

Abstract
The mechanism of alternative activation of antigen-presenting cells (APCs) is largely unknown. Lacto-N-fucopentaose III (LNFPIII) is a biologically conserved pentasaccharide that contains the Lewisx trisaccharide. LNFPIII conjugates and schistosome egg antigens, which contain the Lewisx trisaccharide, drive alternative activation of APCs and induce anti-inflammatory responses in vivo, preventing inflammation-based diseases, including psoriasis, transplant organ rejection, and metabolic disease. In this study, we show that LNFPIII conjugates and schistosome egg antigens interact with APCs via a receptor-mediated process, requiring internalization of these molecules through a clathrin/dynamin-dependent but caveolus-independent endocytic pathway. Using inhibitors/small interfering RNA (siRNA) against dynamin and clathrin, we show for the first time that endocytosis of Lewisx-containing glycans is required to drive alternative maturation of antigen-presenting cells and Th2 immune responses. We identified mouse SIGNR-1 as a cell surface receptor for LNFPIII conjugates. Elimination of SIGNR-1 showed no effect on uptake of LNFPIII conjugates, suggesting that other receptors bind to and facilitate uptake of LNFPIII conjugates. We demonstrate that disruption of actin filaments partially prevented the entry of LNFPIII conjugates into APCs and that LNFPIII colocalizes with both early and late endosomal markers and follows the classical endosomal pathway leading to lysosome maturation. The results of this study show that the ability of LNFPIII to induce alternative activation utilizes a receptor-mediated process that requires a dynamin-dependent endocytosis. Thus, key steps have been defined in the previously unknown mechanism of alternative activation that ultimately leads to induction of anti-inflammatory responses.
INTRODUCTION
The prevalence of inflammation-based diseases, including autoimmune, cardiovascular, and obesity/metabolic diseases, has increased dramatically over the past 2 decades (1, 2). This rapid increase in inflammatory disease prevalence has prompted research to define new means to control inflammation, including identifying mechanisms for induction and regulation of anti-inflammatory responses. Antigen-presenting cells (APCs), macrophages (Mphs), and dendritic cells (DCs) not only play key roles in the initiation of and maintenance of inflammation but also function as regulators of inflammation. Thus, “classically” activated, proinflammatory APCs contribute to inflammation, whereas “alternatively” activated (AA) Mphs and DC2s play roles in control or resolution of inflammation (3). These two differentially activated APCs influence inflammation directly and indirectly, via their roles on T cell maturation into Th1 and Th17 types for proinflammatory responses and into Th2 and T regulatory cells for anti-inflammatory responses (4, 5).
The mechanisms for classical activation of APCs are well defined and minimally include pathogen molecule ligation of pattern recognition receptors (PRRs) and activation via inflammasomes or by danger signals (6). In addition, classical activation can be initiated or enhanced via cytokines, specifically interleukin-12 (IL-12), gamma interferon (IFN-γ), and tumor necrosis factor alpha (TNF-α) (4). In contrast to classical activation, little is known about the factors/mechanisms that promote alternative activation of APCs, other than cytokines (IL-4, IL-13, or IL-10). Little is known regarding the nature of pathogen or self molecules that induce alternative activation of APCs or the mechanism of activation (7, 8).
In this regard, we and others demonstrated that glycans found on pathogens in human milk and in certain foods are capable of inducing alternative activation of APCs (9–11). Glycans on glycoproteins and glycolipids are known to play important roles not only for binding of glycoproteins to receptors on APCs but also in increasing phagocytosis (12–15). Upon internalization, glycans can protect or enhance antigen processing and may influence intracellular trafficking, including endocytosis (16, 17). Endocytosis is a phenomenon that not only regulates nutrient uptake, migration, and pathogen entry but also largely controls immune responses by regulating signaling cascades.
Glycan activation/maturation of APCs has largely been studied in the context of immune modulatory helminth parasites that drive Th2-type and anti-inflammatory responses (18, 19). For the human helminth parasite Schistosoma mansoni, glycans were shown to be critical in order for soluble egg antigens (SEA) to activate APCs that drive Th2 responses (20). Similarly, nematodes or nematode extracts utilize glycans to mature APCs that drive CD4+ T cells to a Th2 type (20, 21). Recently, the ability of the schistosome Th2-driving egg antigen omega-1 was shown to require glycan binding to, and subsequent internalization by, APCs to drive APC maturation to cells that drive CD4+ Th2-type responses (22).
Lewisx is a glycan expressed on multiple stages of S. mansoni and is present on omega-1, which carries tandem repeats of Lewisx, LacNac(LN), and LacdiNAcLDN motifs (23). In addition, schistosomes also express GalNAcb1-4(Fucα1-3) GlcNAc (LDNF)-, Fucα1-3Galb1-4(Fucα1-3)GlcNAc (pseudo-LeY)-, and high-mannose-containing glycoproteins, all of which have been shown to modulate host immune responses (24, 25). The Lewisx trisaccharide is also found within the structure of the human milk pentasaccharide lacto-N-fucopentaose III (LNFPIII), and neoglycoconjugates (NGC) with multiple molecules of LNFPIII (7 to 10) linked to a carrier molecule via linker spacer technology (LNFPIII-NGC) are immune modulatory in vivo, showing dramatic therapeutic effects on autoimmune diseases and metabolic disease (26–29). Further, LNFPIII-NGCs induce AA macrophages and facilitate expansion of IL-10-producing B1-B cells in vivo (10, 30).
Helminth glycans and LNFPIII-NGC require interaction with PRRs to modulate APC function and drive Th2 responses (19, 31–33). Lewisx-containing glycans appear to use both C-type lectin receptors (CLRs) (DC-SIGN)- and Toll-like receptor (TLR)-mediated signaling to modulate APC function and induction of Th2 responses (34). Other lectins such as MGL-1, mannose receptor (MR), scavenger receptor, and galectins have also been reported to bind to schistosome egg antigen glycans (35–37). Thus, helminth glycans bind to multiple receptors on the surface of APCs, yet it is not known if uptake of these helminth glycans by APCs occurs through endocytosis- or phagocytosis-mediated pathways. Although C-type lectins have been shown to play predominant roles in phagocytosis of glycan-expressing antigens/pathogens (12, 38, 39), it is not known if glycan-mediated phagocytosis of antigens/pathogens affects or is required for alternative activation/maturation of APCs.
We report here that Lewisx glycans in S. mansoni SEA and LNFPIII-NGC are in fact not phagocytosed but are endocytosed by APCs through a dynamin- and clathrin-mediated pathway. We identified mouse SIGNR-1 as one of the receptors for LNFPIII conjugates. However, downregulating the expression of SIGNR-1 had no effect on the uptake of glycans. Mechanistically, we show that SEA- or LNFPIII-NGC-induced APC activation and maturation into cells that drive CD4+ Th2 maturation require endocytosis and intracellular processing mediated via dynamin/clathrin. This study presents a new mechanism for the process of glycan-mediated functional activation and maturation of “alternative” APC phenotype. The findings presented in this study along with additional dissection of glycan-induced signaling pathways will identify new approaches and targets to improve therapy for autoimmune and inflammation-based diseases.
MATERIALS AND METHODS
Cell culture.
RAW 264.7 cells (ATCC) were grown in Dulbecco's modified Eagle medium (DMEM) (HyClone) supplemented with 10% fetal calf serum (Atlanta Biotech), 100 U/ml penicillin, 100 μg/ml streptomycin (HyClone), and 2 mM glutamate L. Cells were plated in 12- and 24-well plates and then cultured in a humidified incubator at 37°C with 5% CO2 until they reached 70% confluence.
Bone marrow-derived macrophages (BMDMs) were obtained by flushing bone marrow cells from tibia and femurs with medium and culturing them in Dulbecco's modified Eagle medium supplemented with 10 ng/ml of macrophage colony-stimulating factor (MCSF; PeproTech, Rocky Hill, NJ) essentially as described previously (40). Medium was replaced every 2nd day with fresh MCSF, and cells were used on day 6.
Bone marrow-derived dendritic cells (BMDCs) were obtained by flushing bone marrow cells in RPMI 1640 and culturing the cells with 20 ng/ml of granulocyte-macrophage colony-stimulating factor (GM-CSF; PeproTech, Rocky Hill, NJ) essentially as described previously (10). On days 3 and 5, fresh medium containing GM-CSF was added to the cells. On day 7, nonadherent cells were used for experiments. Cells were >90% CD11c+ dendritic cells as determined by flow cytometry.
DC/CD4+ T cell coculture.
BMDCs (7 × 104/well in triplicate wells) were left untreated or were pretreated with 40 μM dynasore for 1 h and then stimulated with LNFPIII-NGC and cultured for 48 h at 37°C. BMDCs were then placed into coculture with 5-fold excess of ovalbumin (OVA)-specific CD4+ T cells in the presence of 3 μM OVA peptide 323-339 for 72 h. Coculture supernatants were collected and screened for IFN-γ, IL-4, and IL-13 by enzyme-linked immunosorbent assay (ELISA) using kits from BD-Biosciences and eBiosciences. Plates were read on a SpectraMax 190 (Molecular Devices).
Antibodies.
LNFPIII-NGC was stained with monoclonal antibody E.5 (IgM) that recognizes LNFPIII/Lewisx (41). Primary antibodies for Rab5, mannose 6-phosphate receptor (MPR), Lamp-1, and cathepsins were purchased from AbCam. Polyclonal anticlathrin antibody was purchased from Santa Cruz and Cell signaling. Polyclonal EEA-1 antibody was purchased from Abcam. Anti-mSIGNR1 (CD209b) was from R&D systems. Lysotracker-Red DND-99 and Alexa Fluor 488/594-conjugated secondary antibodies were purchased from Molecular Probes, NY, USA. Actin was stained with Alexa Fluor 594-conjugated phalloidin (Invitrogen, NY, USA). Anti-TLR4 antibodies were purchased from Santa Cruz Biotechnology, TX, USA.
