

Abstract
Cronobacter sakazakii is an opportunistic pathogen that causes neonatal meningitis and necrotizing enterocolitis. Its interaction with intestinal epithelium is important in the pathogenesis of enteric infections. In this study, we investigated the involvement of the inv gene in the virulence of C. sakazakii ATCC 29544 in vitro and in vivo. Sequence analysis of C. sakazakii ATCC 29544 inv revealed that it is different from other C. sakazakii isolates. In various cell culture models, an Δinv deletion mutant showed significantly lowered invasion efficiency, which was restored upon genetic complementation. Studying invasion potentials using tight-junction-disrupted Caco-2 cells suggested that the inv gene product mediates basolateral invasion of C. sakazakii ATCC 29544. In addition, comparison of invasion potentials of double mutant (ΔompA Δinv) and single mutants (ΔompA and Δinv) provided evidence for an additive effect of the two putative outer membrane proteins. Finally, the importance of inv and the additive effect of putative Inv and OmpA were also proven in an in vivo rat pup model. This report is the first to demonstrate two proteins working synergistically in vitro, as well as in vivo in C. sakazakii pathogenesis.
INTRODUCTION
Cronobacter sakazakii is a Gram-negative, rod-shaped, non-spore-forming opportunistic pathogen in the family Enterobacteriaceae (1). C. sakazakii is known to transmit through reconstituted infant formula (2) and causes necrotizing enterocolitis, bacteremia, and meningitis (3–5). The mortality rate by C. sakazakii infection has been reported to be 33 to 80% (6–9), and the survivors often experience developmental, chronic, and neurological disorders (10). As a causative agent of neurological disorders after oral ingestion, C. sakazakii must be equipped with the virulence factors necessary to penetrate the intestinal epithelium, to survive the host defense mechanism, and to cross the blood-brain barrier. Production of enterotoxins (11) and the expression of genus specific cell bound zinc metalloprotease (12) were reported to play a role in Cronobacter pathogenesis. A putative sod gene (superoxide dismutase gene) (13) and C. sakazakii plasmid encoding cpa (Cronobacter plasminogen activator) (14) were also identified as potential virulence factors involving survival of C. sakazakii in in vitro human macrophages (U937) and contributing to serum tolerance, respectively.
Several genes are known to be crucial in the interaction of C. sakazakii with epithelial cells and its translocation of C. sakazakii through the epithelial tissue barrier. A study on C. sakazakii adhesion to host cells identified two distinct adherent patterns, and the authors suggested that specific adhesins may be involved in the process (15). Putative lysR-type transcriptional regulator (LTTR) was reported to be important in adhesion to and invasion of human intestinal cells by C. sakazakii (16). Outer membrane protein A (OmpA) of C. sakazakii was identified as playing an essential role in the adhesion and invasion of host cells (17, 18, 19) and was further revealed as important in the transcytosis of C. sakazakii across tight monolayers of epithelial cells on transwell cultures (20). In addition, outer membrane protein OmpX was reported as a virulence determinant (18) that is responsible for the invasion of C. sakazakii in Caco-2 cells. These proteins are assumed to be involved in the recognition of and binding to specific receptors of cells (21), which in turn may facilitate the successful colonization and subsequent translocation of the pathogen to blood circulation.
Previously, inv was reported to be involved in pathogenesis of Yersinia and Salmonella spp. (22–27). In the present study, we confirmed the presence of inv homolog in C. sakazakii ATCC BAA-894 by blast search and in C. sakazakii ATCC 29544 by PCR using the primers derived from C. sakazakii ATCC BAA-894 genome sequence. We further demonstrate that putative Inv is essential for apical and basolateral invasion of the pathogen to host epithelial cells. In addition, for the first time in C. sakazakii, we show that the putative inv gene product exhibits additive effect with OmpA in in vitro and in vivo virulence models.
MATERIALS AND METHODS
Bacterial strains, primers, plasmids, and media.
The bacterial strains and plasmids and the primers used in the present study are listed in Table 1 and Table 2, respectively. C. sakazakii ATCC 29544 and the mutants were routinely grown in half-concentrated brain heart infusion broth (BHI1/2; Difco, Franklin Lakes, NJ) at 37°C with constant shaking unless otherwise indicated. Escherichia coli and C. sakazakii strains harboring the various plasmids were grown on Luria-Bertani (LB; Difco) media at 37°C. When needed, antibiotics were added at concentrations of 25 (chloramphenicol) or 50 μg/ml (ampicillin or kanamycin).
