

Abstract
Chlamydia trachomatis is an obligate intracellular human pathogen responsible for ocular and genital infections. To establish its membrane-bound intracellular niche, the inclusion, C. trachomatis relies on a set of effector proteins that are injected into the host cells or inserted into the inclusion membrane. We previously proposed that insertion of the C. trachomatis effector protein IncD into the inclusion membrane contributes to the recruitment of the lipid transfer protein CERT to the inclusion. Due to the genetically intractable status of C. trachomatis at that time, this model of IncD-CERT interaction was inferred from ectopic expression of IncD and CERT in the host cell. In the present study, we investigated the impact of conditionally expressing a FLAG-tagged version of IncD in C. trachomatis. This genetic approach allowed us to establish that IncD-3×FLAG localized to the inclusion membrane and caused a massive recruitment of the lipid transfer protein CERT that relied on the PH domain of CERT. In addition, we showed that the massive IncD-dependent association of CERT with the inclusion led to an increased recruitment of the endoplasmic reticulum (ER)-resident protein VAPB, and we determined that, at the inclusion, CERT-VAPB interaction relied on the FFAT domain of CERT. Altogether, the data presented here show that expression of the C. trachomatis effector protein IncD mediates the recruitment of the lipid transfer protein CERT and the ER-resident protein VAPB to the inclusion.
INTRODUCTION
Chlamydia species are obligate intracellular Gram-negative bacterial pathogens that infect genital, ocular, and pulmonary epithelial surfaces. Chlamydiae are characterized by a biphasic developmental cycle that occurs exclusively in the host cell. The bacteria alternate between an infectious form called the elementary body (EB), which is characterized by a condensed nucleoid, and an intracellular replicative form named the reticulate body (RB). Once internalized, Chlamydia resides in a membrane-bound compartment termed the inclusion. Shortly after uptake, an uncharacterized switch occurs, leading to differentiation of EBs into RBs. The RBs then start to replicate until the inclusion occupies a large part of the cytosol of the host cell. Midway through, the developmental cycle becomes asynchronous, and RBs start to differentiate back into EBs. At the end of the cycle, which lasts 2 to 3 days depending on the species, EBs are released from the host cell, allowing infection of neighboring cells (1, 2).
To establish and maintain their intracellular niche, Chlamydia species have evolved sophisticated mechanisms to manipulate the host cellular machinery (3). Type III secretion effector proteins are injected into the host cell to target various cellular processes. Some effectors are released into the cytosol, while others, such as the Incs, are inserted into the inclusion membrane (4). Type III effectors were identified through the use of bacterial heterologous systems, followed by confirmation of the secretion of the endogenous proteins during infection. In vitro systems, expression in mammalian cells, and identification of interacting partners suggested that effector proteins play important roles in entry, interaction of the inclusion with Rab GTPases, SNARES, lipid transfer protein, and components of the cytoskeleton, and modulation of signaling pathways (reviewed in reference 5). However, the proposed function(s) of these effectors remains to be validated in the context of the infection process, when they are expressed from the bacteria. Moreover, the actual functions of many effectors remain to be uncovered.
We recently proposed a model in which the Chlamydia trachomatis effector protein IncD is involved in recruitment of the lipid transfer protein CERT to the inclusion membrane, at zones of close apposition with endoplasmic reticulum (ER) tubules that are positive for VAPA and VAPB (vesicle-associated membrane protein-associated protein). We named these structures ER-inclusion membrane contact sites (MCSs) (6). CERT is a functional component of ER-Golgi membrane contact sites (7, 8) involved in the nonvesicular transfer of ceramide from the ER to the Golgi apparatus (9). In addition to the carboxy-terminal START domain (10) that binds ceramide, the ER-to-Golgi transfer process requires a central FFAT motif (11), which binds the ER-resident proteins VAPA and VAPB (12), and an amino-terminal PH domain (13), which recognizes determinants, such as PI4P (phosphatidylinositol 4-phosphate), on the Golgi membrane (14, 15). Our model of IncD-dependent CERT localization to ER-inclusion MCSs was based on the following observations: (i) endogenous IncD and CERT both localized to the inclusion membrane, (ii) IncD interacted with the PH domain of CERT in vitro or when the proteins were coexpressed in mammalian cells, and (iii) CERT localization to the inclusion membrane correlated with the association of VAPA/B-positive tubules in close proximity to the inclusion membrane. The lack of C. trachomatis genetic systems at that time prevented further validation of our model by demonstration of the role of IncD expressed from bacteria in CERT recruitment to the inclusion.
Major advances have occurred in the Chlamydia field with the recent development of genetic tools. Chemical mutagenesis combined with the use of the mismatch-specific endonuclease CEL I (16) and genome sequencing with a system of DNA exchanges among Chlamydia strains (17) led to the isolation of targeted C. trachomatis null mutants and a collection of mutants with distinct phenotypes, respectively. In addition, targeted gene inactivation was very recently achieved using a group II intron (18). Chlamydia transformation was achieved using electroporation (19, 20), dendrimers (21–23), and a calcium-based method (24). Escherichia coli-C. trachomatis shuttle plasmids were successfully introduced and maintained in C. trachomatis, leading to C. trachomatis strains that were resistant to β-lactams and expressed green fluorescent protein (GFP) (24). Recent studies also described that chloramphenicol and blasticidin resistance cassettes can also serve as selection markers to select transformants (25, 26). Cloning vectors were developed (27, 28), and the calcium transformation method was used to study the role of Chlamydia plasmid-carried open reading frames (ORFs) (28–31). Transformants displaying constitutive expression of the fluorescent proteins GFP, cyan fluorescent protein (CFP), mCherry, and mKate2 (24, 27, 32) and the β-galactosidase enzyme (28) were described, and conditional expression of GFP, mCherry, and a subset of Chlamydia secreted proteins was achieved in C. trachomatis by using the Tet system (32, 33).
In the present study, we took advantage of the recent advances in C. trachomatis genetics to demonstrate the role of the inclusion membrane protein IncD expressed from C. trachomatis. We generated a C. trachomatis strain that expressed a 3×FLAG-tagged inducible version of IncD from the Chlamydia plasmid. By controlling the amount of IncD produced by bacteria, we demonstrated the role of IncD in CERT and VAPB recruitment to the inclusion membrane. This genetic approach can be generalized to any C. trachomatis effector protein and will tremendously influence future studies of the molecular mechanisms that support C. trachomatis infection.
MATERIALS AND METHODS
Ethics statement.