Chemicals and reagents.
Inhibitors against dynamin (dynasore), clathrin (chlorpromazine and monodansylcadaverine), and caveoli (filipin and methyl-β-cyclodextrin) were purchased from Sigma-Aldrich, St. Louis, MO, USA. Ultrapure Escherichia coli lipopolysaccharide (LPS) was purchased from Invivogen, San Diego, CA, USA. Recombinant IL-4 was purchased from R&D systems (Minneapolis, MN, USA).
Recombinant GM-CSF and M-CSF used for generating BMDCs and BMDMs were purchased from PeproTech, Rocky Hill, NJ. Transfecting reagent Lipofectamine was purchased from Invitrogen, NY, USA, and Opti-MEM medium from Gibco, NY, USA. Small interfering RNAs (siRNAs) against clathrin and SIGNR1 were purchased from Dharmacon. Chloroquine was purchased from Sigma-Aldrich.
Mice.
C57BL/6J and OT-II OVA TCR transgenic mice were purchased from Jackson Laboratory (Maine, USA). All animals were maintained at the University of Georgia Animal Facilities according to institutional guidelines incorporating an animal welfare policy. The guide for the Care and Use of Laboratory Animals was in accordance with the American Association for Accreditation of Laboratory Animal Care (AALAC) as well as the Animal Welfare Act (AWA) and other applicable federal and state guidelines. All animal work presented here was approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Georgia.
Endocytosis.
For immunofluorescence experiments, cells were seeded at 0.5 × 106 on sterile coverslips for 24 h in 24-well plates. Initially, cells were incubated at 37°C overnight and then starved for 2 to 3 h in incomplete DMEM. For endocytosis experiments, cells were kept on ice for 10 min and then stimulated with 50 μg/ml of LNFPIII-NGC while on ice. Endocytosis was induced by incubating the stimulated cells at 37°C for 10, 20, and 30 min. Endocytosis was stopped by placing the cells back on ice. Adherent cells on coverslips were further processed for immunofluorescence staining.
Immunofluorescence.
Cells were fixed with freshly prepared 3% paraformaldehyde (Sigma-Aldrich) for 10 min at room temperature and then permeabilized with 0.05% Triton X-100 in phosphate-buffered saline (PBS). Fixed cells were blocked in goat serum or 10% bovine serum albumin (BSA) for 20 min at room temperature and then incubated with primary antibodies followed by Alexa Fluor 488/594-conjugated secondary antibodies for 1 h at room temperature. Cells were counterstained with Hoechst dye (blue) for nuclear visualization and mounted in ProLong Gold mounting medium (Molecular Probes). Images were acquired using a Nikon Eclipse Ti A1R confocal microscope system or a Zeiss LSM 510 META confocal microscope (Carl Zeiss) under a 60× objective.
RT-PCR.
RNA was extracted from cells using an RNA isolation kit (Qiagen, CA, USA). RNA concentration and purity were estimated using a Nanodrop spectrophotometer (Thermo Scientific, GA, USA) followed by cDNA synthesis using a reverse transcription (RT)-PCR kit (Bio-Rad). Expression of different genes was quantitated by using 6-carboxyfluorescein (FAM)-labeled primers (Applied Biosystems). VIC-labeled primers for actin or GAPDH (glyceraldehyde-3-phosphate dehydrogenase) were used as housekeeping genes. Real-time PCR was performed using the Applied Biosciences StepOne Plus machine.
Flow cytometric analysis.
For quantitative assessment of LNFPIII-NGC internalization, cells were washed with ice-cold PBS after induction of endocytosis. Washed cells were fixed and permeabilized for 30 min at 4°C. Cells were then washed in permeabilizing buffer and then incubated with buffer containing Fc block for 15 min. Cells were then stained with E.5 (anti-Lewisx) and counterstained with fluorescein isothiocyanate (FITC)-labeled secondary antibody for 1 h each on ice. Cells initially treated with dextran and/or only stained with the secondary antibody were used as background staining controls. Cells were kept on ice throughout the procedure. Quantitation of endocytosis was done by measuring mean fluorescence intensity (MFI) on a BD LSRII instrument using DIVA or FloJo software for analysis.
Transfection.
siRNA transfections were performed using Lipofectamine (Invitrogen) in Opti-MEM medium (Invitrogen) and incubated overnight at 37°C. Medium was changed 12 h posttransfection, and cells were harvested for experimental analysis 36 h posttransfection. Gene knockdown was confirmed by looking at protein expression by Western blotting using antibody specific to the downregulated protein.
Treatment with inhibitors.
When indicated, cells were pretreated with the dynamin inhibitor dynasore (40 or 80 μM for 40 min), clathrin inhibitors monodansylcadaverine (300 or 450 μM) and chlorpromazine (10 and 40 μg/ml) for 40 min, caveolus inhibitors methyl-β-cyclodextrin (2.5 and 5 μM) and filipin (2.5 and 5 μg/ml) for 45 min, calcium chelation with EGTA (3 and 5 mM for 15 min), and hyperosmotic sucrose (0.45 M for 15 min) (all reagents were obtained from Sigma, St. Louis, MO).
Statistical significance.
Statistical calculations were performed using Student's t test analysis. Results are representative of at least 3 independent experiments. For each experiment, samples were in triplicate. For flow cytometry, cells were acquired on a BD LSRII flow cytometer and results were analyzed by FlowJo software (Tree Star Inc.).
RESULTS
Lewisx/LNFPIII conjugates are endocytosed by antigen-presenting cells.
To determine if LNFPIII-NGCs are internalized postbinding by macrophages, we incubated dextran-conjugated LNFPIII with RAW 264.7 macrophages. Cells were washed, fixed, and immunolabeled using anti-Lewisx antibody E.5 (41), followed by incubation with Alexa Fluor 488-conjugated secondary antibody and visualization using confocal microscopy. As shown in Fig. 1a, LNFPIII-dextran conjugates were internalized by macrophages. Similar to the observations made via confocal microscopy, flow cytometry showed specific binding and internalization of LNFPIII-NGC by APCs (Fig. 1b), whereas control FITC-labeled 40-kDa dextran was not significantly internalized (Fig. 1a and b). We also observed macrophage internalization of LNFPIII conjugated to human serum albumin (HSA), showing that internalization of the LNFPIII-dextran and LNFPIII-HSA conjugates is due to the presence of LNFPIII (see Fig. S1c in the supplemental material). Lewisx motifs are abundant in schistosome eggs; therefore, we asked if Lewisx molecules in schistosome soluble egg antigen (SEA) would be internalized by macrophages, similar to what we observed for LNFPIII-NGC. As shown in Fig. 1c, molecules in SEA expressing Lewisx are internalized by macrophages. We next asked if internalization of LNFPIII-NGC is a common feature to APCs in general, tested for internalization of LNFPIII-NGC by bone marrow-derived dendritic cells, and found that LNFPIII-NGCs were internalized by BMDCs (see Fig. S5b in the supplemental material).
FIG 1.

LNFPIII/Lewisx binds to and is internalized by antigen-presenting cells. (a and c) Raw 264.7 murine macrophages were incubated with 50 μg/ml dextran-conjugated LNFPIII-NGC or 25 μg/ml Schistosoma soluble egg antigen (SEA) at 4°C for 10 min followed by incubation at 37°C for 30 min. Cells were harvested, fixed with 3% paraformaldehyde at room temperature, stained with anti-Lewisx antibody (E.5), and counterprobed with Alexa Fluor 488-conjugated secondary antibody for visualization. FITC-labeled 40-kDa dextran was taken as backbone control (panel B). Confocal images were obtained using a Nikon A1R confocal microscope under a 60× objective. (b) Using a similar setup as for panels a and c, uptake assay was also performed by flow cytometry. RAW 264.7 macrophages were incubated with LNFPIII-NGC for 10 min at 4°C followed by incubation at 37°C for 30 min. Cells were harvested, fixed, and probed with E.5 monoclonal antibody followed by FITC-labeled secondary antibody. Thick line, LNFPIII; filled histogram, 40-kDa dextran; thick dotted line, E.5 together with secondary antibody; thin dotted line, secondary antibody only). Mean fluorescence intensity (MFI) for FITC was measured for live cells gated as side scatter (SSC)- and forward scatter (FSC)-positive populations. Cells were acquired on a BD LSRII flow cytometer and analyzed by FlowJo software. Data shown represent 1 of the 3 independent experiments performed. Experimental samples were in triplicate. Statistical calculation was performed using Student's t test analysis (****, P < 0.0001).
Having demonstrated LNFPIII-NGC internalization by APCs, we then asked if endocytotic processes mediate entry of LNFPIII-NGC into macrophages. Dynamin is a 96-kDa twisting molecular motor with GTPase activity, crucial to the formation of endocytotic vesicles. The role of dynasore, a specific inhibitor of dynamin, and its ability to block endocytosis have been widely studied (42). Therefore, we used dynasore to determine if endocytosis of LNFPIII-NGC occurs. We observed that macrophages pretreated with dynasore (40 μM and 80 μM) for 1 h prior to incubation with LNFPIII-NGC showed significant reduction in the uptake of LNFPIII-NGC compared to cells treated with the carrier dimethyl sulfoxide (DMSO) (Fig. 2). Similarly, uptake of Lewisx-containing antigens in SEA was inhibited when cells were pretreated with dynasore (see Fig. S2a in the supplemental material). To rule out cell death due to exposure to DMSO, we simultaneously carried out cell viability tests using similar concentrations of DMSO or dynasore (see Fig. S2b and c in the supplemental material).
FIG 2.
