

Abstract
Hemolytic uremic syndrome (HUS) is mainly induced by Shiga toxin 2 (Stx2)-producing Escherichia coli. Proteinuria can occur in the early phase of the disease, and its persistence determines the renal prognosis. Stx2 may injure podocytes and induce proteinuria. Human serum amyloid P component (SAP), a member of the pentraxin family, has been shown to protect against Stx2-induced lethality in mice in vivo, presumably by specific binding to the toxin. We therefore tested the hypothesis that SAP can protect against Stx2-induced injury of human podocytes. To elucidate the mechanisms underlying podocyte injury in HUS-associated proteinuria, we assessed Stx2-induced activation of mitogen-activated protein kinases (MAPKs) and apoptosis in immortalized human podocytes and evaluated the impact of SAP on Stx2-induced damage. Human podocytes express Stx2-binding globotriaosylceramide 3. Stx2 applied to cultured podocytes was internalized and then activated p38α MAPK and c-Jun N-terminal kinase (JNK), important signaling steps in cell differentiation and apoptosis. Stx2 also activated caspase 3, resulting in an increased level of apoptosis. Coincubation of podocytes with SAP and Stx2 mitigated the effects of Stx2 and induced upregulation of antiapoptotic Bcl2. These data suggest that podocytes are a target of Stx2 and that SAP protects podocytes against Stx2-induced injury. SAP may therefore be a useful therapeutic option.
INTRODUCTION
Shiga toxin (Stx)-associated hemolytic uremic syndrome (HUS) is a life-threatening disease characterized by hemolytic anemia, thrombocytopenia, and renal failure (1). It is the most frequent cause of acute renal failure in childhood and is usually caused by Stx2-producing enterohemorrhagic Escherichia coli (EHEC), but to a lesser extent by Stx1-producing EHEC (2). Stx1 and Stx2 induce apoptosis and activate stress response pathways in endothelial cells (3, 4), causing the clinical picture of thrombotic microangiopathy (TMA) (1). A direct toxic effect of Stx2 on nonendothelial cells has recently been suggested. Stx2 binds to susceptible cells via the unique toxin-binding glycosphingolipid receptor galactose-α1,4-glucose-ceramide (Gb3), which is generated by Gb3 synthase (5, 6). After internalization, Stx2 activates various intracellular stress pathways, such as the endoplasmic stress response or ribotoxic stress response (3). This may lead to apoptosis via activation of various cell death signals (7), such as downregulation of antiapoptotic Bcl2 (8), activation of mitogen-activated protein kinases (MAPKs) p38α and c-Jun N-terminal kinase (JNK) (7, 9), and caspase 3-dependent apoptosis (10, 11).
In endothelial cells, this leads to TMA, which can result in acute renal failure and neurological injury (1, 12). It has been suggested that neurological involvement may be due to deficiency or impaired function of an Stx2-neutralizing factor (13) in affected patients, in which case plasma supply and exchange might be beneficial. Various candidates for such a neutralizing factor have been investigated, and serum amyloid P component (SAP) appears to be the most promising (14, 15). SAP is one member of the pentraxin family; the other member is C-reactive protein (CRP) (16). Although the gene-coding, amino acid sequences, homopentameric molecular assembly, and calcium-dependent ligand-binding properties of SAP are phylogenetically conserved, the baseline plasma concentration, acute-phase behavior, and binding affinity of SAP proteins vary between species. No deficiencies or structural variants of human SAP protein have yet been described, and its physiological functions are therefore not completely understood. Nevertheless, there is experimental evidence that SAP can contribute to host defense against certain bacterial infections (17). SAP neutralizes Stx2, but not Stx1, in vitro, and human SAP protects mice from Stx2-induced disease (14). The Stx2-neutralizing effect is not present in the sera of nonhuman species tested so far (15), consistent with the much higher binding avidity of human SAP than mouse SAP to other known ligands (18).
Microalbuminuria is seen in rats soon after intraperitoneal injection of Stx2 (19), and it is also an early sign of renal involvement in patients with EHEC-HUS. Following recovery from the acute phase of the disease, proteinuria may persist; such persistence correlates with a poor long-term renal prognosis (20–22). Injury of podocytes has emerged as the key mechanism underlying this glomerular dysfunction (23) and may have a critical impact on the course of the disease and renal outcome. Severe podocyte injury results in impaired actin cytoskeleton dynamics, foot process effacement, and proteinuria (24, 25). There is increasing evidence that both Stx1 and -2 injure podocytes. Injection of Stx1 results in foot process effacement in animal models (26). In one child with EHEC-induced HUS, Stx1 was detected in mesangial and endothelial cells, as well as in podocytes (27). Stx1 binds to Gb3 in podocytes, resulting in inhibition of protein synthesis and cell death (28). Stx2 results in activation of mitogen-activated protein kinases and actin remodeling in podocytes (29, 30).
We therefore hypothesized that human SAP neutralizes some of the harmful effects of Stx2 on human podocytes. The rationale was to gain detailed information on the glomerular damage caused by binding of Stx2 to podocytes early in HUS.
MATERIALS AND METHODS
Cell culture and media.
