBiotech mouse inflammatory cytokine glass chip array (RayBiotech, Inc., Norcross, GA), according to the manufacturer's protocol. For detailed methods, see the supplemental material.
Statistical analysis.
The alveolar bone resorption and antibody analysis data are presented as means ± standard deviations (Prism 4; GraphPad Software, San Diego, CA). One-way analysis of variance (ANOVA) with a Bonferroni posttest was done to compare multiple groups. P values of <0.05 were considered statistically significant. For the detailed analysis, see the supplemental material.
RESULTS
Oral colonization of T. denticola.
The infected mouse oral cavities were swabbed 72 h after each week of infection, and colony PCR was performed on these samples to test for T. denticola genomic DNA. Following the second infection, 12 out of 24 mice (50%) were colonized with T. denticola, and by the seventh infection, 90% of the mice were colonized with T. denticola (see Table S1 in the supplemental material). As no stable chronic infection was observed in mouse oral cavities, periodic reinfection was performed in the study to simulate conditions tantamount to chronic infection.
T. denticola-induced systemic antibody response.
Serum IgG antibody analysis of T. denticola orally infected mice at 12 and 24 weeks demonstrated robust generation of the humoral antibody response against the T. denticola infection. IgG levels in infected mice were significantly higher than those in control mice (Fig. 1B; P < 0.01). IgM levels in infected mice at 12 and 24 weeks were approximately higher than those in control mice.
T. denticola oral infection induces alveolar bone resorption and intrabony defects.
To assess whether chronic oral infection had induced alveolar bone resorption and intrabony defects, histomorphometry was performed to determine horizontal alveolar bone loss between the alveolar bone crest (ABC) and the cementoenamel junction (CEJ) in infected and control mouse mandibles and maxillae. Mice at both 12 and 24 weeks had a statistically significant increase (Fig. 2A, B, C, and E) in alveolar bone loss relative to that of control mice (Fig. 2D and F). Further, we observed intrabony defect in 23% of the sites analyzed in the infected mice at 12 weeks compared to 9% in controls and in 19% of infected mice at 24 weeks compared to 5% in controls (Fig. 2G and H).
FIG 2.

Horizontal alveolar bone resorption of ApoE−/− mice following T. denticola infection. (A and B) Morphometric analysis of 12- and 24-week total horizontal alveolar bone resorption in mice (n = 12) (***, P < 0.001; **, P < 0.005). Each bar indicates the mean horizontal alveolar bone resorption. Measurements were made between the cementoenamel junction (CEJ) and alveolar bone crest (ABC) of three molar teeth by three independent individuals blinded to the treatment group. Error bars indicate standard deviations. Panels C to F are representative images of measureable bone resorption denoted by the area inside the red line. Mandible-lingual view of T. denticola-infected (C) and sham-infected (D) mice at 12 weeks; mandible lingual view of 24-week T. denticola-infected (E) and sham-infected (F) mice; (G) representative images of mandible palatal surfaces showing extensive interproximal intrabony defects (shown by black arrows) in mice infected with T. denticola at 24 weeks. M1, first molar; M2, second molar; M3, third molar; H, sham-infected mice showing no visible intrabony defects.
T. denticola infection accelerated atherosclerotic plaque.
Histologic cross sections at the aorta in the infected mice at the level of the aortic valve and in the ascending aorta of mice after 12 weeks of T. denticola infection demonstrated significant increases in plaque area (Fig. 3A; P < 0.05), intimal thickness (Fig. 3B), and intimal/medial thickness ratios (Fig. 3C; P < 0.05) compared to those of sham-infected groups. As expected, there was no significant difference in medial thickness (Fig. 3D). T. denticola-infected mice had highly significant increases in plaque areas, with the characteristic presence of foam cells and invading inflammatory mononuclear cells (Fig. 3E and G), compared to those of sham-infected mice (Fig. 3F and H). Further, at 24 weeks, T. denticola-infected mice also had significantly larger (P < 0.01) plaque areas (Fig. 3I) and intimal/medial thickness ratios (P < 0.05) (Fig. 3K) than sham-infected mice. There was a trend toward increased aortic intimal layer thickness (Fig. 3J) and medial layer thickness (Fig. 3L) between the T. denticola-infected mice and sham-infected mice at 24 weeks, but this trend did not reach statistical significance. Twenty-four weeks of infection demonstrated plaque progression and larger plaques in the ascending aorta at the level of the aortic valve.
