

Abstract
We report that mice deficient for the hematopoietic-specific, actin-bundling protein L-plastin (LPL) succumb rapidly to intratracheal pneumococcal infection. The increased susceptibility of LPL−/− mice to pulmonary pneumococcal challenge correlated with reduced numbers of alveolar macrophages, consistent with a critical role for this cell type in the immediate response to pneumococcal infection. LPL−/− mice demonstrated a very early clearance defect, with an almost 10-fold-higher bacterial burden in the bronchoalveolar lavage fluid 3 h following infection. Clearance of pneumococci from the alveolar space in LPL−/− mice was defective compared to that in Rag1−/− mice, which lack all B and T lymphocytes, indicating that innate immunity is defective in LPL−/− mice. We did not identify defects in neutrophil or monocyte recruitment or in the production of inflammatory cytokines or chemokines that would explain the early clearance defect. However, efficient alveolar macrophage regeneration following irradiation required LPL. We thus identify LPL as being key to alveolar macrophage development and essential to an effective antipneumococcal response. Further analysis of LPL−/− mice will illuminate critical regulators of the generation of alveolar macrophages and, thus, effective pulmonary innate immunity.
INTRODUCTION
Pneumococcal pneumonia remains a major cause of morbidity and mortality worldwide (1). Identification of regulators of pulmonary antipneumococcal immunity is critical to the design of new therapies to modulate the outcome of this disease. Initially, alveolar macrophages and alveolar epithelial cells respond to pulmonary pneumococcal invasion (2, 3) by triggering the production of inflammatory cytokines, such as interleukin-1 (IL-1) and tumor necrosis factor alpha (TNF-α). Alveolar macrophages also eliminate pathogens by engulfing and then killing pneumococci. Alveolar macrophages can effectively clear small numbers of pneumococci and prevent pneumonia. If, however, pneumococci are not cleared within 2 to 4 h, neutrophils and then monocytes are recruited (3). Neutrophils contain the dissemination of pneumococci through phagocytic killing, while peripheral monocytes clear still-viable pneumococci and dead neutrophils, controlling infection and mitigating inflammation.
Alveolar macrophages thus appear to be critical for early responses leading to good outcomes in pneumococcal pneumonia. The depletion of alveolar macrophages in mice using clodronate hindered pneumococcal clearance and diminished survival (4, 5). The influenza virus shifts the activation state of alveolar macrophages, which could contribute to increased postinfluenza susceptibility to pneumococcal pneumonia (6). Morphine and alcohol impair alveolar macrophages, which could explain the increased susceptibility of intravenous drug and alcohol users to pneumonia (4, 7). Elucidation of the molecular mechanisms that regulate alveolar macrophages is therefore crucial to an understanding of host-pathogen interactions during the critical first few hours of pneumococcal lung invasion.
L-Plastin (LPL) is an actin-bundling protein specifically expressed in hematopoietic cells (8, 9). LPL colocalizes with polymerized actin in macrophage podosomes, membrane ruffles, and phagocytic cups (10, 11). The critical roles of LPL in neutrophil integrin signaling, T and B lymphocyte motility, and T cell activation have been established (12–19), but a requirement for LPL in macrophage function has not. Here we show that LPL−/− mice challenged intratracheally (i.t.) with pneumococci were defective in early pathogen clearance and succumbed rapidly to infection despite directed antimicrobial therapy. Defective pathogen clearance correlated with reduced numbers of alveolar macrophages in the bronchoalveolar space, and regeneration of alveolar macrophages from bone marrow derived from LPL−/− mice was impaired. LPL is thus essential for the normal localization of alveolar macrophages in the bronchoalveolar space and, by extension, for the early clearance of pneumococci.
MATERIALS AND METHODS
Mice.
LPL−/− mice backcrossed to a C57BL/6 background were described previously (12, 14). Cohoused control wild-type (WT) mice were age matched (8 to 12 weeks) and gender matched. Unless otherwise specified, all mice were male. All experiments were approved by the Washington University Institutional Animal Care and Use Committee.
Antibodies and reagents.
Commercial antibodies to the following murine antigens were used: CD11b-fluorescein isothiocyanate (FITC), CD11c-phycoerythrin (PE), T cell receptor β (TCRβ)-FITC, CD3-PE, CD8-peridinin chlorophyll protein (PerCP)/Cy5.5, CD4-allophycocyanin (APC), Ly6C-APC (eBioscience); CD115-biotin, CD103-biotin, TCRγ/δ-biotin, B220-PE, streptavidin-APC/Cy7, CD45.2-PE/Cy7, CD45.1-PE/Cy7, I-Ak–APC, Ly6G-Pacific Blue, CD31-PE/Cy7, F4/80-PerCP/Cy5.5, annexin V-APC, gamma interferon (IFN-γ)-PE, Dx5-Pacific Blue (BioLegend), and NK1.1-APC (BD Biosciences). All samples were blocked with 1 μg Fc-block (2.4G2 hybridoma; ATCC). Cells were acquired with a Becton, Dickinson (Franklin Lakes, NJ) FACScan flow cytometer with DxP multicolor upgrades by Cytek Development Inc. (Woodland Park, NJ) and analyzed by using FlowJo software (TreeStar Inc., Ashland, OR).
Infection.
Streptococcus pneumoniae (ATCC 6303; serotype 3) cells from a frozen stock were grown overnight at 35°C on blood agar plates in a 5% CO2 incubator. Colonies were scraped from the plate and suspended in Dulbecco's phosphate-buffered saline (PBS) (DPBS). The suspension was then adjusted to a density of an A600 of 0.1 and diluted 1:20.
Mice were anesthetized with isoflurane. The trachea was surgically exposed, and 20 μl of the bacterial suspension (corresponding to approximately 3 × 104 CFU/animal) or DPBS was injected intratracheally. Mice were held upright for 10 s to drain the inoculum into the lungs. After incision closure, mice received 0.05 mg/kg of body weight buprenorphine (Hospira Inc., Lake Forest, IL) in 1 ml 0.9% saline via subcutaneous injection for fluid resuscitation and pain control (20, 21). During prolonged (>24-h) infections, a single dose of ceftriaxone (1 mg; 50 mg/kg body weight) was given 24 h after infection to mimic clinical therapy of pneumococcal pneumonia (22). Mice were monitored at least twice daily for signs/symptoms of distress.
For harvesting of tissues, mice were sacrificed by deep anesthesia with isoflurane. Blood was obtained by direct cardiac puncture and diluted for quantitative culture. For bronchoalveolar lavage (BAL), animals were placed supine, and the trachea was exposed by dissection. A 26-gauge intravenous (i.v.) catheter was inserted into the trachea, and 1 ml of sterile DPBS was injected and immediately withdrawn. The fluid was diluted from equal volumes for quantitative culture (21).
Pathology.
Lungs were perfused with 10 ml of sterile PBS to remove blood and then fixed in formalin and embedded in paraffin, and sections were stained with hematoxylin and eosin by the Washington University Histology core. Images were captured by using an Olympus BX60 microscope with an Olympus U Plan Fl 20× objective equipped with a Zeiss AxioCam and AxioVision software. The histopathological findings and grading of alveolar involvement were performed in a blind fashion. Each lung section was divided into quarters, and two randomly selected separate regions in each quarter were graded for inflammatory cell infiltrates involving the alveoli, airways, and pulmonary vasculature.
For staining of neutrophils, sections were rehydrated, and an antigen retrieval step was performed by using citric acid buffer. Sections were stained by using a rat anti-mouse neutrophil antibody (NIMP-R14; Abcam, Cambridge, MA), visualized by using a Vectastain ABC AP kit (Vector Laboratories, Burlingame, CA) with Alkaline Phosphatase Substrate kit III (Vector Laboratories), and counterstained with nuclear fast red stain. Images were obtained with an Olympus NanoZoomer 2.0-HT system. NDP.scan 2.5 and NDP.view were used for image acquisition and image viewing, respectively (Hamamatsu). The number of polymorphonuclear leukocytes (PMNs) was quantified by using Image ProPlus (Media Cybernetics, Bethesda, MD).
Cell isolation.
Lungs were perfused with 10 ml of sterile PBS and homogenized (23). Dead cells (Yellow Live/Dead Fixable Dead Cell Stain kit; Invitrogen) and nonhematopoietic (CD45neg) cells were excluded from the analysis. Cells were identified as indicated (Table 1) (23).
TABLE 1.