All genetic manipulations and containment work were approved by the Yale Biological Committee and are in compliance with section III-D-1-a of the National Institutes of Health guidelines for research involving recombinant DNA molecules.
Cell lines and bacterial strains.
HeLa cells were obtained from the ATCC (CCL-2) and cultured at 37°C with 5% CO2 in high-glucose Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Invitrogen). C. trachomatis lymphogranuloma venereum (LGV) type II was obtained from the ATCC (L2/434/Bu VR-902B). Chlamydia propagation and infection were performed as previously described (34).
Plasmid construction.
Restriction enzymes and T4 DNA ligase were obtained from New England BioLabs (Ipswich, MA). PCR was performed using Herculase DNA polymerase (Stratagene). PCR primers were obtained from Integrated DNA Technologies, and their sequences are listed in Table S1 in the supplemental material.
(i) Construction of p2TK2-SW2 mCh(Gro).
The p2TK2-SW2 mCh(Gro) plasmid is a derivative of our cloning vector p2TK2-SW2 (27). It expresses mCherry under the control of the groESL operon promoter and terminator. DNA fragments corresponding to the intergenic regions upstream (Gro Prom) and downstream (Gro Term) of the groESL operon were amplified by PCR from C. trachomatis LGV L2 genomic DNA by using primers GroESLProm Age 5 plus GroESL mCherry START 3 (Gro Prom) and GroESL mCherry STOP 5 plus GroESLTerm Age 3 (Gro Term), respectively. A DNA fragment corresponding to mCherry was amplified using primers GroESL mCherry START 5 and GroESL mCherry STOP 3. A DNA fragment corresponding to Gro Prom-mCherry-Gro Term was then amplified by overlapping PCR using the primers GroESLProm Age 5 and GroESLTerm Age 3 and cloned into the AgeI sites of p2TK2-SW2. The orientation of the insert was such that mCherry was transcribed toward the multiple-cloning site of p2TK2-SW2.
(ii) Amplification of the IncD-3×FLAG-IncDTerm fragment.
A DNA fragment corresponding to IncD-3×FLAG was amplified by PCR from C. trachomatis LGV L2 genomic DNA by using primers TetAP-IncD3F 3 and IncD 3×FLAG 3. The resulting DNA fragment was then used as a template to amplify an IncD-3×FLAG DNA fragment with a 3′ tail corresponding to the first 20 nucleotides of the intergenic region downstream of the incDEFG operon, using primers TetAP-IncD3F 3 and D FLAG STOP 3. A DNA fragment corresponding to the intergenic region downstream of the incDEFG operon (IncDTerm) was amplified by PCR from C. trachomatis LGV L2 genomic DNA by using primers D FLAG STOP 5 and IncD Term NotI. A DNA fragment corresponding to IncD-3×FLAG-IncDTerm was then amplified by overlapping PCR using primers TetAP-IncD3F 3 and IncD Term NotI.
(iii) Construction of p2TK2-SW2 mCh(Gro) TetIncD3F.
The p2TK2-SW2 mCh(Gro) TetIncD3F plasmid is a derivative of p2TK2-SW2 mCh(Gro) (see description above). It expresses mCherry, under the control of the groESL operon promoter and terminator; the TetR repressor; and IncD-3×FLAG, under the control of the tetA gene promoter and the incDEFG operon terminator. A DNA fragment corresponding to the TetR repressor and the tetA promoter was amplified by PCR from the Coxiella plasmid pMiniTn7T-CAT::TetRA-icmDJB (35) by using primers TetR STOP 5 Kpn and TetAP-IncD3F 3. A DNA fragment corresponding to IncD-3×FLAG-IncDTerm was amplified as described above. A DNA fragment corresponding to TetR-TetAP-IncD-3×FLAG-IncDTerm was then amplified by overlapping PCR using primers TetR STOP 5 Kpn and IncD Term NotI, and the fragment was cloned into the KpnI and NotI sites of p2TK2-SW2 mCh(Gro).
C. trachomatis transformation.
Our calcium-based transformation protocol was adapted from the work of Wang et al. (24) and was described previously (27).
Two C. trachomatis transformed strains were used in this study. The CtL2 mCh(Gro) strain harbored the p2TK2-SW2 mCh(Gro) plasmid and expressed mCherry under the control of the groESL operon promoter and terminator. The CtL2 mCh(Gro) TetIncD3F strain harbored the p2TK2-SW2 mCh(Gro) TetIncD3F plasmid and expressed mCherry under the control of the groESL operon promoter and terminator and IncD3×FLAG under the control of an aTc (anhydrotetracycline)-inducible promoter.
Immunofluorescence and microscopy.
At the indicated times, cells seeded onto glass coverslips were fixed for 30 min in phosphate-buffered saline (PBS) containing 4% paraformaldehyde. Immunostaining was performed at room temperature. Antibodies were diluted in PBS containing 0.1% bovine serum albumin (BSA) and 0.1% Triton X-100. Samples were washed with PBS and examined under an epifluorescence or spinning disc confocal microscope.
Infectious progeny production.
HeLa cells were collected at the indicated times postinfection and lysed with water, and dilutions of the lysate were used to infect fresh HeLa cells. The cells were fixed at 24 h postinfection, and the number of inclusion-forming units (IFUs) was determined after assessment of the number of infected cells by immunolabeling.
Antibodies.
The following primary antibodies were used: rabbit polyclonal anti-C. trachomatis IncA (1:200; kindly provided by T. Hackstadt, Rocky Mountain Laboratories), mouse monoclonal anti-FLAG (1:1,000 for immunofluorescence [IF] and 1:20,000 for Western blotting; Sigma), rabbit polyclonal anti-actin (1:1,000; Sigma), and rabbit polyclonal anti-tRFP (red fluorescent protein) (1:2,000; Evrogen). The following secondary antibodies were used: peroxidase-conjugated goat anti-rabbit IgG (1:10,000; Jackson ImmunoResearch), peroxidase-conjugated goat anti-mouse IgG (1:10,000; Jackson ImmunoResearch), Alexa Fluor 514-conjugated goat anti-rabbit antibody (1:1,000 for IF; Molecular Probes), and Alexa Fluor 488-, Alexa Fluor 514-, or Pacific Blue-conjugated goat anti-mouse antibody (1:1,000 for IF; Molecular Probes).
Immunoblotting.
Immunoblots were quantified using Odyssey software.
DNA transfection.
DNA transfection was performed using Fugene 6 according to the manufacturer's recommendations.
Quantification of the fraction of the inclusion membrane covered by a given marker.