Conditionally immortalized human podocytes were produced by transfection with the temperature-sensitive simian virus 40 (SV40) T gene (31). Cells were seeded on collagen type I-coated plates (BD Bioscience, Franklin Lakes, NJ, USA) at 32°C. RPMI 1640 culture medium (PAA, Pasching, Austria) was supplemented with 100 U/ml penicillin, 0.1 mg/ml streptomycin, glutamine, and insulin, transferrin, and sodium selenite mixture (ITS) (Sigma, St. Louis, MO, USA) and with 10% fetal bovine serum (Calbiochem via Merck, Darmstadt, Germany). To induce growth arrest and differentiation, podocytes were maintained under nonpermissive conditions at 37°C for 2 to 3 weeks at about 70% confluence.
Stx2.
Stx2 was provided courtesy of H. Karch, Institute of Hygiene, University of Münster, Münster, Germany. Purification of Stx2 was performed as described previously (32). To investigate the effects of Stx2 on human podocytes, cell cultures were exposed to medium containing Stx2 at a final concentration of 1.5 or 15 ng/ml for up to 48 h. Optimal working concentrations of Stx2 were identified with a dose-mortality curve (data not shown). All studies were performed before cell layers exceeded 80% confluence. RPMI 1640 was used as the incubation medium and was supplemented with 10% fetal calf serum (FCS), which does not contain human SAP, with or without the addition of purified SAP, or with 10% human serum instead of FCS. The medium was provided to the cells before Stx2 was added.
Coincubation with SAP.
Isolated <99.9% pure, intact, fully functional human SAP from healthy blood donors was purified and characterized as described previously (33) and added to FCS-free incubation medium at a final concentration of 3 mg/liter. Alternatively, the medium was supplemented with 10% (vol/vol) serum from one healthy donor with a known SAP concentration of 47 mg/liter (final concentration of SAP, 4.7 mg/liter during incubation) or from a pool of normal human sera (NHS) from healthy laboratory staff.
SAP-free incubation.
SAP-free incubations were performed with 10% FCS or SAP-depleted NHS from the individual mentioned above. Depletion of SAP in NHS was performed using sepharose-phosphoethanolamine (34).
Lipid analysis for detection of Gb3.
Neutral and acidic lipids were separated by anion-exchange chromatography using DEAE-Sephadex according to published methods (35). About 107 differentiated podocytes were homogenized in 1 ml of water using the Homogenizator Precellys 24 (Peqlab, Erlangen, Germany) at 6,500 rpm for 30 s. After extraction of lipids and separation of cell debris by solvent filtration, the sample was evaporated in a stream of nitrogen. Interfering glycerolipids were degraded by alkaline hydrolysis with 2.5 ml of 100 mM sodium hydroxide in methanol for 2 h at 37°C. This was followed by neutralization with 15 μl of acetic acid, and the lipid extract was desalted with reversed-phase C18 (RP-18) chromatography. High-performance thin-layer chromatography (TLC) Silica Gel 60 plates (Merck) were washed and dried before application of the lipid fraction. Chloroform-methanol-water (70:30:4 [vol/vol/vol]) was used as the solvent system. Standard lipids (Neutral Glycosphingolipid Mix; Biotrend, Cologne, Germany) were applied to the TLC plates as markers. The plates were sprayed with a phosphoric acid-copper sulfate reagent {15.6 g of CuSO4(H2O)5 and 9.4 ml of H3PO4 (85% [wt/vol]) in 100 ml of water} and charred at 180°C for 10 min to detect lipid bands (36). Quantification was achieved with densitometry using the TLC-Scanner 3 (Camag, Berlin, Germany) at a wavelength of 595 nm.
Immunofluorescence for detection of Gb3 and Hoechst staining for assessment of apoptosis.
Podocytes were cultured on glass slides, fixed with 4% paraformaldehyde, and permeabilized with 0.05% Triton X-100. Immunofluorescence staining was performed with an anti-Gb3 primary antibody (BD Pharmingen, Heidelberg, Germany) and DyLight 649-conjugated AffiniPure donkey anti-mouse IgM (Jackson ImmunoResearch, West Grove, PA, USA) as the secondary antibody. Slides were then mounted with a commercially available antifade kit (Invitrogen, Darmstadt, Germany) and examined with immunofluorescence microscopy (Axiovert 200 with Apotome System; Zeiss, Jena, Germany). No staining was observed with an isotype-matched naive primary antibody (data not shown). Apoptosis was identified with Hoechst 33258 staining (Calbiochem via Merck) according to the manufacturer's instructions. The percentage of apoptotic cells was determined by counting condensed nuclei in 10 different visual fields. Images were taken, and the cells were counted by blinded observers.
Stx2 internalization and morphological analysis with confocal microscopy.
Podocytes were cultured in 8-well chambered cover glasses (Nalge-Nunc International, Rochester, NY, USA). Oyster 488-labeled Stx2 (200 ng/ml) was added to the cells. Stx2 labeling was performed with an Oyster 488 antibody labeling kit (Luminartis, Münster, Germany). Staining with Oyster 488-labeled isotype-matched primary antibody showed only minor background staining (data not shown). Confocal laser scanning microscopy was performed with a TCS SP5 microscope (Leica, Mannheim, Germany) equipped with an HCX PL APO lambda blue 63.0/1.20 water UV objective. Images were processed with LAS AF software (version 2.4.1; Leica, Mannheim, Germany).
Western blot analysis.