Panels of surface markers used to characterize populations of hematopoietic cellsa
| Cell type | Marker detected by: |
||||||
|---|---|---|---|---|---|---|---|
| FITC | PE | PerCP-Cy5.5 | PE-Cy7 | APC | APC-Cy7 | Pacific Blue | |
| PMNs | CD11b+ | B220− | CD8− | CD45+ | Ly6C− | CD115− | Ly6G+ |
| Monocytes | CD11b+ | B220− | CD8− | CD45+ | Ly6C+ | CD115+ | Ly6G− |
| B cells | CD11b− | B220+ | CD8− | CD45+ | Ly6C− | CD115− | Ly6G− |
| Macrophages | CD11b− | CD11chigh | Autofluor+ | CD45+ | I-Ak− | CD103− | Ly6G− |
| CD11b+ DCs | CD11b+ | CD11chigh | Autofluor− | CD45+ | I-Ak+ | CD103− | Ly6G− |
| CD103+ DCs | CD11b− | CD11chigh | Autofluor− | CD45+ | I-Ak+ | CD103+ | Ly6G− |
| CD4+ T cells | TCRβ+ | CD3+ | CD8− | CD45+ | CD4+ | TCRγ/δ− | Dx5− |
| CD8+ T cells | TCRβ+ | CD3+ | CD8+ | CD45+ | CD4− | TCRγ/δ− | Dx5− |
| γ/δ T cells | TCRβ− | CD3+ | CD8− | CD45+ | CD4− | TCRγ/δ+ | Dx5− |
| NK cells | CD3− | IFN-γ+/− | CD45+ | NK1.1+ | |||
| Progenitors | CD31+ | Ly6Cmid | |||||
| Peritoneal monocytes | B220− | F4/80− | CD115+ | ||||
| Peritoneal macrophages | B220− | F4/80+ | CD115+ | ||||
See reference 23. Dead cells were excluded from the analysis. Positive markers are in boldface type. CD45 was not included for cells isolated from bone marrow (progenitors) or from peritoneal wash specimens (monocytes and macrophages), as virtually all cells isolated from these sites are of hematopoietic origin. Abbreviations: PMNs, polymorphonuclear cells (neutrophils); DCs, dendritic cells; Autofluor, autofluorescence.
Determination of chemokine and cytokine concentrations.
BAL fluid recovered from infected WT and LPL−/− mice was assayed for selected cytokines and chemokines by using the Bio-Plex 23-plex mouse cytokine assay (Bio-Rad, Valencia, CA) according to the manufacturer's instructions.
In vivo phagocytosis assays.
WT and LPL mice were infected intratracheally with FITC-labeled pneumococci. Three hours following infection, BAL fluid was obtained. Fluorescence of extracellular FITC was quenched by washing with trypan blue. Alveolar macrophages were identified as CD45+ CD11chigh, and phagocytosis of pneumococci was measured by the shift in FITC fluorescence. We have confirmed by using additional markers (CD64, MerTK, and Siglec-F) that CD45+ CD11chigh cells in BAL fluid are primarily macrophages (data not shown). Cells in BAL fluid were quantified by flow cytometry.
Determination of intracellular IFN-γ production.
NK cells were assayed for IFN-γ production by using intracellular flow cytometry (24). For in vitro stimulation, splenocytes were cultured with either IL-12 (10 ng/ml; Peprotech) plus IL-15 (100 ng/ml; MBL International) or medium alone for a total of 4 h, with BD GolgiPlug (BD Biosciences) being added for the last 3 h. For assessment of IFN-γ production following pneumococcal infection, cell suspensions were generated from lungs as described above. After staining for surface markers, cells were fixed and permeabilized (Cytofix/Cytoperm; BD Biosciences) and then incubated with IFN-γ–PE.
Generation of bone marrow chimeras.
Twenty-four hours after irradiation with 900 rads, CD45.1+ WT mice were reconstituted with bone marrow from CD45.2+ WT or CD45.2+ LPL−/− mice. For competitive (mixed) bone marrow chimeras, CD45.1+/CD45.2+ WT recipients were irradiated and reconstituted with mixed bone marrow from congenically marked WT and LPL−/− mice. Three weeks after reconstitution, recipient mice were sacrificed, and tissues were analyzed for engrafted donor cells by flow cytometry.
Proliferation and cell death assays.
Proliferation of alveolar macrophages was determined by measuring bromodeoxyuridine (BrdU) incorporation (17). BrdU incorporation was assessed (BrdU-APC kit; BD Biosciences) 24 h after mice were injected intraperitoneally (i.p.) with 2 mg BrdU in PBS (10 mg/ml; BD Biosciences and Sigma-Aldrich, Saint Louis, MO). Cell viability was by determined using annexin V-APC staining according to the manufacturer's instructions (BioLegend) (25). The use of the APC fluorophore avoided overlap of the intrinsic autofluorescence of macrophages.
Statistics.
The mortality rates of infected WT and LPL−/− mice were compared with a Kaplan-Meier survival curve, and the P value to assess the significance of differences was determined by using a log-rank test. All other P values were determined by using the unpaired, two-tailed Mann-Whitney test. Data were analyzed and graphed by using GraphPad Prism 5 (GraphPad Software Inc., La Jolla, CA).
RESULTS
LPL−/− mice rapidly succumb to infection and fail to clear pneumococci from alveoli.
As no requirement for LPL expression by immune cells during infection has been reported previously, we first determined if LPL−/− mice were more susceptible to pneumococcal infection. We modeled pneumococcal pulmonary infection as this is the most common physiological route of infection and the most common organ affected by pneumococci. WT and LPL−/− mice were challenged intratracheally (i.t.) with S. pneumoniae serotype 3 cells. Mice were given a single dose of ceftriaxone (1 mg) 24 h after inoculation to mimic clinical therapy of patients presenting with pneumococcal pneumonia (22). Over 70% of LPL−/− mice rapidly succumbed to infection, while 80% of WT mice survived (Fig. 1A). Expression of LPL is thus essential for survival following pulmonary challenge with pneumococci.
FIG 1.
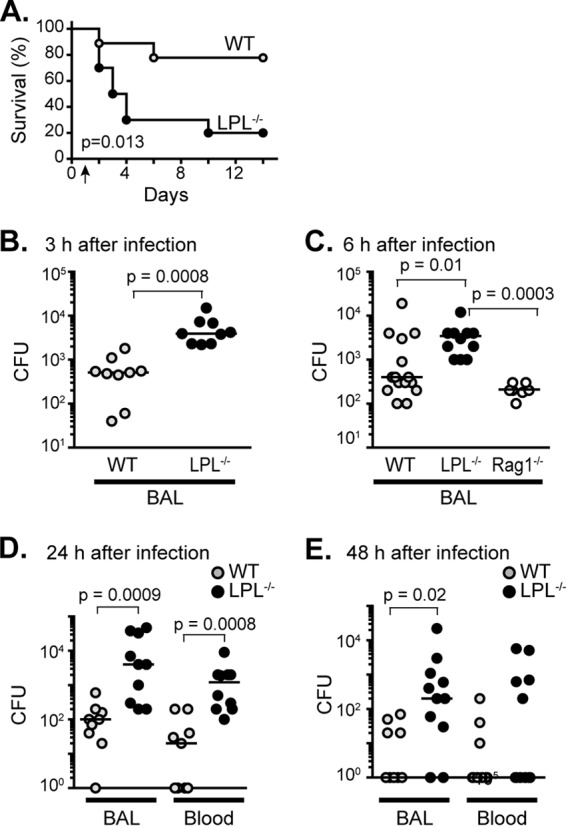
LPL−/− mice are profoundly susceptible to infection with pneumococci and exhibit a very early pneumococcal clearance defect. (A) Age- and gender-matched WT (n = 9; 5 female and 4 male) and LPL−/− (n = 10; 5 female and 5 male) mice were infected i.t. with S. pneumoniae serotype 3 strain ATCC 6303 at time zero. A single, nonsterilizing dose of ceftriaxone was given at 24 h (arrow). Mice were monitored for 2 weeks for morbidity and mortality. Data were pooled from 2 independent experiments. P values were determined by a log-rank test. (B to E) Colony counts from BAL fluid (B to E) or blood (D and E) of the indicated mice 3 h (B), 6 h (C), 24 h (D), and 48 h (E) after i.t. instillation of pneumococci. No antibiotic was given for panels B, C, and D, but a single nonsterilizing dose of ceftriaxone was given at 24 h for panel E, to correlate with the treatment data in panel A. Each point represents a single animal, lines represent median values, and P values were determined by using a two-tailed Mann-Whitney test. Data were from 2 (B to E) or 3 (C) independent experiments. For panels B to D, all mice were male. For panel E, mice of each genotype were male (n = 5) and female (n = 5).
The mortality of LPL−/− mice was associated with a failure to clear pneumococci from the alveolar space: at 3, 6, 24, and 48 h after challenge, LPL−/− mice had 10-fold more pneumococci in BAL fluid than did WT mice (Fig. 1B to E). The increased number of CFU/ml in the BAL fluid of LPL−/− mice at 3 h after challenge indicates a very early defect in pathogen clearance. Early in infection (3 and 6 h following challenge), we did not find dissemination of pneumococci to the bloodstream in either WT or LPL−/− mice (data not shown). However, by 24 and 48 h after infection, there was significantly increased dissemination into the bloodstream of LPL−/− mice compared to WT mice, likely reflecting the increased pneumococcal burden in BAL fluid (Fig. 1D and E). The pneumococcal burden in LPL−/− mice remained higher despite a single dose of ceftriaxone given 24 h after challenge (Fig. 1E), indicating that directed antibiotic therapy cannot rescue animals when very early pathogen clearance is defective.
The increased susceptibility to pneumococcal infection in LPL−/− mice appeared to result from a defect in innate immunity. Normal expression of LPL is restricted to hematopoietic cells (8, 9, 26–29). Thus, defective immunity in LPL−/− mice against pneumococcal challenge must lie within cells of hematopoietic origin. Furthermore, mice deficient for all T and B cells, and, thus, deficient for antibody production, but sufficient for all innate immune components (Rag1−/− mice) cleared pneumococci from the BAL fluid as effectively as WT mice 6 h after infection (Fig. 1C). Therefore, as the pneumococcal burden was significantly higher in LPL−/− mice than in Rag1−/− mice (Fig. 1C), the early defect in the host response of LPL−/− mice lies within innate immunity.
