

Abstract
A long-standing challenge in developing vaccines against enterotoxigenic Escherichia coli (ETEC), the most common bacteria causing diarrhea in children of developing countries and travelers to these countries, is to protect against heat-stable toxin type Ib (STa or hSTa). STa and heat-labile toxin (LT) are virulence determinants in ETEC diarrhea. LT antigens are often used in vaccine development, but STa has not been included because of its poor immunogenicity and potent toxicity. Toxic STa is not safe for vaccines, but only STa possessing toxicity is believed to be able to induce neutralizing antibodies. However, recent studies demonstrated that nontoxic STa derivatives (toxoids), after being fused to an LT protein, induced neutralizing antibodies and suggested that different STa toxoids fused to an LT protein might exhibit different STa antigenic propensity. In this study, we selected 14 STa toxoids from a mini-STa toxoid library based on toxicity reduction and reactivity to anti-native STa antibodies, and genetically fused each toxoid to a monomeric double mutant LT (dmLT) peptide for 14 STa-toxoid-dmLT toxoid fusions. These toxoid fusions were used to immunize mice and were characterized for induction of anti-STa antibody response. The results showed that different STa toxoids (in fusions) varied greatly in anti-STa antigenicity. Among them, STaN12S, STaN12T, and STaA14H were the top toxoids in inducing anti-STa antibodies. In vitro neutralization assays indicated that antibodies induced by the 3×STaN12S-dmLT fusion antigen exhibited the greatest neutralizing activity against STa toxin. These results suggested 3×STaN12S-dmLT is a preferred fusion antigen to induce an anti-STa antibody response and provided long-awaited information for effective ETEC vaccine development.
INTRODUCTION
Diarrhea remains a leading cause of death in children younger than 5 years who live in developing countries (1). Enterotoxigenic Escherichia coli (ETEC) strains (i.e., E. coli strains producing enterotoxins) are the most common bacteria causing diarrhea, particularly in children younger than 1 year from developing countries (2). ETEC diarrhea is responsible for an estimated annual death rate of 300,000 to 500,000, with the vast majority being children younger than 5 years (3, 4). ETEC strains are also the leading cause of diarrhea in children and adults who travel from developed countries to countries or regions where ETEC is endemic and in military personnel deployed in these areas and is also a threat to immunocompromised patients (3, 5–7). The virulence determinants of ETEC in diarrhea are bacterial adhesins and enterotoxins. Adhesins mediate initial ETEC bacteria attachment to host epithelial cells and subsequent colonization at host small intestines. Attachment and colonization bring ETEC bacteria in close proximity, which allows ETEC to deliver enterotoxins to host epithelial cells. Enterotoxins, mainly heat-labile toxin (LT) and heat-stable toxin type Ib (human-type STa or hSTa, which differs from type Ia STa or pSTa associated with ETEC diarrhea in animals and also from heat-stable toxin type II or STb), disrupt fluid homeostasis in host small intestinal epithelial cells to cause electrolyte-rich fluid hypersecretion through activation of intracellular adenylate cyclase (by LT) or guanylate cyclase (by STa), which directly leads to diarrhea (8). Fluid hypersecretion disarrays the mucin layer over host small intestinal epithelial cells and alters microvilli tight junction, which in return enhances ETEC bacterial colonization at host small intestines (9–11). An ideal ETEC vaccine should induce antiadhesin immunity to block ETEC attachment and to prevent bacterial colonization at host small intestines and also antitoxin immunity to neutralize both LT and STa toxins (12–14). However, there are currently no vaccines available to effectively protect against ETEC diarrhea.
Key technical challenges would have to be overcome to develop an effective ETEC vaccine. These include the immunological heterogeneity among ETEC strains or virulence determinants, the potent toxicity of LT and STa toxins, and the poor immunogenicity of the STa molecule. Progress has been made in developing antiadhesin vaccines by targeting multiple CFA adhesins which are expressed by the most prevalent and the most virulent ETEC strains (15, 16) and also in inducing anti-LT immunity protecting against LT by using the nontoxic LTB subunit, a homologous cholera toxin (CT) B subunit, or LT toxoids (17, 18). However, the development of anti-STa immunity or vaccines against STa has been much hampered (12, 14, 19). Indeed, due to its potent toxicity and poor immunogenicity, this small STa (19 amino acid long for human-type STa; 18 amino acid long for porcine-type STa) has been considered unsafe and unsuitable as a vaccine component (20). Nontoxic STa peptides would be safe as vaccine antigens, but they were found unable to induce neutralizing anti-STa antibodies (20). Thus, it has been suggested that STa toxicity and antigenicity are associated and that only toxic STa antigens are able to induce neutralizing antibodies (20).
Data from recent studies, however, indicated that nontoxic STa antigens can induce neutralizing antibodies against STa toxin. Full-length STa, of human-type or porcine-type, were shown to be less toxic or nontoxic after a single amino acid was substituted, and they became immunogenic and elicited neutralizing anti-STa antibodies after being genetically fused to a nontoxic monomeric LT (1A:1B; not 1A:5B holotoxin structured protein) peptide (21, 22). It was also suggested that STa mutated at different amino acid residues or at the same residue but with different replacement amino acids differed not only in toxicity reduction and antigenic structure but also likely in stimulation the of anti-STa antibody response when fused to an LT toxoid peptide (23). More recently, a study indicated that additional copies of an STa toxoid carried by an LT-STa toxoid fusion additively enhanced induction of anti-STa antibody response (24).
To further characterize STa toxoids in the stimulation of neutralizing anti-STa antibody response and to examine the potential application of STa toxoids in antitoxin vaccine development, we first constructed a mini-STa toxoid library and selected a panel of STa toxoids based on their toxicity, anti-STa antigenic propensity, and biochemical properties. Each STa toxoid was then genetically fused to a monomeric double mutant LT (dmLT; LTR192G/L211A) peptide to create different STa-toxoid-dmLT toxoid fusions. These STa-toxoid-dmLT toxoid fusions were used in mouse immunization and were examined for the induction of neutralizing anti-STa antibody responses. To further enhance the stimulation of anti-STa antibody response, we genetically fused three copies of each STa toxoid to one monomeric dmLT peptide in this study.
MATERIALS AND METHODS
Bacterial strains and plasmids.
The E. coli strains and plasmids used in the present study are listed in Table 1. Toxoid fusion recombinant strains 8751, 8752, and 8753, which were constructed previously (22), were used as templates to construct STa-toxoid-dmLT toxoid fusions. Vector pUC19 (Promega, Madison, WI) was used to clone STa toxoids, and vector pET28α (Novagen, Madison, WI) was used to clone and express toxoid fusion genes. E. coli BL21 (GE Healthcare, Piscataway, NJ) was used as the host strain. Recombinant E. coli strains were cultured in Luria broth supplemented with ampicillin (100 μg/ml) or kanamycin (30 μg/ml).
TABLE 1.
E. coli strains and plasmids used in this study
| Strain | Relevant properties | Plasmid | Source or reference |
|---|---|---|---|
| BL21 | F− ompT hsdS (rB− mB−), gal dcm | GE Healthcare | |
| 8751 | Fusion 2b construct, BL21/pfusion-2b | LT192-L-STa13/pET28α | 22 |
| 8752 | Fusion 3b construct, BL21/pfusion-3b | STa13-gly-pro-LT192/pET28α | 22 |
| 8753 | Fusion 4b construct, BL21/pfusion-4b | LT192A1-gly-pro-STa13-LTA2-B/pET28α | 22 |
| 9064 | STa recombinant strain, BL21 | p8835, STa in pUC19 | This study |
| 9355 | 3×STaS3F-dmLT strain, BL21 | p3×STaS3F-dmLT in pET28α | This study |
| 9330 | 3×STaE8Q-dmLT strain, BL21 | p3×STaE8Q-dmLT in pET28α | This study |
| 9357 | 3×STaL9A-dmLT strain, BL21 | p3×STaL9A-dmLT in pET28α | This study |
| 9359 | 3×STaN12I-dmLT strain, BL21 | p3×STaN12I-dmLT in pET28α | This study |
| 9331 | 3×STaN12S-dmLT strain, BL21 | p3×STaN12S-dmLT in pET28α | This study |
| 9361 | 3×STaN12T-dmLT strain, BL21 | p3×STaN12T-dmLT in pET28α | This study |
| 9332 | 3×STaP13A-dmLT strain, BL21 | p3×STaP13A-dmLT in pET28α | This study |
| 9333 | 3×STaP13F-dmLT strain, BL21 | p3×STaP13F-dmLT in pET28α | This study |
| 9363 | 3×STaA14H-dmLT strain, BL21 | p3×STaA14H-dmLT in pET28α | This study |
| 9334 | 3×STaA14Q-dmLT strain, BL21 | p3×STaA14Q-dmLT in pET28α | This study |
| 9335 | 3×STaA14T-dmLT strain, BL21 | p3×STaA14T-dmLT in pET28α | This study |
| 9336 | 3×STaA14V-dmLT strain, BL21 | p3×STaA14V-dmLT in pET28α | This study |
| 9337 | 3×STaT16K-dmLT strain, BL21 | p3×STaT16K-dmLT in pET28α | This study |
| 9374 | 3×STaT16 M-dmLT strain, BL21 | p3×STaT16 M-dmLT in pET28α | This study |
| 9353 | 3×STa-dmLT strain, BL21 | p3×STa-dmLT in pET28α | This study |
| 8955 | Negative control, BL21/pET28α | pET28α | 23 |
Cloning and mutation of STa gene estA.