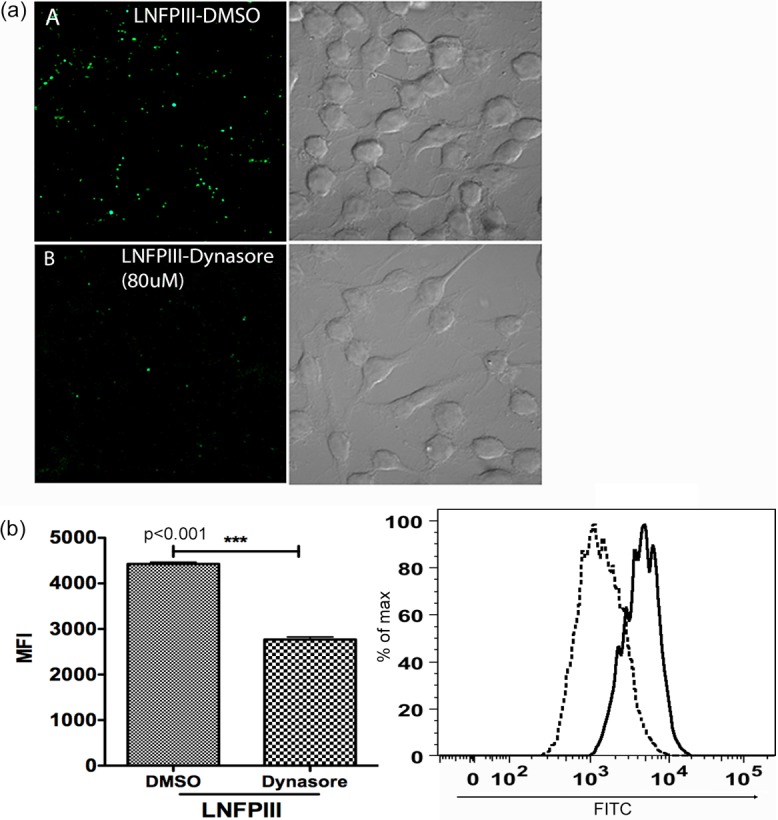
Endocytosis of LNFPIII-NGC is dynamin mediated. (a) Raw 264.7 cells were pretreated with 80 μM dynasore (panel B) or an equal volume of carrier molecule DMSO (panel A) for 40 min at 37°C and then incubated with 50 μg/ml LNFPIII-NGC for 30 min at 37°C. Cells were harvested, fixed, and stained for internalized LNFPIII-NGC (green) using E.5 antibody followed by incubation with Alexa Fluor 488-labeled secondary antibody. Cells were visualized, and images were taken with a Zeiss LSM 510 META inverted confocal microscope under a 60× objective. (b) Raw 264.7 cells were pretreated with 40 μM dynasore for 40 min and were then incubated with 50 μg/ml of LNFPIII-NGC at 4°C for 10 min. Endocytosis of LNFPIII was induced for 20 min at 37°C. Cells were harvested, fixed, and stained for internalized LNFPIII as described before. Cells were acquired on a BD LSRII flow cytometer and analyzed by FlowJo software. Mean fluorescence intensity (MFI) for FITC was measured for live cells gated as SSC- and FSC-positive populations. Data are from three independent experiments performed. Treated and nontreated samples were in triplicate in each independent experiment. Black solid line, LNFPIII (DMSO); black dotted line, LNFPIII (dynasore). Statistical analysis was performed using Student's t test (***, P < 0.001).
LNFPIII-NGC is processed via the clathrin-mediated endocytotic pathway.
Dynamin affects both clathrin- and caveolus-mediated endocytosis (43). Therefore, to determine which endocytic pathway was being utilized for LNFPIII internalization into APCs, we tested specific inhibitors against clathrin and caveoli. Macrophages were incubated with different concentrations of the clathrin inhibitors chlorpromazine (10 μg/ml or 40 μg/ml) and monodansyl cadaverine (300 μM or 450 μM) or the caveolus inhibitors methyl-β-cyclodextrin (2.5 μM or 5 μM) and filipin (2.5 μg/ml or 5 μg/ml) followed by incubation with LNFPIII-NGC as described above. As shown in Fig. 3a, internalization of LNFPIII-NGC was significantly disrupted in macrophages pretreated with inhibitors of the clathrin endocytic pathway. In contrast, caveolus inhibitors did not alter endocytosis of LNFPIII-NGC by macrophages (Fig. 3b). Similar to our observations using confocal microscopy, quantitative flow cytometric analysis via mean fluorescence intensity of cells treated with clathrin inhibitors showed reduced LNFPIII internalization compared to cells treated with caveolus inhibitors (Fig. 3c). The concentrations of inhibitors used here were based on previous reports and did not interfere with cell viability (see Fig. S2b and c in the supplemental material). To further validate the role of clathrin in LNFPIII uptake, we knocked down the endogenous expression of clathrin using siRNA specific to clathrin. Thirty-six hours posttransfection with siRNA, macrophages were incubated with LNFPIII-NGC and endocytosis was induced for 30 min at 37°C. Cells were then immunolabeled for intracellular LNFPIII-NGC. Uptake was quantitated by measuring the mean fluorescence intensity of Alexa Fluor 488-labeled LNFPIII-NGC on a BD LSRII instrument (Fig. 3d). To confirm that the expression of clathrin was successfully knocked down, we performed Western blot analysis using anticlathrin antibody. Figure 3e shows that clathrin expression was significantly knocked down in cells treated with clathrin-specific siRNA compared to cells treated with nonspecific (NT) siRNA. As expected, cells treated with clathrin-specific siRNA had significantly reduced uptake of LNFPIII-NGC, whereas there were no differences in uptake in cells transfected with nonspecific siRNA (Fig. 3d). Importantly, we observed that endocytosed LNFPIII-NGC colocalized with endogenous clathrin in antigen-presenting cells. We performed this experiment using two different time points (10 and 20 min) and observed significant colocalization at both times (Fig. 3f). Overall, these studies confirm that macrophage internalization of LNFPIII-NGC occurs via a dynamin/clathrin-mediated endocytotic pathway and does not require the caveolus pathway.
FIG 3.

Endocytosis of LNFPIII-NGC is clathrin mediated. (a and b) Raw 264.7 cells were pretreated with clathrin inhibitors chlorpromazine (10 and 40 μg/ml) and monodansylcadaverine (300 and 450 μM) or caveolus inhibitors methyl-β-cyclodextrin (2.5 and 5 μM) and filipin (2.5 and 5 μg/ml) for 40 min at 37°C. The cells were further incubated with 50 μg/ml of LNFPIII-NGC. Endocytosis was induced for 30 min at 37°C, and then the cells were fixed and stained for internalized LNFPIII-NGC as described in Materials and Methods. Nuclei were stained with Hoechst dye (blue). (c) Under similar conditions as for panels a and b, FACS analysis of LNFPIII-NGC uptake by cells treated with clathrin and caveolus inhibitors was performed. Different histograms indicate LNFPIII-NGC + DMSO in solid blue line, LNFPIII + MBD (methyl-beta cyclodextrin) as a filled green histogram, LNFPIII + filipin in red dotted line, LNFPIII + monodansyl cadaverine as a filled gray histogram, and LFPIII + Chlrph (chlorpromazine) in a thin black line. FITC mean fluorescence intensity (MFI) was measured for live cells gated as SSC- and FSC-positive populations. Cells were acquired on a BD LSRII flow cytometer and analyzed by FlowJo software (Tree Star Inc.). Statistical calculation was performed using Student's t test analysis (***, P < 0.001; **, P < 0.01). Inhibitor-treated and -nontreated samples were in triplicate, and data represent 3 independent experiments performed. (d) siRNA-mediated knockdown of endogenous expression of clathrin reduced LNFPIII-NGC uptake by APCs. RAW 264.7 cells were transfected with clathrin-specific siRNA or nontarget siRNA for 36 h. Transfected cells were incubated with LNFPIII-NGC at 37°C for 30 min and immunolabeled with E.5, followed by incubation with Alexa Fluor 488-conjugated secondary antibody. Cells were then acquired and analyzed via flow cytometry as described before. The data are representative of 3 independent experiment with n = 4. Statistical analysis was performed using Student's t test (*, P < 0.05). (e) Western blot showing downregulated expression of clathrin protein. Thirty-six hours posttransfection with clathrin siRNA, cell lysates were run on SDS-PAGE, and the corresponding Western blots were probed with anticlathrin antibody. Total Erk was used as a loading control. (f) LNFPIII-NGC colocalizes with endogenous clathrin. Endocytosis of LNFPIII-NGC was induced in RAW 264.7 macrophages for 10 and 20 min at 37°C. Cells were fixed and double stained for LNFPIII-NGC and clathrin protein as described in Materials and Methods. Images were captured using a Nikon Eclipse Ti A1R confocal microscope system. Endocytosed LNFPIII-NGC is in green, clathrin is in red, and an overlay image of colocalized LNFPIII-NGC and clathrin is in yellow. The zoomed-in image of part of a cell is kept as an inset for better representation.
LNFPIII-NGC follows the early to late endosomal pathway.