Diminished numbers of alveolar macrophages in BAL fluid of LPL−/− mice.
Alveolar macrophages provide the first line of defense against pulmonary pneumococcal infection. The very early pneumococcal clearance defect observed in LPL−/− mice suggested a defect in an innate immune cell type critical in the first hours following pneumococcal challenge, such as alveolar macrophages. We therefore quantified alveolar macrophages in the BAL fluid of LPL−/− mice using flow cytometric analysis of CD45+ CD11chigh Ly6Gneg cells, excluding neutrophils as Ly6G+. In uninfected animals and in animals infected for only a few hours, there were significantly fewer alveolar macrophages in the BAL fluid of LPL−/− mice (Fig. 2A and B). By 24 h after infection, numbers of alveolar macrophages in the BAL fluid of WT and LPL−/− mice were similar (data not shown), although the pneumococcal burden in LPL−/− BAL fluid was 10-fold higher. LPL−/− mice also had fewer alveolar macrophages than did Rag1−/− mice (Fig. 2B), correlating with the ability to clear pneumococci from the alveolar space at 6 h after infection (Fig. 1C). Diminished numbers of alveolar macrophages in LPL−/− lungs early in infection would explain the defective clearance of pneumococci.
FIG 2.

Decreased numbers of alveolar macrophages recovered from BAL fluid of LPL−/− mice. Phagocytosis of pneumococci by alveolar macrophages was unimpaired in LPL−/− mice. (A) Total numbers of alveolar macrophages isolated from BAL fluid of WT (gray circles) and LPL−/− (filled circles) mice, uninfected or infected for the indicated periods of time. (B) Percentages of CD45+ cells that were CD11chigh isolated from the BAL fluid of WT (gray circles), LPL−/− (filled circles), and Rag1−/− (open circles) mice 6 h after i.t. instillation of pneumococci. For panels A and B, each symbol represents data from one mouse, lines represent median values, P values were determined by a Mann-Whitney test, and data were pooled from at least 2 independent experiments per time interval. (C) Representative flow cytometric analysis of numbers of alveolar macrophages (left) and phagocytosis of FITC-labeled S. pneumoniae (SP-FITC) by gated macrophages (right). Alveolar macrophages were assessed by using the phenotypic markers CD45+ and CD11c+; neutrophils were excluded based on Ly6G+ CD11c− staining. A control panel for a WT mouse challenged with PBS is shown at the top. (D) Percentages of alveolar macrophages isolated from BAL fluid of WT or LPL−/− mice that were positive for FITC-labeled pneumococci 3 h after challenge. Each symbol represents one mouse, lines indicate median values, P values were determined by a Mann-Whitney test, and data were from 2 independent experiments.
We next determined whether LPL is required for phagocytosis of pneumococci by alveolar macrophages (Fig. 2C and D) by infecting mice with FITC-labeled pneumococci and quantifying a shift in fluorescence by flow cytometry. Trypan blue was used to quench the signal from labeled pneumococci that externally adhered to macrophages. While there were fewer alveolar macrophages present in the BAL fluid of LPL−/− mice (Fig. 2C), we found no difference in the percentages of LPL−/− and WT alveolar macrophages that ingested FITC-labeled pathogens (Fig. 2D). We also did not observe any difference in the mean fluorescence intensity of FITC-positive macrophages (Fig. 2C and data not shown), indicating that alveolar macrophages from WT and LPL−/− mice ingested similar quantities of pneumococcal particles. To confirm that LPL was dispensable for phagocytosis, we also measured the ingestion of opsonized fluorescent beads by peritoneal macrophages harvested from thioglycolate-injected mice. There was no difference in uptake between WT and LPL−/− peritoneal macrophages (data not shown).
Pulmonary inflammatory infiltrates in lungs from WT and LPL−/− mice are similar 24 h after infection.
To determine whether there were additional defects in the recruitment of innate immune cells in LPL−/− mice during pneumococcal infection, we assessed pulmonary inflammatory infiltrates via histology. Histological sections 24 h after infection demonstrated a few mononuclear cell nodules in the air space and adjacent to bronchi, with no significant difference between lungs from WT and lungs from LPL−/− mice (Fig. 3). No endobronchial involvement was noted. Significant neutrophil infiltrates were not observed in sections stained with hematoxylin and eosin. Specific staining for neutrophils revealed a trend toward a greater number of neutrophils present in LPL−/− lungs (Fig. 3B), but the increase across multiple sections was not statistically significant (data not shown). The relative lack of congestion or consolidation in the lungs of LPL−/− mice suggests that a failure to limit pneumococcal replication and dissemination, rather than overt pneumonia, caused lethality.
FIG 3.
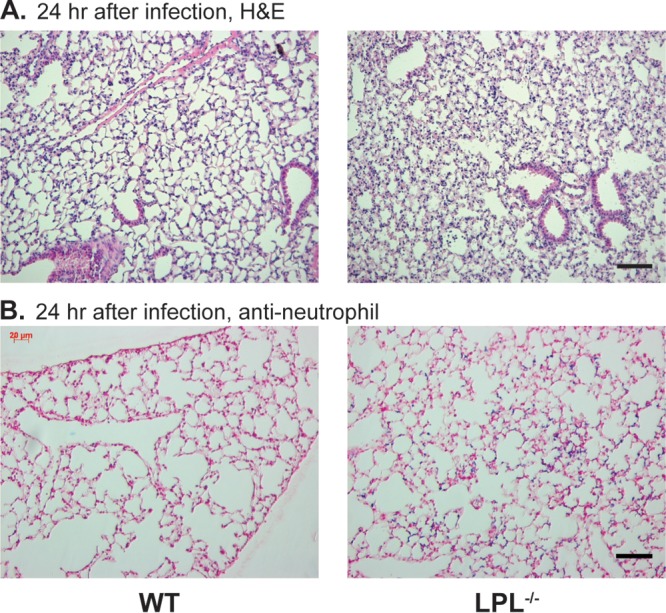
There is no dramatic increase in consolidation in lungs from infected LPL−/− mice. (A) Hematoxylin and eosin (H&E) staining of formalin-fixed sections of lungs from infected WT and LPL−/− mice 24 h after challenge. (B) Specific staining for neutrophils in lung sections from infected WT and LPL−/− mice 24 h after challenge. In panels A and B, the black scale bar indicates 50 μm. Images are representative of 9 to 10 mice.
Neutrophil influx into lungs of LPL−/− mice following pneumococcal infection is intact.
Neutrophils and inflammatory monocytes are rapidly recruited into the lungs as part of the innate immune response to pneumococcal infection. We further quantified pulmonary inflammatory infiltrates through flow cytometric analysis of hematopoietic cells from perfused, homogenized lungs from naive and infected WT and LPL−/− mice (Fig. 4). Total numbers of hematopoietic cells were increased in both WT and LPL−/− mice following infection, with no differences between WT and LPL−/− mice. The trend toward a greater neutrophilic infiltrate in the lungs of LPL−/− mice observed by histology (Fig. 3B) correlated with the quantification obtained by flow cytometry (Table 1) (neutrophils defined as CD45+ CD11b+ Ly6C+ Ly6G+ cells), although the increase was not significantly different. There was also no difference in numbers of recruited inflammatory monocytes (defined as CD45+ CD11b+ Ly6C+ Ly6Gneg CD115+ cells). NK cell recruitment, using Dx5 as a marker, was also unimpaired in LPL−/− mice. Thus, the recruitment of several innate immune cell populations into the lung parenchyma 24 h following pneumococcal infection did not appear to be impaired in LPL−/− mice (Fig. 4).
FIG 4.

Enumeration of immune cells in lung tissue from uninfected and infected WT and LPL−/− mice. Homogenates of perfused right lungs from either uninfected or infected WT or LPL−/− mice were analyzed by flow cytometry to determine the distribution of CD45+ cells. Cell types were determined by using markers described in Materials and Methods (Table 1). Data shown are means ± standard errors of the means, and P values were determined by a Mann-Whitney test (n = 9 or 10 mice for each column, with data pooled from at least 2 independent experiments).
To further characterize the pulmonary immune response of LPL−/− mice, we determined the numbers of dendritic cells (DCs), B cells, and T cells in the lungs of uninfected and infected WT and LPL−/− mice (Fig. 4). In uninfected animals, the numbers of CD8+ T cells and B cells present in the lung interstitium were reduced in LPL−/− mice compared to WT mice. Following infection, the numbers of CD11b+ dendritic cells and γ/δ T cells increased in both WT and LPL−/− mice, although there were significantly fewer cells of both of these populations in LPL−/− mice. Infection also resulted in a loss of CD8+ T cells in both groups, while the numbers of CD4+ T cells and B cells were unchanged. Rag1−/− mice lack all B and T cells but effectively clear pneumococci 6 h after challenge (Fig. 1C), so it is unlikely that the reduced numbers of lung B, CD8+, and γ/δ T cells contribute significantly to defective pneumococcal clearance in LPL−/− mice. Thus, although we found selective decreases in certain populations of immune cells in the lungs of LPL−/− mice at 24 h after infection, the recruitment of neutrophils and monocytes appeared to be intact.