The human-type STa gene (estA) encoding hSTa toxin was isolated from E. coli H10407 genomic DNA and was cloned into vector pUC19 as described previously (23). Briefly, the estA gene was PCR amplified with the primers hSTapUCHindIII-F (5′-GCGCAAAGCTTCTGATTTTGATAGCCACGGCGGATCCAAATATAAAGGG-3′; the BamHI restriction site is underlined). PCR products were purified using gel electrophoresis, digested with HindIII and BamHI restriction enzymes (New England BioLabs, Ipswich, MA), and ligated into vector pUC19. After the verification of STa protein expression and enterotoxicity, the cloned native STa gene had residue 3, 4, 8, 9, 12, 13, 14, 16, or 17 mutated (but not residues 6, 7, 10, 11, 15, and 18, which are involved in disulfide bond formation). Replacement amino acid residues were selected, respectively, based on biochemistry properties, such as molecule weight, accessible surface area, and hydrophobicity or hydrophilicity.
Splice overlapping extensions (SOE) PCRs with specifically designed PCR primers were used to mutate the estA gene as described previously (23). Mutated genes were verified initially by DNA sequencing. After sequencing verification, these STa mutated genes were examined for protein expression by measuring reactivity to anti-STa antibodies using STa competitive ELISA and for toxicity by examining stimulation of cyclic GMP levels in T-84 cells using an EIA cGMP enzyme-linked immunosorbent assay (ELISA; Assay Design, Ann Arbor, MI) as described previously (21).
STa-toxoid-dmLT toxoid fusion construction.
A group of STa toxoids that had their toxicity reduced or eliminated but retained reactivity to polyclonal antibodies derived against native STa were selected to be genetically fused to a monomeric dmLT peptide. In contrast to the 1A:5B holotoxin-structured dmLT (LTR192G/L211A) (25), this monomeric dmLT molecule is a single peptide that consisted of only one copy of the A subunit and one copy of the B subunit. It was derived by modifying the native LT eltAB genes with disruption of the cistron gene structure (between the eltA gene and eltB gene) and the removal of nucleotides coding the B subunit signal leading peptide.
Three copies of each STa toxoid were genetically fused to a monomeric dmLT at its N terminus, at its C terminus, and between the A1 segment and the A2 segment, respectively, using an SOE PCR described previously (24). However, instead of using a monomeric tmLT (LTS63K/R192G/L211A) peptide (24), we used a monomeric dmLT (LTR192G/L211A) peptide as the carrier protein. Briefly, three nucleotide segments, which were generated from three separate SOE PCRs, were fused together to form a single open reading frame coding an STa-toxoid-dmLT toxoid fusion (Fig. 1A). The first nucleotide segment was produced by overlapping two regular PCR products: one was amplified with the primers T7-F (5′-TAATACGACTCACTATAGGG-′3) and STa-toxoid-R, and the other was amplified with the primers STa-toxoid-F and LT73-R (5′-AGACTGTCCTGCTAAGTGAGCACT-′3). Since plasmid p8752 (STa13-gly-pro-LT192/pET28α) was used as the DNA template, the first segment consisted of nucleotides coding the first copy of a STa toxoid (at the N terminus of the fusion) and the first 75 amino acids of LT (N terminus). The second nucleotide segment was generated by connecting of another set of PCR products: one was amplified with the primers LT73-F (5′-).
FIG 1.
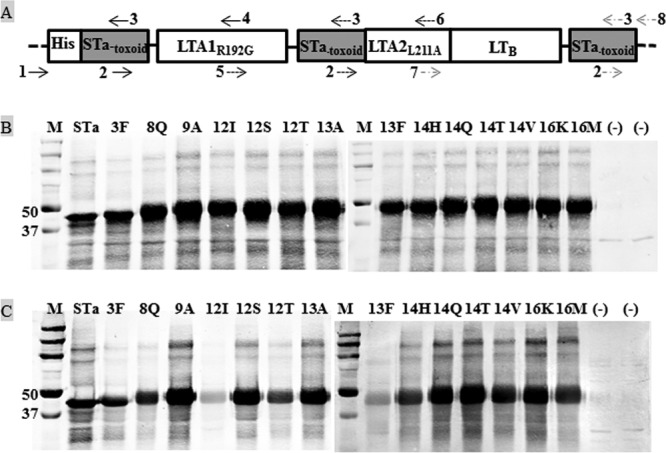
Construction and detection of STa-toxoid-dmLT toxoid fusions. (A) Illustration of an STa-toxoid-dmLT toxoid fusion gene. Three copies of each STa-toxoid gene were genetically fused to the 5′ end, between A1 and A2 of LTA, and the 3′ end of a monomeric dmLT peptide gene (LTR192G/L211A; as a single open reading frame) using splicing overlap extension PCRs. Primers: 1, T7-F; 2, STa-toxoid-F; 3, STa-toxoid-R; 4, LT73-R; 5, LT73-F; 6, LT211-R; 7, LT211-F; 8, T7-R. Primers 2 and 3 were used to mutate the STa gene for different STa toxoids (Table 2). Primers 1 and 3 and primers 2 and 4 were, respectively, used in two PCRs (which generated two nucleotide fragments to be fused) to produce the first segment of the fusion gene, which included the first copy of a STa toxoid and the first 75 amino acids of the LT A subunit (at the N terminus). Primers 5 and 3 and primers 2 and 6 were used in two other PCRs (for two other fragments to be fused) to generate the second segment of the fusion gene, which consisted of the second copy of a STa toxoid and amino acids 68 to 216 of the LT A subunit. Primers 7 and 3 and primers 2 and 8 were used in another two PCRs (for another two fragments to be fused) to create the third segment of the fusion gene, which contains the third copy of a STa toxoid, amino acids 204 to 240 of the LT A subunit and the LT B subunit (1 to 100 amino acids). These three segments were connected in an SOE PCR for a single open reading frame coding a STa-toxoid-dmLT toxoid fusion protein. (B) Western blot to detect each STa-toxoid-dmLT fusion protein with anti-CT antibodies. A fusion protein (100 ng) separated in 10 to 12% PAGE gel was detected using rabbit anti-CT antiserum (1:3,300; Sigma) and IRDye-labeled goat anti-rabbit IgG (1:5,000; LI-COR). (C) Western blot to detect toxoid fusion proteins with anti-STa antibodies. Fusion proteins (100 ng each) were detected with protein A-purified rabbit anti-STa antiserum (1:5,000) and IRDye-labeled goat anti-rabbit IgG (1:5,000; LI-COR). The total protein extracted from host strain 8955 was used as the negative control (−). Lane M is the protein marker (in kilodaltons; Precision Plus Protein Pre-stained standards; Bio-Rad).
TABLE 2.
STa-toxoid forward and reverse (STa-toxoid-F, STa-toxoid-R) PCR primers designed to mutate the STa gene (estA) for STa toxoids which were fused to a modified LT toxoid gene for STa-toxoid-dmLT toxoid fusionsa
| STa toxoid | Primer | Nucleotide sequence (5′-3′) |
|---|---|---|
| 3F | hSTaS3F-F | ATG AAT AGT TTC AAT TAC TGC TGT |
| hSTaS3F-R | ACA GCA GTA ATT GAA ACT ATT CAT | |
| 8Q | hSTaE8Q-F | AAT TAC TGC TGT CAA TTG TGT TGT |
| hSTaE8Q-R | ACA ACA CAA TTG ACA GCA GTA ATT | |
| 9A | hSTaL9A-F | TAC TGC TGT GAA GCA TGT TGT AAT |
| hSTaL9A-R | TGG ATT ACA ACA TGC TTC ACA GCA | |
| 12I | hSTaN12I-F | GAA TTG TGT TGT ATC CCT GCT TGT |
| hSTaN12I-R | ACA AGC AGG GAT ACA ACA CAA TTC | |
| 12S | hSTaN12S-F | GAA TTG TGT TGT AGC CCT GCT TGT |
| hSTaN12S-R | ACA AGC AGG GCT ACA ACA CAA TTC | |
| 12T | hSTaN12T-F | GAA TTG TGT TGT ACC CCT GCT TGT |
| hSTaN12T-R | ACA AGC AGG GGT ACA ACA CAA TTC | |
| 13A | hSTaP13A-F | TTG TGT TGT AAT GCA GCT TGT ACC |
| hSTaP13A-R | GGT ACA AGC TGC ATT ACA ACA CAA | |
| 13F | hSTaP13F-F | TTG TGT TGT AAT TTT GCT TGT ACC |
| hSTaP13F-R | GGT ACA AGC AAA ATT ACA ACA CAA | |
| 14H1 | hSTaA14H-F | TGT TGT AAT CCT CAT TGT ACC GGG |
| hSTaA14H-R | CCC GGT ACA ATG AGG ATT ACA ACA | |
| 14Q | hSTaA14Q-F | TGT TGT AAT CCT CAG TGT ACC GGG |
| hSTaA14Q-R | CCC GGT ACA CTG AGG ATT ACA ACA | |
| 14T | hSTaA14T-F | TGT TGT AAT CCT ACA TGT ACC GGG |
| hSTaA14T-R | CCC GGT ACA TGT AGG ATT ACA ACA | |
| 14V | hSTaA14V-F | TGT TGT AAT CCT GTA TGT ACC GGG |
| hSTaA14V-R | CCC GGT ACA TAC AGG ATT ACA ACA | |
| 16K | hSTaT16K-F | AAT CCT GCT TGT AAG GGG TGC TAT |
| hSTaT16K-R | ATA GCA CCC CTT ACA AGC AGG ATT | |
| 16M | hSTaT16 M-F | AAT CCT GCT TGT ATG GGG TGC TAT |
| hSTaT16 M-R | ATA GCA CCC CAT ACA AGC AGG ATT |
Nucleotides that are underlined indicate mutations.