Quantification was done using Volocity software on images acquired with a spinning disc confocal microscope. For each inclusion, the quantification was performed on a stack of x-y planes corresponding to a 2.2-μm slice located in the middle of the inclusions of interest. The volume corresponding to the signal of the marker of interest associated with the inclusion membrane was measured and normalized to the length of the inclusion membrane. The inclusion boundary was determined using the fluorescence signal of the mCherry-tagged bacteria. Full coverage was set to 100%.
RESULTS
IncD-3×FLAG expression from C. trachomatis.
To investigate the role of the inclusion membrane protein IncD expressed from C. trachomatis, we engineered a C. trachomatis strain that harbored the plasmid p2TK2-SW2-mCh(Gro)-TetIncD3F, encoding mCherry, under the control of the groESL operon promoter and terminator; the TetR repressor; and IncD-3×FLAG, under the control of an aTc-inducible promoter (see Materials and Methods). This strain is referred to as CtL2 mCh(Gro) TetIncD3F.
We first followed the production of IncD-3×FLAG by immunoblotting during a time course of aTc induction. HeLa cells infected with CtL2 mCh(Gro) TetIncD3F for 24 h were grown in the absence or presence of increasing doses of aTc for the indicated times. The corresponding lysates were probed using antibodies against FLAG, mCherry, and actin (Fig. 1A). The intensities of the signals corresponding to mCherry and FLAG were quantified and normalized to the intensity of the actin signal (Fig. 1B). As expected, mCherry levels increased slightly after 8 h and 24 h of induction, reflecting bacterial replication between the 24-h and 48-h time points of infection. In the absence of aTc, the levels of IncD-3×FLAG expression were negligible, even at 48 h postinfection. In the presence of 2 ng/ml aTc, IncD-3×FLAG expression displayed a 2- to 3-fold increase during the first hour of induction and kept increasing over time, with 98- and 175-fold increases at 8 h and 24 h postinduction, respectively. In the presence of 20 ng/ml aTc, IncD-3×FLAG was detected as early as 15 min postinduction, with a 10-fold increase in expression levels. The amount of IncD-3×FLAG produced increased over time, with 50-, 190-, and 340-fold increases at 1 h, 8 h, and 24 h postinduction, respectively. A similar result was obtained in the presence of 200 ng/ml aTc. The expression level of IncD-3×FLAG was increased 90-, 200-, and 380-fold at 1 h, 8 h, and 24 h postinduction, respectively.
FIG 1.

Time course of IncD-3×FLAG induction. (A) Immunoblots of cell lysates from cells infected with the CtL2 mCh(Gro) TetIncD3F strain. The cells were infected for 24 h to allow for inclusion formation before addition of 2, 20, or 200 ng/ml aTc. The cells were collected at 15, 30, or 45 min or 1, 8, or 20 h post-aTc induction, and immunoblots of the corresponding lysates were probed using antibodies. (B) Quantification of three independent experiments as presented in panel A. The mCherry (left) and FLAG (right) signal intensities were quantified using Odyssey software and normalized to the actin signal intensity, and protein levels (arbitrary units) were plotted. Please note the split y axis.
Altogether, these results indicate that the level of expression of IncD-3×FLAG is time and aTc dose dependent, allowing for precise regulation of the amount of protein produced.
IncD-3×FLAG localization during a time course of aTc induction.
We next investigated the precise localization of IncD-3×FLAG during a time course of aTc induction. HeLa cells were infected with the CtL2 mCh(Gro) TetIncD3F strain for 24 h in the absence of aTc to allow for the formation of inclusions in the absence of expression of the IncD-3×FLAG construct. The infected cells were then incubated in the absence or presence of the indicated aTc concentration (2, 20, or 200 ng/ml). At the indicated time post-aTc induction (5, 15, 30, or 45 min or 1 or 20 h), the infected cells were fixed, immunostained with anti-FLAG antibodies, and processed for imaging using a confocal microscope. Representative micrographs of an extended-focus view combining all the confocal planes spanning the inclusion and of a single plane across the middle of the inclusion (x-y view) are presented in Fig. 2. In the absence of aTc, there was no detectable FLAG signal on the inclusion (see Fig. S1 in the supplemental material). In the presence of 2 ng/ml aTc, IncD-3×FLAG was detectable at 45 min post-aTc induction, whereas with 20 ng/ml aTc, it was detectable as early as 15 min postinduction. Five minutes of induction was sufficient to detect IncD-3×FLAG with 200 ng/ml aTc. Regardless of the aTc concentration used, the IncD-3×FLAG construct was first detected in the lumen of the inclusion and then accumulated at the inclusion membrane. After 20 h of aTc induction, most of the IncD-3×FLAG signal localized exclusively to the inclusion membrane. The amount of IncD-3×FLAG present in the inclusion membrane appeared to be proportional to the aTc concentration (Fig. 2, compare panels for 20 h and 2 ng/ml to those for 20 h and 20 to 200 ng/ml).
FIG 2.

IncD-3×FLAG localization during a time course of aTc induction. Confocal micrographs show inclusions of the CtL2 mCh(Gro) TetIncD3F strain. The cells were infected for 24 h to allow for inclusion formation before addition of 2 ng/ml (left panels), 20 ng/ml (middle panels), or 200 ng/ml (right panels) aTc. The cells were fixed at 5, 15, 30, or 45 min or 1 or 20 h post-aTc induction, immunostained with anti-FLAG antibodies, and imaged using a confocal microscope. The same imaging settings were used for all conditions, and the images were not manipulated after acquisition, so the intensities of the signals on the micrographs truly reflect the expression levels of the IncD-3×FLAG construct. For each combination of aTc concentration and time, the left panels correspond to the FLAG signal of the IncD-3×FLAG construct (IncD3F; green) and the middle panels to the bacteria (mCherry; red). Merged images are shown on the right. The top and bottom panels correspond to an extended-focus view combining all the confocal planes (Ext.Foc.) and a single plane crossing the middle of the inclusion (XYView), respectively. Bar, 10 μm.
We concluded, in agreement with the results obtained by immunoblot analysis (Fig. 1), that the levels of IncD-3×FLAG expression observed at the inclusion membrane by immunofluorescence analysis were time and dose dependent. Moreover, our analysis established that as previously described for the endogenous IncD protein (36), the IncD-3×FLAG construct encoded by the transformed plasmid localized to the inclusion membrane when expressed from C. trachomatis. As a side note, we observed the formation of multiple IncD-3×FLAG- and IncA-positive fibers that originated from the inclusion of the CtL2 mCh(Gro) TetIncD3F strain upon expression of the IncD-3×FLAG construct (37, 38) (see Fig. S2 in the supplemental material). We have ruled out the possibility that fiber formation resulted from a side effect of aTc treatment (see Fig. S3 in the supplemental material). Additional studies will be required to determine the exact significance of this observation.