The enumeration of hematopoietic cells in the blood and spleen of WT and LPL−/− mice 24 h after pneumococcal infection reflected the sicker status of LPL−/− mice (Fig. 5). Severe sepsis causes apoptotic loss of lymphocyte populations (30), and we found fewer lymphocytes in the blood and spleens of infected LPL−/− mice (Fig. 5 and data not shown). Numbers of blood monocytes were reduced in infected WT and LPL−/− mice, probably because of recruitment to peripheral tissues. The numbers of blood and splenic monocytes in WT and LPL−/− mice were similar (Fig. 5). The number of neutrophils in the blood and spleen of infected LPL−/− mice was increased, correlating with the increased pneumococcal burden at this time point (Fig. 1D and 5). Thus, we found no defect in the peripheral mobilization of neutrophils or monocytes to explain the increased susceptibility of LPL−/− mice to pneumococcal infection.
FIG 5.

Increased numbers of neutrophils in the blood (A) and spleens (B) of LPL−/− mice infected with pneumococci. Twenty-four hours after i.t. instillation of pneumococci, blood and spleens were harvested from WT and LPL−/− mice. Cell types were determined by flow cytometric analysis as described in Materials and Methods (Table 1). Data shown are means ± standard errors of the means, and P values were determined by a Mann-Whitney test (n = 8 to 10 mice for each column, with data pooled from at least 2 independent experiments).
Production of inflammatory cytokines and chemokines in LPL−/− mice following pneumococcal infection is intact.
To further characterize the pulmonary immune response to pneumococcal infection in LPL−/− mice, we determined if the production of inflammatory cytokines or chemokines (e.g., IL-6, TNF-α, IL-1β, granulocyte colony-stimulating factor [G-CSF], CCL5, CCL4, CXCL1, and CCL2) (31) was defective in LPL−/− mice by assaying the BAL fluid of WT and LPL−/− mice 24 h after pneumococcal challenge (Fig. 6). The concentrations of many of these factors were significantly higher in the BAL fluid of LPL−/− mice, consistent with a higher burden of pneumococcal disease (Fig. 1B to E). While levels of IL-6 and other cytokines were significantly elevated in the BAL fluid of LPL−/− mice, the level of IL-1β was not. The lack of elevated levels of IL-1β is perhaps reflective of the decreased numbers of alveolar macrophages. In some systems, IFN-γ production has been associated with increased survival following pneumococcal challenge (31–35). However, we did not detect significant levels of IFN-γ in the BAL fluid from WT or LPL−/− mice 6 or 24 h after pneumococcal infection (data not shown). The use of either a different pneumococcal serotype or a different murine background likely explains the observed differences in IFN-γ production, and indeed, not all investigations found increased IFN-γ levels in the first 24 h following pneumococcal infection (36, 37). Thus, we did not find a significant loss of any measured chemokine or cytokine in LPL−/− mice, and thus, a failure to generate a required inflammatory mediator is not likely to explain pneumococcal susceptibility in LPL−/− mice.
FIG 6.

Increased concentrations of inflammatory cytokines and chemokines in BAL fluid of infected LPL−/− mice. BAL fluid was harvested from WT (gray circles) and LPL−/− (filled circles) mice 24 h after pneumococcal infection. Concentrations of the indicated inflammatory chemokines or cytokines were determined by using the Bio-Rad Bio-Plex assay. Each symbol represents data from one animal, lines represent mean values, P values were determined by using a Mann-Whitney test, and data were pooled from two independent experiments. MCP-1, monocyte chemoattractant protein 1.
NK cell recruitment is intact in LPL−/− mice following pneumococcal infection.
NK cells constitute a significant proportion of lung-resident innate immune cells (38). To further clarify whether defects in NK cell recruitment could underlie the susceptibility of LPL−/− mice to pneumococcal infection, we analyzed the presence of NK cells in the lungs of WT and LPL−/− mice using NK1.1 (Fig. 7A). While the percentage of NK cells increased in the lung following infection with Staphylococcus aureus or Klebsiella pneumoniae within 24 h (38), we did not find an increase in NK cell percentages in WT mice following serotype 3 pneumococcal infection (Fig. 7A). The observed decrease in percentages is likely due to the influx of other immune cells types, such as neutrophils and monocytes (Fig. 4). There was also no difference in the total number of NK1.1+ cells isolated from the lungs of WT and LPL−/− mice 24 h after pneumococcal infection (data not shown). Thus, we did not find a defect in NK cell numbers to explain the susceptibility of LPL−/− mice to pneumococci. NK cells can serve as a source of IFN-γ following cytokine activation (39). In vitro cytokine stimulation of WT and LPL−/− splenocytes did not reveal defective IFN-γ induction in LPL−/− NK cells (Fig. 7B). Finally, we did not see a significant intracellular signal for IFN-γ in NK1.1+ cells in the lungs of infected WT and LPL−/− mice (Fig. 7C), consistent with the undetectable levels of IFN-γ in the BAL fluid (data not shown). Thus, we did not find a deficiency in the numbers or IFN-γ production of lung LPL−/− NK cells to explain the early pneumococcal clearance defect.
FIG 7.
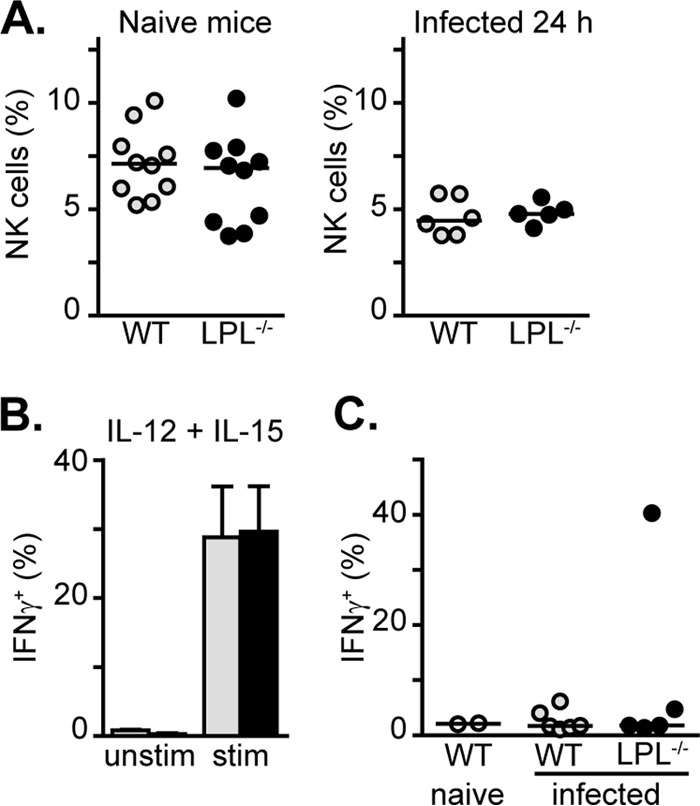
NK cell recruitment is unimpaired in infected LPL−/− mice. (A) Percentages of NK cells found in the lungs of either naive WT (gray circles) and LPL−/− (filled circles) mice or mice 24 h after pneumococcal infection. Each symbol represents data from one mouse, lines represent median values, and data are from at least two independent experiments. (B) Percentages of splenic NK cells positive for IFN-γ following in vitro stimulation with IL-12 and IL-15. Cells were from WT (gray bars) or LPL−/− (filled bars) mice; data shown are means ± standard errors of the means (n = 3 mice in each group). (C) Percentages of lung NK cells positive for IFN-γ 24 h after pneumococcal infection. Each symbol represents data from one animal (open circles, uninfected WT mice; gray circles, infected WT mice; filled circles, infected LPL−/− mice), lines represent median values, and data are from two independent experiments.
Evaluation of the intrinsic requirement for LPL in regeneration of myeloid cells following irradiation.
The most significant defect identified in our analysis of the early inflammatory response to pneumococcal infection in LPL−/− mice is the reduced numbers of alveolar macrophages (Fig. 2A and B). Reduced numbers of alveolar macrophages would be predicted to reduce the ability to initially clear pneumococci from the alveolar space, consistent with the early clearance defect (Fig. 1B). To confirm that LPL is required to establish or maintain alveolar macrophages within the alveolar space, we analyzed the regeneration of alveolar macrophages in bone marrow chimeras following whole-body irradiation (Fig. 8A to D). WT (CD45.1+) recipient mice were reconstituted with bone marrow from CD45.2+ WT or CD45.2+ LPL−/− mice (Fig. 8A to C). Three weeks later, cells from blood, lungs, and BAL fluid were assessed for replacement by donor-derived cells. As expected, radiosensitive cell types, such as circulating neutrophils, were completely replaced by donor-derived cells in recipients receiving bone marrow from either WT or LPL−/− mice, indicating equivalent irradiation of recipient mice and equivalent engraftment of donor-derived bone marrow (Fig. 8A and B). In the lung parenchyma, CD11b+ dendritic cells (CD45+ CD11chigh autofluorescentlow I-Akhigh CD11b+ CD103neg Ly6Gneg) were also completely replaced by donor-derived cells. Equivalent numbers of recipient (CD45.2+) alveolar macrophages were still present in the BAL fluid of all recipient mice (Fig. 8C), recapitulating previous reports that macrophages are relatively radioresistant, with a half-life of 2 to 4 weeks following irradiation (40, 41). In mice receiving bone marrow from LPL−/− mice, the percentages and numbers of alveolar macrophages derived from donor bone marrow were significantly reduced (Fig. 8A to C). We confirmed a cell-intrinsic requirement for LPL in the repopulation of alveolar macrophages using mixed, competitive bone marrow chimeras (Fig. 8D), in which residual recipient cells could be identified as CD45.1+/CD45.2+. Neutrophils in the blood were almost completely replaced by donor marrow (approximately 65% WT derived and 30% LPL−/− derived). The percentage of alveolar macrophages derived from donor WT marrow was the same as that of neutrophils. However, the percentage of alveolar macrophages derived from LPL−/− bone marrow was significantly reduced, indicating a relative impairment in the regeneration of alveolar macrophages from LPL−/− bone marrow (Fig. 8D). LPL is therefore essential for normal regeneration of alveolar macrophages following irradiation.