FIG 3.

T. denticola infection induced an increase in atherosclerotic plaque growth in the ascending aorta of ApoE−/− mice in comparison to sham-infected mice. (A) Bar graph of total aortic plaque area (mm2) in T. denticola-infected ApoE−/− mice at 12 weeks (*, P < 0.05); (B) intimal thickness (mm) (*, P < 0.05); (C) intimal/medial thickness ratios in T. denticola-infected mice; (D) medial thickness (mm); (E) representative cross section of T. denticola-infected mouse aorta at 12 weeks at the level of the aortic valve showing a larger plaque; (F) ascending aortic cross section at the level of aortic valve from a sham-infected mouse at 12 weeks; (G) ascending aorta with large globular plaque in a T. denticola-infected mouse at 24 weeks with increased macrophage infiltration and large cholesterol crystals; (H) ascending aortic cross section from a control mouse at 24 weeks with smaller aortic plaque. Black arrowheads indicate plaque margins; thin arrows indicate plaque with cholesterol crystals. The adventitia is indicated by A, media by M, intima by I, and lumen by L. (I) Total aortic plaque area (mm2) in T. denticola-infected ApoE−/− mice at 24 weeks (**, P < 0.01); (J) intimal thickness (mm); (K) intimal/medial thickness ratios (*, P < 0.05); (L) medial thickness (mm) in the 24-week T. denticola-infected mice. Six animals were examined in each group at both 12 and 24 weeks of infection. Multiple cross sections were stained for measurement, and analysis was performed three independent times (2 to 3 sections per aortic area per mouse) by two individuals blinded to the treatment group. Magnification, ×100 for panels E and F and ×200 for panels G and H.
Localization of T. denticola in tissues by FISH.
A T. denticola species-specific probe was hybridized with the spirochetes present in gingival tissue and demonstrated spirochete morphology by a bright-orange fluorescence. The detection of spirochetes in gingival tissue of infected mice at 12 weeks demonstrated the primary localization of the spirochetes in the gingival tissue after infection (Fig. 4A to C). Further, bacteria were also detected in the aortic tissues of infected mice at 12 and 24 weeks, and the intercellular localization of the bacteria was confirmed by confocal microscopy (Fig. 4D to I). The intercellular localization of T. denticola in aortic tissues by FISH reveals its invasiveness and its dissemination from the oral cavities into systemic circulation and subsequent penetration into vascular tissues.
FIG 4.

Fluorescent in situ hybridization (FISH) of T. denticola-infected mouse gingival and aortic tissue sections. (A) T. denticola-infected mouse gingival connective tissue at 12 weeks; (B) T. denticola-infected mouse gingival epithelium at 12 weeks; (C) T. denticola-infected gingival epithelial matrix at 12 weeks; (D) T. denticola-infected heart tissue (endothelium and smooth muscle) at 12 weeks; (E) T. denticola-infected aorta at 12 weeks; (F) T. denticola-infected aortic plaque at 24 weeks; (G) fluorescent image of T. denticola-infected aortic plaque tissue; (H) bright-field view (same location as panel G); (I) overlaid image (panels G and H) of T. denticola-infected aortic plaque tissue. P, plaque; L, lumen; RBC, red blood cells in the lumen. White arrows point to the presence of T. denticola in the infected aortic plaque tissue. Brightly fluorescent bacteria (white arrows) are seen with the rRNA species-specific T. denticola probe conjugated to Alexa Fluor 568. The procedure was repeated several times on different sections for confirmation, and a representative few were chosen.
Effect of oral infection on inflammatory marker SAA.
ELISA was used to assess the acute-phase reactant SAA at 24 weeks after oral bacterial infection. SAA levels were not changed significantly between the infected mice and sham-infected mice (Fig. 5A).
FIG 5.