Having determined that LNFPIII-NGC is endocytosed via a dynamin/clathrin-mediated pathway, we next examined the intracellular endocytotic trafficking of LNFPIII-NGC. Bone marrow-derived macrophages were stimulated with 50 μg/ml of LNFPIII-dextran conjugates, and then endocytosis was induced for 10, 20, and 30 min at 37°C. Cells were washed, fixed, and immunolabeled for LNFPIII-NGC and for vesicle-specific proteins expressed during different phases of the endosomal pathway. Endosomal vesicle-specific proteins were detected using Alexa Fluor 488/594-conjugated secondary antibodies, and images were obtained using a confocal microscope. Ten minutes following initiation of endocytosis, we observed colocalization of LNFPIII-NGC (green) with the early endosomal marker EEA-1 (red) as shown in Fig. 4a, panel A. To demonstrate that this process was LNFPIII dependent, cells were incubated with 40-kDa FITC-labeled dextran. As shown in Fig. 4a, panel B, we did not observe any colocalization of FITC-dextran with EEA-1, demonstrating that internalization of LNFPIII-dextran conjugates is LNFPIII specific. Examination of LNFPIII-NGC incubated with BMDM cells at 20 and 30 min after initiation of endocytosis showed significant colocalization of LNFPIII-NGC with the late endosomal marker MPR and, at subsequent time points, colocalization with the lysosomal marker Lamp-1 (Fig. 4b). To extend and confirm our observation of lysosomal maturation following LNFPIII-NGC internalization, we introduced Lysotracker Red DND-99 dye after 15 min of endocytosis. Using Lysotracker Red, we observed acidification of intracellular vesicles/lysosomal maturation in LNFPIII-NGC-stimulated RAW 264.7 cells (Fig. 4c). The effect was not observed in dextran-treated samples. Lastly, we wanted to determine the intracellular fate of LNFPIII-NGC at longer times postinternalization. Here, we incubated LNFPIII-dextran conjugate with RAW 264.7 macrophages at 4°C and induced endocytosis for 30 min. Cells were then washed and further incubated at 37°C for 1, 3, 8, and 16 h. Confocal examination of cells showed a significant reduction in the ability to detect endocytosed LNFPIII-NGC by 8 h postincubation compared to cells 1 h postincubation (Fig. 4d). At 16 h postincubation, endocytosed LNFPIII-NGC was almost undetectable (Fig. 4d). Therefore, we conclude that LNFPIII conjugates are endocytosed in an LNFPIII-specific manner, with intracellular trafficking through early and late endosomes and then lysosomes. Further, by 16 h postinternalization, LNFPIII-NGC has been largely degraded and/or processed and is undetectable in the cytoplasm of antigen-presenting cells. To determine if degradation of LNFPIII-NGC occurs in the acidic endosomal compartments, we treated cells with chloroquine for 1, 3, 8, and 16 h following LNFPIII-NGC internalization. Chloroquine is a chemical inhibitor that blocks acidification of lysosomal or endosomal compartments. As expected, chloroquine inhibition of vesicle acidification resulted in our ability to detect greater accumulation of LNFPIII-NGC in the cytoplasm than in untreated cells. We were unable to monitor the accumulation of LNFPIII-NGC at 16 h postincubation due to the toxic nature of chloroquine to cells at this time point. These observations suggest that LNFPIII-NGC degradation is indeed induced by acidification inside the vesicles (Fig. 4d), which strengthens our observation that LNFPIII-NGC gets degraded in lysosomes postinternalization.
FIG 4.

Endocytosed LNFPIII-NGC follows classical endosomal pathway and induces lysosomal acidification. (a) LNFPIII-NGC was incubated with BMDMs for 10 min at 37°C. Cells were fixed and double stained for LNFPIII-NGC (green) and the early endosomal marker, EEA-1 (red in panel A), as described before. FITC-labeled 40-kDa dextran was taken as a control (panel B). The zoomed-in image of part of a cell is kept as an inset for better visualization. (b) BMDMs were incubated with LNFPIII-NGC for 20 min at 37°C. Cells were fixed and stained for both internalized LNFPIII-NGC and the late endosomal marker MPR (red in panel A) and lysosomal marker LAMP-1 (red in panel B) as described in Materials and Methods. Internalized LNFPIII-NGC is green in panels A and B. The zoomed-in image of part of a cell is kept in an inset for better representation. (c) Acidification of endosomal compartment was observed in RAW 264.7 incubated with LNFPIII-NGC. Lysotracker Red-DND-99 (0.2 μM) was added to the cells 15 min postendocytosis to visualize acidic compartments. Cells were harvested and fixed, and images were acquired on a confocal microscope. Cells incubated with 40-kDa dextran were taken as a control. (d) RAW 264.7 cells were pretreated or left untreated with chloroquine for 10 min and then incubated with LNFPIII-NGC (50 μg/ml) for 30 min at 37°C. Medium was replaced by fresh medium to wash off excess LNFPIII-NGC, and cells were further incubated for 1, 3, 8, and 16 h at 37°C. Cells were then fixed with 3% paraformaldehyde, permeabilized, and immunolabeled for internalized LNFPIII-NGC (green) as described before. Results shown represent one of the two independent experiments performed.
Actin filaments partially regulate LNFPIII-NGC endocytosis.
Actin plays a key role in phagocytosis (44, 45). Importantly, there are actin-independent and -dependent examples of clathrin-mediated endocytosis (46). The actin cytoskeletal system not only helps in the formation of, but also facilitates transportation of, intracellular vesicles, thereby forming an important regulator of endocytosis. We investigated the role of actin in endocytosis of LNFPIII-NGC by disrupting actin filaments via treatment with 1 μM latrunculin A for 10 min at 37°C and then induced endocytosis for 20 min. We observed a significant reduction in the uptake of LNFPIII by confocal microscopy and FACS analysis in cells treated with latrunculin (Fig. 5a and b), demonstrating that actin filaments play an important role in endocytosis of LNFPIII-NGC. In addition, we performed this experiment with another actin inhibitor, cytochalasin D (3 μM), and observed similar results (Fig. 5b; see Fig. S3a in the supplemental material). We titrated concentrations of cytochalasin D and latrunculin A and examined the cells at different times posttreatment to measure disruption of the actin cytoskeleton as well as to test for any impact of inhibitor treatment on cell viability (see Fig. S3b in the supplemental material).
FIG 5.

Actin partially regulates receptor-mediated internalization of LNFPIII-NGC. (a) RAW 264.7 cells were treated with 1 μM of latrunculin A for 10 min at 37°C to depolymerize actin filaments. Cells were then incubated with 50 μg/ml LNFPIII-NGC for 20 min at 37°C. Actin filaments were stained with Alexa Fluor 594-conjugated phalloidin (red), and LNFPIII-NGC was immunolabeled with E.5 antibody and then incubated with Alexa Fluor 488-conjugated secondary antibody (green). Carrier solvent DMSO was used as a control. (b) Under similar conditions, endocytosis of LNFPIII-NGC in the presence of latrunculin A was performed, cells were acquired on a BD LSRII flow cytometer, and data were analyzed by FlowJo software (TreeStar Inc.). MFI was calculated as described above (Fig. 1), and statistical significance was calculated by Student's t test (*, P < 0.05). Data represent one of the three independent experiments performed, and samples were in triplicate (n = 3). (c) Endocytosis of LNFPIII-NGC was reduced in the presence of EGTA. Cells were preincubated with 3 mM or 5 mM EGTA for 15 min, and endocytosis was induced for 30 min at 37°C. (Panel A) Cells are in normal DMEM medium. Green staining is for endocytosed LNFPIII-NGC. Cells were counterstained with Hoechst dye for nucleus (blue). (d) Cell viability assay showing no cell death on RAW 264.7 cells pretreated with 3 and 5 mM EGTA. Samples were in triplicate, and data represent 4 independent experiments. Statistical analysis was done using Student's t test. (e) Cells pretreated with 3 mM EGTA were taken for FACS analysis for endocytosed LNFPIII-NGC. Data were acquired on a BD LSRII instrument and analyzed by FlowJo software (TreeStar Inc.). Solid black line, LNFPIII; filled gray histogram, EGTA. Statistical calculation and MFI measurement were performed as described for Fig. 1 (****, P < 0.0001). Data represent at least three independent experiments.
Endocytosis of LNFPIII-NGC is receptor mediated.
Based on observations that LNFPIII-NGC gets endocytosed in antigen-presenting cells, we next wanted to test if LNFPIII-NGC uptake occurs via passive diffusion or by receptor-mediated endocytosis. To test for receptor-mediated endocytosis, we preincubated RAW 264.7 cells in 0.45 M sucrose (hypertonic solution) for 15 min followed by incubation with LNFPIII-NGC for 20 min at 37°C. Hypertonic medium has been widely used to study receptor-mediated endocytosis. Hypertonic medium disrupts clathrin-coated pit formation and also K+ ion channels. We observed significantly reduced uptake of LNFPIII-NGC in cells placed in hypertonic medium (see Fig. S3c in the supplemental material) compared to macrophages that were not pretreated with hypertonic medium. This confirms that internalization of LNFPIII-NGC is receptor mediated and is not due to passive diffusion. Further support for this observation comes from studies in which we observed less internalization of LNFPIII-NGC when the cells were pretreated with a mild acidic solution (20 mM acetic acid) (see Fig. S3d in the supplemental material).
C-type lectin receptors (CLRs) have been shown to bind glycans through carbohydrate binding domains (CRDs) (47, 48). The Lewisx motif present in the LNFPIII pentasaccharide can bind to several different C-type lectins, including DC-SIGN, MGL-1, and mannose receptor (MR). Extracellular calcium is crucial for CLRs to bind glycans (38). Therefore, we next tested if receptor-mediated endocytosis of LNFPIII-NGC requires calcium. RAW 264.7 macrophages were pretreated with different concentrations of EGTA for 15 min, and then endocytosis of LNFPIII-NGC was induced for 30 min. We found that chelation of calcium ions in medium led to a dramatic reduction of RAW 264.7 cell binding and uptake of LNFPIII-NGC (Fig. 5c). Further, we quantitated internalization of LNFPIII-NGC after treatment with EGTA by flow cytometry and observed similar results (Fig. 5e). To show that EGTA is not toxic to the cells, we performed viability assays with cells treated with EGTA under similar conditions and found no cell death due to EGTA (Fig. 5d). Taken together, these studies demonstrate that LNFPIII-NGC entry into APCs is receptor mediated and requires divalent calcium, suggesting that C-type lectins are likely receptors.
Endocytosis is required for LNFPIII-NGC activation of APCs that drive CD4+ Th2 maturation.