FIG 8.
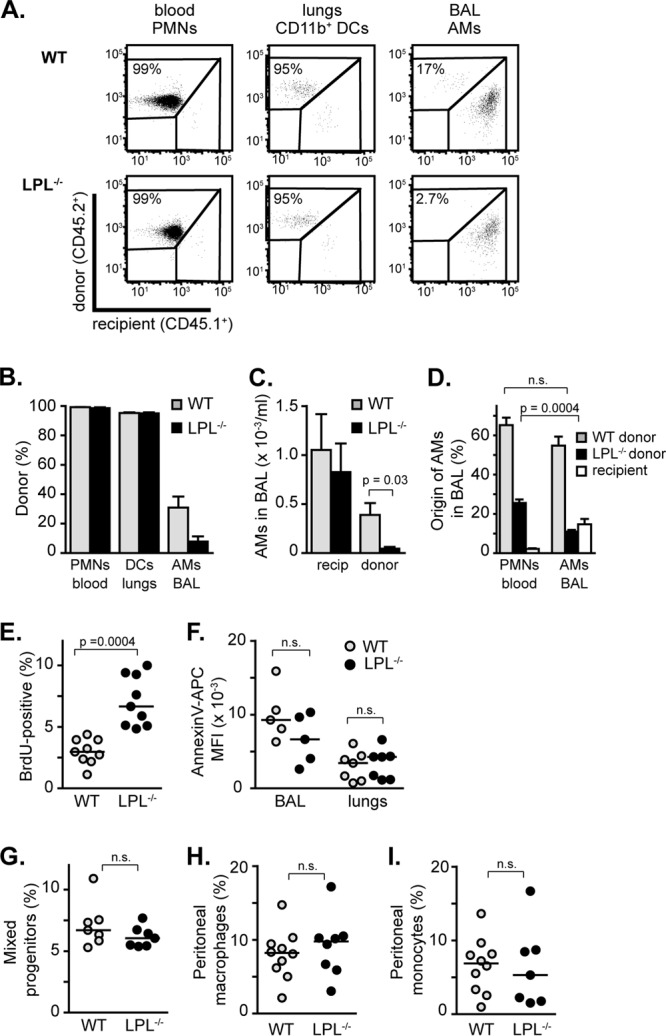
Production of alveolar macrophages requires LPL. (A to D) Regeneration of alveolar macrophages is delayed when donor bone marrow is derived from LPL−/− mice. (A) Representative flow cytometric analysis of bone marrow chimeras showing the percentages of the indicated cells derived from either donor (CD45.2+) or recipient (CD45.1+) bone marrow. Populations of neutrophils (PMNs), CD11b+ dendritic cells (DCs), and alveolar macrophages (AMs) were determined as described in Materials and Methods (Table 1). (B) Percentages of each indicated cell type derived from either WT or LPL−/− donor bone marrow. (C) Numbers of alveolar macrophages isolated from BAL fluid of recipient mice that were endogenous (CD45.1+; recipients) or derived from donor bone marrow (CD45.2+). For panels B and C, data shown are means ± standard errors of the means, and P values were determined by a Mann-Whitney test (n = 5 [LPL−/− reconstituted] or 6 [WT reconstituted], with data pooled from 2 independent experiments). (D) Percentages of neutrophils (PMNs) and alveolar macrophages derived from CD45.1+ WT donor bone marrow, CD45.2+ LPL−/− donor bone marrow, or CD45.1+/CD45.2+ recipient cells. Data shown are means ± standard errors of the means, and P values were determined by a Mann-Whitney test (n = 9 mice, with data pooled from 2 independent experiments). (E) Percentages of alveolar macrophages positive for BrdU incorporation. (F) Mean fluorescence intensities (MFI) of annexin V-APC staining on alveolar macrophages either present in the BAL fluid or isolated from lungs. (G) Percentages of mixed progenitor cells found in bone marrow. (H) Percentages of total cells from peritoneal wash specimens that were macrophages. (I) Percentages of total cells from peritoneal wash specimens that were monocytes. For panels E to I, each symbol represents data from one mouse, lines represent median values, P values were determined by a Mann-Whitney test, and data were pooled from at least two independent experiments. n.s., not significant.
To further analyze the requirement for LPL in alveolar macrophage homeostasis, we explored the self-renewal and cell death of alveolar macrophages in naive WT and LPL−/− mice. Under homeostatic conditions, alveolar macrophages are not repopulated from blood-derived monocytes (42). Rather, alveolar macrophages are thought to seed the lungs in late fetal development and then self-renew through proliferation (43). Alveolar macrophages in LPL−/− mice exhibited increased BrdU incorporation, revealing that LPL is not required for the proliferation or self-renewal of alveolar macrophages (Fig. 8E). The increased proliferative rate of alveolar macrophages in LPL−/− mice may well be a compensatory upregulation given the overall reduced numbers. We did not find evidence of increased cell death of alveolar macrophages in LPL−/− lungs or BAL fluid using annexin V staining (Fig. 8F), indicating that LPL is dispensable for alveolar macrophage survival in naive animals.
The failure of LPL−/− bone marrow to repopulate the alveolar macrophage niche following total body irradiation along with the increased proliferative rate and normal cell death rate in naive mice suggest that LPL is required for the maturation or production of alveolar macrophages. To determine whether LPL was required for the maintenance of precursor cells required to generate monocytes and macrophages, we analyzed the percentages of CD31+ Ly6Cmid cells in bone marrow (Fig. 8G) (44). We found no significant difference in the percentages of progenitor cells in the bone marrow of WT and LPL−/− mice (Fig. 8G). We also did not find significant differences in the percentages of peritoneal macrophages or peritoneal monocytes isolated from WT and LPL−/− mice (Fig. 8H and I). Thus, we did not find evidence that LPL is required for general myeloid development. Rather, LPL appears to be specifically required for either alveolar macrophage maturation or migration of alveolar macrophage precursors into the lungs. LPL is thus required for the generation of a full complement of alveolar macrophages. LPL−/− mice are extraordinarily sensitive to pulmonary pneumococcal challenge, identifying LPL as a critical protein in the pulmonary host immune response.
DISCUSSION
The rapid mortality of LPL−/− mice following intratracheal pneumococcal challenge reveals a previously undiscovered role for LPL in host defense during the early hours of infection. The early postchallenge mortality of LPL−/− mice correlated with diminished numbers of alveolar macrophages in uninfected mice and in mice infected for short durations (3 and 6 h). Our results suggest that impaired maturation of alveolar macrophages leads to increased susceptibility of LPL−/− mice to pulmonary pneumococcal challenge, consistent with previous reports that the depletion of alveolar macrophages impairs the ability to eliminate pneumococci from the alveolar space early in infection (4, 5).
The increased pneumococcal burden in the BAL fluid of LPL−/− mice as early as 3 h after infection distinguishes our model from previous models of pneumococcal susceptibility (45–48). In a recent report, neutrophilic defects in antipneumococcal killing increased the pulmonary pneumococcal burden 12 and 24 h after challenge, but differences were not observed at 6 h, a point in infection when alveolar macrophages would be expected to dominate the initial immune response (46). Further analysis of pneumococcal susceptibility in LPL−/− mice should therefore delineate critical determinants of an effective host immune response that are essential in the first few hours following challenge.
Alveolar macrophages serve multiple functions during pneumococcal pneumonia. Engagement of Toll-like receptors by pathogen-associated molecular patterns induces IL-1 and TNF-α (31, 36). The absence of significantly reduced IL-1 or TNF-α levels in the BAL fluid of LPL−/− mice at either 6 or 24 h (Fig. 6 and data not shown) suggests that either LPL−/−-derived alveolar macrophages retain the ability to generate these critical inflammatory cytokines or other cell types also produce these cytokines during pneumococcal infection. Alveolar epithelial cells are thought to produce significant levels of proinflammatory cytokines such as IL-1β, TNF-α, macrophage inflammatory protein 2 (MIP-2), and keratinocyte-derived chemokine (KC) during pulmonary infection (49–51). Because LPL is normally expressed only in hematopoietic cells, the function of non-hematopoietically derived epithelial cells would be expected to be fully intact in LPL−/− mice. Indeed, the increased concentrations of many inflammatory cytokines noted during pneumococcal infection of LPL−/− mice (Fig. 6) may be due to an increased activation of unaffected cell types due to the higher pneumococcal burden. Future work will compare the activations of WT and LPL−/− alveolar macrophages and other cell types in greater detail.