Cells were harvested using tissue protein extraction buffer (Thermo Scientific 78510) supplemented with complete Mini Inhibitor Cocktail (Roche, Basel, Switzerland), 1 mM calyculin A (Cell Signaling Technologies, Danvers, MA, USA), 1 mM NaF, and 1 mM NaVO3. After centrifugation, the supernatant was collected and supplemented with 4× NuPAGE LDS Sample Buffer (Invitrogen) and 0.1 M dithiothreitol (DTT). After boiling, the samples were loaded on NuPAGE Novex 4% to 12% Bis-Tris Gel (1.0 mm; Invitrogen). IRDye (Li-Cor, Lincoln, NE, USA) was used as a marker. Proteins were transferred onto Immobilon-FL polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA, USA) and electrophoresed with an XCell SureLock Mini-Cell electrophoresis system (Invitrogen). The membranes were incubated with blocking buffer (Li-Cor) and primary antibody overnight at 4°C. Primary antibodies against phospho-p38α (p-p38α), p-JNK, Bcl2, extracellular signal-regulated kinase 1/2 (ERK1/2), and cleaved caspase 3 were purchased from Cell Signaling Technologies; GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was from Santa Cruz Biotechnology (Santa Cruz, CA, USA); and p38β was from R&D Systems (Wiesbaden, Germany). The following secondary antibodies were used: IRDye 800CW against mouse IgG and goat or IRDye 680RD against rat and rabbit (Li-Cor). Fluorescence was measured with an Odyssey Clx Blot Scanner and Image Studio software (Li-Cor). All experiments were performed at least three times in duplicate. Blots were quantified with ImageJ 1.47v. Graphs were made and statistical analysis was performed with Prism 5.0b.
Conventional PCR.
Cells were harvested using complete medium and trypsin-EDTA and then centrifuged at 300 × g for 10 min at 15°C. The pellet was washed with phosphate-buffered saline (PBS) and centrifuged, and RNA was then isolated using an innuPrep RNA Mini Kit (Analytik Jena, Biometra, Jena, Germany). The RNA concentration was measured with a NanoDrop ND-1000 spectrophotometer (Thermo Scientific). Total RNA (100 ng/μl) was used for cDNA synthesis. PCR was accomplished using the Mesa Green qPCR MasterMix Plus for SYBR Assay (Eurogentec, Cologne, Germany) with an Applied Biosystems StepOnePlus system and StepOnePlus 2.0 software for evaluation. The primer sequences for Gb3 synthase were 5′-CACGACGCACGAGGCCATGA-3′ and 5′-CCCGGGCCCTCAATCTTGCC-3′ (Life Technologies via Invitrogen). The PCR product was loaded onto a 1.5% agarose gel for size determination. A Biomol 100-bp DNA Ladder (Biomol GmbH, Hamburg, Germany) was used as a marker.
Caspase 3 assay.
An EnzChek Caspase 3 Assay Kit 2 (Invitrogen) was used according to the manufacturer's instructions. Caspase 3 activity was measured by the absorbance at 535 nm using a microplate reader (Genios Plus; Tecan, Maennedorf, Switzerland). The protein content was measured by the bicinchoninic acid method (Thermo Scientific). The amount of converted enzyme was related to the total amount of protein, and a ratio of sample and untreated control was calculated to allow better comparability of each experiment. Controls were normalized to a ratio of 1.
Statistical analysis and images.
Statistical analyses were done for caspase 3 activity, the densitometry of Western blots, and counting of nuclei in Hoechst staining, using a paired Wilcoxon test (with Prism 5) or a paired t test (with SPSS). P values of <0.05 were regarded as significant. Controls were normalized to a ratio of 1. All experiments were performed at least three times in duplicate. Images were arranged in Microsoft PowerPoint, and minimal processing of brightness, contrast, and color balance was carried out.
RESULTS
Gb3 expression by human podocytes.
We started to investigate the effects of Stx2 on podocytes by determining whether podocytes express Stx2-binding Gb3 and the corresponding synthase. We therefore performed PCR for Gb3 synthase mRNA, Gb3 immunofluorescence staining, and lipid analyses. Our mRNA studies found that human podocytes show transcription of the Gb3 synthase gene (Fig. 1A). Lipid analyses and immunofluorescence staining confirmed that podocytes can synthesize Gb3, which was mostly localized along the cell membrane and in the cytosol (Fig. 1B and C).
FIG 1.

Expression of Gb3 synthase and Gb3 in human podocytes. (A) Expression of Gb3 synthase in untreated human podocytes (huPo) by conventional PCR (lane 3). (B) Lipid analysis for the presence of Gb3 in untreated human podocytes. (C) Localization of Gb3 in differentiated untreated podocytes along the membrane (arrows) and within the cytoplasm by immunofluorescence (pink) and in cell nuclei (blue). All experiments were repeated at least four times in independent experiments (scale bars = 15 μm).
Internalization of Stx2 by human podocytes.
To test whether Stx2 could be internalized by human podocytes, we performed Stx2 stimulation experiments. Confocal microscopy revealed binding and immediate uptake of fluorescence-labeled Stx2 by differentiated human podocytes. After 40 min of exposure, the majority of the Stx2 was already located within the cell, where it was widely distributed throughout the cytoplasm (Fig. 2).
FIG 2.

Internalization of labeled Stx2 (white) in podocytes after 40 min of incubation. Distribution is predominantly within the cytoplasm. No Stx2 staining was found in the cell nucleus. The cell outline (dotted lines) and nucleus (dashed line) are marked (scale bar = 15 μm). The experiment was repeated at least three times independently.