TABLE 1.
Bacterial strains and plasmids
| Strain or plasmid | Relevant characteristicsa | Source or reference |
|---|---|---|
| Strains | ||
| C. sakazakii | ||
| ATCC 29544 | Wild-type strain | 17 |
| ES1001 | 29544 harboring pKD46(Apr) | 17 |
| ES2005 | ΔompA | 17 |
| ES2010 | Δinv::Kanr | This study |
| ES2011 | Δinv | This study |
| ES2012 | pACYC184-inv | This study |
| ES2013 | ΔompA::Kanr | This study |
| ES2014 | ΔompA Δinv | This study |
| E. coli | ||
| DH5α | λ− ϕ80dlacZΔ(lacZYA-argF)U169 recA1 endA1 hsdR17(rK− mK−) supE44 thi-1 gyrA relA1 | 17 |
| Top10 | F− mcrA Δ(mrr-hsdRMS-mcrBC) 80lacZΔM15 ΔlacX74 recA1 ΔaraD139 Δ(ara-leu)7697 galU galK rpsL (StrR) endA1 nupG | Invitrogen |
| Plasmids | ||
| pKD13 | oriR6Kγ Apr FRT Kanr FRT | 16 |
| pKD46 | oriR101 repA101t,s Apr araBAD pgam-bet-exo | 16 |
| pCP20 | oripSC101t,s Apr Cmr cI857λ PRflp | 16 |
| pACYC184 | Tetr Cmr; p15A ori | 16 |
| pBAD202/D-TOPO | Kanr | Invitrogen |
| pBADD202-r-inv | Harboring C. sakazakii inv gene reverse complement | This study |
Apr, ampicillin resistance; Kanr, kanamycin resistance; Cmr, chloramphenicol resistance; Tetr, tetracycline resistance.
TABLE 2.
Oligonucleotide primers used for PCR amplification and sequencing
| Primer | Sequence (5′–3′)a | Reference or source |
|---|---|---|
| OmpA-F | GTG CTC ATC AAT AGA CCG ACA | 17 |
| OmpA-R | ACT ACA TGC AGC AGA GAA ATT | |
| Inv_ FP | ATG CAT GAG CAA TCC ATA ATG | This study |
| Inv_RP | TTA AAG CAA ATA ATC TCT GGT | |
| Inv_F1 | ATG CTG TCT GGT CTG GCA TCG | This study |
| Inv_R1 | CCA CGT TTA ATC ACT GCT GCG | |
| Inv_F2 | GGG TCA GCG TCA CCT GGA ACG | This study |
| Inv_R2 | CGT CAT ATC CGC CAC CGC TTT | |
| Inv_F3 | CGG TGA TAA CGT GGC GTT GTT | This study |
| Inv_R3 | CAC CGG CAC CGC GGT AAA GAC | |
| Inv_Kan_F | CAT CAT TAA GGA TAC CTG TAG CAC GGG TGT TGA TTG AGA ATT T TGT AGG CTG GAG CTG CTT CG* | This study |
| Inv_Kan_R | ATA CGG AGC CGA AAT CAG GGG AGT TAA TAA GAT GCC ATG A ATT CCG GGG ATC CGT CGA CC* | This study |
| inv HindIII-F | AGA GAG GCA TGC GCT TTC AGA CTC** | This study |
| inv SphI-R | AGA GAG AAG CTT TTA AAG CAA ATA** | This study |
| Inv_pBAD_F | ATG CAT GAG CAA TCC ATA ATG | This study |
| Inv_pBAD_R | CAC CAA GCA AAT AAC TTC TGG TAT TGG T*** | This study |
| Primer_2_F inV | GCA ACA TTG GCT TAG GGG TA | This study |
| Primer_2_R inV | AGC TAT TGG CGG AAA GTT TG | This study |
*, Nucleotide sequences that originated from the C. sakazakii ATCC 29544 inv gene are shown in italics, and those from pKD13 are underlined; **, artificially added restriction enzyme recognition sites are underlined; ***, an artificially added four-base sequence is underlined.
Identification and sequencing of inv gene in C. sakazakii ATCC 29544.