The regulation of alveolar macrophage development remains to be fully elucidated (42, 43, 52, 53). Compelling evidence that alveolar macrophages are derived from hematopoietic precursors that migrate into the lung during late fetal development has been presented. Under homeostatic conditions, alveolar macrophages self-renew through proliferation and are not replaced from circulating monocytes (43). Self-renewal via increased proliferation without repopulation from blood monocyte precursors was also found in parabiotic experiments examining alveolar macrophage repopulation following pneumonectomy (42). We found increased proliferation of alveolar macrophages in LPL−/− mice under homeostatic conditions, suggesting that the existing pool of alveolar macrophages in LPL−/− mice is compensating for decreased numbers by increased self-renewal. However, alveolar macrophages were regenerated from peripheral blood monocytes via a lung macrophage intermediary after alveolar macrophages were depleted via diphtheria toxin treatment in CD11c:DTR mice (52). Alveolar macrophages can also be regenerated from blood monocytes following pneumococcal infection (54). Our results indicate that LPL is essential for the regeneration of alveolar macrophages following depletion via irradiation, consistent with those previous results. The maintained populations of bone marrow progenitor cells, peritoneal macrophages, and peritoneal monocytes in LPL−/− mice suggest that LPL is not required for myeloid development in general. Taken as a whole, our data suggest that LPL is required either for the specific maturation of alveolar macrophages or possibly for the migration of alveolar macrophage precursors into the lung tissue. The requirement for LPL for the development of normal numbers of alveolar macrophages provides a model system that we will use in future work to illuminate a clearer understanding of the ontogeny of alveolar macrophages.
Because LPL localizes to the macrophage phagocytic cup (55), we were surprised that it was not required for phagocytosis. LPL was also dispensable for neutrophil-mediated phagocytosis of Staphylococcus aureus (12). Either actin bundles are not required for phagocytosis or other proteins compensate for LPL. We are currently assessing if pneumococcal killing is disrupted by an LPL deficiency. Even if alveolar macrophage function is otherwise normal in LPL−/− mice, the relative reduction of alveolar macrophage numbers would be predicted to allow pneumococcal replication and increased susceptibility.
We have not yet identified defects in LPL−/− neutrophils that contribute to pneumococcal susceptibility. We found no defects in the recruitment of neutrophils into the lungs following infection, consistent with previous observations (12). The only defect in LPL−/− neutrophils is integrin-mediated signaling to the generation of the oxidative burst, although other stimuli can successfully trigger the oxidative burst (12). Neutrophil killing of streptococci is thought to occur independently of the oxidative burst (46, 56). Finally, the outgrowth of pneumococci in the alveolar space of LPL−/− mice occurs in the first few hours of infection, a time during which neutrophils are yet to be recruited in substantial numbers to eliminate pneumococci (3, 46). Taken together, these considerations additionally suggest that alveolar macrophages are the defective cells in early antipneumococcal defense in LPL−/− mice.
Interestingly, at baseline and after pneumococcal challenge, there are reduced numbers of other populations of immune cells (CD11b+ and CD103+ DCs and CD4+ T, CD8+ T, and γ/δ T and B cells) (Fig. 4) in the lungs of LPL−/− mice. The functions of conventional pulmonary CD4+ and CD8+ T cells, B cells, NK cells, and dendritic cells during acute pneumococcal infection, if any, are unclear (38, 57–62). It is unlikely that the diminished numbers of pulmonary T and B cells contribute to the extreme susceptibility of LPL−/− mice to pneumococcal challenge, given that Rag1−/− mice clear pneumococci effectively at 6 h after infection (Fig. 1C). NK cells may contribute to pneumococcal clearance by producing IFN-γ and activating monocytes and macrophages when the pneumococcal challenge dose is very high (4 × 108 CFU/mouse); no requirement for NK cells was found at lower doses (5 × 107 CFU/mouse) at early time points in infection (61). It remains possible that there are other NK cell defects in LPL−/− mice that could contribute to increased susceptibility at later time points during infection, which will be investigated in future work. The role for lung-resident dendritic cells during pneumococcal pneumonia is currently unclear and in fact may vary under different experimental conditions. CD103+ dendritic cells played a crucial role in protecting mice from death by producing IFN-γ and IL-17 and in activating invariant NKT cells following pretreatment of mice with α-galactosylceramide (34). In contrast, increasing pulmonary dendritic cell populations via treatment with FMS-like tyrosine kinase 3 ligand (Flt3L) exacerbated inflammation and mortality during pulmonary pneumococcal infection (63), and depleting all dendritic cells prior to infection actually promoted survival in another report (64). Thus, it is possible that reduced recruitment of dendritic cells in LPL−/− mice contributes to the susceptibility of the LPL−/− mice by decreasing production of needed inflammatory mediators such as IL-17. It is also possible that diminished numbers of dendritic cells are protective in LPL−/− mice but are ultimately insufficient for survival. LPL−/− mice will thus offer a new model in which to study the specific contributions of other lung-resident immune cells in future work.
In summary, LPL−/− mice are profoundly susceptible to intratracheal infection with pneumococci. Early mortality correlates with increased bacterial counts at very early time points following infection and with diminished numbers of alveolar macrophages recovered from BAL fluid of LPL−/− mice. We report a previously unrecognized requirement for LPL in the establishment of normal numbers of alveolar macrophages. LPL−/− mice provide a novel model system which we will use in future work to dissect the requirements for alveolar macrophage maturation and for effective early defense against pneumococcal lung infection. Defining host determinants of early antipneumococcal defense is critical for developing new therapies to prevent or ameliorate this devastating disease.
ACKNOWLEDGMENTS
S.C.M. is supported by the Children's Discovery Institute in Saint Louis (grant number MD-FR-2010-83), by the National Institute of Allergy and Infectious Diseases at the National Institutes of Health (grant number K08AI081751-01), by the Center for Women's Infectious Disease Research (cWIDR) at Washington University School of Medicine, and by a Basil O'Connor Starter Scholar research award (March of Dimes). S.C.M. is also a scholar of the Child Health Research Center of Excellence in Developmental Biology at Washington University School of Medicine (grant number K12-HD01487). J.T.M. was supported by the National Institute of General Medical Sciences at the National Institutes of Health (grant number K08GM084143-04). B.T.E. is a recipient of a Burroughs Wellcome Fund career award for medical scientists, an American Society of Hematology scholar award, a grant from the Edward Mallinckrodt, Jr., Foundation, and a Basil O'Connor starter scholar research award. M.A.C. is supported by the Children's Discovery Institute and St. Louis Children's Hospital in Saint Louis, MO. This work was supported in part by the Hope Center Alafi Neuroimaging Laboratory and a P30 Neuroscience Blueprint Interdisciplinary Center Core award to Washington University (grant number P30 NS057105).
We thank Greg Storch, Phil Tarr, and David A. Hunstad for critical reading of the manuscript. We thank Darren Kreamalmeyer for technical assistance with maintenance of the mouse colony.