Schistosome soluble egg antigen (SEA) and LNFPIII-NGC stimulation of APCs induces maturation of APCs that induce CD4+ T cells to the Th2 type (20, 26, 49). Based on our observation that LNFPIII-NGCs are endocytosed by APCs in a clathrin- and dynamin-dependent process, we asked if endocytosis of LNFPIII-NGC is required to induce alternative maturation of APCs. We tested this using a dendritic cell CD4+ T cell coculture in which bone marrow-derived DCs were treated or not treated with the dynamin inhibitor dynasore (40 μM) followed by incubation with LNFPIII-NGC at 37°C for 48 h. Medium was replaced, and OVA peptide and OVA-specific purified CD4+ T cells were added to DCs as described above and cultured for 72 h at 37°C. Culture supernatants were harvested then analyzed for IFN-γ, IL-4, and IL-13 by ELISA. As shown in Fig. 6, production of the Th2-type cytokine IL-13 was completely inhibited in cultures in which the DCs were treated with dynasore prior to addition of LNFPIII-NGC. We did not see any differences in IFN-γ production, and IL-4 was not in detectable range by ELISA. SEA was a positive control, having been previously shown to mature APCs that drive CD4+ cells to a Th2-type response in DC-T cell coculture. We did not see any effect of dynasore on DC viability (see Fig. S4 in the supplemental material). This finding shows that LNFPIII-NGC must be endocytosed via the dynamin/clathrin pathway in order for the initiation of the alternative maturation pathway for APCs, which in this case produce DC2s.
FIG 6.
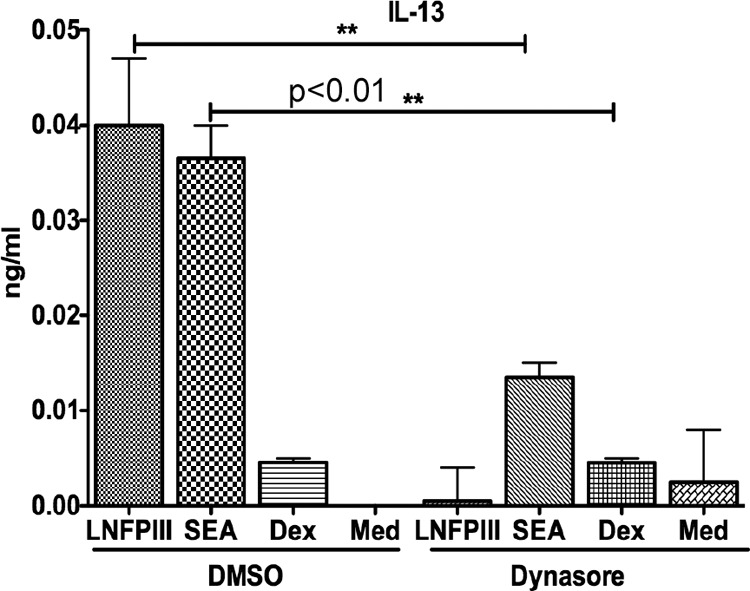
Endocytosis is required for LNFPIII-NGC to drive DC2 maturation. BMDCs were pretreated with 40 μM dynasore for 1 h and then stimulated with SEA (25 μg/ml), LNFPIII-NGC (50 μg/ml), or dextran (50 μg/ml) for 48 h at 37°C. Naive CD4+T cells isolated from spleens of OT-II mice were added to BMDCs at 1:5 ratios and 3 μM OVA peptide in fresh medium. Supernatants were collected after 72 h of coculture and analyzed for IL-13 production using ELISA. Treated and untreated samples were in triplicate. Statistical analysis was done using GraphPad Quick Calcs Student's t test (**, P < 0.01). Results represent at least 3 independent experiments.
LNFPIII-NGC binds to C-type lectin receptor mouse SIGNR-1.
We next evaluated the ability of several C-type lectins to bind to LNFPIII-NGC. Lewisx- or Lewisx motif-containing glycans are known to bind to the human C-type lectin receptor DC-SIGN (50). In mouse, there are at least 3 functional homologues of DC-SIGN: SIGNR1, SIGNR3, and SIGNR5. Other than DC-SIGN, Lewisx also binds MGL-1. To evaluate which murine C-type lectins bind LNFPIII, we took advantage of the observation that LNFPIII-NGC binds to APCs but fails to bind NIH 3T3 fibroblast cells. Therefore, we utilized overexpression of C-type lectin receptors (CLRs) cloned in fusion with green fluorescence protein (GFP) in NIH 3T3 fibroblast cells to study LNFPIII-NGC binding. As shown in Fig. 7a, we observed clear binding of LNFPIII-NGC to NIH 3T3 cells overexpressing GFP-SIGNR1 protein. In contrast, we were unable to detect direct binding of LNFPIII-NGC to NIH 3T3 cells overexpressing GFP-SIGNR3, GFP-SIGNR5, or GFP-MGL1 receptors, suggesting that LNFPIII-NGC binding is mediated via mouse SIGNR1 receptor (Fig. 7a). To further validate direct binding of LNFPIII to mouse SIGNR1, we performed an ELISA using recombinant purified SIGNR1 (rSIGNR1). Recombinant SIGNR1 protein-coated ELISA plates were incubated with LNFPIII-NGC, and binding of LNFPIII was determined by probing with anti-HSA antibody. As shown in Fig. 7b, LNFPIII-NGC strongly binds to recombinant SIGNR1 protein but not to the carrier molecule HSA, validating that binding of LNFPIII to SIGNR1-overexpressing NIH 3T3 cells is due to direct interaction between LNFPIII and the SIGNR1 receptor (Fig. 7b).
FIG 7.

LNFPIII-NGC binds to mouse SIGNR1 in vitro. (a) NIH 3T3 cells were transfected with green fluorescent protein (GFP)-fused constructs of mouse SIGNR1, SIGNR3, SIGNR5, or MGL (green). Cells were then incubated with LNFPIII-NGC (50 μg/ml), and endocytosis was induced for 30 min as described before. Cells were fixed, stained, and then analyzed under a confocal microscope. LNFPIII-NGCs are stained in red, SIGNRs in green, and overlay/colocalization in yellow. The above-described experiment represents 4 independent experiments repeated and reproduced 3 or 4 times. (b) LNFPIII-HSA directly binds to recombinant SIGNR1. ELISA was performed to confirm the direct interaction between SIGNR1 and LNFPIII-NGCs. Ninety-six-well plates were coated with recombinant SIGNR1 (10 μg/ml) overnight followed by incubation with LNFPIII-HSA conjugate (10 μg/ml) for 1 h at room temperature. Binding was detected using anti-HSA antibody followed by horseradish peroxidase (HRP)-conjugated secondary antibody. Recombinant HSA was used as a control. The data represent 4 independent experiment with n = 4. Statistical significance was calculated by Student's t test (****, P < 0.0001). O.D. 450 nm, optical density at 450 nm. (c) No change in LNFPIII-NGC uptake in macrophages lacking SIGNR1 expression. Endogenous SIGNR1 protein expression was downregulated in peritoneal macrophages by transfection with SIGNR1-specific siRNA for 36 h. Nonspecific siRNA was used as a control. Cells were then incubated with LNFPIII-NGC (50 μg/ml) for 30 min at 37°C as described previously. LNFPIII-NGC is stained in green. Data represent two individual experiments performed, giving similar results. (d) Validation of SIGNR1 protein expression knockdown by quantitative real-time PCR (qRT-PCR). Total RNAs from cells transfected with siRNA or nonspecific controls were used to measure expression of signr1 gene by the qRT-PCR method. Transfected samples were in triplicate, and the data shown are from 1 of 2 independent experiments. Statistical significance was calculated by Student's t test analysis (***, P < 0.001).
Knockdown of SIGNR1 expression in macrophages does not affect LNFPIII-NGC uptake in APCs.
Next, we wanted to evaluate if uptake of LNFPIII is dependent on SIGNR1 expression in macrophages. Using siRNA, we knocked down the expression of SIGNR1 in mouse peritoneal macrophages, and uptake of LNFPIII was studied using confocal microscopy. Although we saw strong binding of LNFPIII-NGC to SIGNR1, macrophages with reduced expression of mSIGNR1 did not show any defect in uptake of LNFPIII-NGC. This may be due to macrophages binding LNFPIII via a different receptor or more than one receptor being required for LNFPIII-NGC binding (Fig. 7c and d). Further, it may be that knocking down SIGNR1 expression by 50% was not sufficient to impact uptake of LNFPIII.
DISCUSSION
Antigen-presenting cells play key roles in driving and directing adaptive T cell responses as well as regulating the inflammatory/anti-inflammatory balance. Mechanisms defining classical or proinflammatory activation of APCs, from ligation of APC PRRs such as the Toll-like receptors and subsequent downstream signaling, are well known. In contrast, little is known regarding the mechanisms of alternative or anti-inflammatory maturation of APCs. The goal of this study was to define the required mechanisms for alternative activation of APCs using the defined glycan LNFPIII and schistosome egg extracts, both known to induce alternative activation of APCs (19, 21, 51).
The Lewisx trisaccharide is abundantly expressed on schistosome egg antigens and is the terminal trisaccharide of the LNFPIII pentasaccharide that we have used to study pathogen/self glycan activation of innate immune responses (10, 26). LNFPIII neoglycoconjugates (LNFPIII-NGC) comprised of multiple (7 to 10) LNFPIII moieties conjugated via linker-spacer technology to carrier molecules have been employed to determine how LNFPIII/Lewisx activates and induces alternative activation of APCs and drives anti-inflammatory responses in vivo.