T. denticola infection induced changes in serum of ApoE−/− mice. (A) Effects of oral infection with T. denticola on serum levels of serum amyloid A (SAA) (n = 6 mice); (B) alteration in serum lipoprotein (chylomicrons, VLDL [**, P < 0.01], LDL, HDL) levels in T. denticola-infected mice compared to those in controls (n = 6 mice); (C) significant increase in serum total cholesterol (**, P < 0.01) and serum total triglycerides levels in T. denticola-infected mice (n = 6); (D) significant changes in serum nitric oxide levels (*, P < 0.05) in T. denticola-infected mice compared to control mice (n = 6 mice in each group); (E) changes in serum-oxidized low-density lipoprotein (OxyLDL) levels after infection with T. denticola and sham-infected ApoE−/− mice (n = 6 in each group). All the tests were run in triplicates. Data points and error bars represent means and standard deviations for infected compared to control mice.
Effect of oral infection on atherosclerosis risk factors.
We determined the effect of T. denticola chronic oral infection (24 weeks) on atherosclerosis risk factors by analyzing serum lipoprotein and NO levels. T. denticola infection induced an increase in very low-density lipoprotein (VLDL) levels (Fig. 5B; P < 0.01). There was a significant difference in the level of total cholesterol between the T. denticola-infected (Fig. 5C; P < 0.01) and sham-infected mice. In addition, significant reductions in plasma NO (μM) levels were detected in T. denticola-infected mice (4.76 ± 0.25) compared to those of sham-infected (9.45 ± 0.24) mice (Fig. 5D; n = 6 mice per group; P < 0.05), suggesting endothelial dysfunction during active T. denticola infection. Further, when we measured the effect of chronic T. denticola infection on serum-oxidized LDL levels, the infected mice showed significantly higher levels (Fig. 5E; n = 6 mice per group; P < 0.05) than the sham-infected control mice.
T. denticola oral infection induced changes in atherosclerosis development-related gene expression.
We observed a great decrease in the expression of genes encoding blood clotting/coagulation cascade molecules fibrinogen α chain (Fga; 4.28-fold), fibrinogen β chain (Fgb; 3.29-fold), SerpinB2 (2.12-fold), and neuropeptide Y (Npy; 17-fold). In contrast, the Serpin1 gene showed a 3-fold increase in expression. Among the genes involved in endothelial cell adhesion, the connective tissue growth factor gene (Ctgf) was upregulated 3-fold and the selectin-E gene (Sele) by 4-fold, while expression of the thrombospondin-4 gene showed a 5-fold decrease. Among genes involved in the inflammatory response, expression of the chemokine ligand 5 gene (Ccl5) was decreased by 2-fold and IL1r2 was increased in expression by 2.7-fold. Antiapoptotic regulator gene Birc3 expression was increased 9-fold, while proapoptotic gene Fas expression was moderately decreased by 1.6-fold. The expression of the ATP-binding cassette subfamily A member 1 gene (Abca1) involved in lipid transport and metabolism was increased by 2.5-fold, while that of the apolipoprotein A1 gene (ApoA1) was decreased by 3.5-fold (Table 1).
TABLE 1.
Atherosclerosis-related gene expression changes to T. denticola infectiona
| Gene grouping | Gene (protein) | Fold change |
|---|---|---|
| Apoptosis | Birc3 (baculoviral IAP repeat containing 3) | 8.9 |
| Fas (tumor necrosis factor receptor superfamily, member 6) | −1.6 | |
| Blood clotting/coagulation cascade | Serpin1 | 2.9 |
| SerpinB2 | −2.12 | |
| Npy (neuropeptide Y) | −16.95 | |
| Fga (fibrinogen α chain) | −4.28 | |
| Fgb (fibrinogen β chain) | −3.29 | |
| Immune response | Ccl5 | −2.04 |
| IL1r2 | 2.7 | |
| Leukocyte/endothelial cell adhesion | Thbs4 (thrombospondin 4) | −5.29 |
| Ctgf (connective tissue growth factor) | 3.2 | |
| Sele (selectin E) | 3.9 | |
| Lipid transport and metabolism | Abca1 (ATP-binding cassette, subfamily A, member 1) | 2.52 |
| Apoa1 (apolipoprotein A-1) | −3.46 |
T. denticola-infected and sham-infected control aortic tissue samples at 24 weeks were processed and analyzed as described in Materials and Methods. Genes in each group that are physiologically related and are significantly altered are listed. Values represent the means of the data obtained from three independent experiments (n = 3) in each group. Bolding indicates atherosclerotic genes that are highly significantly up- or downregulated after T. denticola infection.