PCR for nucleotide sequencing was performed using Pfu DNA polymerase (Enzynomix, South Korea). In order to PCR amplify complete inv gene homolog in C. sakazakii ATCC 29544, the primers Inv_F1 and Inv_R1 were designed based on the nucleotide sequence of the genome of C. sakazakii ATCC-BAA 894 (NCBI accession number NC_009778.1) (Table 2). PCR was performed under the thermal cycling conditions of a hot start at 95°C for 5 min, followed by amplification for 30 cycles of 95°C for 60 s, 63°C for 60 s, and 72°C for 4 min, and with a final extension at 72°C for 10 min. After agarose gel electrophoresis and gel extraction, the PCR amplicon was sequenced using Inv_F1, Inv_R1, Inv_F2, Inv_R2, Inv_F3, and Inv_R3 (Table 2) in Macrogen, Inc. (Seoul, South Korea). Sequence information was submitted to the GenBank (NCBI accession number KC602378).
Bioinformatics.
Protein similarity search was performed using the BLASTP algorithm (National Center for Biotechnology Information [http://www.ncbi.nlm.nih.gov]). TMPred (28) and HMMTOP (29) were used to predict transmembrane domains and subcellular localization of the putative Inv protein. The secondary structure of the protein was determined using the online program Phyre2, and conserve protein family and domains were identified using InterProScan program (Pfam database [http://www.ebi.ac.uk]).
Mutant construction.
In-frame deletion mutants of C. sakazakii ATCC 29544 were generated by the lambda red recombination method, as described by Datsenko and Wanner (16). Briefly, to construct the inv deletion mutant, a kanamycin resistance (Kanr) cassette from plasmid pKD13 was amplified using the primers, Inv_Kan_F (forward primer) and Inv_Kan_R (reverse primer) (Table 2). The PCRs were performed (GeneAmp PCR System 9700; Applied Biosystems, Foster City, CA) under the thermal cycling conditions of a hot start at 95°C for 5 min, followed by amplification for 30 cycles of 95°C for 60 s, 63°C for 60 s, and 72°C for 4 min, and with a final extension at 72°C for 10 min. The PCR products were analyzed by electrophoresis in 1% agarose gel and examined on a UV transilluminator after staining with ethidium bromide. The resulting PCR products were electroporated (Gene Pulser, 2.5 V, 200 Ω, 25 μFD capacity; Bio-Rad, Hercules, CA) into the wild-type strain (WT), C. sakazakii ATCC 29544, harboring the pKD46 plasmid as described previously (18). Kanr transformants (inv::Kan) were selected on LB agar plates containing 50 μg of kanamycin/ml. Finally, the Kanr cassette was removed by using the pCP20 plasmid, as described previously (16). The ΔompA Δinv double mutant was constructed by using the ΔompA mutant (17) as a parental strain and the primers Inv-FP and Inv-RP (Table 2).
Complementation.
For construction of complement strains, complete inv open reading frame (ORF) plus promoter region of C. sakazakii ATCC 29544 inv (350 bp upstream from the start codon) was amplified using the forward primer inv HindIII-F and reverse primer, inv SphI-R (Table 2). The thermal cycling conditions consisted of a hot start at 95°C, for 5 min, followed by amplification for 30 cycles of 95°C for 60 s, 62°C for 60 s, and 72°C for 4 min, with a final extension at 72°C for 10 min. The PCR products were digested with HindIII and SphI and cloned into pACYC184 vector (30), followed by transformation into single (Δinv or ΔompA) and double (ΔompA Δinv) mutants.
Cell culture.
Human intestinal epithelial Caco-2 (HTB 37 [derived from human colon adenocarcinoma]; American Type Culture Collection [ATCC], Manassas, VA), human intestinal epithelial INT-407 (ATCC CCL6 [derived from human embryonic jejunum and ileum]), and Hep-2 (ATCC CCL-23 [derived via HeLa contamination]) cells were maintained in Dulbecco modified Eagle medium (DMEM; Lonza, MD) containing 10% fetal bovine serum (FBS) (Lonza). Trypsin-treated cells were seeded (approximately 5 × 104 cells per well) into 24-well cell culture plates (Sarstedt, Newton, NC) and grown at 37°C in the presence of 5% CO2. The cells formed monolayers after 3 to 4 days. The medium was replaced every 2 days, and cell viability was determined by trypan blue staining.
EGTA treatment.