Despite the immune modulatory and in vivo therapeutic properties of LNFPIII-NGC, it was not known how LNFPIII-NGC interacts with antigen-presenting cells or whether active internalization and intracellular processing of LNFPIII-NGC are key to modulating APC functions until this study. Here we observed that LNFPIII-NGC was endocytosed by APCs whereas the 40-kDa dextran backbone conjugated to FITC or Texas Red was not internalized (see Fig. S1a in the supplemental material). Reducing the size of dextran to 10 kDa did show some internalization in macrophages (see Fig. S1a and b in the supplemental material). The effect of size of dextran molecules on internalization was reported by Pellegrin et al., noting that internalization of dextran is size dependent, with smaller sizes being internalized more efficiently (52). In contrast to our observation that 40-kDa dextran is not internalized, a few studies have reported internalization of 40-kDa dextran. However, in all these studies the dextran concentrations ranged from 1 to 5 mg/ml, 20- to 100-fold higher than the 50-μg/ml concentration used in this study (52, 53).
In regard to the mechanism of internalization, we observed that LNFPIII-NGC interacts with APCs via cell surface receptors, which leads to active endocytosis of LNFPIII-NGC via dynamin- and clathrin-mediated pathways. We found that the ability of LNFPIII-NGC to alternatively activate APCs that drive CD4+ Th2 responses is dependent on APC endocytosis of LNFPIII-NGC, as APCs treated with dynasore failed to drive CD4+ Th2 responses. These results are similar to an earlier study suggesting that DC2s generated by activation with SEA had a distinct phenotype that included upregulation of clathrin and the endosomal marker Rab5 (54). The study by Ferret-Bernard et al. strengthens our observation that dynamin/clathrin-mediated endocytosis of LNFPIII-NGC induces Th2-type immune responses (54).
We also found that blocking endocytosis abrogated the ability of LNFPIII-NGC to alternatively activate APCs, suggesting that signaling pathways initiated by LNFPIII-NGC cell surface receptor interactions are not sufficient to induce alternative maturation of APCs. The finding that LNFPIII/Lewisx induces the alternative maturation pathway downstream of cell surface signaling is an important concept.
To examine receptor-mediated uptake of LNFPIII-NGC, we treated APCs with hypertonic medium or medium depleted of extracellular calcium to demonstrate that LNFPIII conjugate binding to APCs is receptor mediated and likely occurs via Ca2+-dependent C-type lectins. Additionally, we observed LNFPIII-NGC colocalization with TLR4 (see Fig. S5a in the supplemental material). While earlier studies suggested a requirement for TLR4 for LNFPIII-NGC activation of DCs, here we observed that TLR4 was not required for APC endocytosis of LNFPIII-NGC as TLR4-deficient APCs endocytosed LNFPIII-NGC at levels similar to those of wild-type APCs (see Fig. S5b in the supplemental material). In regard to possible cross talk between CLR and TLR pathways, an earlier study reported that Lewisx-containing glycans bound to human DCs via DC-SIGN and subsequently modulated TLR4-mediated signaling (55). Therefore, we attempted to determine if individual murine DC-SIGN homologues (SIGN-R1, SIGN-R3, or SIGN-R5) would bind LNFPIII-NGC. We observed LNFPIII-NGC binding to the surface of cells transfected with SIGN-R1 and demonstrated binding of LNFPIII-NGC to SIGNR1 by ELISA. However, we could not detect expression of SIGNR1 in BMDCs (data not shown), suggesting that there are other receptors that LNFPIII utilizes to interact and enter into APCs to modulate immune functions. These results are similar to those of an earlier study showing that glycans in SEA require at least three C-type lectins, MglI, mannose receptor 1 (MR1), and DC-SIGN, to mature APCs into cells that drive Th2 immune responses (22, 56). In mice, the presence of multiple C-type lectin receptors and multiple copies of the SIGNR1, a homologue of human DC-SIGN, makes it difficult to selectively study the role of lectins in LNFPIII-mediated alternative activation of APCs. However, our finding that LNFPIII binds to SIGNR1 suggests that LNFPIII may trigger anti-inflammatory responses in vivo via mouse SIGNR-1-expressing macrophages (57). Several reports show that LNFPIII-NGC's ability to prevent onset of autoimmune or proinflammatory diseases occurs in part via modulating macrophage effector functions (29).
Our finding that endocytosis of LNFPIII-NGC is required to drive alternative activation of APCs may be a general phenomenon for Lewisx-expressing molecules, as Lewisx-expressing molecules in SEA required endocytosis in a dynamin-dependent manner to drive alternative activation of APCs (see Fig. S2a in the supplemental material). Further support for this observation comes from Everts et al. (22), who demonstrated that glycans on the S. mansoni egg protein omega-1 were required for omega-1 binding to and uptake by APCs, confirming the requirement of helminth glycans in initiating APC maturation that leads to CD4+ T cell biasing to Th2 type. However, while Everts et al. (22) confirmed the requirement for glycans, they suggested that the glycans on omega-1 were utilized only for omega-1 uptake by dendritic cells and were not directly involved in driving DC maturation toward DC2s. They suggested that because Lewisx and other glycan structures found on omega-1 were not multivalent, they were unable to drive DCs toward a DC2 phenotype. Everts et al. (22) stated that the loss of total ribosomal RNAs in APCs due to omega-1 RNase activity is what primes DCs to induce CD4+ Th2-type maturation. An alternative explanation for omega-1 activity is that the RNase activity kills APCs, releasing danger-associated molecular patterns into the environment, which then activates viable APCs to embark on an alternative maturation process. This fits with previously published findings on the hepatotoxic properties of omega-1 (58).
Similar to the findings reported here, several studies demonstrate that fucose- or mannose-containing glycans by themselves drive anti-inflammatory or Th2 immune responses as well as modulate TLR (LPS)-induced inflammation via various signaling cascades (26, 55, 59). As suggested earlier, the ability of glycans to directly drive alternative activation of APCs may lie in the nature of the glycans. The majority of the studies demonstrating that glycans by themselves activate and mature APCs into anti-inflammatory and Th2-driving phenotypes employed multivalent neoglycoconjugates or naturally occurring β-glucans (9, 60, 61). The results presented here showing that LNFPIII-NGC are taken up and processed via the classical endosomal pathway suggest that signaling originating from endosomes may be the requisite intracellular signaling pathways that multivalent glycans activate to initiate APC polarization to anti-inflammatory and/or Th2-type cells. Our studies also suggest that glycans may regulate uptake, processing, and presentation of lipids and proteins and also act as adjuvants by strengthening the immune responses elicited by these molecules. It has been shown that treatment of macrophages with carbohydrates changes their phagocytic index (62–64). Further to this point, cross-linking glycans to proteins increases their immunogenicity (26, 65), resulting in better antigen-specific humoral and memory responses (65).
Carbohydrates have been largely classified as T cell-independent antigens. However, bacterial capsular zwitterionic polysaccharides have been shown to be presented by major histocompatibility complex class II (MHC-II) and elicit T cell immune modulatory responses (66). Interestingly, zwitterionic carbohydrates conjugated to proteins enhance vaccine-specific responses to the proteins by increasing endosomal acidity, leading to enhanced acid-dependent antigen processing (67). Avci et al. showed carbohydrate-specific T cell and B cell responses against carbohydrates covalently attached to protein antigens (61). Similar to zwitterionic carbohydrates, LNFPIII-NGC endocytosis also enhances endosome acidification. However, we were unable to show that endocytosed LNFPIII-NGC colocalizes with MHC-II. The absence of conclusive MHC-II colocalization, along with our time course internalization experiments, suggests that unlike bacterial capsular zwitterionic carbohydrates, endocytosed LNFPIII-NGC follows the complete endosomal pathway, eventually leading to degradation of LNFPIII-NGC.
In conclusion, our studies demonstrate that Lewisx-containing glycans in schistosoma egg antigens or as LNFPIII-NGC are taken up by APCs in a cell surface receptor-mediated process leading to a classical endocytic pathway and phagosomal maturation. Importantly, the endocytic pathway is required for both SEA and LNFPIII-NGC to drive alternative APC maturation such that these APCs will mature CD4+ T cells toward anti-inflammatory and Th2-type cells. The results presented here contribute to our overall understanding of the mechanism of glycan-mediated induction of Th2 and anti-inflammatory immune responses and are likely to lead to additional studies directed at improving glycan-mediated therapeutic approaches against inflammation-based diseases (29).
Supplementary Material
ACKNOWLEDGMENTS
These studies were supported by an NIH grant (AI056484) and GRA and UGARF start-up funds awarded to D.H. We thank the CVM Cytometry Core Facility for using the confocal microscope.
We declare that we have no conflicts of interests.
Footnotes
Published ahead of print 24 February 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01293-13.