Each STa-toxoid-dmLT fusion gene was further amplified with the PCR primers T7-F and T7-R and purified by using gel electrophoresis. Purified PCR products were digested with NheI and EagI restriction enzymes (BioLab) and cloned into expression vector pET28α (Novagen). Toxoid fusions were expressed as 6×His-tagged proteins in E. coli BL21 by following standard protocols (26). In addition, the native STa gene was fused to the modified dmLT gene for fusion STa-dmLT.
Expression and detection of toxoid fusion proteins.
The expression of each toxoid fusion protein in E. coli BL21 was examined in a standard sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) as described previously (24). Briefly, each toxoid fusion recombinant strain was cultured at 37°C in 500 ml of Luria-Bertani medium supplemented with kanamycin (30 μg/ml). Overnight-grown bacteria were induced with isopropyl-1-thio-β-d-galactoside (IPTG; 100 μM) for 4 h after the culture optical density (OD) reached 0.5. Bacterial culture was centrifuged at 5,000 × g for 20 min, and pellets were suspended with 5 ml of B-PER (bacterial protein extraction reagent in phosphate buffer; Pierce, Rockford, IL) for total protein extraction (with a majority of proteins as inclusion bodies; in denaturing buffer). Recombinant 6×His-tagged toxoid fusion proteins were further extracted from total insoluble proteins to a purity of >90% with Ni-nitrilotriacetic acid agarose according to the manufacturer's protocol (Qiagen, Valencia, CA). Extracted 6×His-tagged proteins were refolded using a Novagen protein refolding kit according to the manufacturer's protocol (Novagen).
A 100-ng portion of refolded 6×His-tagged protein from each toxoid fusion strain was analyzed using 10 to 12% SDS-PAGE gels and an immunoblot assay. Rabbit anti-CT (1:3,300; Sigma) and protein A-purified anti-STa antisera (1:5,000; Robertson Laboratory) were used as primary antibodies, respectively. IRDye-labeled goat anti-rabbit IgG (1:5,000; LI-COR, Lincoln, NE) were used as the secondary antibody to detect each fusion protein with a LI-COR Odyssey premium infrared gel imaging system.
In addition, the expression of each toxoid fusion protein was examined in an enzyme-linked immunosorbent assay (ELISA). One hundred nanograms of each toxoid fusion protein was coated to each well of an Immulon 2HB plate (Thermo Scientific, Rochester, NY). Rabbit anti-CT (1:3,000) and anti-STa (1:3,000) sera were used as primary antibodies, and horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (1:3,000; Sigma) was used as the secondary antibody. OD values were measured with a plate reader at a 405-nm wavelength, after 25 min of incubation with 100 μl of 3,3′,5,5′-tetramethylbenzidine (TMB; Microwell Peroxidase Substrate System [2-C]; KPL, Gaithersburg, MD).
Toxicity detection of toxoid fusion proteins.
The in vitro enterotoxic activity of each toxoid fusion was determined by measuring stimulation of intracellular cyclic AMP (cAMP) and cGMP in T-84 cells with EIA cAMP and cGMP ELISA kits (Assay Designs). Toxoid fusions would stimulate an increase of intracellular cAMP and cGMP level if they retain LT and STa toxicity. As described previously (21, 22, 24), 100 ng of each toxoid fusion protein (in 150 μl of phosphate-buffered saline [PBS]), 10 ng of cholera toxin (CT [Sigma]; in 150 μl of PBS, as an LT positive control, which is highly homologous to LT structurally and functionally), or 2 ng of purified STa toxin (from the Clements laboratory, in 150 μl of PBS, as an STa positive control) was added to each microplate well (in duplicate) that was seeded with 105 T-84 cells. After incubation in a CO2 incubator for 1 h (for cGMP) or 3 h (for cAMP), T-84 cells were gently washed with 1 ml of PBS (2×) and then lysed. Cell lysates were collected and measured for intracellular cAMP or cGMP levels (pmol/ml) using EIA ELISAs according to the manufacturer's protocol and with Assay Blaster software (Enz Life Sciences, Farmingdale, NY).
Mouse immunization with each toxoid fusion.
Five 6- to 8-week-old female adult BALB/c mice (Charles River Laboratories International, Inc., Wilmington, MA) as a group were immunized intraperitoneally with each refolded toxoid fusion protein. Each mouse was primarily injected with 200 μg of refolded fusion protein (in 200 μl) and 200 μl of Freund complete adjuvant (Sigma). Five mice in the control group were each intraperitoneally injected with 200 μl of Freund complete adjuvant and 200 μl of protein buffer (0.02 M Tris-HCl). Two booster injections at the same dose as the primary injection but with Freund incomplete adjuvant followed at biweekly intervals. Serum and fecal suspension samples (1 g of feces was suspended in 5 ml of fecal reconstitution buffer [10 mM Tris, 100 mM NaCl, 0.05% Tween 20, 5 mM sodium azide, pH 7.4; 1:6 dilution]) (22, 27) were collected from each mouse prior to immunization and 10 to 12 days after each immunization and were then stored at −80°C until use. On day 37, mice were anesthetized with CO2 and exsanguinated. Animal studies complied with the Animal Welfare Act according to National Research Council guidelines (28) and were approved and supervised by state veterinarians and university Institutional Animal Care and Use Committees.
Anti-LT and anti-STa antibody titration.
Anti-LT and anti-STa IgG antibodies in serum and IgA antibodies in fecal suspension samples of each mouse were examined in ELISAs as described previously (21, 22, 24). Briefly, 100 ng of CT (an LT homologue which has been commonly used as a coating antigen to titrate anti-LT antibodies) in 100 μl of antigen coating buffer (15 mM Na2CO3, 35 mM NaHCO3; pH 9.6) was used to coat each well of an Immulon 2HB plate to titrate anti-LT IgG and IgA antibodies, whereas 10 ng of STa-ovalbumin conjugates in 100 μl of STa ELISA buffer (29) was coated at each well of a Costar plate (Corning, Inc., Corning, NY) to titrate anti-STa IgG and IgA antibodies. After incubation at 37°C for 1 h and then at 4°C overnight, the plates were washed with PBST (PBS plus 0.05% Tween 20) and blocked with 5% (for anti-STa) or 10% (for anti-LT) nonfat milk-PBST (150 μl per well) at 37°C for 1 h. After washes with PBST (3× for anti-STa and 5× for anti-LT), wells were incubated with 2-fold serial dilutions of serum samples (100 μl; at an initial dilution of 1:200 in 5% milk-PBST for anti-LT or of 1:25 dilution in 1% milk-PBST for anti-STa) or fecal suspension samples (1:20 dilution for anti-LT or at 1:15 to 1:20 for anti-STa in 5% milk-PBST) for 1 h at 37°C. All samples were examined in triplicate. Wells were washed (five times with PBST) and incubated with HRP-conjugated goat anti-mouse IgG (a 1:5,000 dilution for anti-LT or a 1:3,000 dilution for anti-STa) or IgA (1:1,000 dilution; Sigma) at 100 μl per well for 1 h at 37°C. The plates were washed again (three times with PBST, followed by two times with PBS) and incubated with a TMB microwell peroxidase substrate system (2-C; KPL) at 100 μl per well for 20 min at room temperature. The OD was measured with a plate reader at a 405-nm wavelength. Antibody titers were calculated as the highest dilution that produced OD readings of >0.3 above the background readings and were displayed in a log10 scale as previously described (21, 22, 24).
Anti-LT and anti-STa antibody neutralization assays.
Serum and fecal suspension samples pooled from mice in each group were examined for in vitro antibody neutralization activities against STa and CT toxins by using EIA cAMP and cGMP kits (Assay Design) and T-84 cells. CT and STa toxins stimulate increases in cAMP and cGMP, respectively, in T-84 cells. Neutralizing antibodies would neutralize toxins and prevent CT or STa from stimulating an increase of intracellular cAMP or cGMP level in cells; therefore, neutralizing activity against CT and STa from antibodies in serum and fecal suspension samples of the immunized mice can be measured. As described previously (21, 24), serum (30 μl of serum pooled from each group, 6 μl per mouse; a 1:33.3 dilution in a final volume of 1 ml) or fecal (30 μl, 1:6 diluted fecal suspension, a 1:200 final dilution) samples were incubated with 2 ng of STa toxin or 10 ng of CT (diluted in 150 μl of Dulbecco modified Eagle medium [DMEM]/F-12 medium) for 1 h at room temperature. Incubated solution was brought up to 300 μl with DMEM/F-12 medium and transferred to T-84 cells (105 cells in 700 μl of culture medium), which was pretreated with 1 mM IBMX (3-isobutyl-1-methylxanthine; Sigma). After incubation of 1 h (STa in cGMP) to 3 h (CT in cAMP) at 37°C in a CO2 incubator, the cells were washed and lysed. Cell lysates were collected and measured for intracellular cAMP or cGMP levels (pmol/ml) using cAMP and cGMP ELISA kits, respectively. CT or STa, alone or mixed with a serum or fecal suspension of the control group to show enterotoxicity in the stimulation of cAMP or cGMP, and cell culture medium (without toxin or antibodies) to show a baseline of ELISA were used as controls.