To disrupt the tight junction of Caco-2 cells, EGTA (Sigma, St. Louis, MO) treatment was used. The Caco-2 monolayer was incubated with DMEM containing 5 mM EGTA (pH 7.5) or phosphate-buffered saline (PBS; pH 7.5) as a control for 60 min before bacterial infection (17). The cells were then washed twice with PBS (pH 7.4) and used for further studies.
Invasion assay.
To test the ability of C. sakazakii to invade the cultured mammalian cells (Caco-2, INT-407, and Hep-2), invasion assay was performed as described previously (17, 18). Briefly, C. sakazakii was prepared by transferring overnight culture (2% inoculum) to fresh prewarmed BHI1/2 medium and was incubated at 37°C for 3 h with constant shaking (160 rpm; optical density at 600 nm [OD600] of 0.6). Cells were collected by centrifugation at 10,000 × g for 5 min, and the cell pellet was suspended in 1 ml of DMEM (10% FBS). The mammalian cells grown in 24-well tissue culture plates were infected with 2 × 107 CFU of bacteria (multiplicity of infection [MOI] of 40) and incubated for 1.5 h. Monolayers were washed three times with PBS and further incubated for another 1.5 h with fresh medium containing gentamicin (100 μg/ml; Sigma) to kill the bacteria outside the cell. The cells were washed three times with PBS and treated with Triton X-100 (0.25% in PBS) for 10 min (Triton X-100 treatment [0.25%, 10 min] of bacteria has no effect on bacterial viability). Internalized C. sakazakii cells were enumerated by plating on BHI1/2 agar medium in duplicate. The results were expressed as an average of log CFU/well or log relative percentage of invasion compared to 4- to 7-day-old EGTA untreated Caco-2 cells from at least three independent experiments.
Invasion assay and TEER measurement using transwell culture of Caco-2 cells.
Caco-2 cells grown in upper chamber (12-mm insert in diameter) of transwell system (0.4-μm-pore-size polycarbonate filters; Corning Life Sciences, Inc.) for 14 days were treated with either EGTA (5 mM) for 1 h, purified Salmonella lipopolysaccharide (LPS; 10 ng/ml; Sigma) (31), or PBS (pH 7.5) for 3 h (20). The cells were washed three times with PBS and used for an invasion assay (MOI of 100) as described above. The 0.4-μm-pore-size inserts efficiently prevented C. sakazakii from crossing the filter (0.08% translocation rate in the absence of Caco-2). In order to evaluate disruption of tight junction, transepithelial electrical resistance (TEER) across the transwell filter was measured before and after the treatment using a Millicell-ERS meter (Millipore, Inc., Billerica, MA) (20).
Blocking assay.
Overexpression and purification of OmpA recombinant protein was carried out as previously described (32) with modifications. Briefly, the overnight culture of E. coli Top10 harboring pBAD202/D-TOPO::ompA (17) was transferred into fresh LB medium, and culture was induced with 2% l-arabinose when it reached an OD600 of 0.5. The induced culture further incubated for 4 h, and the cells were recovered by centrifugation at 10,000 × g for 5 min at 4°C, followed by suspension in denaturation buffer B (100 mM Tris-HCl, 300 mM NaCl, 8 M urea [pH 8.0]) supplemented with 0.01% Triton X-100, 10 mM 2-mercaptoethanol, and 1% glycerol. After the cells were lysed by gentle vortexing at room temperature, lysate was centrifuged at 8,800 × g for 30 min at room temperature. The supernatant was loaded onto an Ni-NTA Superflow affinity column (Qiagen, Valencia, CA), and recombinant protein was eluted four times with 0.5 ml of buffer D (100 mM Tris-HCl, 300 mM NaCl, 8 M urea [pH 5.9]), followed by four elutions with 0.5 ml of buffer E (100 mM Tris-HCl, 300 mM NaCl, 8 M urea [pH 4.5]). Peak fractions were determined by SDS-PAGE, pooled, and dialyzed against PBS (pH 7.4) overnight at 4°C. Protein concentration was analyzed using NanoDrop (Biotek, South Korea) and stored at −20°C for further experiments. For blocking assay, Caco-2 monolayers were preincubated with purified OmpA protein (50 μg/well) for 1 h, followed by three washes with PBS (17). C. sakazakii invasion was determined as described above.
Inhibition of inv translation by introduction of antisense inv.