REFERENCES
- 1.Hirabara SM, Gorjao R, Vinolo MA, Rodrigues AC, Nachbar RT, Curi R. 2012. Molecular targets related to inflammation and insulin resistance and potential interventions. J. Biomed. Biotechnol. 2012:379024. 10.1155/2012/379024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tundup S, Srivastava L, Harn DA., Jr 2012. Polarization of host immune responses by helminth-expressed glycans. Ann. N. Y. Acad. Sci. 1253:E1–E13. 10.1111/j.1749-6632.2012.06618.x [DOI] [PubMed] [Google Scholar]
- 3.Gordon S. 2003. Alternative activation of macrophages. Nat. Rev. Immunol. 3:23–35. 10.1038/nri978 [DOI] [PubMed] [Google Scholar]
- 4.Sica A, Mantovani A. 2012. Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest. 122:787–795. 10.1172/JCI59643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cook PC, Jones LH, Jenkins SJ, Wynn TA, Allen JE, MacDonald AS. 2012. Alternatively activated dendritic cells regulate CD4+ T-cell polarization in vitro and in vivo. Proc. Natl. Acad. Sci. U. S. A. 109:9977–9982. 10.1073/pnas.1121231109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinez FO, Helming L, Gordon S. 2009. Alternative activation of macrophages: an immunologic functional perspective. Annu. Rev. Immunol. 27:451–483. 10.1146/annurev.immunol.021908.132532 [DOI] [PubMed] [Google Scholar]
- 7.Semnani RT, Mahapatra L, Moore V, Sanprasert V, Nutman TB. 2011. Functional and phenotypic characteristics of alternative activation induced in human monocytes by interleukin-4 or the parasitic nematode Brugia malayi. Infect. Immun. 79:3957–3965. 10.1128/IAI.05191-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reyes JL, Terrazas LI. 2007. The divergent roles of alternatively activated macrophages in helminthic infections. Parasite Immunol. 29:609–619. 10.1111/j.1365-3024.2007.00973.x [DOI] [PubMed] [Google Scholar]
- 9.Kawashima S, Hirose K, Iwata A, Takahashi K, Ohkubo A, Tamachi T, Ikeda K, Kagami S, Nakajima H. 2012. beta-glucan curdlan induces IL-10-producing CD4+ T cells and inhibits allergic airway inflammation. J. Immunol. 189:5713–5721. 10.4049/jimmunol.1201521 [DOI] [PubMed] [Google Scholar]
- 10.Atochina O, Da'dara AA, Walker M, Harn DA. 2008. The immunomodulatory glycan LNFPIII initiates alternative activation of murine macrophages in vivo. Immunology 125:111–121. 10.1111/j.1365-2567.2008.02826.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Voehringer D, van Rooijen N, Locksley RM. 2007. Eosinophils develop in distinct stages and are recruited to peripheral sites by alternatively activated macrophages. J. Leukoc. Biol. 81:1434–1444. 10.1189/jlb.1106686 [DOI] [PubMed] [Google Scholar]
- 12.Ma J, Becker C, Lowell CA, Underhill DM. 2012. Dectin-1-triggered recruitment of light chain 3 protein to phagosomes facilitates major histocompatibility complex class II presentation of fungal-derived antigens. J. Biol. Chem. 287:34149–34156. 10.1074/jbc.M112.382812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doyle SE, O'Connell RM, Miranda GA, Vaidya SA, Chow EK, Liu PT, Suzuki S, Suzuki N, Modlin RL, Yeh WC, Lane TF, Cheng G. 2004. Toll-like receptors induce a phagocytic gene program through p38. J. Exp. Med. 199:81–90. 10.1084/jem.20031237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shao H, Lee S, Gae-Scott S, Nakata C, Chen S, Hamad AR, Chakravarti S. 2012. Extracellular matrix lumican promotes bacterial phagocytosis, and Lum-/- mice show increased Pseudomonas aeruginosa lung infection severity. J. Biol. Chem. 287:35860–35872. 10.1074/jbc.M112.380550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blander JM, Medzhitov R. 2004. Regulation of phagosome maturation by signals from toll-like receptors. Science 304:1014–1018. 10.1126/science.1096158 [DOI] [PubMed] [Google Scholar]
- 16.Ryan SO, Cobb BA. 2012. Host glycans and antigen presentation. Microbes Infect. 11:894–903. 10.1016/j.micinf.2012.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Kooyk Y, Unger WW, Fehres CM, Kalay H, Garcia-Vallejo JJ. 2013. Glycan-based DC-SIGN targeting vaccines to enhance antigen cross-presentation. Mol. Immunol. 2:143–145. 10.1016/j.molimm.2012.10.031 [DOI] [PubMed] [Google Scholar]
- 18.Barron L, Wynn TA. 2011. Macrophage activation governs schistosomiasis-induced inflammation and fibrosis. Eur. J. Immunol. 41:2509–2514. 10.1002/eji.201141869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harn DA, McDonald J, Atochina O, Da'dara AA. 2009. Modulation of host immune responses by helminth glycans. Immunol. Rev. 230:247–257. 10.1111/j.1600-065X.2009.00799.x [DOI] [PubMed] [Google Scholar]
- 20.Okano M, Satoskar AR, Nishizaki K, Abe M, Harn DA., Jr 1999. Induction of Th2 responses and IgE is largely due to carbohydrates functioning as adjuvants on Schistosoma mansoni egg antigens. J. Immunol. 163:6712–6717 [PubMed] [Google Scholar]
- 21.Tawill S, Le Goff L, Ali F, Blaxter M, Allen JE. 2004. Both free-living and parasitic nematodes induce a characteristic Th2 response that is dependent on the presence of intact glycans. Infect. Immun. 72:398–407. 10.1128/IAI.72.1.398-407.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Everts B, Hussaarts L, Driessen NN, Meevissen MH, Schramm G, van der Ham AJ, van der Hoeven B, Scholzen T, Burgdorf S, Mohrs M, Pearce EJ, Hokke CH, Haas H, Smits HH, Yazdanbakhsh M. 2012. Schistosome-derived omega-1 drives Th2 polarization by suppressing protein synthesis following internalization by the mannose receptor. J. Exp. Med. 209:1753–1767, S1. 10.1084/jem.20111381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meevissen MH, Wuhrer M, Doenhoff MJ, Schramm G, Haas H, Deelder AM, Hokke CH. 2010. Structural characterization of glycans on omega-1, a major Schistosoma mansoni egg glycoprotein that drives Th2 responses. J. Proteome Res. 9:2630–2642. 10.1021/pr100081c [DOI] [PubMed] [Google Scholar]
- 24.Nyame AK, Kawar ZS, Cummings RD. 2004. Antigenic glycans in parasitic infections: implications for vaccines and diagnostics. Arch. Biochem. Biophys. 426:182–200. 10.1016/j.abb.2004.04.004 [DOI] [PubMed] [Google Scholar]
- 25.Hokke CH, Deelder AM, Hoffmann KF, Wuhrer M. 2007. Glycomics-driven discoveries in schistosome research. Exp. Parasitol. 117:275–283. 10.1016/j.exppara.2007.06.003 [DOI] [PubMed] [Google Scholar]
- 26.Okano M, Satoskar AR, Nishizaki K, Harn DA., Jr 2001. Lacto-N-fucopentaose III found on Schistosoma mansoni egg antigens functions as adjuvant for proteins by inducing Th2-type response. J. Immunol. 167:442–450 [DOI] [PubMed] [Google Scholar]
- 27.Velupillai P, dos Reis EA, dos Reis MG, Harn DA. 2000. Lewis(x)-containing oligosaccharide attenuates schistosome egg antigen-induced immune depression in human schistosomiasis. Hum. Immunol. 61:225–232. 10.1016/S0198-8859(99)00136-6 [DOI] [PubMed] [Google Scholar]
- 28.Wang Y, Da'Dara AA, Thomas PG, Harn DA. 2010. Dendritic cells activated by an anti-inflammatory agent induce CD4(+) T helper type 2 responses without impairing CD8(+) memory and effector cytotoxic T-lymphocyte responses. Immunology 129:406–417. 10.1111/j.1365-2567.2009.03193.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bhargava P, Li C, Stanya KJ, Jacobi D, Dai L, Liu S, Gangl MR, Harn DA, Lee CH. 2012. Immunomodulatory glycan LNFPIII alleviates hepatosteatosis and insulin resistance through direct and indirect control of metabolic pathways. Nat. Med. 18:1665–1672. 10.1038/nm.2962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Velupillai P, Secor WE, Horauf AM, Harn DA. 1997. B-1 cell (CD5+B220+) outgrowth in murine schistosomiasis is genetically restricted and is largely due to activation by polylactosamine sugars. J. Immunol. 158:338–344 [PubMed] [Google Scholar]
- 31.Thomas PG, Carter MR, Atochina O, Da'Dara AA, Piskorska D, McGuire E, Harn DA. 2003. Maturation of dendritic cell 2 phenotype by a helminth glycan uses a Toll-like receptor 4-dependent mechanism. J. Immunol. 171:5837–5841 [DOI] [PubMed] [Google Scholar]
- 32.Ritter M, Gross O, Kays S, Ruland J, Nimmerjahn F, Saijo S, Tschopp J, Layland LE, Prazeres da Costa C. 2010. Schistosoma mansoni triggers Dectin-2, which activates the Nlrp3 inflammasome and alters adaptive immune responses. Proc. Natl. Acad. Sci. U. S. A. 107:20459–20464. 10.1073/pnas.1010337107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Liempt E, van Vliet SJ, Engering A, Garcia Vallejo JJ, Bank CM, Sanchez-Hernandez M, van Kooyk Y, van Die I. 2007. Schistosoma mansoni soluble egg antigens are internalized by human dendritic cells through multiple C-type lectins and suppress TLR-induced dendritic cell activation. Mol. Immunol. 44:2605–2615. 10.1016/j.molimm.2006.12.012 [DOI] [PubMed] [Google Scholar]