Growth characteristics of the C. trachomatis strain expressing IncD-3×FLAG.
We next investigated the effect of IncD-3×FLAG expression on C. trachomatis growth. For this purpose, we compared the growth of the CtL2 mCh(Gro) TetIncD3F strain to that of a control strain, CtL2 mCh(Gro), that expressed mCherry only, in the absence or presence of increasing doses of aTc. To follow the progression of the infection, one set of infected cells was fixed at 24, 48, 72, and 96 h postinfection and analyzed by fluorescence microcopy. The second set of infected cells was processed to determine the numbers of infectious progeny recovered at 48, 72, and 96 h postinfection.
Except for that at the highest aTc concentration (200 ng/ml), the replication of the control strain [CtL2 mCh(Gro)] was not affected (Fig. 3). The cells harbored primary inclusions whose size increased between 24 h and 48 h (Fig. 3A). Secondary inclusions were visible at 72 h postinfection, and by 96 h, the whole monolayer was infected (Fig. 3A). The apparently normal progression of the developmental cycle was confirmed by the increasing numbers of infectious particles recovered at 48, 72, and 96 h postinfection (Fig. 3B).
FIG 3.
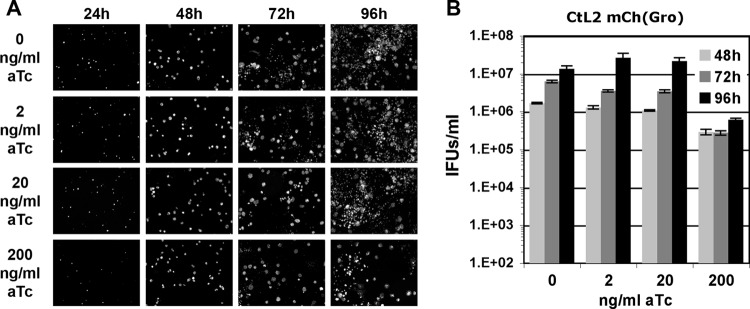
Growth characteristics of C. trachomatis CtL2 mCh(Gro) strain. (A) Black and white micrographs of confluent HeLa cells infected with a strain of C. trachomatis expressing mCherry under the control of the groESL operon regulatory element [CtL2 mCh(Gro)]. The infected cells were incubated with the indicated concentrations of aTc (0, 2, 20, and 200 ng/ml) at 1 h postinfection and imaged at the indicated times postinfection (24, 48, 72, and 96 h). The channel displaying the mCherry bacteria is shown. (B) Quantification of the numbers of infectious bacteria produced (IFUs/ml) at 48 h, 72 h, and 96 h postinfection (light gray, dark gray, and black bars, respectively) in HeLa cells infected with a strain of C. trachomatis expressing mCherry under the control of the groESL operon regulatory element [CtL2 mCh(Gro)] and incubated in the presence of 0, 2, 20, or 200 ng/ml aTc at 1 h postinfection.
The developmental cycle of the CtL2 mCh(Gro) TetIncD3F strain also progressed normally in the absence of aTc (Fig. 4A). However, it was affected in the presence of 2 ng/ml aTc (Fig. 4A). Although large primary inclusions were visible at 48 h postinfection, these inclusions failed to produce large amounts of infectious bacteria, as few secondary inclusions were observed at 72 h and 96 h postinfection. The direct quantification of infectious bacteria produced confirmed a 10-fold reduction in progeny production (Fig. 4B, 2 ng/ml). A similar result was observed in the presence of 20 ng/ml aTc (Fig. 4A), with a more dramatic impact on the production of infectious bacteria (Fig. 4B, 20 ng/ml). In the presence of 200 ng/ml aTc, a concentration that partially affects C. trachomatis growth (Fig. 3), the CtL2 mCh(Gro) TetIncD3F strain formed primary inclusions (Fig. 4A, mCherry panels for 24 and 48 h) that failed to produce infectious bacteria (Fig. 4A, panels for 72 and 96 h, and Fig. 4B, 200ng/ml). Finally, at all time points and aTc concentrations tested, both primary and secondary inclusions were positive for IncD-3×FLAG, confirming that the protein was produced under these conditions (Fig. 4A, IncD3FLAG panels).
FIG 4.

Growth characteristics of the C. trachomatis CtL2 mCh(Gro) TetIncD3F strain. (A) Black and white micrographs of confluent HeLa cells infected with a strain of C. trachomatis expressing mCherry under the control of the groESL operon regulatory element and IncD-3×FLAG under the control of an aTc-inducible promoter [CtL2 mCh(Gro) TetIncD3F]. The infected cells were incubated with the indicated concentrations of aTc (0, 2, 20, and 200 ng/ml) at 1 h postinfection and imaged at the indicated times postinfection (24, 48, 72, and 96 h). The channels displaying the mCherry bacteria and the IncD-3×FLAG construct are shown in the upper (mCherry) and lower (IncD3FLAG) panels, respectively. (B) Quantification of the numbers of infectious bacteria produced (IFUs/ml) at 48 h, 72 h, and 96 h postinfection (light gray, dark gray, and black bars, respectively) in HeLa cells infected with a strain of C. trachomatis expressing mCherry under the control of the groESL operon regulatory element and IncD-3×FLAG under the control of an aTc-inducible promoter [CtL2 mCh(Gro) TetIncD3F] and incubated in the presence of 0, 2, 20, or 200 ng/ml aTc at 1 h postinfection. N.D., none detected.
Altogether, these results indicate that the CtL2 mCh(Gro) TetIncD3F strain is fully infectious when grown in the absence of aTc. However, aTc concentrations that allowed for the growth of the CtL2 mCh(Gro) control strain affected the optimal progression of the developmental cycle and the production of infectious bacteria of the CtL2 mCh(Gro) TetIncD3F strain.
IncD-3×FLAG expression leads to a massive recruitment of CERT to the inclusion membrane.