Statistical analysis.
Data were analyzed by using the SAS for Windows, version 8 (SAS Institute, Cary, NC). The results are presented as means ± the standard deviations. A Student t test was used to compare the different treatment groups. Calculated P values of <0.05 were regarded as significant when treatments were compared using two-tailed distribution and two-sample equal or unequal variance.
RESULTS
Fourteen STa toxoids were identified for toxoid fusion construction.
By substituting amino acid 3 (S), 4 (N), 8 (E), 9 (L), 12 (N), 13 (P), 14 (A), 16 (T), or 17 (G) of STa with different residues, we generated 47 STa toxoid candidates for a mini-STa toxoid library. These STa toxoid candidates were examined for toxicity (stimulation of the cGMP level) by using a cGMP EIA kit with T-84 cells and were assessed for antigenic structure (reactivity to anti-STa antibodies) by using an STa competitive ELISA with antiserum against native STa. The data showed that these STa derivatives differed from each other in toxicity reduction and also activity, reacting to anti-STa antibodies. Some mutants, such as STaN4Y and STaT16S, were as toxic as the native STa in the stimulation of intracellular cGMP in T-84 cells. In contrast, other mutants, for example, STaA14Q, STaA14L, STaN12K, and STaE8Q, exhibited no or little activity in stimulating cGMP levels in T-84 cells. The data from the STa competitive ELISA showed that some mutants, such as STaT16R, STaT16M, STaT16Q, and STaA14T, retained a similar level of activity reacting to anti-STa antiserum as the native STa. However, others, including STaE8G, STaL9K, STaL9Q, and STaN12K, showed no or low reactivity to anti-STa antiserum.
Fourteen STa mutants—STaS3F, STaE8Q, STaL9A, STaN12I, STaN12S, STaN12T, STaP13A, STaP13F, STaA14H, STaA14Q, STaA14T, STaA14V, STaT16K, and STaT16M—that showed no or little enterotoxicity but a level of reactivity similar to anti-STa antiserum as the native STa were selected for toxoid fusion construction. Three copies of each of these 14 STa toxoids were fused to a monomeric dmLT peptide. DNA sequencing verified that each toxoid fusion carried three copies of a STa toxoid gene and one copy of a monomeric dmLT gene and formed a single open reading frame encoding a single peptide (Fig. 1A). In addition, a fusion with three copies of the native STa and one copy of the monomeric dmLT was also constructed and was verified from DNA sequencing.
Each toxoid fusion protein was expressed and showed no enterotoxicity.
Fourteen STa-toxoid-dmLT toxoid fusions and the native STa fusion protein 3×STa-dmLT were examined in Western blot with anti-CT and anti-STa antiserum. The detectable proteins from all fusion strains had a molecular mass about 48 kDa, the expected size of all fusion proteins (Fig. 1B and C). In addition, the ELISA data indicated that all fusion proteins were recognized by the anti-LT and anti-STa antiserum. This finding confirmed that toxoid fusion proteins were expressed by each recombinant fusion strain (Table 3).
TABLE 3.
ELISA OD values obtained with anti-CT and anti-STa antisera to verify the expression of each STa-toxoid-dmLT fusion protein
| Fusion proteina | Mean OD ± SD |
|---|---|
| Anti-CTb | |
| STa | 1.05 ± 0.03 |
| 3F | 1.4 ± 0.02 |
| 8Q | 1.32 ± 0.0 |
| 9A | 1.5 ± 0.03 |
| 12I | 1.5 ± 0.07 |
| 12S | 1.4 ± 0.03 |
| 12T | 1.36 ± 0.03 |
| 13A | 1.41 ± 0.01 |
| 13F | 1.49 ± 0.07 |
| 14H | 1.34 ± 0.03 |
| 14Q | 1.28 ± 0.02 |
| 14T | 1.32 ± 0.04 |
| 14V | 1.45 ± 0.07 |
| 16K | 1.36 ± 0.06 |
| 16M | 1.29 ± 0.02 |
| (–) | 0.11 ± 0.05 |
| Anti-STac | |
| STa | 1.95 ± 0.07 |
| 3F | 1.94 ± 0.02 |
| 8Q | 1.69 ± 0.0 |
| 9A | 1.97 ± 0.03 |
| 12I | 1.84 ± 0.05 |
| 12S | 1.95 ± 0.04 |
| 12T | 1.71 ± 0.0 |
| 13A | 1.86 ± 0.05 |
| 13F | 1.71 ± 0.06 |
| 14H | 1.93 ± 0.07 |
| 14Q | 1.89 ± 0.03 |
| 14T | 1.91 ± 0.05 |
| 14V | 1.89 ± 0.02 |
| 16K | 2.1 ± 0.01 |
| 16M | 2.04 ± 0.01 |
| (–) | 0.34 ± 0.11 |
Fusion proteins: STa, fusion 3×STa-dmLT; 3F, 3×STaS3F-dmLT; 8Q, 3×STaE8Q-dmLT; 9A, 3×STaL9A-dmLT; 12I, 3×STaN12I-dmLT; 12S, 3×STaN12S-dmLT; 12T, 3×STaN12T-dmLT; 13A, 3×STaP13A-dmLT; 13F, 3×STaP13F-dmLT; 14H, 3×STaA14H-dmLT; 14Q, 3×STaA14Q-dmLT; 14T, 3×STaA14T-dmLT; 14V, 3×STaA14V-dmLT; 16K, 3×STaT16K-dmLT; 16M, 3×STaT16 M-dmLT. Each purified fusion protein (100 ng per well) was used to coat ELISA 2HB plate (in triplicate). PBS was used as the negative control (–).
Rabbit anti-CT (1:3,000; Sigma) and HRP-conjugated goat-anti-rabbit IgG (1:3,000; Sigma) were used as the primary and secondary antibodies to detect the LT peptide of each fusion.
Protein A-purified rabbit anti-STa serum (1:3,000; Robertson laboratory) and HRP-conjugated goat anti-rabbit IgG (1:3,000) were used to detect the STa toxoid of each fusion protein.
In vitro toxicity assays showed that enterotoxicity was not detected with any of the fusion proteins (including the native STa fusion). T-84 cells incubated with 100 ng of each fusion protein showed no increase in intracellular cAMP levels (an average of 0.96 pmol/ml) or cGMP levels (an average of 8.31 pmol/ml). These levels are similar to those observed in cells incubated with the negative controls: cell culture medium (0.98 pmol/ml of cAMP, 7.8 pmol/ml of cGMP) and protein buffer (1.06 pmol/ml of cAMP, 6.8 pmol/ml of cGMP). In contrast, T-84 cells incubated with 2 ng of STa showed a significant increase in cGMP (72 ± 2.83 pmol/ml; P < 0.01) and showed a significant increase in cAMP when incubated with 10 ng of CT (>200 pmol/ml; P < 0.01) (Table 4).
TABLE 4.
Detection of in vitro enterotoxic activity of each fusion protein, measured by the stimulation of intracellular cAMP and cGMP (pmol/ml) in T-84 cells with EIA cAMP and cGMP ELISA kits (Assay Designs)
| Fusion proteina | Mean pmol of cAMP or cGMP/ml ± SD |
|---|---|
| cAMPb | |
| STa | 5.4 ± 0.56 |
| 3F | 5.2 ± 0.07 |
| 8Q | 10 ± 0.7 |
| 9A | 9.2 ± 0.85 |
| 12I | 10.5 ± 1.56 |
| 12S | 10.4 ± 0.84 |
| 12T | 6.4 ± 0.57 |
| 13A | 6.8 ± 0.28 |
| 13F | 8.6 ± 1.41 |
| 14H | 11 ± 0.28 |
| 14Q | 8.4 ± 3.39 |
| 14T | 6.8 ± 1.13 |
| 14V | 6.2 ± 0.85 |
| 16K | 9.3 ± 1.56 |
| 16M | 10.4 ± 0.57 |
| CT(+) | >200 |
| Medium (–) | 7.8 ± 0.99 |
| Buffer (–) | 6.8 ± 1.41 |
| cGMPc | |
| STa | 1.05 ± 0.01 |
| 3F | 1.26 ± 0.34 |
| 8Q | 1.10 ± 0.14 |
| 9A | 1.0 ± 0.56 |
| 12I | 1.05 ± 0.07 |
| 12S | 0.84 ± 0.05 |
| 12T | 1.2 ± 0.14 |
| 13A | 1.05 ± 0.07 |
| 13F | 0.62 ± 0.59 |
| 14H | 0.88 ± 0.04 |
| 14Q | 0.96 ± 0.13 |
| 14T | 1.05 ± 0.14 |
| 14V | 0.81 ± 0.30 |
| 16K | 0.88 ± 0 |
| 16M | 0.7 ± 0.42 |
| STa(+)d | 72 ± 2.83 |
| Medium (–) | 0.98 ± 0.25 |
| Buffer (–) | 1.06 ± 0.20 |
Fusion proteins are labeled as in Table 3. Each fusion protein (100 ng) was used to stimulate cAMP or cGMP in T-84 cells for toxicity detection (in duplicate). The intracellular cAMP and cGMP levels serve as indicators of LT and STa enterotoxicity, respectively.