Blockage by SAP of Stx2-induced phosphorylation of MAPK p38α and JNK.
SAP has emerged as a potential blocker of Stx2 effects on various cell types. We therefore tested whether SAP had beneficial effects on Stx2-stimulated human podocytes by investigating the extent to which MAPK phosphorylation was evident in cell lysates. Stx2 (15 ng/ml) induced phosphorylation of MAPK p38α at 60 min. Activation was highest under the influence of 15 ng/ml; phosphorylation was maximal at 180 min and was still present at 480 min. Human SAP, which was added at the same time in physiological concentrations (3 mg/liter), significantly inhibited Stx2-dependent phosphorylation of p38α at 180 and 480 min (Fig. 3A). Similarly, Stx2 induced phosphorylation of JNK at 60, 180, and 480 min. SAP significantly inhibited this phosphorylation at 60 and 180 min (Fig. 3B). In contrast, MAPK p38β, MAPK, and ERK1/2 were not activated at any stage of incubation (data not shown).
FIG 3.

Western blot and densitometry of phosphorylation of MAPK p38α and JNK. P-p38α (A) and p-JNK (B) after treatment with 15 ng/ml Stx2 and in combination with SAP (3 mg/liter) after 60, 180, or 480 min. Below are shown loading controls with GAPDH. *, P < 0.05. Means and standard errors of the means of five independent experiments, performed in duplicate, are shown.
Inhibition of Stx2-induced caspase 3 activation by SAP.
To measure apoptosis, we determined caspase 3 activity with an enzyme-linked immunosorbent assay (ELISA). After 24 h and 48 h, Stx2 (1.5 ng/ml) induced significantly greater caspase 3 activity in podocytes than in control cells (10.5 ± 11.1 arbitrary units [AU] versus 1.3 ± 0.9 AU; P < 0.05) (Fig. 4A). When the medium was supplemented with SAP, the Stx2-induced increase in caspase 3 activity was significantly diminished (3.7 ± 3.5 AU versus 10.5 ± 11.1 AU; P < 0.05) (Fig. 4A) but was still higher than in the control group (1.3 ± 0.9; P < 0.05) (Fig. 4A). Consistent with these results, the quantity of cleaved caspase 3 was elevated and reached a peak after 24 h of incubation with Stx2. When SAP was added to medium containing Stx2, cleaved caspase 3 was not detected after 24 or 48 h (Fig. 4B).
FIG 4.
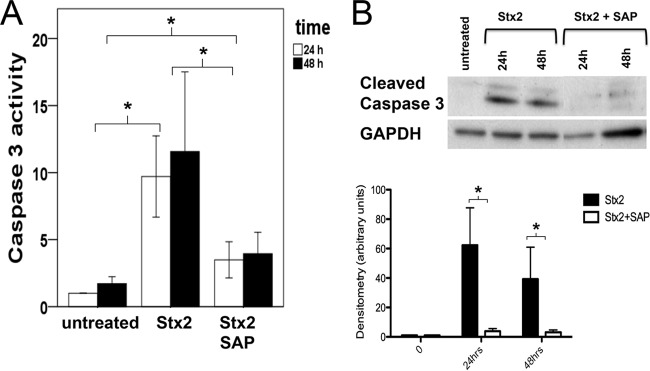
Caspase 3 activity and cleaved caspase 3. (A) Caspase 3 activity (in AU) after Stx2 incubation (1.5 ng/ml) for 24 or 48 h compared to untreated cells and after coincubation with SAP (3 mg/liter) as evaluated by ELISA. For statistical analysis, the values at 24 and 48 h were compared. (B) Amounts of cleaved caspase 3 after Stx2 incubation with or without addition of SAP after 24 and 48 h. Below is shown loading control with GAPDH. *, P < 0.05. Means and standard errors of the means of four independent experiments, performed in duplicate, are shown.
Prevention of Stx2-induced condensation of nuclei by SAP.
Following Stx2 incubation (1.5 ng/ml) for 24 h, Hoechst staining demonstrated significantly enhanced formation of fragmented nuclei and condensed chromatin (39.5% ± 16.4% versus 14.0% ± 5.8%; P < 0.05). Addition of SAP inhibited the Stx2-induced apoptosis, so that only 16.7% ± 7.4% of podocyte nuclei showed condensed chromatin (P < 0.05 versus Stx2; P > 0.05 versus control) (Fig. 5).
FIG 5.

Apoptosis, measured via the amount of condensed or apoptotic nuclei after Stx2 incubation. (A) Untreated control. (B) Nuclei of podocytes after treatment with 1.5 ng/ml Stx2. (Left) Typical enclosures/cysts (arrowheads) and indentations (arrows). (Right) An apoptotic body. (C) Content of condensed nuclei after Stx2 treatment compared to control or coincubation of Stx2 and SAP (3 mg/liter). *, P < 0.05. Means and standard deviations of three independent experiments are shown.
Upregulation of Bcl2 expression by SAP.
Endoplasmic stress enhances the expression of CHOP, which in turn diminishes anti-apoptotic Bcl2, so we investigated this effector molecule, which is critical for cell survival. We observed significantly enhanced Bcl2 expression after coincubation of Stx2 with SAP at 60 and 180 min (Fig. 6, top row on right) compared to incubation without SAP (Fig. 6, top row on left). There was no significant increase in Bcl2 at any time point after incubation with Stx alone.