To determine whether IncD-3×FLAG would affect the association of CERT with the inclusion membrane, HeLa cells expressing yellow fluorescent protein (YFP)-CERT were infected with the CtL2 mCh(Gro) TetIncD3F strain. At 1 h postinfection, infected cells were incubated in the absence or presence of increasing aTc concentrations. The cells were fixed at 23 h postinduction, immunostained with anti-FLAG antibodies, and imaged by confocal microscopy. Representative micrographs of an extended-focus view combining all the confocal planes spanning the inclusion and of a single plane across the inclusion (x-y view) are presented in Fig. 5. IncD-3×FLAG was not detectable in the absence of aTc, and YFP-CERT displayed discrete patches at the surface of the inclusion. In the presence of 2 ng/ml aTc, IncD-3×FLAG was expressed and localized to the inclusion membrane. Under the same conditions, YFP-CERT still localized to patches on the inclusion membrane, but the YFP-CERT signal appeared to cover a larger area of the inclusion membrane. In the presence of 20 ng/ml aTc, the IncD-3×FLAG signal on the inclusion membrane appeared to be more intense than that with 2 ng/ml aTc (Fig. 5, compare the IncD3×FLAG signals between 2 and 20 ng/ml in both the extended-focus and x-y views). The increased amount of IncD-3×FLAG at the inclusion membrane correlated with a massive recruitment of YFP-CERT, and instead of localizing to discrete patches, YFP-CERT appeared to cover the entire inclusion (Fig. 5, compare the YFP-CERT signals between 2 and 20 ng/ml in both the extended-focus and x-y views). A similar result was observed in the presence of 200 ng/ml aTc. We confirmed that the presence of aTc, even at the highest concentration, did not affect the patchy localization of YFP-CERT on the inclusion of the CtL2 mCh(Gro) control strain (see Fig. S4 in the supplemental material).
FIG 5.

IncD-3×FLAG expression leads to massive recruitment of CERT to the inclusion membrane. Confocal fluorescence micrographs show inclusions of the CtL2 mCh(Gro) TetIncD3F strain (mCherry; red) in HeLa cells expressing YFP-CERT (YFP-CERT; yellow). At 1 h postinfection, the infected cells were incubated in the absence (0 ng/ml aTc) or presence (2, 20, or 200 ng/ml aTc) of the indicated aTc concentration. The infected cells were fixed at 23 h post-aTc induction, coimmunostained using anti-FLAG (IncD3FLAG; blue), and imaged using a confocal microscope. The same imaging settings were used for all conditions, and the images were not manipulated after acquisition, so the intensities of the signals on the micrographs truly reflect the expression levels of the IncD-3×FLAG construct. The left and right groups of panels correspond to an extended-focus view combining all the confocal planes spanning an entire inclusion (Ext.Foc.) and a single plane crossing the middle of the inclusion (XY View), respectively. Merged images are shown in the right column for each group. Bar, 10 μm.
Altogether, these results indicate that the amount of YFP-CERT recruited to the inclusion membrane correlates with the amount of IncD-3×FLAG present on the surface of the inclusion, which validates our previously proposed model in which IncD recruits CERT to the inclusion membrane (6).
The massive recruitment of CERT to the inclusion membrane depends on the PH domain of CERT.
At ER-Golgi MCSs, the PH domain of CERT mediates the association of CERT with the Golgi apparatus. To determine whether the PH domain of CERT mediates the massive recruitment of CERT to the inclusion membrane, as displayed in Fig. 5, HeLa cells expressing YFP-CERT (full length) or YFP-CERTNoPH, a construct lacking the PH domain of CERT, were infected with the CtL2 mCh(Gro) TetIncD3F strain. In the absence of aTc, IncD-3×FLAG expression was not detectable (Fig. 6A and B), and CERT localized to discrete patches (Fig. 6A) which covered about 30% of the inclusion (Fig. 6B). In contrast, in the presence of 20 ng/ml aTc, IncD-3×FLAG expression was clearly detectable (Fig. 6A and B), and CERT covered 80% of the inclusion membrane (Fig. 6B). In the same experimental setup, a CERT construct lacking the PH domain failed to associate with the inclusion membrane, despite the presence of IncD-3×FLAG on the inclusion membrane (Fig. 6C). Quantification of the fractions of inclusion membrane associated with the respective markers established that CERT lacking the PH domain covered less than 10% of the inclusion, even when IncD-3×FLAG covered 80% of the inclusion membrane (Fig. 6D).
FIG 6.

The massive recruitment of CERT to the inclusion membrane depends on IncD and the PH domain of CERT. (A and C) Confocal fluorescence micrographs of a single plane crossing the middle of inclusions of a C. trachomatis strain, expressing mCherry (red) and IncD3×FLAG (blue) under the control of an aTc-inducible promoter, in HeLa cells expressing wild-type YFP-CERT (YFP-CERT; yellow) (A) or a truncated version lacking the PH domain (YFP-CERT NoPH; yellow) (C). At 1 h postinfection, the infected cells were incubated in the absence (No aTc) or presence (+ aTc) of 20 ng/ml aTc. The cells were fixed at 23 h post-aTc induction, immunostained with anti-FLAG antibodies (IncD3FLAG; blue), and imaged using a confocal microscope. Merged images are shown on the left. Bar, 10 μm. (B and D) Quantifications of the proportions of the inclusion membrane covered by the indicated markers. The quantification method is described in Materials and Methods.
Altogether, these experiments demonstrate that expression of the C. trachomatis inclusion membrane protein IncD mediates the PH domain-dependent recruitment of CERT to the inclusion.
IncD-3×FLAG expression leads to the massive CERT-dependent recruitment of VAPB to the inclusion membrane.
At ER-Golgi MCSs, CERT interacts with the ER-resident protein VAPB. To further explore the possibility that the recruitment of CERT to C. trachomatis inclusions may lead to the recruitment of VAPB, HeLa cells coexpressing CFP-VAPB and wild-type YFP-CERT were infected with the CtL2 mCh(Gro) TetIncD3FLAG strain. At 1 h postinfection, the cells were incubated in the absence or presence of 20 ng/ml aTc. The cells were fixed at 23 h post-aTc induction and imaged using a confocal microscope. Representative micrographs of an extended-focus view combining the confocal planes spanning each inclusion are presented (Fig. 7A). In the absence of IncD-3×FLAG (no aTc), the CFP-VAPB and wild-type YFP-CERT constructs colocalized to small patches distributed throughout the inclusion membrane, as previously described (6). In the presence of IncD-3×FLAG (+ aTc), CFP-VAPB no longer localized to discrete patches on the surface of the inclusion membrane but instead appeared to cover most of the inclusion. A similar result was observed when different aTc concentrations were used to induce IncD-3×FLAG expression (see Fig. S5A in the supplemental material). We also confirmed that the extensive association of VAPB with the inclusion membrane was not due to aTc treatment (see Fig. S5B in the supplemental material). Moreover, in the absence of expression of the YFP-CERT construct, VAPB localized to discrete patches on the surfaces of inclusions displaying IncD-3×FLAG (see Fig. S5C in the supplemental material), showing that the level of VAPB recruitment to the inclusion membrane relied on CERT expression.