Intracellular cAMP levels in T-84 cells were used to measure LT enterotoxicity. A total of 10 ng of CT (cholera toxin) was used as the positive (toxic) control; cell culture medium and protein buffer were used as the negative (nontoxic) controls.
Intracellular cGMP levels in T-84 cells were used to measure STa enterotoxicity. A total of 10 ng of STa was used as the positive (toxic) control; cell culture medium and protein buffer were used as the negative (nontoxic) control.
Purified STa toxin was used as the positive control.
All toxoid fusions elicited anti-LT and anti-STa antibodies, but anti-STa antibodies varied among and within immunized groups.
Among immunized mice, anti-LT and anti-STa IgG antibodies were detected in serum, and IgA antibodies were detected in fecal suspension samples. Anti-LT IgG antibodies in serum samples were detected at an average titer of 3.17 ± 0.10 (in log10) among all immunized groups, but not in the control mice (Table 5). Anti-LT IgG antibody titers in mice immunized with the toxoid fusion were not significantly different than titers in mice immunized with 3×STa-dmLT fusion (P > 0.05) (Table 5). In contrast, anti-STa IgG antibody titers in the serum samples varied greatly among groups immunized with different fusion antigens, ranging from 0.73 ± 1.03 to 3.43 ± 0.29 (Fig. 2). Compared to the unimmunized control group, all immunization groups except those immunized with 3×STa-dmLT, 3×STaS3F-dmLT, 3×STaP13F-dmLT, 3×STaA14T-dmLT, and 3×STaA14V-dmLT were significantly different in detection of anti-STa IgG antibodies in serum samples. However, compared to the anti-STa IgG titer in mice immunized with 3×STa-dmLT, only titers in serum samples of the mice immunized with 3×STaN12S-dmLT, 3×STaA14H-dmLT, 3×STaN12T-dmLT, and 3×STaN12I-dmLT were significantly different (P < 0.05) (Fig. 2). Among them, mice immunized with 3×STaN12S-dmLT, 3×STaA14H-dmLT, and 3×STaN12T-dmLT had the greatest anti-STa serum IgG titers detected (Fig. 2). The differences among these three groups, however, were not significant (P = 0.78 for 3×STaN12S-dmLT versus 3×STaN12T-dmLT; P = 0.24 for 3×STaN12S-dmLT versus 3×STaA14H-dmLT; P = 0.44 for 3×STaN12T-dmLT versus 3×STaA14H-dmLT).
TABLE 5.
Mouse serum anti-LT IgG antibody titers
| Mouse immunization group (fusion antigen)a | Mean IgG titer (log10) ± SDb | Pc |
|---|---|---|
| STa | 3.15 ± 0.05 | |
| 3F | 3.13 ± 0.09 | 0.58 |
| 8Q | 3.16 ± 0.14 | 0.84 |
| 9A | 3.17 ± 0.14 | 0.74 |
| 12I | 3.15 ± 0.10 | 0.99 |
| 12S | 3.17 ± 0.11 | 0.71 |
| 12T | 3.27 ± 0.09 | 0.10 |
| 13A | 3.21 ± 0.08 | 0.15 |
| 13F | 3.15 ± 0.06 | 0.92 |
| 14H | 3.18 ± 0.11 | 0.69 |
| 14Q | 3.22 ± 0.07 | 0.14 |
| 14T | 3.15 ± 0.11 | 0.96 |
| 14V | 3.16 ± 0.22 | 0.14 |
| 16K | 3.24 ± 0.06 | 0.08 |
| 16M | 3.15 ± 0.12 | 0.99 |
| Control | 0 ± 0 | <0.001 |
Groups of mice (n = 5 animals per group) immunized with different fusion antigens: STa, mice immunized with fusion 3×STa-dmLT; 3F, 3×STaS3F-dmLT; 8Q, 3×STaE8Q-dmLT; 9A, 3×STaL9A-dmLT; 12I, 3×STaN12I-dmLT; 12S, 3×STaN12S-dmLT; 12T, 3×STaN12T-dmLT; 13A, 3×STaP13A-dmLT; 13F, 3×STaP13F-dmLT; 14H, 3×STaA14H-dmLT; 14Q, 3×STaA14Q-dmLT; 14T, 3×STaA14T-dmLT; 14V, 3×STaA14V-dmLT; 16K, 3×STaT16K-dmLT; 16M, 3×STaT16M-dmLT; and control, mice injected with protein buffer and Freund adjuvant. Anti-LT IgG in serum sample of each immunized or control mouse was titrated by ELISA using CT (100 ng per well of H2B plates) as the coating agent and HRP-conjugated goat-anti-mouse IgG (1:5,000) as the secondary antibodies.
Means and standard deviations of the anti-LT IgG titers in each immunization group and the control group are shown. Antibody titers were calculated from the highest dilution of each serum sample that produced an ELISA OD of 0.3 above the background and are presented in log10 scale.
P values were calculated by using a Student t test comparing mouse antibody titers in each group to titers in the STa group, mice immunized with fusion 3×STa-dmLT, which was used as a reference for toxoid fusions.
FIG 2.
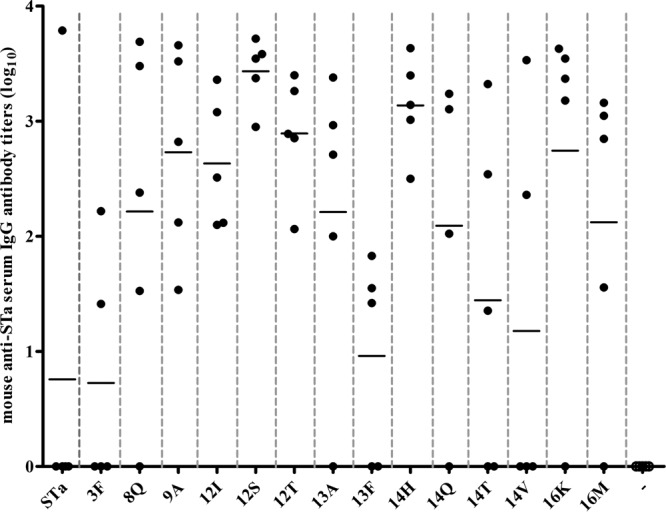
Mouse serum anti-STa IgG antibody titers. Anti-STa IgG antibodies in serum samples of the immunization and control groups (five mice per group) were titrated in ELISA using STa-ovalbumin conjugates (10 ng per well of Costar plates; coating agent) and HRP-conjugated goat anti-mouse IgG (1:3,000; the secondary antibodies). The anti-STa antibody titer was calculated from the highest dilution of a serum sample that produced an ELISA OD of >0.3 (above the background). Each dot represented a mouse IgG titer, and the bar indicated the mean titer of the group. Mouse group labels (on the x axis) indicated the immunization groups (immunized with each different fusion antigen) and the control group (−). The mean antibody titer (mean ± the standard deviation) and P value (compared to titers of mice in the group immunized with fusion 3×STa-dmLT) were calculated for the groups immunized with different fusions—STa, 3×STa-dmLT (0.76 ± 1.69); 3F, 3×STaS3F-dmLT (0.73 ± 1.03, P = 0.97); 8Q, 3×STaE8Q-dmLT (2.22 ± 1.52, P = 0.19); 9A, 3×STaL9A-dmLT (2.73 ± 0.91, P = 0.051); 12I, 3×STaN12I-dmLT (2.63 ± 0.57, P = 0.047); 12S, 3×STaN12S-dmLT (3.43 ± 0.29, P = 0.008); 12T, 3×STaN12T-dmLT (2.89 ± 0.52, P = 0.027); 13A, 3×STaP13A-dmLT (2.21 ± 1.46, P = 0.17); 13F, 3×STaP13F-dmLT (0.96 ± 0.89, P = 0.82); 14H, 3×STaA14H-dmLT (3.14 ± 0.43, P = 0.016); 14Q, 3×STaA14Q-dmLT (2.1 ± 1.46, P = 0.26); 14T, 3×STaA14T-dmLT (1.44 ± 1.49, P = 0.52); 14V, 3×STaA14V-dmLT (1.18 ± 1.67, P = 0.70); 16K, 3×STaT16K-dmLT (2.74 ± 1.54, P = 0.089); 16M, 3×STaT16M-dmLT (2.12 ± 1.35, P = 0.35)—and for the control group (0 ± 0, P = 0.37).
For every mouse in these three groups and in the groups immunized with 3×STaL9A-dmLT and 3×STaN12I-dmLT, an anti-STa IgG antibody response was detected. In contrast, mice in the remaining groups showed a wide range of intragroup variations upon the induction of an anti-STa antibody response (Fig. 2). For example, while for one mouse anti-STa IgG was detected at a titer as great as 3.8 log10, for the remaining four mice from the same group no anti-STa IgG was detected in their serum samples.