T. denticola oral infection induces serum inflammatory mediators.
There were important differences in the kinetics of cytokine and chemokine production between the T. denticola-infected mice and the control groups in 12- and 24-week infection periods. Twelve weeks of T. denticola infection demonstrated the highest expressions of IL-12 p40/p70, CD30L, and IL-4, followed by increased, but to a lesser extent, expression of the macrophage colony-stimulating factor (MCSF), monocyte chemoattractant protein (MCP), interferon-inducible T-cell alpha chemoattractant (ITAC), IL-1α, IL-3, and lymphotactin. In contrast, B-lymphocyte chemoattractant (BLC) was significantly decreased (Table 2). At 24 weeks of infection, there was an increase in expression of cytokines IL-4, IL-13, and tissue inhibitor of metalloproteinase-1 (TIMP-1) and a decrease in expression of eotaxin, keratinocyte chemoattractant (KC), lipopolysaccharide-induced CXC chemokine (Lix), eotaxin 2, and regulated on activation normal T cell expressed and secreted (RANTES) (Table 2).
TABLE 2.
Alteration of serum inflammatory markers after T. denticola infectiona
| Cytokine grouping | 12 wks |
24 wks |
||
|---|---|---|---|---|
| Cytokine | Fold difference | Cytokine | Fold difference | |
| Cell activation and proliferation | CD 30L | 245 | CD 30L | 2.8 |
| IL-1α | 2.9 | IL-1α | 2.5 | |
| IL-1β | 2.1 | IL-1β | 4.3 | |
| IL-3 | 2.8 | IL-4 | 172 | |
| IL-4 | 192 | IL-13 | 19.7 | |
| IL-6 | 2.1 | IL-17 | 1.6 | |
| IL-10 | 2.0 | |||
| IL12 p40/p70 | 3,436 | |||
| IL-17 | 2.1 | |||
| IL-12 p70 | 2.4 | |||
| Leukocyte chemoattractants | BLC | −2.8 | BLC | −1.7 |
| MCP-1 | 3.2 | Eotaxin | −2.3 | |
| KC | −2.2 | |||
| LIX | −6.3 | |||
| T-cell chemoattractants | I-TAC | 3.2 | Eotaxin 2 | −1.5 |
| MCSF | 5.0 | Fractalkine | 1.3 | |
| Lymphotactin | 2.4 | Lymphotactin | 2.2 | |
| RANTES | −1.6 | |||
| MMP inhibitor | TIMP-1 | −1.8 | TIMP- 1 | 1.9 |
Sera from T. denticola-infected mice (n = 6) and control mice (n = 6) at both 12 and 24 weeks were analyzed as described in Materials and Methods. Samples for inflammatory cytokine analysis were prepared by pooling equal volumes of sera from 10 mice per group. The data represent the means ± SD from the three independent experiments. Cytokines in each group that have physiological functions related to those cytokines which are significantly altered are listed. Bolding indicates serum cytokines that are highly altered after T. denticola infection.
DISCUSSION
The present study is designed to test the hypothesis that chronic oral infection with an established periodontal pathogen, specifically the highly prevalent T. denticola spirochete, directly contributes to the development and progression of atherosclerosis in a susceptible mouse model. The investigation of a chronic oral infection is based on the rationale that patients with periodontitis are chronically exposed to nonsymptomatic bacteremias at increased levels, for longer duration, and with greater microbial diversity of infections, which increase with periodontal disease severity (17). Dental procedures, including dental extraction, periodontal surgery, tooth scaling, and even tooth brushing and flossing, seed oral bacteria into the systemic circulation (24).We employed intraoral inoculation as the route of infection in the current study, as it fully mimics the natural route of physiological infection of the pathogen. Further, we chose to examine a chronic infection model of mice rather than a short-term infection, as this provides the best index for observing the natural means by which periodontal infection can affect tissues and organs involved in atherogenesis.