Footnotes
Published ahead of print 4 March 2014
REFERENCES
- 1.Rudan I, El Arifeen S, Bhutta ZA, Black RE, Brooks A, Chan KY, Chopra M, Duke T, Marsh D, Pio A, Simoes EA, Tamburlini G, Theodoratou E, Weber MW, Whitney CG, Campbell H, Qazi SA. 2011. Setting research priorities to reduce global mortality from childhood pneumonia by 2015. PLoS Med. 8:e1001099. 10.1371/journal.pmed.1001099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Calbo E, Garau J. 2010. Of mice and men: innate immunity in pneumococcal pneumonia. Int. J. Antimicrob. Agents 35:107–113. 10.1016/j.ijantimicag.2009.10.002 [DOI] [PubMed] [Google Scholar]
- 3.Smith AM, McCullers JA, Adler FR. 2011. Mathematical model of a three-stage innate immune response to a pneumococcal lung infection. J. Theor. Biol. 276:106–116. 10.1016/j.jtbi.2011.01.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang J, Barke RA, Charboneau R, Schwendener R, Roy S. 2008. Morphine induces defects in early response of alveolar macrophages to Streptococcus pneumoniae by modulating TLR9-NF-kappa B signaling. J. Immunol. 180:3594–3600 http://www.jimmunol.org/content/180/5/3594.long [DOI] [PubMed] [Google Scholar]
- 5.Xu F, Droemann D, Rupp J, Shen H, Wu X, Goldmann T, Hippenstiel S, Zabel P, Dalhoff K. 2008. Modulation of the inflammatory response to Streptococcus pneumoniae in a model of acute lung tissue infection. Am. J. Respir. Cell Mol. Biol. 39:522–529. 10.1165/rcmb.2007-0328OC [DOI] [PubMed] [Google Scholar]
- 6.Chen WH, Toapanta FR, Shirey KA, Zhang L, Giannelou A, Page C, Frieman MB, Vogel SN, Cross AS. 2012. Potential role for alternatively activated macrophages in the secondary bacterial infection during recovery from influenza. Immunol. Lett. 141:227–234. 10.1016/j.imlet.2011.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burnham EL, Phang TL, House R, Vandivier RW, Moss M, Gaydos J. 2011. Alveolar macrophage gene expression is altered in the setting of alcohol use disorders. Alcohol. Clin. Exp. Res. 35:284–294. 10.1111/j.1530-0277.2010.01344.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin CS, Aebersold RH, Kent SB, Varma M, Leavitt J. 1988. Molecular cloning and characterization of plastin, a human leukocyte protein expressed in transformed human fibroblasts. Mol. Cell. Biol. 8:4659–4668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin CS, Park T, Chen ZP, Leavitt J. 1993. Human plastin genes. Comparative gene structure, chromosome location, and differential expression in normal and neoplastic cells. J. Biol. Chem. 268:2781–2792 [PubMed] [Google Scholar]
- 10.Messier JM, Shaw LM, Chafel M, Matsudaira P, Mercurio AM. 1993. Fimbrin localized to an insoluble cytoskeletal fraction is constitutively phosphorylated on its headpiece domain in adherent macrophages. Cell Motil. Cytoskeleton 25:223–233. 10.1002/cm.970250303 [DOI] [PubMed] [Google Scholar]
- 11.Evans JG, Correia I, Krasavina O, Watson N, Matsudaira P. 2003. Macrophage podosomes assemble at the leading lamella by growth and fragmentation. J. Cell Biol. 161:697–705. 10.1083/jcb.200212037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen H, Mocsai A, Zhang H, Ding RX, Morisaki JH, White M, Rothfork JM, Heiser P, Colucci-Guyon E, Lowell CA, Gresham HD, Allen PM, Brown EJ. 2003. Role for plastin in host defense distinguishes integrin signaling from cell adhesion and spreading. Immunity 19:95–104. 10.1016/S1074-7613(03)00172-9 [DOI] [PubMed] [Google Scholar]
- 13.Lin SL, Chien CW, Han CL, Chen ES, Kao SH, Chen YJ, Liao F. 2010. Temporal proteomics profiling of lipid rafts in CCR6-activated T cells reveals the integration of actin cytoskeleton dynamics. J. Proteome Res. 9:283–297. 10.1021/pr9006156 [DOI] [PubMed] [Google Scholar]
- 14.Morley SC, Wang C, Lo WL, Lio CW, Zinselmeyer BH, Miller MJ, Brown EJ, Allen PM. 2010. The actin-bundling protein L-plastin dissociates CCR7 proximal signaling from CCR7-induced motility. J. Immunol. 184:3628–3638. 10.4049/jimmunol.0903851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wabnitz GH, Lohneis P, Kirchgessner H, Jahraus B, Gottwald S, Konstandin M, Klemke M, Samstag Y. 2010. Sustained LFA-1 cluster formation in the immune synapse requires the combined activities of L-plastin and calmodulin. Eur. J. Immunol. 40:2437–2449. 10.1002/eji.201040345 [DOI] [PubMed] [Google Scholar]
- 16.Wang C, Morley SC, Donermeyer D, Peng I, Lee WP, Devoss J, Danilenko DM, Lin Z, Zhang J, Zhou J, Allen PM, Brown EJ. 2010. Actin-bundling protein L-plastin regulates T cell activation. J. Immunol. 185:7487–7497. 10.4049/jimmunol.1001424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Todd EM, Deady LE, Morley SC. 2011. The actin-bundling protein L-plastin is essential for marginal zone B cell development. J. Immunol. 187:3015–3025. 10.4049/jimmunol.1101033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wabnitz GH, Michalke F, Stober C, Kirchgessner H, Jahraus B, van den Boomen DJ, Samstag Y. 2011. L-plastin phosphorylation: a novel target for the immunosuppressive drug dexamethasone in primary human T cells. Eur. J. Immunol. 41:3157–3169. 10.1002/eji.201041366 [DOI] [PubMed] [Google Scholar]
- 19.Freeley M, O'Dowd F, Paul T, Kashanin D, Davies A, Kelleher D, Long A. 2012. L-plastin regulates polarization and migration in chemokine-stimulated human T lymphocytes. J. Immunol. 188:6357–6370. 10.4049/jimmunol.1103242 [DOI] [PubMed] [Google Scholar]
- 20.Schreiber T, Swanson PE, Chang KC, Davis CC, Dunne WM, Karl IE, Reinhart K, Hotchkiss RS. 2006. Both gram-negative and gram-positive experimental pneumonia induce profound lymphocyte but not respiratory epithelial cell apoptosis. Shock 26:271–276. 10.1097/01.shk0000225856.32260.0d [DOI] [PubMed] [Google Scholar]
- 21.Muenzer JT, Davis CG, Chang K, Schmidt RE, Dunne WM, Coopersmith CM, Hotchkiss RS. 2010. Characterization and modulation of the immunosuppressive phase of sepsis. Infect. Immun. 78:1582–1592. 10.1128/IAI.01213-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McConnell KW, Fox AC, Clark AT, Chang NY, Dominguez JA, Farris AB, Buchman TG, Hunt CR, Coopersmith CM. 2011. The role of heat shock protein 70 in mediating age-dependent mortality in sepsis. J. Immunol. 186:3718–3725. 10.4049/jimmunol.1003652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edelson BT, KC W, Juang R, Kohyama M, Benoit LA, Klekotka PA, Moon C, Albring JC, Ise W, Michael DG, Bhattacharya D, Stappenbeck TS, Holtzman MJ, Sung SS, Murphy TL, Hildner K, Murphy KM. 2010. Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8alpha+ conventional dendritic cells. J. Exp. Med. 207:823–836. 10.1084/jem.20091627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keppel MP, Yang L, Cooper MA. 2013. Murine NK cell intrinsic cytokine-induced memory-like responses are maintained following homeostatic proliferation. J. Immunol. 190:4754–4762. 10.4049/jimmunol.1201742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Todd EM, Deady LE, Morley SC. 2013. Intrinsic T- and B-cell defects impair T-cell-dependent antibody responses in mice lacking the actin-bundling protein L-plastin. Eur. J. Immunol. 43:1735–1744. 10.1002/eji.201242780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goldstein D, Djeu J, Latter G, Burbeck S, Leavitt J. 1985. Abundant synthesis of the transformation-induced protein of neoplastic human fibroblasts, plastin, in normal lymphocytes. Cancer Res. 45:5643–5647 [PubMed] [Google Scholar]
- 27.de Arruda MV, Watson S, Lin CS, Leavitt J, Matsudaira P. 1990. Fimbrin is a homologue of the cytoplasmic phosphoprotein plastin and has domains homologous with calmodulin and actin gelation proteins. J. Cell Biol. 111:1069–1079. 10.1083/jcb.111.3.1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pacaud M, Derancourt J. 1993. Purification and further characterization of macrophage 70-kDa protein, a calcium-regulated, actin-binding protein identical to L-plastin. Biochemistry 32:3448–3455. 10.1021/bi00064a031 [DOI] [PubMed] [Google Scholar]
- 29.Wu C, Orozco C, Boyer J, Leglise M, Goodale J, Batalov S, Hodge CL, Haase J, Janes J, Huss JW, III, Su AI. 2009. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 10:R130. 10.1186/gb-2009-10-11-r130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hotchkiss RS, Karl IE. 2003. The pathophysiology and treatment of sepsis. N. Engl. J. Med. 348:138–150. 10.1056/NEJMra021333 [DOI] [PubMed] [Google Scholar]
- 31.McConnell KW, McDunn JE, Clark AT, Dunne WM, Dixon DJ, Turnbull IR, Dipasco PJ, Osberghaus WF, Sherman B, Martin JR, Walter MJ, Cobb JP, Buchman TG, Hotchkiss RS, Coopersmith CM. 2010. Streptococcus pneumoniae and Pseudomonas aeruginosa pneumonia induce distinct host responses. Crit. Care Med. 38:223–241. 10.1097/CCM.0b013e3181b4a76b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rubins JB, Pomeroy C. 1997. Role of gamma interferon in the pathogenesis of bacteremic pneumococcal pneumonia. Infect. Immun. 65:2975–2977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun K, Salmon SL, Lotz SA, Metzger DW. 2007. Interleukin-12 promotes gamma interferon-dependent neutrophil recruitment in the lung and improves protection against respiratory Streptococcus pneumoniae infection. Infect. Immun. 75:1196–1202. 10.1128/IAI.01403-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ivanov S, Fontaine J, Paget C, Macho Fernandez E, Van Maele L, Renneson J, Maillet I, Wolf NM, Rial A, Leger H, Ryffel B, Frisch B, Chabalgoity JA, Sirard JC, Benecke A, Faveeuw C, Trottein F. 2012. Key role for respiratory CD103(+) dendritic cells, IFN-gamma, and IL-17 in protection against Streptococcus pneumoniae infection in response to alpha-galactosylceramide. J. Infect. Dis. 206:723–734. 10.1093/infdis/jis413 [DOI] [PubMed] [Google Scholar]