- 34.Dennehy KM, Ferwerda G, Faro-Trindade I, Pyz E, Willment JA, Taylor PR, Kerrigan A, Tsoni SV, Gordon S, Meyer-Wentrup F, Adema GJ, Kullberg BJ, Schweighoffer E, Tybulewicz V, Mora-Montes HM, Gow NA, Williams DL, Netea MG, Brown GD. 2008. Syk kinase is required for collaborative cytokine production induced through Dectin-1 and Toll-like receptors. Eur. J. Immunol. 38:500–506. 10.1002/eji.200737741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coombs PJ, Graham SA, Drickamer K, Taylor ME. 2005. Selective binding of the scavenger receptor C-type lectin to Lewisx trisaccharide and related glycan ligands. J. Biol. Chem. 280:22993–22999. 10.1074/jbc.M504197200 [DOI] [PubMed] [Google Scholar]
- 36.Garcia-Vallejo JJ, van Liempt E, da Costa Martins P, Beckers C, van het Hof B, Gringhuis SI, Zwaginga JJ, van Dijk W, Geijtenbeek TB, van Kooyk Y, van Die I. 2008. DC-SIGN mediates adhesion and rolling of dendritic cells on primary human umbilical vein endothelial cells through LewisY antigen expressed on ICAM-2. Mol. Immunol. 45:2359–2369. 10.1016/j.molimm.2007.11.001 [DOI] [PubMed] [Google Scholar]
- 37.van den Berg TK, Honing H, Franke N, van Remoortere A, Schiphorst WE, Liu FT, Deelder AM, Cummings RD, Hokke CH, van Die I. 2004. LacdiNAc-glycans constitute a parasite pattern for galectin-3-mediated immune recognition. J. Immunol. 173:1902–1907 [DOI] [PubMed] [Google Scholar]
- 38.Kerrigan AM, Brown GD. 2009. C-type lectins and phagocytosis. Immunobiology 214:562–575. 10.1016/j.imbio.2008.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Napoletano C, Zizzari IG, Rughetti A, Rahimi H, Irimura T, Clausen H, Wandall HH, Belleudi F, Bellati F, Pierelli L, Frati L, Nuti M. 2012. Targeting of macrophage galactose-type C-type lectin (MGL) induces DC signaling and activation. Eur. J. Immunol. 42:936–945. 10.1002/eji.201142086 [DOI] [PubMed] [Google Scholar]
- 40.Thomas PG, Carter MR, Da'dara AA, DeSimone TM, Harn DA. 2005. A helminth glycan induces APC maturation via alternative NF-kappa B activation independent of I kappa B alpha degradation. J. Immunol. 175:2082–2090 [DOI] [PubMed] [Google Scholar]
- 41.Ko AI, Drager UC, Harn DA. 1990. A Schistosoma mansoni epitope recognized by a protective monoclonal antibody is identical to the stage-specific embryonic antigen 1. Proc. Natl. Acad. Sci. U. S. A. 87:4159–4163. 10.1073/pnas.87.11.4159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oh P, Horner T, Witkiewicz H, Schnitzer JE. 2012. Endothelin induces rapid, dynamin-mediated budding of endothelial caveolae rich in ET-B. J. Biol. Chem. 287:17353–17362. 10.1074/jbc.M111.338897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ferguson SM, De Camilli P. 2012. Dynamin, a membrane-remodelling GTPase. Nat. Rev. Mol. Cell Biol. 13:75–88. 10.1038/nrm3266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goodridge HS, Underhill DM, Touret N. 2012. Mechanisms of Fc receptor and dectin-1 activation for phagocytosis. Traffic 13:1062–1071. 10.1111/j.1600-0854.2012.01382.x [DOI] [PubMed] [Google Scholar]
- 45.Mooren OL, Galletta BJ, Cooper JA. 2012. Roles for actin assembly in endocytosis. Annu. Rev. Biochem. 81:661–686. 10.1146/annurev-biochem-060910-094416 [DOI] [PubMed] [Google Scholar]
- 46.Boulant S, Kural C, Zeeh JC, Ubelmann F, Kirchhausen T. 2011. Actin dynamics counteract membrane tension during clathrin-mediated endocytosis. Nat. Cell Biol. 13:1124–1131. 10.1038/ncb2307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Feinberg H, Taylor ME, Weis WI. 2007. Scavenger receptor C-type lectin binds to the leukocyte cell surface glycan Lewis (x) by a novel mechanism. J. Biol. Chem. 282:17250–17258. 10.1074/jbc.M701624200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Feinberg H, Park-Snyder S, Kolatkar AR, Heise CT, Taylor ME, Weis WI. 2000. Structure of a C-type carbohydrate recognition domain from the macrophage mannose receptor. J. Biol. Chem. 275:21539–21548. 10.1074/jbc.M002366200 [DOI] [PubMed] [Google Scholar]
- 49.Atochina O, Daly-Engel T, Piskorska D, McGuire E, Harn DA. 2001. A schistosome-expressed immunomodulatory glycoconjugate expands peritoneal Gr1(+) macrophages that suppress naive CD4(+) T cell proliferation via an IFN-gamma and nitric oxide-dependent mechanism. J. Immunol. 167:4293–4302 [DOI] [PubMed] [Google Scholar]
- 50.van Die I, van Vliet SJ, Nyame AK, Cummings RD, Bank CM, Appelmelk B, Geijtenbeek TB, van Kooyk Y. 2003. The dendritic cell-specific C-type lectin DC-SIGN is a receptor for Schistosoma mansoni egg antigens and recognizes the glycan antigen Lewis x. Glycobiology 13:471–478. 10.1093/glycob/cwg052 [DOI] [PubMed] [Google Scholar]
- 51.Pearce EJ, Kane CM, Sun J. 2006. Regulation of dendritic cell function by pathogen-derived molecules plays a key role in dictating the outcome of the adaptive immune response. Chem. Immunol. Allergy 90:82–90. 10.1159/000088882 [DOI] [PubMed] [Google Scholar]
- 52.Pellegrin P, Fernandez A, Lamb NJ, Bennes R. 2002. Macromolecular uptake is a spontaneous event during mitosis in cultured fibroblasts: implications for vector-dependent plasmid transfection. Mol. Biol. Cell 13:570–578. 10.1091/mbc.01-06-0280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Czerkies M, Borzecka K, Zdioruk MI, Plociennikowska A, Sobota A, Kwiatkowska K. 2013. An interplay between scavenger receptor A and CD14 during activation of J774 cells by high concentrations of LPS. Immunobiology 218:1217–1226. 10.1016/j.imbio.2013.04.005 [DOI] [PubMed] [Google Scholar]
- 54.Ferret-Bernard S, Castro-Borges W, Dowle AA, Sanin DE, Cook PC, Turner JD, MacDonald AS, Thomas JR, Mountford AP. 2012. Plasma membrane proteomes of differentially matured dendritic cells identified by LC-MS/MS combined with iTRAQ labelling. J. Proteomics 75:938–948. 10.1016/j.jprot.2011.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gringhuis SI, den Dunnen J, Litjens M, van Het Hof B, van Kooyk Y, Geijtenbeek TB. 2007. C-type lectin DC-SIGN modulates Toll-like receptor signaling via Raf-1 kinase-dependent acetylation of transcription factor NF-kappaB. Immunity 26:605–616. 10.1016/j.immuni.2007.03.012 [DOI] [PubMed] [Google Scholar]
- 56.Meevissen MH, Driessen NN, Smits HH, Versteegh R, van Vliet SJ, van Kooyk Y, Schramm G, Deelder AM, Haas H, Yazdanbakhsh M, Hokke CH. 2012. Specific glycan elements determine differential binding of individual egg glycoproteins of the human parasite Schistosoma mansoni by host C-type lectin receptors. Int. J. Parasitol. 42:269–277. 10.1016/j.ijpara.2012.01.004 [DOI] [PubMed] [Google Scholar]
- 57.Taylor PR, Brown GD, Herre J, Williams DL, Willment JA, Gordon S. 2004. The role of SIGNR1 and the beta-glucan receptor (dectin-1) in the nonopsonic recognition of yeast by specific macrophages. J. Immunol. 172:1157–1162 [DOI] [PubMed] [Google Scholar]
- 58.Dunne DW, Lucas S, Bickle Q, Pearson S, Madgwick L, Bain J, Doenhoff MJ. 1981. Identification and partial purification of an antigen (omega 1) from Schistosoma mansoni eggs which is putatively hepatotoxic in T-cell deprived mice. Trans. R. Soc. Trop. Med. Hyg. 75:54–71. 10.1016/0035-9203(81)90013-4 [DOI] [PubMed] [Google Scholar]
- 59.Collins HL, Schaible UE, Kaufmann SH. 1998. Early IL-4 induction in bone marrow lymphoid precursor cells by mycobacterial lipoarabinomannan. J. Immunol. 161:5546–5554 [PubMed] [Google Scholar]
- 60.Khoo KH, Dell A. 2001. Glycoconjugates from parasitic helminths: structure diversity and immunobiological implications. Adv. Exp. Med. Biol. 491:185–205. 10.1007/978-1-4615-1267-7_14 [DOI] [PubMed] [Google Scholar]
- 61.Avci FY, Li X, Tsuji M, Kasper DL. 2011. A mechanism for glycoconjugate vaccine activation of the adaptive immune system and its implications for vaccine design. Nat. Med. 17:1602–1609. 10.1038/nm.2535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fuentes AL, Millis L, Sigola LB. 2011. Laminarin, a soluble beta-glucan, inhibits macrophage phagocytosis of zymosan but has no effect on lipopolysaccharide mediated augmentation of phagocytosis. Int. Immunopharmacol. 11:1939–1945. 10.1016/j.intimp.2011.08.005 [DOI] [PubMed] [Google Scholar]
- 63.Chen H, Yuan B, Zheng Z, Liu Z, Wang S. 2011. Lewis X oligosaccharides-heparanase complex targeting to DCs enhance antitumor response in mice. Cell. Immunol. 269:144–148. 10.1016/j.cellimm.2011.03.021 [DOI] [PubMed] [Google Scholar]
- 64.Maeda R, Ida T, Ihara H, Sakamoto T. 2012. Immunostimulatory activity of polysaccharides isolated from Caulerpa lentillifera on macrophage cells. Biosci. Biotechnol. Biochem. 76:501–505. 10.1271/bbb.110813 [DOI] [PubMed] [Google Scholar]
- 65.Ahlen G, Strindelius L, Johansson T, Nilsson A, Chatzissavidou N, Sjoblom M, Rova U, Holgersson J. 2012. Mannosylated mucin-type immunoglobulin fusion proteins enhance antigen-specific antibody and T lymphocyte responses. PLoS One 7:e46959. 10.1371/journal.pone.0046959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cobb BA, Kasper DL. 2005. Zwitterionic capsular polysaccharides: the new MHCII-dependent antigens. Cell. Microbiol. 7:1398–1403. 10.1111/j.1462-5822.2005.00591.x [DOI] [PubMed] [Google Scholar]
- 67.Lewis CJ, Cobb BA. 2010. Carbohydrate oxidation acidifies endosomes, regulating antigen processing and TLR9 signaling. J. Immunol. 184:3789–3800. 10.4049/jimmunol.0903168 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.