Anti-STa IgA antibody titers detected in fecal suspension samples varied among immunized groups as well. Greater anti-STa IgA antibody titers were detected in mice in groups immunized with 3×STaT16M-dmLT (1.51 ± 0.62), 3×STaL9A-dmLT (1.14 ± 0.82), and 3×STaN12S-dmLT (1.11 ± 0.66). Compared to anti-STa IgA detected in the fecal samples of mice immunized with 3×STa-dmLT (0.88 ± 0.74), however, the titers of these three groups were not significantly different (P = 0.19, 0.62, and 0.62). Indeed, compared to 3×STa-dmLT, none of the toxoid fusions, except 3×STaS3F-dmLT, which had no anti-STa IgA detected, showed a significant difference in the stimulation of anti-STa IgA antibodies (P > 0.05). However, compared to the IgA titer of the control group, anti-STa IgA titers in mice immunized with 3×STaT16M-dmLT (P = 0.006), 3×STaT16M-dmLT (P = 0.02), 3×STaA14Q-dmLT (P = 0.01), 3×STaA14H-dmLT (P = 0.048), 3×STaN12T-dmLT (P = 0.03), 3×STaN12S-dmLT (P = 0.02), and 3×STaL9A-dmLT (P = 0.02) were significantly different. Only the group immunized with the 3×STaN12S-dmLT or the 3×STaT16M-dmLT toxoid fusion displayed an anti-STa IgA antibody response that was detected in all five mice (Fig. 3). No anti-LT or anti-STa IgG or IgA antibodies were detected in serum or fecal suspension samples of the control mice.
FIG 3.
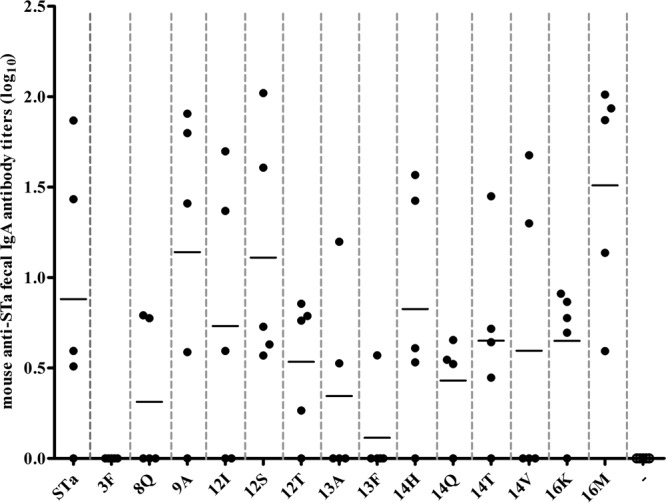
Mouse anti-STa IgA antibody titers, detected in fecal suspension samples of each immunized or control mouse. Anti-STa IgA in fecal suspension samples of the immunization groups and the control group (five mice per group) were titrated in ELISA with STa-ovalbumin conjugates (10 ng per well, Costar plates) as the coating agent and HRP-conjugated goat anti-mouse IgA (1:1,000) as secondary antibodies. The antibody titer was calculated from the highest dilution of a fecal suspension sample that produced an ELISA OD of >0.3 above the background, and results are presented in a log10 scale. Each dot represented a mouse IgA antibody titer, and the bar was the mean titer of the group. Groups (labeled on the x axis) were mouse groups immunized with each fusion antigen and the control group (“−” column), of which fecal suspension samples were titrated for anti-STa IgA antibodies.
Elicited anti-STa antibodies exhibit in vitro neutralizing activities against STa toxin.
In vitro antibody neutralization assays indicated that serum samples pooled from each immunized group showed neutralizing activity against STa toxin but with variations among groups. Serum samples pooled from the groups immunized with 3×STa12S-dmLT, 3×STa14H-dmLT, 3×STa16K-dmLT, and 3×STa-dmLT showed stronger neutralizing activities against STa toxins, since serum samples from these groups prevented STa toxin from stimulating cGMP levels in T-84 cells (Fig. 4). The intracellular cGMP levels in T-84 cells incubated with 2 ng STa toxin and the pooled serum samples of mice immunized with these four fusions were 1.65 ± 0.21, 4.65 ± 0.21, 5 ± 0.14, and 7.7 ± 0.42 pmol/ml, respectively. These cGMP levels differed significantly from the cGMP in cells incubated with STa and serum of control mice (62 ± 0 pmol/ml; P < 0.01) or STa alone (72 ± 2.83 pmol/ml; P < 0.01). When the STa toxin dose was increased to 5 or 10 ng, only serum sample pooled from the 3×STaN12S-dmLT-immunized group maintained strong neutralizing activity, showing cGMP levels of 0.28 ± 0.03 and 1.16 ± 0.18 pmol/ml, respectively, in the incubated T-84 cells.
FIG 4.
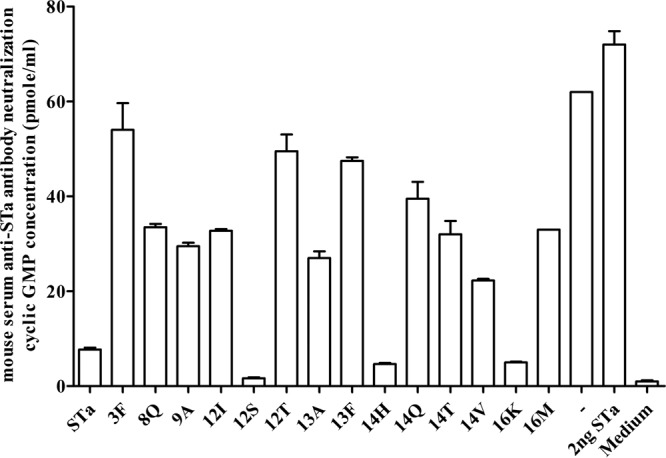
Mouse serum in vitro antibody neutralizing activity against STa toxin. Intracellular cGMP levels in T-84 cells measured with an EIA cGMP ELISA kit (Assay Design) were used to show anti-STa antibody neutralization activity. If antibodies in serum (or fecal suspension) are neutralizing against STa toxin, these antibodies will prevent the toxin from stimulating intracellular cGMP in T-84 cells, which will result in a low cGMP level (lower than that of the control group [the “−” column]). The serum sample (30 μl; in a final dilution of 1:33.3) pooled from each immunization group or the control group (−) was incubated with STa toxin (2 ng, in 150 μl of cell culture medium) for 1 h at room temperature, and the serum-toxin mixture was added to T-84 cells (1 ml of final volume with cell culture medium). The intracellular cGMP level in the T-84 cells was measured after 1 h of incubation, with the mean cGMP level and standard deviation (from two replicates) of each group indicated as columns and lines. Labels on the x axis indicate the mouse groups immunized with different fusion antigens and the control group (“−”), from which serum samples were used for the antibody neutralization assay. STa toxin (without serum sample) was used as the control to show STa toxicity in stimulation of cGMP, and cell culture medium (without STa toxin and serum) was used as the control to show baseline cGMP level in T-84 cells.
Pooled fecal samples (in a dilution of 1:200; versus 1:33.3 in serum) were also analyzed but showed mild or moderate neutralizing activity against STa toxin (Fig. 5). Lower cGMP levels were detected in T-84 cells incubated with STa and the fecal samples of mice immunized with 3×STaN12I-dmLT (13.5 ± 0.71 pmol/ml), 3×STaP13A-dmLT (14 ± 0 pmol/ml), 3×STaN12T-dmLT (16.3 ± 1.1 pmol/ml), 3×STaE8Q-dmLT (16.5 ± 0.71 pmol/ml), or 3×STaN12S-dmLT (18.8 ± 0.35 pmol/ml). The mean cGMP level in T-84 cells incubated with STa toxin and the fecal samples of the control mice was 62 ± 0 pmol/ml, and the mean cGMP level in cells incubated with cell culture medium was 0.98 ± 0.25 pmol/ml (Fig. 5).
FIG 5.
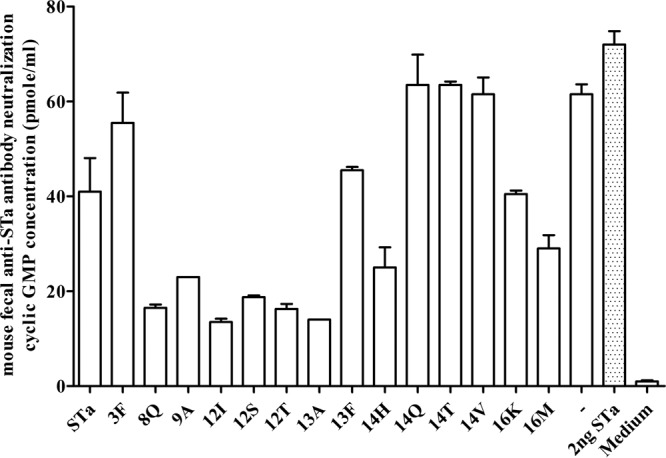
Mouse fecal suspension in vitro antibody neutralization activity against STa toxin, with intracellular cGMP levels in T-84 cells used to measure neutralizing activity of antibodies in fecal suspension samples against STa toxin. A fecal suspension sample (30 μl; in a final dilution of 1:200) pooled from each immunized group or the control group (5 mice per group) was incubated with STa toxin (2 ng; in 150 μl cell culture medium), and the fecal-toxin mixture was added to T-84 cells. The intracellular cGMP level in T-84 cells was measured by using an EIA cGMP ELISA kit (Assay Design), with columns and lines indicating the mean cGMP levels and standard deviations. A low cGMP level (lower than that of the control group) indicated antibody neutralizing activity against STa toxin. Group labels (on the x axis) indicate mice immunized with different fusion antigens and the control mice, for which fecal suspension samples were examined for STa neutralizing activity. STa toxin only (toxin without fecal sample) to show enterotoxicity in the stimulation of cGMP and cell culture medium (no STa toxin and anti-STa antibodies) to show the baseline cGMP level in T-84 cells were included as controls.