- 35.Marques JM, Rial A, Munoz N, Pellay FX, Van Maele L, Leger H, Camou T, Sirard JC, Benecke A, Chabalgoity JA. 2012. Protection against Streptococcus pneumoniae serotype 1 acute infection shows a signature of Th17- and IFN-gamma-mediated immunity. Immunobiology 217:420–429. 10.1016/j.imbio.2011.10.012 [DOI] [PubMed] [Google Scholar]
- 36.Lauw FN, Branger J, Florquin S, Speelman P, van Deventer SJ, Akira S, van der Poll T. 2002. IL-18 improves the early antimicrobial host response to pneumococcal pneumonia. J. Immunol. 168:372–378 http://www.jimmunol.org/content/168/1/372.long [DOI] [PubMed] [Google Scholar]
- 37.Ferreira DM, Moreno AT, Cianciarullo AM, Ho PL, Oliveira ML, Miyaji EN. 2009. Comparison of the pulmonary response against lethal and non-lethal intranasal challenges with two different pneumococcal strains. Microb. Pathog. 47:157–163. 10.1016/j.micpath.2009.05.005 [DOI] [PubMed] [Google Scholar]
- 38.Wang J, Li F, Zheng M, Sun R, Wei H, Tian Z. 2012. Lung natural killer cells in mice: phenotype and response to respiratory infection. Immunology 137:37–47. 10.1111/j.1365-2567.2012.03607.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cooper MA, Fehniger TA, Ponnappan A, Mehta V, Wewers MD, Caligiuri MA. 2001. Interleukin-1beta costimulates interferon-gamma production by human natural killer cells. Eur. J. Immunol. 31:792–801. 10.1002/1521-4141(200103)31:3<792::AID-IMMU792>3.0.CO;2-U [DOI] [PubMed] [Google Scholar]
- 40.Matute-Bello G, Lee JS, Frevert CW, Liles WC, Sutlief S, Ballman K, Wong V, Selk A, Martin TR. 2004. Optimal timing to repopulation of resident alveolar macrophages with donor cells following total body irradiation and bone marrow transplantation in mice. J. Immunol. Methods 292:25–34. 10.1016/j.jim.2004.05.010 [DOI] [PubMed] [Google Scholar]
- 41.Hahn I, Klaus A, Maus R, Christman JW, Welte T, Maus UA. 2011. Dendritic cell depletion and repopulation in the lung after irradiation and bone marrow transplantation in mice. Am. J. Respir. Cell Mol. Biol. 45:534–541. 10.1165/rcmb.2010-0279OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chamoto K, Gibney BC, Ackermann M, Lee GS, Lin M, Konerding MA, Tsuda A, Mentzer SJ. 2012. Alveolar macrophage dynamics in murine lung regeneration. J. Cell. Physiol. 227:3208–3215. 10.1002/jcp.24009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, Deswarte K, Malissen B, Hammad H, Lambrecht BN. 2013. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J. Exp. Med. 210:1977–1992. 10.1084/jem.20131199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trottier MD, Irwin R, Li Y, McCabe LR, Fraker PJ. 2012. Enhanced production of early lineages of monocytic and granulocytic cells in mice with colitis. Proc. Natl. Acad. Sci. U. S. A. 109:16594–16599. 10.1073/pnas.1213854109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schabbauer G, Matt U, Gunzl P, Warszawska J, Furtner T, Hainzl E, Elbau I, Mesteri I, Doninger B, Binder BR, Knapp S. 2010. Myeloid PTEN promotes inflammation but impairs bactericidal activities during murine pneumococcal pneumonia. J. Immunol. 185:468–476. 10.4049/jimmunol.0902221 [DOI] [PubMed] [Google Scholar]
- 46.Hahn I, Klaus A, Janze AK, Steinwede K, Ding N, Bohling J, Brumshagen C, Serrano H, Gauthier F, Paton JC, Welte T, Maus UA. 2011. Cathepsin G and neutrophil elastase play critical and nonredundant roles in lung-protective immunity against Streptococcus pneumoniae in mice. Infect. Immun. 79:4893–4901. 10.1128/IAI.05593-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pittet LA, Quinton LJ, Yamamoto K, Robson BE, Ferrari JD, Algul H, Schmid RM, Mizgerd JP. 2011. Earliest innate immune responses require macrophage RelA during pneumococcal pneumonia. Am. J. Respir. Cell Mol. Biol. 45:573–581. 10.1165/rcmb.2010-0210OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van der Windt GJ, Hoogendijk AJ, de Vos AF, Kerver ME, Florquin S, van der Poll T. 2011. The role of CD44 in the acute and resolution phase of the host response during pneumococcal pneumonia. Lab. Invest. 91:588–597. 10.1038/labinvest.2010.206 [DOI] [PubMed] [Google Scholar]
- 49.Skerrett SJ, Liggitt HD, Hajjar AM, Ernst RK, Miller SI, Wilson CB. 2004. Respiratory epithelial cells regulate lung inflammation in response to inhaled endotoxin. Am. J. Physiol. Lung Cell. Mol. Physiol. 287:L143–L152. 10.1152/ajplung.00030.2004 [DOI] [PubMed] [Google Scholar]
- 50.Koppe U, Hogner K, Doehn JM, Muller HC, Witzenrath M, Gutbier B, Bauer S, Pribyl T, Hammerschmidt S, Lohmeyer J, Suttorp N, Herold S, Opitz B. 2012. Streptococcus pneumoniae stimulates a STING- and IFN regulatory factor 3-dependent type I IFN production in macrophages, which regulates RANTES production in macrophages, cocultured alveolar epithelial cells, and mouse lungs. J. Immunol. 188:811–817. 10.4049/jimmunol.1004143 [DOI] [PubMed] [Google Scholar]
- 51.Yamamoto K, Ferrari JD, Cao Y, Ramirez MI, Jones MR, Quinton LJ, Mizgerd JP. 2012. Type I alveolar epithelial cells mount innate immune responses during pneumococcal pneumonia. J. Immunol. 189:2450–2459. 10.4049/jimmunol.1200634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Landsman L, Jung S. 2007. Lung macrophages serve as obligatory intermediate between blood monocytes and alveolar macrophages. J. Immunol. 179:3488–3494 http://www.jimmunol.org/content/179/6/3488.long [DOI] [PubMed] [Google Scholar]
- 53.Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SE, Pollard JW, Frampton J, Liu KJ, Geissmann F. 2012. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336:86–90. 10.1126/science.1219179 [DOI] [PubMed] [Google Scholar]
- 54.Taut K, Winter C, Briles DE, Paton JC, Christman JW, Maus R, Baumann R, Welte T, Maus UA. 2008. Macrophage turnover kinetics in the lungs of mice infected with Streptococcus pneumoniae. Am. J. Respir. Cell Mol. Biol. 38:105–113. 10.1165/rcmb.2007-0132OC [DOI] [PubMed] [Google Scholar]
- 55.Ohsawa K, Imai Y, Sasaki Y, Kohsaka S. 2004. Microglia/macrophage-specific protein Iba1 binds to fimbrin and enhances its actin-bundling activity. J. Neurochem. 88:844–856. 10.1046/j.1471-4159.2003.02213.x [DOI] [PubMed] [Google Scholar]
- 56.Standish AJ, Weiser JN. 2009. Human neutrophils kill Streptococcus pneumoniae via serine proteases. J. Immunol. 183:2602–2609. 10.4049/jimmunol.0900688 [DOI] [PubMed] [Google Scholar]
- 57.Walker J, Lee R, Mathy N, Doughty S, Conlon J. 1996. Restricted B-cell responses to microbial challenge of the respiratory tract. Vet. Immunol. Immunopathol. 54:197–204. 10.1016/S0165-2427(96)05709-1 [DOI] [PubMed] [Google Scholar]
- 58.GeurtsvanKessel CH, Willart MA, van Rijt LS, Muskens F, Kool M, Baas C, Thielemans K, Bennett C, Clausen BE, Hoogsteden HC, Osterhaus AD, Rimmelzwaan GF, Lambrecht BN. 2008. Clearance of influenza virus from the lung depends on migratory langerin+CD11b− but not plasmacytoid dendritic cells. J. Exp. Med. 205:1621–1634. 10.1084/jem.20071365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.LeMessurier K, Hacker H, Tuomanen E, Redecke V. 2010. Inhibition of T cells provides protection against early invasive pneumococcal disease. Infect. Immun. 78:5287–5294. 10.1128/IAI.00431-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Teijaro JR, Verhoeven D, Page CA, Turner D, Farber DL. 2010. Memory CD4 T cells direct protective responses to influenza virus in the lungs through helper-independent mechanisms. J. Virol. 84:9217–9226. 10.1128/JVI.01069-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Elhaik-Goldman S, Kafka D, Yossef R, Hadad U, Elkabets M, Vallon-Eberhard A, Hulihel L, Jung S, Ghadially H, Braiman A, Apte RN, Mandelboim O, Dagan R, Mizrachi-Nebenzahl Y, Porgador A. 2011. The natural cytotoxicity receptor 1 contribution to early clearance of Streptococcus pneumoniae and to natural killer-macrophage cross talk. PLoS One 6:e23472. 10.1371/journal.pone.0023472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weber SE, Tian H, Pirofski LA. 2011. CD8+ cells enhance resistance to pulmonary serotype 3 Streptococcus pneumoniae infection in mice. J. Immunol. 186:432–442. 10.4049/jimmunol.1001963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Winter C, Taut K, Langer F, Mack M, Briles DE, Paton JC, Maus R, Srivastava M, Welte T, Maus UA. 2007. FMS-like tyrosine kinase 3 ligand aggravates the lung inflammatory response to Streptococcus pneumoniae infection in mice: role of dendritic cells. J. Immunol. 179:3099–3108 http://www.jimmunol.org/content/179/5/3099.long [DOI] [PubMed] [Google Scholar]
- 64.Rosendahl A, Bergmann S, Hammerschmidt S, Goldmann O, Medina E. 2013. Lung dendritic cells facilitate extrapulmonary bacterial dissemination during pneumococcal pneumonia. Front. Cell. Infect. Microbiol. 3:21. 10.3389/fcimb.2013.00021 [DOI] [PMC free article] [PubMed] [Google Scholar]