To construct the plasmid transcribing reverse complement of inv gene, first complete inv gene of C. sakazakii ATCC 29544 was PCR amplified using the forward primer Inv_pBAD_F and the reverse primer Inv_pBAD_R (Table 2). The PCR conditions consisted of a hot start at 95°C for 5 min, followed by amplification for 30 cycles of 95°C for I min, 62°C for 1 min, and 72°C for 5 min and a final extension at 72°C for 10 min. The PCR products were inserted into the TOPO recognition site of pBAD202/D-TOPO and transformed into E. coli Top10 cells according to the manufacturer's instructions (Invitrogen). Insertion of inv in required orientation was confirmed by sequencing (Macrogen). The construct was transformed into C. sakazakii ATCC 29544 ΔompA and Δinv single mutants. The transformants were selected on LB agar plates containing 50 μg of kanamycin/ml.
To confirm the production of antisense mRNA from pBAD::r-inv construct, reverse transcriptase PCR was performed. Total RNA was isolated from cultures of WT, Δinv, WT(pBAD::r-inv), and Δinv(pBAD::r-inv) strains at mid-exponential phase (OD600 = 0.6). After removal of genomic DNA by DNase treatment, cDNA was synthesized by using 1 μg of RNA (reverse transcription master mix; Qiagen). For amplification of the region from bp 436 to 589 of C. sakazakii ATCC 29544 inv gene or its reverse complement, PCR was performed using the forward primer Primer_2_F inV and the reverse primer Primer_2_R inV (Table 2) under the following thermocycling conditions: a hot start at 95°C for 5 min, followed by amplification for 30 cycles of 95°C for I min, 54°C for 1 min, and 72°C for 3 min, with final extension at 72°C for 10 min. The PCR products were analyzed by electrophoresis in 1% agarose gel and examined on a UV transilluminator after staining with ethidium bromide.
In vivo animal study.
Four-day-old pathogen-free Sprague-Dawley rat pups (DBL, South Korea) (17) were used. To prepare the bacteria, a 2% overnight culture of C. sakazakii was inoculated into fresh BHI1/2 medium, and late exponential-phase cells (OD600 = 1.5) were collected. Cells were washed once with PBS and resuspended in PBS (2 × 108 CFU/ml). Fifty-microliter portions (107 CFU) of four different 1:1 (vol/vol) mixtures (WT–ΔompA::Kanr, WT–Δinv::Kanr, ΔompA Δinv–ΔompA::Kanr, and ΔompA Δinv–Δinv::Kanr) were fed to each group of five rat pups (n = 5) by oral gavage. Rat pups were euthanized at 24 h postinfection by carbon dioxide inhalation. For analysis of the bacterial colonization in organs, spleens and livers were removed aseptically and homogenized in 1 ml of ice-cold PBS, and the bacterial loads in each tissue were enumerated by plating on BHI1/2 agar and BHI1/2 agar containing 50 μg of kanamycin/ml. The competitive index (CI) was calculated by as follows: (the CFU of the reference strain – the CFU of the test strain)/(the CFU of the test strain). The results were expressed as the CI, where 1 indicates that two strains invade the tissue at equal rates in vivo. All experiments were performed according to Chonbuk National University Animal Care and Use Committee guidelines (approval number CBU 2013-0016).
Statistical analysis.
All experiments were performed at least in triplicate. The data from cell culture experiments were analyzed by one-way analysis of variance with Duncan's post test with a 95% confidence interval using SAS software (v9.1.3; SAS Institute, Cary, NC). P values of <0.05 were considered significant.
RESULTS
The inv homolog of C. sakazakii ATCC 29544 encodes a putative outer membrane protein.
PCR amplification of the C. sakazakii ATCC 29544 inv gene homolog using primers (Inv_FP and Inv_RP; Table 2) originating from C. sakazakii BAA-894 resulted in a 2.8-kp product which was different from that of C. sakazakii BAA-894 (3,083 bp, GenBank accession number NC_009778.1) (Fig. 1A). Sequencing of the PCR amplicon showed that the inv gene of C. sakazakii ATCC 29544 (GenBank accession number, KC602378) is 2,796 nucleotides long and that a nucleotide sequence from nucleotides 1951 to 2239 in C. sakazakii BAA-894 is absent in C. sakazakii ATCC 29544. Further sequence analysis showed that the homolog exhibits 97% identity at the nucleotide level and 92% identity at the protein level (93% of sequence coverage) with inv in other C. sakazakii strains (ESA_00987 in C. sakazakii BAA-894 and ES15_1237 in C. sakazakii ES15, 3,066 bp, NC_017933.1). The deduced inv gene product of the C. sakazakii ATCC 29544 is composed of 931 amino acids and is predicted to contain an N terminus transmembrane domain in TMpred and InterproScan analysis (data not shown). In addition, a series of C terminus intimin and invasin domains which localize on the bacterial cell surface and allow the bacteria to adhere (32) and to penetrate mammalian cells (22) were also found (see the Discussion).