Neutralization activity against CT toxin was detected in the serum and fecal samples of the immunized groups. The average cAMP level in T-84 cells incubated with 10 ng of CT and the serum sample pooled from each immunized group was 17 pmol/ml, whereas the average cAMP level in cells incubated with the toxin and the serum of the control group was 37 pmol/ml. When fecal suspensions were used in neutralization assays, an average of cAMP level in T-84 cells incubated with CT and samples of each immunized group was detected at 10.3 pmol/ml, but the cAMP level in T-84 cells incubated with toxin and the fecal suspension of the control group was 21 pmol/ml.
DISCUSSION
Recent studies indicated that nontoxic STa, after being genetically fused to a monomeric LT toxoid peptide, can elicit moderately neutralizing anti-STa antibodies (21, 22, 24). That indicates a previously suggested STa dilemma, of which STa has to possess enterotoxicity (which is not safe as a vaccine component) to be able to induce neutralizing antibody response (20), can be solved. These findings led to the current effort toward further characterization of STa toxoids for the induction of antibody response and the search for optimal STa toxoid(s) that induce neutralizing antibodies against STa toxin. Shown differences in toxicity reduction and more importantly antigenic structure alteration, individual STa toxoids were thought to be different in induction of neutralizing antibody response (23). A thorough examination of all potential STa toxoids would help to identify toxoids that have maximum reduction in enterotoxicity but minimum alteration in STa antigenic structure. The antigenic propensity and perhaps the toxicity of these optimal STa toxoids, however, could be altered after being genetically fused to or chemically conjugated to a carrier protein, which is required to facilitate anti-STa antibody response. Therefore, toxoid fusions or chemical conjugates have to be examined essentially to identify STa-related antigens that induce neutralizing anti-STa antibodies. However, construction of toxoid fusions and the expression of fusion proteins, or the purification of STa toxoids and then conjugation to a carrier, is labor-intensive and also technically challenging (some STa toxoids cannot be purified based on current protocols [J. D. Clements, unpublished data]), especially if a large number of STa toxoids are to be examined.
In the present study, we preselected a panel of STa toxoids from a mini-STa toxoid library to construct STa-toxoid-dmLT toxoid fusions and then used these fusions for mouse immunization to characterize STa toxoids for the induction of neutralizing antibody response and to identify optimal STa toxoid(s) or more precisely toxoid fusion antigen(s) for future ETEC antitoxin vaccine development. Here, a monomeric dmLT peptide was served as a protein carrier to enhance STa toxoids for anti-STa immunogenicity and also an antigen used to induce an anti-LT antibody response. This monomeric dmLT peptide, however, probably does not possess adjuvant activity as the holotoxin structured dmLT does. When a similar monomeric peptide, tmLT, was used, the toxoid fusion 3×STaA14Q-tmLT alone (without Freund or CT adjuvant) was unable to stimulate anti-STa or anti-LT antibodies (24). It will be interesting to examine currently constructed toxoid fusions for stimulation of anti-STa and anti-LT antibodies when the holotoxin structured dmLT (instead of Freund adjuvant) is used as the adjuvant.
STa enterotoxicity was thought to be tightly associated with antigenic propensity, especially of the toxicity domain (from residues 6 to 18) (30). Changes in the amino acid residues of a small molecule, such as STa, which consists of only 19 amino acids, 6 of which form three disulfide bonds, were considered to have significant effects upon alteration of its antigenic structure and enterotoxicity. It has been noted that STaG17S retains an antigenic structure very similar to that of native STa based on its reactivity to antiserum against native STa and that it also possessed a level of toxicity similar to that of STa (23). After screening 47 mutated STa molecules, we noted that some STa toxoids exhibited an association between the antigenic structure and enterotoxicity, but such an association did not occur with other toxoids. It was found that STaT16K, STaT16M, STaA14V, and STaN12S had levels of reactivity to anti-STa antiserum similar to that of native STa, but they showed no STa toxic activity in vitro. That indicates that STa toxoids can maintain an original antigenic structure or epitope topology after their toxicity is eliminated. More interestingly, the nontoxic STaT16K, STaT16M, and STaN12S molecules seemed to retain their antigenic topology after being fused to the monomeric dmLT peptide, as indicated by the ELISA data (Table 3) and the fact that these fusions induced stronger anti-STa antibody responses in immunized mice (Fig. 2).
It was unexpected that the fusion with native STa, 3×STa-dmLT, was not the top antigen in the induction of an anti-STa antibody response. Indeed, the sera of mice immunized with fusion 3×STa-dmLT had the lowest overall anti-STa IgG antibody titers detected; four of five immunized mice did not develop an anti-STa immune response. That suggests that native STa antigenic topology was substantially altered after being genetically fused to the monomeric dmLT peptide. However, the one mouse that developed anti-STa antibody response had the greatest anti-STa IgG titer detected in the serum sample. It is unknown to us that how the genetic fusion process affected the STa or STa toxoid antigenic propensity and why such a wide range variation occurred in the induction of anti-STa antibody responses among these immunized mice. Regardless, native STa has to be pre-excluded from vaccine development, since unintentional proteolytic cleavage of native STa from toxoid fusions by cellular proteases or dissociation of toxic STa from conjugates raises safety concerns.
Our data showed that variation in the induction of an anti-STa antibody response also existed among groups immunized with different toxoid fusions and among individual mice within the group immunized the same toxoid fusion. Of five mice in an immunized group, only one (3×STa-dmLT), two (3×STaS3F-dmLT, 3×STaA14v-dmLT), three (3×STaP13F-dmLT, 3×STaA14Q-dmLT, 3×STaA14T-dmLT), or four mice (3×STaE8Q-dmLT, 3×STaP13A-dmLT, 3×STaT16K-dmLT, 3×STaT16M-dmLT) had anti-STa IgG detected in the serum samples. Even in the group of which all mice developed anti-STa IgG antibody response, the antibody titers of individual mice varied greatly, except the group immunized with the 3×STaN12S-dmLT. In contrast to the variation in anti-STa antibody response, the anti-LT IgG antibody response was detected in all immunized mice and at a very similar titer among and within the immunized groups (Table 5). The causes of such “hit-or-miss” in the induction of a host anti-STa antibody response by these toxoid fusions are unclear to us. However, the poorly immunogenic nature of STa likely attributes to the inconsistency in inducing anti-STa antibody response. Increasing a sampling size likely will be helpful to further characterize the anti-STa antibody induction from these toxoid fusions. However, the present study was repeated in a separate laboratory, which brought up an overall sampling size of 10 mice per group, and the anti-STa antibody response pattern was not much changed (data not shown).
This study showed that toxoid fusions 3×STaN12S-dmLT, 3×STaA14H-dmLT, and 3×STaT16K-dmLT were the top candidates in eliciting anti-STa IgG antibodies in mice. Serum samples of mice immunized with these fusion antigens exhibited stronger neutralizing activity against the STa toxin. Among them, 3×STaN12S-dmLT was the most effective at inducing a neutralizing anti-STa antibody response. Only the serum sample pooled from mice immunized with the toxoid fusion 3×STaN12S-dmLT was able to neutralize higher doses of STa toxin, since it prevented 5 ng and even 10 ng of STa from stimulating any increase in cGMP in T-84 cells. Even when 20 ng of STa (a 10-fold increase) was incubated with this serum sample, T-84 cells had only 10% cGMP level of those incubated with the toxin and the serum sample of the control mice (data not shown). In addition, in fecal samples from mice immunized with 3×STaN12S-dmLT, more anti-STa IgA antibodies were detected that also displayed better neutralization activity against STa toxin. Future studies of in vivo antibody neutralization assays, such as the infant suckling mouse assay, or even a piglet challenge study, will further define neutralization activity against STa toxin from antibodies induced by this toxoid fusion.
Induction of the strongest anti-STa antibody response by this 3×STaN12S-dmLT was confirmed from the study carried out in the separate laboratory (data not shown). The data from that mouse immunization study, using the same toxoid fusion antigens and the same immunization protocol, showed that the serum samples of the mice immunized with 3×STaN12S-dmLT showed the greatest anti-STa IgG antibody titers and also the strongest neutralizing activity against STa toxin detected (data not shown). Although continued construction of toxoid fusions with different STa toxoids and additional screening of fusions for anti-STa antibody responses likely will reveal new findings, the toxoid fusion 3×STaN12S-dmLT currently should be primarily considered for next-step animal challenge or human volunteer studies in ETEC antitoxin vaccine development.
The effective prevention of ETEC diarrhea requires antitoxin immunity to neutralize the enterotoxicity of both toxins and also anti-adhesin immunity to block bacterial adherence and to prevent ETEC colonization at host small intestines (14). Anti-STa and anti-LT antibody responses induced by this 3×STaN12S-dmLT toxoid fusion or other fusions or conjugates alone may not be sufficient for effective protection against ETEC diarrhea. Future studies should include the 3×STaN12S-dmLT fusion antigen among promising ETEC vaccine candidates that induce anti-adhesin immunity against multiple ETEC adhesins, or coadministration with a product that induces broadly protective anti-adhesin immunity may be desirable for developing effective vaccines against ETEC diarrhea.
ACKNOWLEDGMENTS
Financial support for this study was provided by National Institutes of Health AI083897 (W. Zhang), EVI/PATH STa Toxoid Vaccine Consortium project, and the South Dakota Agricultural Experiment Station. This study was also supported by all members of the STa Toxoid Vaccine Consortium Group: J. P. Nataro (University of Virginia School of Medicine), E. M. Barry (University of Maryland School of Medicine), J. D. Clements (University of Tulane School of Medicine), W. Zhang (South Dakota State University), D. H. Francis (South Dakota State University), H. Sommerfelt (University of Bergen), P. Puntervoll (University of Bergen), A. Louis Bourgeois (PATH), and R. Walker (PATH).