FIG 6.
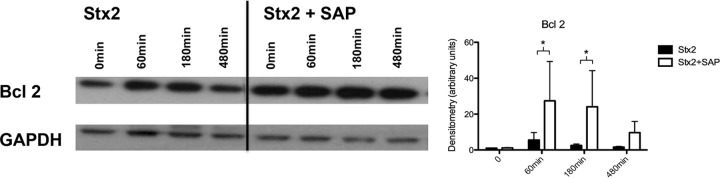
Induction of Bcl2 after Stx2 treatment (15 ng/ml) with or without coincubation with SAP (3 mg/liter) after 60, 180, and 480 min. Below is shown loading control with GAPDH. *, P < 0.05. Means and standard errors of the means of five independent experiments, performed in duplicate, are shown.
DISCUSSION
Our study demonstrates the protective effect of human SAP on Stx2-injured podocytes. Our data are consistent with the results of previous studies that have analyzed the effects of Stx2 on human (29) and murine (29, 30) podocytes. No previously reported studies on the effects of Stx2 on human podocytes have provided any novel therapeutic information. We show here for the first time that direct interactions between Stx2 and human SAP have novel features and important functional effects in vitro. These may prove to be clinically significant in future.
Although proteinuria is not a major symptom in patients with EHEC-HUS, increased glomerular protein loss influences the renal outcome of patients and is often associated with a progressive decline in renal function (20). Even though podocytes may not be the main target of Stx2-induced renal damage, the data of Morigi et al. indicate that Stx2 can induce vasoactive endothelin 1 (ET-1) expression in cultured murine podocytes, which emphasizes the possible important cross talk between endothelial cells and podocytes (30). Vascular endothelial growth factor (VEGF) is a protein secreted by podocytes that is necessary for the survival of endothelial cells, podocytes, and mesangial cells. Stx2 mediated a decline in VEGF release by human podocytes. As decreased podocyte VEGF has been demonstrated to cause glomerular thrombotic microangiopathy in mice and in humans, the mechanism of Stx2-mediated reduction of VEGF may contribute to HUS clinically (37). Substantial deterioration of the glomerular barrier can occur in HUS and is of particular prognostic importance due to the low regenerative capacity of podocytes.
Stx2 is internalized and routed to the endoplasmic reticulum (ER) (3) and induces multiple signaling pathways. Some of these contribute to apoptosis, such as activation of MAPKs and caspase 3 or downregulation of antiapoptotic Bcl2 (7). We confirmed that immortalized human podocytes express Gb3 (which is required for Stx2 binding and uptake) and observed rapid binding to the cell membrane and internalization of Stx2. Once internalized, Stx2 induced phosphorylation of p38α and JNK and apoptosis via the caspase 3 pathway. Such mechanisms have been previously described in endothelial cells (3) and in podocytes (29). In human podocytes, the nonselective caspase inhibitor Q-VD-OPH rescued over 75% of the cytotoxic Stx2 effects (29).
The MAPK family regulates a wide variety of cellular processes (inflammation, cell cycle regulation, apoptosis, differentiation, senescence, and tumorigenesis). Both p38α and JNK contribute to apoptosis (38, 39). Inhibiting the activation of p38α can decrease Stx2-induced apoptosis in intestinal epithelial cells. Apoptosis is mainly initiated by caspases, which are proteases and effector molecules in programmed cell death. Caspase 3 is a key enzyme in this cascade (40), which is activated in the cytoplasm by an increase in calcium concentration. Calcium is released from the ER after prolonged endoplasmic stress (3). Persistent ER stress also induces downregulation of Bcl2. The Bcl2 family comprises important regulators of apoptosis. It prevents apoptosis in various ways, such as stabilization of mitochondria and inhibition of caspase activation (41).
In recent years, investigators have tried to identify the Stx2-neutralizing factor in normal human serum (13). Armstrong and colleagues identified SAP as a potential neutralizing factor (14). SAP is synthesized in the liver and circulates in the blood at levels of 8 to 55 mg/liter (42). It is a universal constituent of amyloid deposits (due to its specific calcium-dependent binding to amyloid fibrils) and contributes to the pathogenesis and persistence of these deposits (43). SAP is also a constituent of the extracellular matrix and is present in the glomerular basement membrane (44) and the microfibrillar mantle of elastic fibers throughout the body (45). However, the function of human SAP is not fully understood. Despite reports that it can opsonize in vitro (46), binding of SAP to bacteria is potently antiopsonic (17). Furthermore, the ubiquity of SAP in amyloid deposits is definitely not opsonic, since these deposits are almost universally ignored by phagocytic cells (43). On the other hand, SAP clearly contributes to host resistance against bacterial infections (17, 44, 45), although the mechanisms are still unknown. However, human SAP is known to bind to Stx2, neutralizing its toxic effects (14, 15). We showed that SAP markedly reduced MAPK activation and apoptosis of Stx2-injured podocytes, implying that SAP may protect podocytes from severe Stx2-induced damage. These observations in cultured podocytes suggest a mechanism by which SAP may confer clinical protection on patients with an EHEC infection.