FIG 1.

Construction of in-frame deletion mutants of the inv gene homolog in C. sakazakii ATCC 29544. (A) PCR amplification of the inv gene ORF. Lane M, nucleotide 1-kb size markers; lane 1, inv of C. sakazakii ATCC 29544; lane 2, inv of C. sakazakii ATCC-BAA 894. (B) Gene arrangement near inv in C. sakazakii and the Δinv mutant. H1 and H2 denote 40-nucleotide homology extensions located immediately downstream from the stop codon and upstream from the start codon of inv, respectively. The nucleotide numbers are adopted from the C. sakazakii ATCC-BAA 894 whole-genome database. (C) Domain analysis of putative Inv of C. sakazakii ATCC BAA894 and ATCC 29544 (the figure is adapted from the Pfam database [http://www.ebi.ac.uk]). Gap (96-amino-acid region in ATCC BAA 894) and domains are indicated in the figure. (D) Confirmation of inv deletion mutant construction. Lane 1, nucleotide size markers (1 kb); lane 2, WT; lane 3, ΔompA single mutant; lane 4, Δinv single mutant; lane 5, ΔompA Δinv double mutant. (E) Confirmation of ompA deletion mutant construction. Lane 1, nucleotide size markers (100 kb); lane 2, WT; lane 3, ΔompA single mutant; lane 4, Δinv single mutant; lane 5, ΔompA Δinv double mutant.
Site-specific deletion mutants (Δinv, ΔompA, and ΔompA Δinv) were constructed and confirmed by PCR.
In order to study a possible role of the inv gene product in the pathogenesis of C. sakazakii ATCC 29544, a site-specific deletion mutant was constructed by using the lambda red recombination system (Fig. 1B). In addition, a double mutant (ΔompA Δinv) was generated using the previously reported ΔompA strain (17) as a parental strain. PCR amplifications of the respective mutant strains using inv-specific primers (Table 2) which were designed 109 bp upstream (Inv_F1) and 119 bp downstream (Inv_R1) of inv showed 3.0-, 3.0-, 0.3-, and 0.3-kb bands for WT, ΔompA, Δinv, and ΔompA Δinv strains, respectively, confirming the inv gene deletion in Δinv and ΔompA Δinv mutants (Fig. 1D). In addition, PCR using the ompA-specific primers OmpA-F and OmpA-R (17) showed 1.4-, 0.3-, 1.4-, and 0.3-kb bands for WT, ΔompA, Δinv, and ΔompA Δinv strains, respectively, confirming the deletion of ompA in ΔompA and ΔompA Δinv strains (Fig. 1E). Growth of the mutants in different media (LB medium, TSB, and DMEM) was comparable to that of the WT (data not shown).
C. sakazakii putative Inv contributes to the invasion of host cells and works synergistically with OmpA for Caco-2 cells.
To determine the ability of C. sakazakii to invade various host cells (Caco-2, INT-407, and Hep-2), an invasion assay was performed (Fig. 2). We compared the invasion efficiency of each strain relative to the number of gentamicin-protected WT after 1.5 h of incubation (100%).
FIG 2.
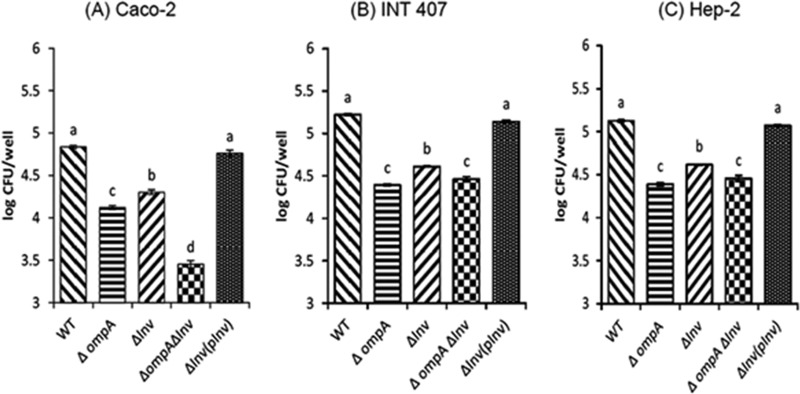
C. sakazakii ATCC 29544 invasion of Caco-2 (A), INT-407 (B) and Hep-2 (C) cells. Confluent cell monolayers were infected with bacteria at an MOI of 40 and incubated for 1.5 h, followed by a gentamicin protection assay. The cells were treated with 0.25% Triton X-100 to release the bacteria, and intracellular bacteria were enumerated. The data represent the average log CFU per well ± the standard deviation (SD) from at least three independent experiments performed in duplicate. Values of P of <0.05 were considered significantly different, and bars with the same lowercase letter are not significantly different from one another.