FIG 7.

VAPB association with the inclusion membrane is dependent on IncD and the FFAT motif of CERT. (A, B, and D) Confocal fluorescence micrographs of inclusions of a C. trachomatis strain, expressing mCherry (red) and IncD3×FLAG under the control of an aTc-inducible promoter, in HeLa cells coexpressing a CFP-VAPB (blue) construct and either a wild-type YFP-CERT construct (YFP-CERT; yellow) (A and B) or a version displaying a mutation in the FFAT motif (YFP-CERT FFATmut; yellow) (D). At 1 h postinfection, the infected cells were incubated in the absence (No aTc) or presence (+ aTc) of 20 ng/ml aTc. The cells were fixed at 23 h post-aTc induction and imaged using a confocal microscope. An extended-focus view combining all the confocal planes spanning an entire inclusion (A) or views of a single plane crossing the middle of the inclusion (B and D) are shown. Merged images are shown on the left. Bar, 10 μm. (C and E) Quantifications of the proportions of the inclusion membrane covered by the indicated markers. The quantification method is described in Materials and Methods.
Altogether, these experiments demonstrate that the expression of the C. trachomatis inclusion membrane protein IncD mediates the CERT-dependent recruitment of VAPB to the inclusion.
VAPB association with the inclusion membrane depends on IncD expression and the FFAT motif of CERT.
We next investigated whether CERT-VAPB interaction occurred through the FFAT motif of CERT, as previously shown for ER-Golgi MCSs (11, 12). The experimental setup was similar to that described above, except that the cells coexpressed CFP-VAPB and YFP-CERT that was either wild type or mutated in the FFAT motif (CERT-FFATmut) (39). Representative micrographs of a single plane across the inclusion are presented in Fig. 7B and D, and the corresponding quantifications of the proportions of inclusion membrane covered by the respective markers are shown in Fig. 7C and E. In the absence of IncD-3×FLAG expression (no aTc), CERT and VAPB localized to discrete patches on the inclusion membrane (Fig. 7B, top panels) which covered 20 to 30% of the inclusion (Fig. 7C, No aTc). When IncD-3×FLAG was expressed, CERT and VAPB were massively recruited to the inclusion (Fig. 7B, bottom panels) and covered 70% of the inclusion membrane (Fig. 7C, + aTc). In the absence of IncD-3×FLAG expression, CERT-FFATmut covered 60% of the inclusion membrane, probably reflecting the interaction of the PH domain of CERT-FFATmut with the endogenous IncD protein, and VAPB covered only 20% of the inclusion membrane (Fig. 7E, no aTc). Upon IncD-3×FLAG expression, CERT-FFATmut associated with the inclusion in a pattern identical to that observed for the wild-type construct (Fig. 7D, bottom panels), and it covered 75% of the inclusion membrane (Fig. 7E, + aTc), but VAPB failed to associate with the inclusion (Fig. 7D, bottom panels) and covered less than 10% of the inclusion membrane (Fig. 7E, + aTc). Altogether, these results demonstrate that the association of VAPB with the inclusion upon IncD-3×FLAG expression depends on the presence of CERT and occurs through the interaction of VAPB with the FFAT motif of CERT.
DISCUSSION
Inducible expression of IncD-3×FLAG in C. trachomatis.
In this study, we took advantage of the recent advances in C. trachomatis genetics and engineered a strain that expressed IncD-3×FLAG under the control of an aTc-inducible promoter. Our time course of induction showed that the aTc-inducible system allowed for a tight dose- and time-dependent regulation of IncD expression (Fig. 1 and 2). Two additional studies recently reported the use of the Tet system in Chlamydia and described time courses of induction of the fluorescent proteins GFP (32) and mCherry (33), using increasing doses of aTc. Despite the differences in overall experimental setup and the nature of the reporter constructs, all three studies showed that high doses of aTc (200 to 400 ng/ml) affect C. trachomatis growth. Low concentrations of aTc, ranging from 0.25 to 30 ng/ml, do not affect C. trachomatis replication and efficiently relieve TetR repression, leading to the detectable expression of the reporter construct within 1 h of aTc induction. Although the induction conditions should be adjusted for each protein of interest, the results reported so far by our group and the Hefty and Hackstadt groups suggest that concentrations in the low ng/ml range should be favored to ensure efficient induction without affecting C. trachomatis replication.
Growth of the CtL2 mCh(Gro) TetIncD3F strain in the presence of aTc.
Given the obligate intracellular nature of the developmental cycle, any toxicity due to the expression of proteins of interest may impair the generation of the corresponding transgenic strains. In fact, we failed in our initial attempt to establish a C. trachomatis strain expressing IncD-3×FLAG under the control of the incDEFG operon promoter. Even though transient expression of IncD-3×FLAG could occasionally be detected during the transformation procedure, a stable strain could not be established. Interestingly, Bauler and Hackstadt reported that strains of C. trachomatis expressing IncD-FLAG and IncD-CyaA constructs from the rpoB promoter grew as well as the wild-type strain (33). Additional experiments will be needed to explain this discrepancy, but our observations are compatible with the idea that a slight increase in IncD expression, due to the copy number of the plasmid, might be sufficient to perturb the developmental cycle. Accordingly, the inducible expression of IncD-3×FLAG led to a reduced production of infectious particles (Fig. 3). We showed that a large amount of IncD-3×FLAG protein on the inclusion membrane led to massive recruitment of the CERT and VAPB proteins to the inclusion membrane (Fig. 5 and 7), which may affect the proper function of the ER-inclusion MCSs and impair bacterial growth. It is also possible that the presence of large amounts of IncD-3×FLAG protein on inclusions may indirectly interfere with the interaction of the inclusions with additional host factors or organelles that are beneficial for C. trachomatis replication.
Thus, our results suggest that misregulation of IncD expression may be detrimental to C. trachomatis. Because of the complex nature of the C. trachomatis developmental cycle, this could be the case for many Chlamydia effector proteins, because of a loss of temporal expression and/or because of misregulation of expression levels. As exemplified here with IncD, the conditional and dose-dependent expression of these factors from C. trachomatis is feasible and will bypass these problems and allow for the dissection of effector functions from the native environment.
IncD-CERT-VAPB interaction at the inclusion.