Numerous epidemiologic observational studies support the association between PD and ASVD (1, 25). Oral treponemes constitute a major bacterial population and are considered the most abundant oral bacterial pathogen group in deep periodontal pockets (26, 27). Due to the abundance of T. denticola at diseased sites, it is considered a prime contributor to periodontal-disease-associated tissue destruction (28). The observation that oral treponemes can penetrate intercellular junctions in vitro (29, 30) and disseminate systemically in humans to various organs, such as heart (13, 14) and brain (15), indicates the pathogenic potential of T. denticola and serves as the basis for our in vivo mouse experimentation. In the current study, using oral inoculations of T. denticola in ApoE−/− mice, we demonstrated the presence of T. denticola in the oral cavity, generation of specific antibody responses, and induction of both alveolar bone resorption and intrabony defects. These features establish development of a periodontitis model in ApoE−/− mice using oral infection of T. denticola.
Furthermore, we have demonstrated that infection of ApoE−/− mice with the oral spirochete T. denticola resulted in accelerated atherosclerosis associated with increased plaque lipid content and elevated serum VLDL, oxidized LDL, and total cholesterol as well as reduced serum NO levels and increased inflammatory mediator molecules compared with those of mock-infected control mice. The observation that serum NO levels were decreased suggests that endothelial function may be impaired in the infected mice. This is in agreement with the report that in periodontal disease patients, a decrease in NO bioavailability may lead to endothelial dysfunction and coronary artery disease (31). Collectively, the above-given data suggest that chronic inflammation caused by T. denticola may lead to production of proinflammatory cytokines, resulting in activation of endothelial cells and excessive induction of adhesion molecules, cytokines, growth factors, and vasoconstrictors.
Serum cytokine analysis revealed a transition of cytokine response from Th1 to Th2. During the first 12 weeks of T. denticola infection, mice exhibited strong production of IL-12, leading to a Th1 response, followed by a return to baseline levels at 24 weeks of infection. During the latter 12 weeks of the 24 weeks of chronic T. denticola infection, a strong induction of IL-13, a Th2 cytokine, was seen. This spirochete infection-induced shift in balance toward T-cell-adaptive response might be orchestrating a cellular environment leading to the development of atherosclerotic plaque, as observed in 24-week-infected mice. Further, our evaluation of atherosclerosis development-related gene expression in infected aorta suggested endothelial dysfunction, reduced apoptotic activity of macrophages, and a decrease in vascular smooth muscle mitogen, which corresponded with an increase in lesion formation associated with T. denticola infection. Identifying T. denticola in the aortic root at the level of the aortic valve from orally infected mice provides important insights into the chronology of spirochete adhesion/colonization, penetration of intracellular junctions, dissemination into the circulating blood, and adhesion in aortic tissues. We employed 16S rRNA to specifically identify T. denticola cells that are actively synthesizing proteins (22), and hence the spirochete we identified in gingival and aortic tissue sections was alive and metabolically active at the time of euthanasia of mice.
Our results in the current study agree with previous studies by other groups studying Chlamydia pneumoniae (32) and P. gingivalis (5), which have highlighted a synergistic association between infection and hyperlipidemia, culminating in the development of atherosclerosis in mice. However, this is the first analysis demonstrating a direct correlation between oral infections and concurrent changes in arterial plaque growth, dyslipidemia, and endothelial dysfunction. Observation of a T. denticola-infected aortic root at the level of the aortic valve leaflets as well as infection in the ascending aorta and aortic arch segments would also suggest that other arteries, such as the carotid or coronary arteries, may also represent a potential target for disease mediated by chronic oral infection with recurrent bacteremia. The endothelium represents a continuum of interacting cells that extend throughout the arterial tree and even across the surface of the aortic valve, and thus the aortic valve may potentially be affected by chronic oral bacterial infections and recurrent bacteremia. Although a contribution of periodontal disease to ASVD is biologically plausible, as evident from the data obtained through observational human studies (1, 25), there are significant gaps in establishing a causal relationship. Using a combinatorial approach consisting of a natural course of infection and a long-term duration of infection within an effective mouse model for evaluating accelerated atherosclerosis, we demonstrate that chronic oral infection with T. denticola can initiate and accelerate atherosclerotic plaque progression.