Our findings raise the question as to whether susceptibility to EHEC-HUS is related to plasma SAP levels and whether new therapeutic approaches may emerge with the availability of recombinant SAP, which has already been administered to patients with pulmonary fibrosis (47). The efficacy of plasma therapy in EHEC-HUS is currently controversial (22). There is evidence that SAP may not be able to prevent thrombotic microangiopathy because Stx2 binds to human neutrophils by a mechanism independent of Gb3 (48), but our data suggest that SAP may be able to attenuate the toxic effects of Stx2 on podocytes. Prevention of proteinuria could have a positive influence on the long-term renal outcome.
In conclusion, Stx2 induces activation of MAPK and apoptosis in human podocytes. Such effects may trigger progressive deterioration of renal function in patients with EHEC-HUS. The protective effect of SAP on Stx2-stressed podocytes may be a potential therapeutic option for EHEC-HUS patients by administration of SAP itself or Stx-binding derivatives.
ACKNOWLEDGMENTS
This study was supported by the Georg und Jürgen Rickertsen Stiftung, Hamburg, Germany.
We thank G. Gstraunthaler (Innsbruck, Austria) for laboratory space and helpful scientific discussions. We thank H. Karch and M. Bielaszewska, Münster, Germany, for providing Stx2. We owe special thanks to A. Rosales and S. Ehrlenbach (Innsbruck, Austria) and S. Brodesser (Cologne, Germany) for supplementary data.
M.P. reports grants from the United Kingdom Medical Research Council and work independent of the National Institute of Health Research. M.P. also has a patent on work done with Pentraxin Therapeutics Ltd. C.E.K. and M.C.L. report grant funding from the Deutsche Forschungsgemeinschaft and Shire HGT during the conduct of this study. The other authors have nothing to disclose.
Footnotes
Published ahead of print 24 February 2014
REFERENCES
- 1.Tarr PI. 2009. Shiga toxin-associated hemolytic uremic syndrome and thrombotic thrombocytopenic purpura: distinct mechanisms of pathogenesis. Kidney Int. Suppl. 112:S29–S32. 10.1038/ki.2008.615 [DOI] [PubMed] [Google Scholar]
- 2.Orth D, Grif K, Khan AB, Naim A, Dierich MP, Wurzner R. 2007. The Shiga toxin genotype rather than the amount of Shiga toxin or the cytotoxicity of Shiga toxin in vitro correlates with the appearance of the hemolytic uremic syndrome. Diagn. Microbiol. Infect. Dis. 59:235–242. 10.1016/j.diagmicrobio.2007.04.013 [DOI] [PubMed] [Google Scholar]
- 3.Tesh VL. 2012. Activation of cell stress response pathways by Shiga toxins. Cell. Microbiol. 14:1–9. 10.1111/j.1462-5822.2011.01684.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tesh VL. 2012. The induction of apoptosis by Shiga toxins and ricin. Curr. Top. Microbiol. Immunol. 357:137–178. 10.1007/82_2011_155 [DOI] [PubMed] [Google Scholar]
- 5.Lingwood CA. 1999. Glycolipid receptors for verotoxin and Helicobacter pylori: role in pathology. Biochim. Biophys. Acta 1455:375–386. 10.1016/S0925-4439(99)00062-9 [DOI] [PubMed] [Google Scholar]
- 6.Lingwood CA. 2003. Shiga toxin receptor glycolipid binding. Pathology and utility. Methods Mol. Med. 73:165–186. 10.1385/1-59259-316-X:165 [DOI] [PubMed] [Google Scholar]
- 7.Kitamura M. 2008. Endoplasmic reticulum stress and unfolded protein response in renal pathophysiology: Janus faces. Am. J. Physiol. Renal Physiol. 295:F323–F334. 10.1152/ajprenal.00050.2008 [DOI] [PubMed] [Google Scholar]
- 8.McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. 2001. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell. Biol. 21:1249–1259. 10.1128/MCB.21.4.1249-1259.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith WE, Kane AV, Campbell ST, Acheson DW, Cochran BH, Thorpe CM. 2003. Shiga toxin 1 triggers a ribotoxic stress response leading to p38 and JNK activation and induction of apoptosis in intestinal epithelial cells. Infect. Immun. 71:1497–1504. 10.1128/IAI.71.3.1497-1504.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zong WX, Li C, Hatzivassiliou G, Lindsten T, Yu QC, Yuan J, Thompson CB. 2003. Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J. Cell Biol. 162:59–69. 10.1083/jcb.200302084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Orrenius S, Zhivotovsky B, Nicotera P. 2003. Regulation of cell death: the calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 4:552–565. 10.1038/nrm1150 [DOI] [PubMed] [Google Scholar]
- 12.Tarr PI, Gordon CA, Chandler WL. 2005. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet 365:1073–1086 [DOI] [PubMed] [Google Scholar]
- 13.Bitzan M, Klemt M, Steffens R, Muller-Wiefel DE. 1993. Differences in verotoxin neutralizing activity of therapeutic immunoglobulins and sera from healthy controls. Infection 21:140–145. 10.1007/BF01710530 [DOI] [PubMed] [Google Scholar]