Our goal was to demonstrate the function of the C. trachomatis effector protein IncD expressed from bacteria, instead of relying on its expression from the mammalian host. We showed that when it was expressed from C. trachomatis as a 3×FLAG-tagged protein, IncD localized to the inclusion membrane (Fig. 2). This result is in agreement with the inclusion membrane localization of the endogenous IncD protein (36). Moreover, while the manuscript was under revision, an elegant study from the Hackstadt group also described that various tagged IncD constructs also localized to the inclusion membrane (33). Because of the lack of a C. trachomatis IncD null mutant strain, we could not definitively prove that IncD is necessary and sufficient for CERT recruitment to the inclusion. However, our data demonstrate a positive correlation between the level of IncD expression and the level of CERT recruitment at the surface of the inclusion (Fig. 5). Our data also demonstrate the importance of the PH domain of CERT in the recruitment process (Fig. 6). Altogether, these results are consistent with our previous findings and bring additional evidence to validate our model of IncD-CERT interaction at the inclusion membrane (6). In addition, our approach has also allowed us to gain insight into the mechanisms supporting the association of VAPB with the inclusion. By modulating the level of IncD-3×FLAG, and therefore the amount of CERT associated with the inclusion membrane, we were able to show a positive correlation between the levels of CERT and VAPB recruited to the inclusion (Fig. 7). Moreover, we showed that VAPB association with the inclusion was dependent on the FFAT motif of CERT. Altogether, these results experimentally validated our proposed model, in which, at ER-inclusion MCSs, the C. trachomatis effector protein IncD recruits CERT to the inclusion membrane through its PH domain, which mediates the FFAT motif-dependent recruitment of the ER-resident protein VAPB.
In conclusion, by using an inducible expression system, we were able to modulate the expression of an effector protein in C. trachomatis in order to investigate its function. This long-needed approach in this previously genetically intractable pathogen can now be applied and generalized to any potential virulence factors and will tremendously benefit future genetic studies of the molecular mechanisms supporting C. trachomatis pathogenesis.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Agaisse, Roy, and Galán laboratories for helpful discussions and T. Hackstadt (Rocky Mountain Laboratories) for anti-IncA antibodies.
This work was supported by NIH grant AI088312 to I.D.
Footnotes
Published ahead of print 4 March 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01530-14.
REFERENCES
- 1.Friis RR. 1972. Interaction of L cells and Chlamydia psittaci: entry of the parasite and host responses to its development. J. Bacteriol. 110:706–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moulder JW. 1991. Interaction of chlamydiae and host cells in vitro. Microbiol. Rev. 55:143–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bastidas RJ, Elwell CA, Engel JN, Valdivia RH. 2013. Chlamydial intracellular survival strategies. Cold Spring Harb. Perspect. Med. 3:a010256. 10.1101/cshperspect.a010256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bannantine JP, Griffiths RS, Viratyosin W, Brown WJ, Rockey DD. 2000. A secondary structure motif predictive of protein localization to the chlamydial inclusion membrane. Cell. Microbiol. 2:35–47. 10.1046/j.1462-5822.2000.00029.x [DOI] [PubMed] [Google Scholar]
- 5.Mueller KE, Plano GV, Fields KA. 2014. New frontiers in type III secretion biology: the Chlamydia perspective. Infect. Immun. 82:2–9. 10.1128/IAI.00917-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Derré I, Swiss R, Agaisse H. 2011. The lipid transfer protein CERT interacts with the Chlamydia inclusion protein IncD and participates to ER-Chlamydia inclusion membrane contact sites. PLoS Pathog. 7:e1002092. 10.1371/journal.ppat.1002092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levine T, Loewen C. 2006. Inter-organelle membrane contact sites: through a glass, darkly. Curr. Opin. Cell Biol. 18:371–378. 10.1016/j.ceb.2006.06.011 [DOI] [PubMed] [Google Scholar]
- 8.Lebiedzinska M, Szabadkai G, Jones AW, Duszynski J, Wieckowski MR. 2009. Interactions between the endoplasmic reticulum, mitochondria, plasma membrane and other subcellular organelles. Int. J. Biochem. Cell Biol. 41:1805–1816. 10.1016/j.biocel.2009.02.017 [DOI] [PubMed] [Google Scholar]
- 9.Hanada K, Kumagai K, Yasuda S, Miura Y, Kawano M, Fukasawa M, Nishijima M. 2003. Molecular machinery for non-vesicular trafficking of ceramide. Nature 426:803–809. 10.1038/nature02188 [DOI] [PubMed] [Google Scholar]
- 10.Ponting CP, Aravind L. 1999. START: a lipid-binding domain in StAR, HD-ZIP and signalling proteins. Trends Biochem. Sci. 24:130–132. 10.1016/S0968-0004(99)01362-6 [DOI] [PubMed] [Google Scholar]
- 11.Loewen CJ, Roy A, Levine TP. 2003. A conserved ER targeting motif in three families of lipid binding proteins and in Opi1p binds VAP. EMBO J. 22:2025–2035. 10.1093/emboj/cdg201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lev S, Ben Halevy D, Peretti D, Dahan N. 2008. The VAP protein family: from cellular functions to motor neuron disease. Trends Cell Biol. 18:282–290. 10.1016/j.tcb.2008.03.006 [DOI] [PubMed] [Google Scholar]
- 13.Lemmon MA. 2008. Membrane recognition by phospholipid-binding domains. Nat. Rev. Mol. Cell Biol. 9:99–111. 10.1038/nrm2328 [DOI] [PubMed] [Google Scholar]
- 14.Balla A, Balla T. 2006. Phosphatidylinositol 4-kinases: old enzymes with emerging functions. Trends Cell Biol. 16:351–361. 10.1016/j.tcb.2006.05.003 [DOI] [PubMed] [Google Scholar]
- 15.Hanada K, Kumagai K, Tomishige N, Yamaji T. 2009. CERT-mediated trafficking of ceramide. Biochim. Biophys. Acta 1791:684–691. 10.1016/j.bbalip.2009.01.006 [DOI] [PubMed] [Google Scholar]