It has been argued that the failure of antimicrobial treatment to alter ASVD outcomes is evidence against the infectious theory of atherosclerosis. However, the failure to improve clinical outcomes observed in intervention studies (33) after treatment with antibiotics does not exclude infection as a potential etiology of acute coronary events in humans. Atheroma development incited by oral bacterial pathogen infection may well be attributed to bacteria entering a resistant phase (34) that renders them resistant to treatment yet capable of inciting inflammation and to the inability of antimicrobials to effectively penetrate the atheromas (35). Thus, antibiotic treatment given at the stage of ASVD when intervention is performed may be too late to be effective, but treatment of oral pathogens may yet have a role in disease prevention. In conclusion, we present data with the first clear evidence that T. denticola can directly accelerate atherosclerosis in vivo and thus provide a rodent model for exploration of synergistic links between periodontal disease and atherosclerotic vascular disease.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH National Institute for Dental and Craniofacial Research (R01DE020820; to L. Kesavalu) and NIH National Center for Research Resources (1U54RR026140-01; to P. Gangula).
Footnotes
Published ahead of print 24 February 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01511-14.
REFERENCES
- 1.Lockhart PB, Bolger AF, Papapanou PN, Osinbowale O, Trevisan M, Levison ME, Taubert KA, Newburger JW, Gornik HL, Gewitz MH, Wilson WR, Smith SC, Jr, Baddour LM, American Heart Association Rheumatic Fever Endocarditis and Kawasaki Disease Committee of the Council on Cardiovascular Disease in the Young Council on Epidemiology and Prevention Council on Peripheral Vascular Disease and Council on Clinical Cardiology 2012. Periodontal disease and atherosclerotic vascular disease: does the evidence support an independent association? A scientific statement from the American Heart Association. Circulation 125:2520–2544. 10.1161/CIR.0b013e31825719f3 [DOI] [PubMed] [Google Scholar]
- 2.Libby P, Egan D, Skarlatos S. 1997. Roles of infectious agents in atherosclerosis and restenosis: an assessment of the evidence and need for future research. Circulation 96:4095–4103. 10.1161/01.CIR.96.11.4095 [DOI] [PubMed] [Google Scholar]
- 3.Haraszthy VI, Zambon JJ, Trevisan M, Zeid M, Genco RJ. 2000. Identification of periodontal pathogens in atheromatous plaques. J. Periodontol. 71:1554–1560. 10.1902/jop.2000.71.10.1554 [DOI] [PubMed] [Google Scholar]
- 4.Lalla E, Lamster IB, Hofmann MA, Bucciarelli L, Jerud AP, Tucker S, Lu Y, Papapanou PN, Schmidt AM. 2003. Oral infection with a periodontal pathogen accelerates early atherosclerosis in apolipoprotein E-null mice. Arterioscler. Thromb. Vasc. Biol. 23:1405–1411. 10.1161/01.ATV.0000082462.26258.FE [DOI] [PubMed] [Google Scholar]
- 5.Gibson FC, III, Hong C, Chou HH, Yumoto H, Chen J, Lien E, Wong J, Genco CA. 2004. Innate immune recognition of invasive bacteria accelerates atherosclerosis in apolipoprotein E-deficient mice. Circulation 109:2801–2806. 10.1161/01.CIR.0000129769.17895.F0 [DOI] [PubMed] [Google Scholar]
- 6.Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL., Jr 1998. Microbial complexes in subgingival plaque. J. Clin. Periodontol. 25:134–144. 10.1111/j.1600-051X.1998.tb02419.x [DOI] [PubMed] [Google Scholar]
- 7.Ishihara K. 2010. Virulence factors of Treponema denticola. Periodontol. 2000 54:117–135. 10.1111/j.1600-0757.2009.00345.x [DOI] [PubMed] [Google Scholar]