Footnotes
Published ahead of print 18 February 2014
REFERENCES
- 1.Black RE, Cousens S, Johnson HL, Lawn JE, Rudan I, Bassani DG, Jha P, Campbell H, Walker CF, Cibulskis R, Eisele T, Liu L, Mathers C. 2010. Global, regional, and national causes of child mortality in 2008: a systematic analysis. Lancet 375:1969–1987. 10.1016/S0140-6736(10)60549-1 [DOI] [PubMed] [Google Scholar]
- 2.Kotloff KF, Nataro JP, Blackwelder WC, Nasrin D, Farag TH, Panchalinam S, Wu Y, Sow SO, Sur D, Breiman RF, Faruque ASG, Zaidi AKM, Saha D, Alonso PL, Tamboura B, Sanoo D, Onwuchekwa U, Manna B, Ramamurth T, Kanungo S, Ochieng JB, Omore R, Oundo JO, Hassain A, Das SK, Ahmed S, Qureshi S, Quadri F, Adegbola RA, Antonio M, Hossain MJ, Akinsola A, Mandomando I, Nhampossa T, Acacio S, Biswas K, O'Reilly CE, Mintz ED, Berkeley LY, Muhsen K, Sommerfelt H, Robins-Browne RM, Levine MM. 2013. Burden and etiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet 382:209–222. 10.1016/S0140-6736(13)60844-2 [DOI] [PubMed] [Google Scholar]
- 3.WHO. 2006. Future directions for research on enterotoxigenic Escherichia coli vaccines for developing countries. Wkly. Epidemiol. Rec. 81:97–107 [PubMed] [Google Scholar]
- 4.WHO. 1999. The World Health Report 1999: making a difference. World Health Organization, Geneva, Switzerland [Google Scholar]
- 5.DuPont HL. 2008. Systematic review: prevention of travellers' diarrhoea. Aliment. Pharmacol. Ther. 27:741–751. 10.1111/j.1365-2036.2008.03647.x [DOI] [PubMed] [Google Scholar]
- 6.Sanders JW, Putnam SD, Riddle MS, Tribble DR. 2005. Military importance of diarrhea: lessons from the Middle East. Curr. Opin. Gastroenterol. 21:9–14 [PubMed] [Google Scholar]
- 7.Wenneras C, Erling V. 2004. Prevalence of enterotoxigenic Escherichia coli-associated diarrhoea and carrier state in the developing world. J. Health Pop. Nut. 22:370–382 [PubMed] [Google Scholar]
- 8.Nataro JP, Kaper JB. 1998. Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 11:142–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berberov EM, Zhou Y, Francis DH, Scott MA, Kachman SD, Moxley RA. 2004. Relative importance of heat-labile enterotoxin in the causation of severe diarrheal disease in the gnotobiotic piglet model by a strain of enterotoxigenic Escherichia coli that produces multiple enterotoxins. Infect. Immun. 72:3914–3424. 10.1128/IAI.72.7.3914-3924.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang W, Berberov EM, Freeling J, He D, Moxley RA, Francis DH. 2006. Significance of heat-stable and heat-labile enterotoxins in porcine colibacillosis in an additive model for pathogenicity studies. Infect. Immun. 74:3107–3114. 10.1128/IAI.01338-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Glenn GM, Francis DH, Danielsen EM. 2009. Toxin-mediated effects on the innate mucosal defenses: implications for enteric vaccines. Infect. Immun. 77:5206–5215. 10.1128/IAI.00712-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boedeker EC. 2005. Vaccines for enterotoxigenic Escherichia coli: current status. Curr. Opin. Gastroenterol. 21:15–19 [PubMed] [Google Scholar]
- 13.Svennerholm AM. 2011. From cholera to enterotoxigenic Escherichia coli (ETEC) vaccine development. Ind. J. Med. Res. 133:188–196 [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang W, Sack DA. 2012. Progress and hurdles in the development of vaccines against enterotoxigenic Escherichia coli in humans. Expert Rev. Vaccines 11:677–684. 10.1586/erv.12.37 [DOI] [PubMed] [Google Scholar]
- 15.Svennerholm AM, Ahren C, Wenneras C, Holmgren J. 1991. Development of an oral vaccine against enterotoxigenic Escherichia coli diarrhea, p 287–294 In Wadstrom T, Makela PH, Svennerholm AM, Wolf-Watz H. (ed), Molecular pathogenesis of gastrointestinal infections, Plenum Press, London, United Kingdom [Google Scholar]
- 16.Turner AK, Stephens JC, Beavis JC, Greenwood J, Gewert C, Randall R, Freeman D, Darsley MJ. 2011. Generation and characterization of a live attenuated enterotoxigenic Escherichia coli combination vaccine expressing six colonization factors and heat-labile toxin subunit B. Clin. Vaccine Immunol. 18:2128–2135. 10.1128/CVI.05345-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clements JD. 1990. Construction of a nontoxic fusion peptide for immunization against Escherichia coli strains that produce heat-labile and heat-stable enterotoxins. Infect. Immun. 1990. 58:1159–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jertborn M, Ahren C, Holmgren J, Svennerholm AM. 1998. Safety and immunogenicity of an oral inactivated enterotoxigenic Escherichia coli vaccine. Vaccine 16:255–260. 10.1016/S0264-410X(97)00169-2 [DOI] [PubMed] [Google Scholar]
- 19.Taxt A, Aasland R, Sommerfelt H, Nataro J, Puntervoll P. 2010. Heat-stable enterotoxin of enterotoxigenic Escherichia coli as a vaccine target. Infect. Immun. 78:1824–1831. 10.1128/IAI.01397-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Svennerholm AM, Holmgren J. 1995. Oral B-subunit whole-cell vaccines against cholera and enterotoxigenic Escherichia coli diarrhoea, p 205–232 In Dlawer AAla', Aldeen A, Hormaeche CE. (ed), Molecular and clinical aspects of bacterial vaccine development. John Wiley & Sons, Ltd, Chichester, United Kingdom [Google Scholar]
- 21.Zhang W, Zhang C, Francis DH, Fang Y, Knudsen D, Nataro JP, Robertson DC. 2010. Genetic fusions of heat-labile (LT) and heat-stable (ST) toxoids of porcine enterotoxigenic Escherichia coli elicit neutralizing anti-LT and anti-STa antibodies. Infect. Immun. 78:316–325. 10.1128/IAI.00497-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu M, Ruan X, Zhang C, Lawson SR, Knudsen DE, Nataro JP, Robertson DC, Zhang W. 2011. Heat-labile- and heat-stable-toxoid fusions (LTR192G-STaP13F) of human enterotoxigenic Escherichia coli elicit neutralizing antitoxin antibodies. Infect. Immun. 79:4002–4009. 10.1128/IAI.00165-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu M, Zhang C, Mateo K, Nataro JP, Robertson DC, Zhang W. 2011. Modified heat-stable toxins (hSTa) of enterotoxigenic Escherichia coli lose toxicity but display antigenicity after being genetically fused to heat-labile toxoid LT(R192G). Toxins (Basel) 3:1146–1162. 10.3390/toxins3091146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang C, Knudsen DE, Liu M, Robertson DC, Zhang W, and the STa Toxoid Vaccine Consortium Group 2013. Toxicity and immunogenicity of enterotoxigenic Escherichia coli heat-labile and heat-stable toxoid fusion 3xSTaA14Q-LTS63K/R192G/L211A in a murine model. PLoS One 8:e77386. 10.1371/journal.pone.0077386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Norton EB, Lawson LB, Freytag LC, Clements JD. 2011. Characterization of a mutant Escherichia coli heat-labile toxin, LT(R192G/L211A), as a safe and effective oral adjuvant. Clin. Vaccine Immunol. 18:546–551. 10.1128/CVI.00538-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ausubel FM, Brent R, Kingston RK, Moore DD, Seidman JG, Smith JA, Struhl K. 1999. Short protocols in molecular biology, 4th ed. John Wiley & Sons Inc,, New York, NY [Google Scholar]
- 27.Ruan X, Liu M, Casey TA, Zhang W. 2011. A tripartite fusion, FaeG-FedF-LT(192)A2:B, of enterotoxigenic Escherichia coli (ETEC) elicits antibodies that neutralize cholera toxin, inhibit adherence of K88 (F4) and F18 fimbriae, and protect pigs against K88ac/heat-labile toxin infection. Clin. Vaccine Immunol. 18:1593–1599. 10.1128/CVI.05120-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.National Research Council. 1996. Guide for the care and use of laboratory animals. National Academy Press, Washington, DC [Google Scholar]
- 29.Zhang W, Robertson DC, Zhang C, Bai W, Zhao M, Francis DH. 2008. Escherichia coli constructs expressing human or porcine enterotoxins induce identical diarrheal diseases in a piglet infection model. Appl. Environ. Microbiol. 74:5832–5837. 10.1128/AEM.00893-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sato T, Shimonishi Y. 2004. Structural features of Escherichia coli heat-stable enterotoxin that activates membrane-associated guanylyl cyclase. J. Peptide Res. 63:200–206. 10.1111/j.1399-3011.2004.00125.x [DOI] [PubMed] [Google Scholar]