- 14.Armstrong GD, Mulvey GL, Marcato P, Griener TP, Kahan MC, Tennent GA, Sabin CA, Chart H, Pepys MB. 2006. Human serum amyloid P component protects against Escherichia coli O157:H7 Shiga toxin 2 in vivo: therapeutic implications for hemolytic-uremic syndrome. J. Infect. Dis. 193:1120–1124. 10.1086/501472 [DOI] [PubMed] [Google Scholar]
- 15.Kimura T, Tani S, Matsumoto Yi Y, Takeda T. 2001. Serum amyloid P component is the Shiga toxin 2-neutralizing factor in human blood. J. Biol. Chem. 276:41576–41579. 10.1074/jbc.M107819200 [DOI] [PubMed] [Google Scholar]
- 16.Pepys MB, Baltz ML. 1983. Acute phase proteins with special reference to C-reactive protein and related proteins (pentaxins) and serum amyloid A protein. Adv. Immunol. 34:141–212. 10.1016/S0065-2776(08)60379-X [DOI] [PubMed] [Google Scholar]
- 17.Noursadeghi M, Bickerstaff MC, Gallimore JR, Herbert J, Cohen J, Pepys MB. 2000. Role of serum amyloid P component in bacterial infection: protection of the host or protection of the pathogen. Proc. Natl. Acad. Sci. U. S. A. 97:14584–14589. 10.1073/pnas.97.26.14584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hawkins PN, Myers MJ, Epenetos AA, Caspi D, Pepys MB. 1988. Specific localization and imaging of amyloid deposits in vivo using 123I-labeled serum amyloid P component. J. Exp. Med. 167:903–913. 10.1084/jem.167.3.903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ochoa F, Oltra G, Gerhardt E, Hermes R, Cohen L, Damiano AE, Ibarra C, Lago NR, Zotta E. 2012. Microalbuminuria and early renal response to lethal dose Shiga toxin type 2 in rats. Int. J. Nephrol. Renovasc. Dis. 5:29–36. 10.2147/IJNRD.S27623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Milford DV, White RH, Taylor CM. 1991. Prognostic significance of proteinuria one year after onset of diarrhea-associated hemolytic-uremic syndrome. J. Pediatr. 118:191–194. 10.1016/S0022-3476(05)80481-0 [DOI] [PubMed] [Google Scholar]
- 21.Yamamoto T, Satomura K, Okada S, Ozono K. 2009. Risk factors for neurological complications in complete hemolytic uremic syndrome caused by Escherichia coli O157. Pediatr. Int. 51:216–219. 10.1111/j.1442-200X.2008.02690.x [DOI] [PubMed] [Google Scholar]
- 22.Rosales A, Hofer J, Zimmerhackl LB, Jungraithmayr TC, Riedl M, Giner T, Strasak A, Orth-Holler D, Wurzner R, Karch H, German-Austrian HUS Study Group ,. 2012. Need for long-term follow-up in enterohemorrhagic Escherichia coli-associated hemolytic uremic syndrome due to late-emerging sequelae. Clin. Infect. Dis. 54:1413–1421. 10.1093/cid/cis196 [DOI] [PubMed] [Google Scholar]
- 23.Patrakka J, Tryggvason K. 2009. New insights into the role of podocytes in proteinuria. Nat. Rev. Nephrol. 5:463–468. 10.1038/nrneph.2009.108 [DOI] [PubMed] [Google Scholar]
- 24.Faul C, Donnelly M, Merscher-Gomez S, Chang YH, Franz S, Delfgaauw J, Chang JM, Choi HY, Campbell KN, Kim K, Reiser J, Mundel P. 2008. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat. Med. 14:931–938. 10.1038/nm.1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oh J, Reiser J, Mundel P. 2004. Dynamic (re)organization of the podocyte actin cytoskeleton in the nephrotic syndrome. Pediatr. Nephrol. 19:130–137. 10.1007/s00467-003-1367-y [DOI] [PubMed] [Google Scholar]
- 26.Taylor FB, Jr, Tesh VL, DeBault L, Li A, Chang AC, Kosanke SD, Pysher TJ, Siegler RL. 1999. Characterization of the baboon responses to Shiga-like toxin: descriptive study of a new primate model of toxic responses to Stx-1. Am. J. Pathol. 154:1285–1299. 10.1016/S0002-9440(10)65380-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chaisri U, Nagata M, Kurazono H, Horie H, Tongtawe P, Hayashi H, Watanabe T, Tapchaisri P, Chongsa-nguan M, Chaicumpa W. 2001. Localization of Shiga toxins of enterohaemorrhagic Escherichia coli in kidneys of paediatric and geriatric patients with fatal haemolytic uraemic syndrome. Microb. Pathog. 31:59–67. 10.1006/mpat.2001.0447 [DOI] [PubMed] [Google Scholar]
- 28.Hughes AK, Stricklett PK, Schmid D, Kohan DE. 2000. Cytotoxic effect of Shiga toxin-1 on human glomerular epithelial cells. Kidney Int. 57:2350–2359. 10.1046/j.1523-1755.2000.00095.x [DOI] [PubMed] [Google Scholar]
- 29.Psotka MA, Obata F, Kolling GL, Gross LK, Saleem MA, Satchell SC, Mathieson PW, Obrig TG. 2009. Shiga toxin 2 targets the murine renal collecting duct epithelium. Infect. Immun. 77:959–969. 10.1128/IAI.00679-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morigi M, Buelli S, Zanchi C, Longaretti L, Macconi D, Benigni A, Moioli D, Remuzzi G, Zoja C. 2006. Shiga toxin-induced endothelin-1 expression in cultured podocytes autocrinally mediates actin remodeling. Am. J. Pathol. 169:1965–1975. 10.2353/ajpath.2006.051331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saleem MA, O'Hare MJ, Reiser J, Coward RJ, Inward CD, Farren T, Xing CY, Ni L, Mathieson PW, Mundel P. 2002. A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J. Am. Soc. Nephrol. 13:630–638 [DOI] [PubMed] [Google Scholar]