- 8.Dashper SG, Seers CA, Tan KH, Reynolds EC. 2011. Virulence factors of the oral spirochete Treponema denticola. J. Dent. Res. 90:691–703. 10.1177/0022034510385242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Visser MB, Ellen RP. 2011. New insights into the emerging role of oral spirochetes in periodontal disease. Clin. Microbiol. Infect. 17:502–512. 10.1111/j.1469-0691.2011.03460.x [DOI] [PubMed] [Google Scholar]
- 10.Kesavalu L, Sathishkumar S, Bakthavatchalu V, Matthews C, Dawson D, Steffen M, Ebersole JL. 2007. Rat model of polymicrobial infection, immunity and alveolar bone resorption in periodontal disease. Infect. Immun. 75:1704–1712. 10.1128/IAI.00733-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Verma RK, Rajapakse S, Meka A, Hamrick C, Pola S, Bhattacharyya I, Nair M, Wallet SM, Aukhil I, Kesavalu L. 2010. Porphyromonas gingivalis and Treponema denticola mixed microbial infection in a rat model of periodontal disease. Interdiscip. Perspect. Infect. Dis. 2010:605125. 10.1155/2010/605125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bakthavatchalu V, Meka A, Sathishkumar S, Lopez MC, Verma RK, Wallet SM, Bhattacharyya I, Boyce BF, Mans JJ, Lamont RJ, Baker HV, Ebersole JL, Kesavalu L. 2010. Molecular characterization of Treponema denticola infection-induced bone and soft tissue transcriptional profiles. Mol. Oral Microbiol. 25:260–274. 10.1111/j.2041-1014.2010.00575.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okuda K, Ishihara K, Nakagawa T, Hirayama A, Inayama Y, Okuda K. 2001. Detection of Treponema denticola in atherosclerotic lesions. J. Clin. Microbiol. 39:1114–1117. 10.1128/JCM.39.3.1114-1117.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cavrini F, Sambri V, Moter A, Servidio D, Marangoni A, Montebugnoli L, Foschi F, Prati C, Di Bartolomeo R, Cevenini R. 2005. Molecular detection of Treponema denticola and Porphyromonas gingivalis in carotid and aortic atheromatous plaques by FISH: report of two cases. J. Med. Microbiol. 54:93–96. 10.1099/jmm.0.45845-0 [DOI] [PubMed] [Google Scholar]
- 15.Riviere GR, Riviere KH, Smith KS. 2002. Molecular and immunological evidence of oral Treponema in the human brain and their association with Alzheimer's disease. Oral Microbiol. Immunol. 17:113–118. 10.1046/j.0902-0055.2001.00100.x [DOI] [PubMed] [Google Scholar]
- 16.Okuda T, Kimizuka R, Miyamoto M, Kato T, Yamada S, Okuda K, Ishihara K. 2007. Treponema denticola induces interleukin-8 and macrophage chemoattractant protein 1 production in human umbilical vein epithelial cells. Microbes Infect. 9:907–913. 10.1016/j.micinf.2007.03.009 [DOI] [PubMed] [Google Scholar]
- 17.Mattila KJ, Valtonen VV, Nieminen MS, Asikainen S. 1998. Role of infection as a risk factor for atherosclerosis, myocardial infarction, and stroke. Clin. Infect. Dis. 26:719–734. 10.1086/514570 [DOI] [PubMed] [Google Scholar]
- 18.Chi B, Qi M, Kuramitsu HK. 2003. Role of dentilysin in Treponema denticola epithelial cell layer penetration. Res. Microbiol. 154:637–643. 10.1016/j.resmic.2003.08.001 [DOI] [PubMed] [Google Scholar]
- 19.Rivera MF, Lee JY, Aneja M, Goswami V, Liu L, Velsko IM, Chukkapalli SS, Bhattacharyya I, Chen H, Lucas AR, Kesavalu LN. 2013. Polymicrobial infection with major periodontal pathogens induced periodontal disease and aortic atherosclerosis in hyperlipidemic ApoE (null) mice. PLoS One 8:e57178. 10.1371/journal.pone.0057178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang SH, Reddick RL, Piedrahita JA, Maeda N. 1992. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science 258:468–471. 10.1126/science.1411543 [DOI] [PubMed] [Google Scholar]