- 16.Kari L, Goheen MM, Randall LB, Taylor LD, Carlson JH, Whitmire WM, Virok D, Rajaram K, Endresz V, McClarty G, Nelson DE, Caldwell HD. 2011. Generation of targeted Chlamydia trachomatis null mutants. Proc. Natl. Acad. Sci. U. S. A. 108:7189–7193. 10.1073/pnas.1102229108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nguyen BD, Valdivia RH. 2012. Virulence determinants in the obligate intracellular pathogen Chlamydia trachomatis revealed by forward genetic approaches. Proc. Natl. Acad. Sci. U. S. A. 109:1263–1268. 10.1073/pnas.1117884109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson CM, Fisher DJ. 2013. Site-specific, insertional inactivation of incA in Chlamydia trachomatis using a group II intron. PLoS One 8:e83989. 10.1371/journal.pone.0083989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tam JE, Davis CH, Wyrick PB. 1994. Expression of recombinant DNA introduced into Chlamydia trachomatis by electroporation. Can. J. Microbiol. 40:583–591. 10.1139/m94-093 [DOI] [PubMed] [Google Scholar]
- 20.Binet R, Maurelli AT. 2009. Transformation and isolation of allelic exchange mutants of Chlamydia psittaci using recombinant DNA introduced by electroporation. Proc. Natl. Acad. Sci. U. S. A. 106:292–297. 10.1073/pnas.0806768106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mishra MK, Gérard HC, Whittum-Hudson JA, Hudson AP, Kannan RM. 2012. Dendrimer-enabled modulation of gene expression in Chlamydia trachomatis. Mol. Pharm. 9:413–421. 10.1021/mp200512f [DOI] [PubMed] [Google Scholar]
- 22.Gérard HC, Mishra MK, Mao G, Wang S, Hali M, Whittum-Hudson JA, Kannan RM, Hudson AP. 2013. Dendrimer-enabled DNA delivery and transformation of Chlamydia pneumoniae. Nanomedicine 9:996–1008. 10.1016/j.nano.2013.04.004 [DOI] [PubMed] [Google Scholar]
- 23.Kannan RM, Gerard HC, Mishra MK, Mao G, Wang S, Hali M, Whittum-Hudson JA, Hudson AP. 2013. Dendrimer-enabled transformation of Chlamydia trachomatis. Microb. Pathog. 65:29–35. 10.1016/j.micpath.2013.08.003 [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, Kahane S, Cutcliffe LT, Skilton RJ, Lambden PR, Clarke IN. 2011. Development of a transformation system for Chlamydia trachomatis: restoration of glycogen biosynthesis by acquisition of a plasmid shuttle vector. PLoS Pathog. 7:e1002258. 10.1371/journal.ppat.1002258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu S, Battaglia L, Bao X, Fan H. 2013. Chloramphenicol acetyltransferase as a selection marker for chlamydial transformation. BMC Res. Notes 6:377. 10.1186/1756-0500-6-377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ding H, Gong S, Tian Y, Yang Z, Brunham R, Zhong G. 2013. Transformation of sexually transmitted infection-causing serovars of Chlamydia trachomatis using blasticidin for selection. PLoS One 8:e80534. 10.1371/journal.pone.0080534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Agaisse H, Derré I. 2013. A C. trachomatis cloning vector and the generation of C. trachomatis strains expressing fluorescent proteins under the control of a C. trachomatis promoter. PLoS One 8:e57090. 10.1371/journal.pone.0057090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y, Kahane S, Cutcliffe LT, Skilton RJ, Lambden PR, Persson K, Bjartling C, Clarke IN. 2013. Genetic transformation of a clinical (genital tract), plasmid-free isolate of Chlamydia trachomatis: engineering the plasmid as a cloning vector. PLoS One 8:e59195. 10.1371/journal.pone.0059195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gong S, Yang Z, Lei L, Shen L, Zhong G. 2013. Characterization of Chlamydia trachomatis plasmid-encoded open reading frames. J. Bacteriol. 195:3819–3826. 10.1128/JB.00511-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song L, Carlson JH, Whitmire WM, Kari L, Virtaneva K, Sturdevant DE, Watkins H, Zhou B, Sturdevant GL, Porcella SF, McClarty G, Caldwell HD. 2013. Chlamydia trachomatis plasmid-encoded Pgp4 is a transcriptional regulator of virulence-associated genes. Infect. Immun. 81:636–644. 10.1128/IAI.01305-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Y, Cutcliffe LT, Skilton RJ, Persson K, Bjartling C, Clarke IN. 2013. Transformation of a plasmid-free, genital tract isolate of Chlamydia trachomatis with a plasmid vector carrying a deletion in CDS6 revealed that this gene regulates inclusion phenotype. Pathog. Dis. 67:100–103. 10.1111/2049-632X.12024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wickstrum J, Sammons LR, Restivo KN, Hefty PS. 2013. Conditional gene expression in Chlamydia trachomatis using the Tet system. PLoS One 8:e76743. 10.1371/journal.pone.0076743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bauler LD, Hackstadt T. 2014. Expression and targeting of secreted proteins from Chlamydia trachomatis. J. Bacteriol. 196:1325–1334. 10.1128/JB.01290-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Derré I, Pypaert M, Dautry-Varsat A, Agaisse H. 2007. RNAi screen in Drosophila cells reveals the involvement of the Tom complex in Chlamydia infection. PLoS Pathog. 3:1446–1458. 10.1371/journal.ppat.0030155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beare PA, Gilk SD, Larson CL, Hill J, Stead CM, Omsland A, Cockrell DC, Howe D, Voth DE, Heinzen RA. 2011. Dot/Icm type IVB secretion system requirements for Coxiella burnetii growth in human macrophages. mBio 2:e00175–11. 10.1128/mBio.00175-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scidmore-Carlson MA, Shaw EI, Dooley CA, Fischer ER, Hackstadt T. 1999. Identification and characterization of a Chlamydia trachomatis early operon encoding four novel inclusion membrane proteins. Mol. Microbiol. 33:753–765. 10.1046/j.1365-2958.1999.01523.x [DOI] [PubMed] [Google Scholar]
- 37.Rockey DD, Heinzen RA, Hackstadt T. 1995. Cloning and characterization of a Chlamydia psittaci gene coding for a protein localized in the inclusion membrane of infected cells. Mol. Microbiol. 15:617–626 [DOI] [PubMed] [Google Scholar]
- 38.Bannantine JP, Stamm WE, Suchland RJ, Rockey DD. 1998. Chlamydia trachomatis IncA is localized to the inclusion membrane and is recognized by antisera from infected humans and primates. Infect. Immun. 66:6017–6021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kawano M, Kumagai K, Nishijima M, Hanada K. 2006. Efficient trafficking of ceramide from the endoplasmic reticulum to the Golgi apparatus requires a VAMP-associated protein-interacting FFAT motif of CERT. J. Biol. Chem. 281:30279–30288. 10.1074/jbc.M605032200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.