- 32.Zhang W, Bielaszewska M, Pulz M, Becker K, Friedrich AW, Karch H, Kuczius T. 2008. New immuno-PCR assay for detection of low concentrations of Shiga toxin 2 and its variants. J. Clin. Microbiol. 46:1292–1297. 10.1128/JCM.02271-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pepys MB, Gallimore JR, Lloyd J, Li Z, Graham D, Taylor GW, Ellmerich S, Mangione PP, Tennent GA, Hutchinson WL, Millar DJ, Bennett G, More J, Evans D, Mistry Y, Poole S, Hawkins PN. 2012. Isolation and characterization of pharmaceutical grade human pentraxins, serum amyloid P component and C-reactive protein, for clinical use. J. Immunol. Methods 384:92–102. 10.1016/j.jim.2012.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hawkins PN, Tennent GA, Woo P, Pepys MB. 1991. Studies in vivo and in vitro of serum amyloid P component in normals and in a patient with AA amyloidosis. Clin. Exp. Immunol. 84:308–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Momoi T, Ando S, Magai Y. 1976. High resolution preparative column chromatographic system for gangliosides using DEAE-Sephadex and a new porus silica, Iatrobeads. Biochim. Biophys. Acta 441:488–497. 10.1016/0005-2760(76)90245-9 [DOI] [PubMed] [Google Scholar]
- 36.Yao JK, Rastetter GM. 1985. Microanalysis of complex tissue lipids by high-performance thin-layer chromatography. Anal. Biochem. 150:111–116. 10.1016/0003-2697(85)90447-6 [DOI] [PubMed] [Google Scholar]
- 37.Eremina V, Jefferson JA, Kowalewska J, Hochster H, Haas M, Weisstuch J, Richardson C, Kopp JB, Kabir MG, Backx PH, Gerber HP, Ferrara N, Barisoni L, Alpers CE, Quaggin SE. 2008. VEGF inhibition and renal thrombotic microangiopathy. N. Engl. J. Med. 358:1129–1136. 10.1056/NEJMoa0707330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cargnello M, Roux PP. 2011. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 75:50–83. 10.1128/MMBR.00031-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davies C, Tournier C. 2012. Exploring the function of the JNK (c-Jun N-terminal kinase) signalling pathway in physiological and pathological processes to design novel therapeutic strategies. Biochem. Soc. Trans. 40:85–89. 10.1042/BST20110641 [DOI] [PubMed] [Google Scholar]
- 40.Vaculova A, Zhivotovsky B. 2008. Caspases: determination of their activities in apoptotic cells. Methods Enzymol. 442:157–181. 10.1016/S0076-6879(08)01408-0 [DOI] [PubMed] [Google Scholar]
- 41.Burlacu A. 2003. Regulation of apoptosis by Bcl-2 family proteins. J. Cell. Mol. Med. 7:249–257. 10.1111/j.1582-4934.2003.tb00225.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nelson SR, Tennent GA, Sethi D, Gower PE, Ballardie FW, Amatayakul-Chantler S, Pepys MB. 1991. Serum amyloid P component in chronic renal failure and dialysis. Clin. Chim. Acta 200:191–199. 10.1016/0009-8981(91)90090-Y [DOI] [PubMed] [Google Scholar]
- 43.Pepys MB. 2006. Amyloidosis. Annu. Rev. Med. 57:223–241. 10.1146/annurev.med.57.121304.131243 [DOI] [PubMed] [Google Scholar]
- 44.Dyck RF, Lockwood CM, Kershaw M, McHugh N, Duance VC, Baltz ML, Pepys MB. 1980. Amyloid P-component is a constituent of normal human glomerular basement membrane. J. Exp. Med. 152:1162–1174. 10.1084/jem.152.5.1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Breathnach SM, Melrose SM, Bhogal B, de Beer FC, Dyck RF, Tennent G, Black MM, Pepys MB. 1981. Amyloid P component is located on elastic fibre microfibrils in normal human tissue. Nature 293:652–654. 10.1038/293652a0 [DOI] [PubMed] [Google Scholar]
- 46.Du Clos TW, Mold C. 2011. Pentraxins (CRP, SAP) in the process of complement activation and clearance of apoptotic bodies through Fcgamma receptors. Curr. Opin. Organ Transplant. 16:15–20. 10.1097/MOT.0b013e32834253c7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dillingh MR, van den Blink B, Moerland M, van Dongen MG, Levi M, Kleinjan A, Wijsenbeek MS, Lupher ML, Jr, Harper DM, Getsy JA, Hoogsteden HC, Burggraaf J. 2013. Recombinant human serum amyloid P in healthy volunteers and patients with pulmonary fibrosis. Pulm. Pharmacol. Ther. 26:672–676. 10.1016/j.pupt.2013.01.008 [DOI] [PubMed] [Google Scholar]
- 48.Griener TP, Mulvey GL, Marcato P, Armstrong GD. 2007. Differential binding of Shiga toxin 2 to human and murine neutrophils. J. Med. Microbiol. 56:1423–1430. 10.1099/jmm.0.47282-0 [DOI] [PubMed] [Google Scholar]