- 21.Bainbridge B, Verma RK, Eastman C, Yehia B, Rivera M, Moffatt C, Bhattacharyya I, Lamont RJ, Kesavalu L. 2010. Role of Porphyromonas gingivalis phosphoserine phosphatase enzyme SerB in inflammation, immune response, and induction of alveolar bone resorption in rats. Infect. Immun. 78:4560–4569. 10.1128/IAI.00703-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moter A, Gobel UB. 2000. Fluorescence in situ hybridization (FISH) for direct visualization of microorganisms. J. Microbiol. Methods 41:85–112. 10.1016/S0167-7012(00)00152-4 [DOI] [PubMed] [Google Scholar]
- 23.Manning-Tobin JJ, Moore KJ, Seimon TA, Bell SA, Sharuk M, Alvarez-Leite JI, de Winther MP, Tabas I, Freeman MW. 2009. Loss of SR-A and CD36 activity reduces atherosclerotic lesion complexity without abrogating foam cell formation in hyperlipidemic mice. Arterioscler. Thromb. Vasc. Biol. 29:19–26. 10.1161/ATVBAHA.108.176644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carroll GC, Sebor RJ. 1980. Dental flossing and its relationship to transient bacteremia. J. Periodontol. 51:691–692. 10.1902/jop.1980.51.12.691 [DOI] [PubMed] [Google Scholar]
- 25.Humphrey LL, Fu R, Buckley DI, Freeman M, Helfand M. 2008. Periodontal disease and coronary heart disease incidence: a systematic review and meta-analysis. J. Gen. Intern. Med. 23:2079–2086. 10.1007/s11606-008-0787-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dewhirst FE, Tamer MA, Ericson RE, Lau CN, Levanos VA, Boches SK, Galvin JL, Paster BJ. 2000. The diversity of periodontal spirochetes by 16S rRNA analysis. Oral Microbiol. Immunol. 15:196–202. 10.1034/j.1399-302x.2000.150308.x [DOI] [PubMed] [Google Scholar]
- 27.Ellen RP, Galimanas VB. 2005. Spirochetes at the forefront of periodontal infections. Periodontol. 2000 38:13–32. 10.1111/j.1600-0757.2005.00108.x [DOI] [PubMed] [Google Scholar]
- 28.Holt SC, Ebersole JL. 2005. Porphyromonas gingivalis, Treponema denticola, and Tannerella forsythia: the “red complex,” a prototype polybacterial pathogenic consortium in periodontitis. Periodontol. 2000 38:72–122. 10.1111/j.1600-0757.2005.00113.x [DOI] [PubMed] [Google Scholar]
- 29.Peters SR, Valdez M, Riviere G, Thomas DD. 1999. Adherence to and penetration through endothelial cells by oral treponemes. Oral Microbiol. Immunol. 14:379–383. 10.1034/j.1399-302X.1999.140609.x [DOI] [PubMed] [Google Scholar]
- 30.Lux R, Miller JN, Park NH, Shi W. 2001. Motility and chemotaxis in tissue penetration of oral epithelial cell layers by Treponema denticola. Infect. Immun. 69:6276–6283. 10.1128/IAI.69.10.6276-6283.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Higashi Y, Goto C, Hidaka T, Soga J, Nakamura S, Fujii Y, Hata T, Idei N, Fujimura N, Chayama K, Kihara Y, Taguchi A. 2009. Oral infection-inflammatory pathway, periodontitis, is a risk factor for endothelial dysfunction in patients with coronary artery disease. Atherosclerosis 206:604–610. 10.1016/j.atherosclerosis.2009.03.037 [DOI] [PubMed] [Google Scholar]
- 32.Blessing E, Campbell LA, Rosenfeld ME, Chough N, Kuo CC. 2001. Chlamydia pneumoniae infection accelerates hyperlipidemia induced atherosclerotic lesion development in C57BL/6J mice. Atherosclerosis 158:13–17. 10.1016/S0021-9150(00)00758-9 [DOI] [PubMed] [Google Scholar]
- 33.Andraws R, Berger JS, Brown DL. 2005. Effects of antibiotic therapy on outcomes of patients with coronary artery disease: a meta-analysis of randomized controlled trials. JAMA 293:2641–2647. 10.1001/jama.293.21.2641 [DOI] [PubMed] [Google Scholar]
- 34.Kalayoglu MV, Libby P, Byrne GL. 2002. Chlamydia pneumoniae as an emerging risk factor in cardiovascular disease. JAMA 288:2724–2731. 10.1001/jama.288.21.2724 [DOI] [PubMed] [Google Scholar]
- 35.Meier CR. 2000. Antibiotics in the prevention and treatment of coronary heart disease. J. Infect. Dis. 181(Suppl 3):S558–S562 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.