When Caco-2 cells were used, 4.83 ± 0.01 log CFU/well (100%) of WT was recovered after 1.5 h of incubation, while significantly lower numbers of the mutants were intracellular (4.12 ± 0.02 [17.72%] and 4.30 ± 0.02 [24.19%] log CFU/well for the ΔompA and Δinv strains, respectively) (Fig. 2A). The complementation strain showed fully restored invasion ability (4.76 ± 0.03 log CFU/well) for the inv deletion mutant.
Interestingly, when the ΔompA Δinv mutant was used, a significantly lower number of bacteria (3.45 ± 0.04 log CFU/ml) was recovered than when WT or single mutants (ΔompA and Δinv) were used. Differences in invasion efficiencies between ΔompA and Δinv mutants and between those of either of the two single mutants and a double mutant were statistically significant, suggesting an additive effect of the putative Inv and OmpA proteins in C. sakazakii invasion to Caco-2 cells.
In the case of INT-407 cells (Fig. 2B), a higher invasion rate than that in Caco-2 was observed, recovering 5.25 ± 0.01 log CFU/ml (100%) for the WT. For the single mutants, significantly lower numbers of the ΔompA (4.39 ± 0.01 log CFU/well) and Δinv (4.68 ± 0.01 log CFU/well) strains were intracellular, representing invasion rates that were 17.49 and 34.01%, respectively, of the WT rate. A similar pattern was observed in Hep-2 cells (Fig. 2C), where WT, ΔompA, and Δinv strains showed 5.19 ± 0.01 (100%), 4.38 ± 0.01 (17.26%), and 4.61 ± 0.01 (32.97%) log CFU/well, respectively. The complementation strain showed completely restored invasion ability for the inv deletion mutant in INT-407 and Hep-2 cells.
In contrast to Caco-2 cells, however, no additive effect was observed in the invasion of the ΔompA Δinv double mutant to INT-407 and Hep-2 cells showing 4.47 ± 0.01 log CFU/well (22.25%) and 4.45 ± 0.03 log CFU/well (20.81%), respectively.
Pretreatment of Caco-2 cells with purified recombinant OmpA significantly decreases invasion efficiency of the Δinv mutant.
To confirm the involvement of inv in invasion of C. sakazakii and the additive effect of putative Inv and OmpA, a blocking assay was carried out using purified recombinant OmpA (Fig. 3). Recombinant OmpA was obtained using a previously reported overexpression construct (17). After purification of His-tagged proteins, two major bands were observed in SDS-PAGE, approximately 55 and 37 kDa (Fig. 3A), that were not found in the control strain, E. coli with pBAD-TOPO (data not shown). Mass spectroscopy analysis showed that the 37-kDa protein was a degradation product of OmpA, which is 55 kDa (data not shown). A purified protein mixture was used for the blocking assay in the present study.
FIG 3.

Attenuation of Δinv mutant invasion of Caco-2 cells pretreated with purified OmpA. (A) C. sakazakii ompA mutant was overexpressed in E. coli Top10 and purified by using metal affinity resin under denaturation conditions as described in Materials and Methods. Lane 1, protein size marker; lane 2, purified recombinant OmpA. (B and C) Invasion of C. sakazakii ATCC 29544 in recombinant OmpA-pretreated Caco-2 (B) and INT-407 (C) cell monolayers. Purified recombinant OmpA protein was added to Caco-2 or INT-407 cell monolayers at 5 μg/well and incubated for 1 h, followed by three washes with PBS and invasion assay with WT, ΔompA, Δinv, and ΔompA Δinv strains. The data represent the relative percentage invasion ± the SD from at least three independent experiments performed in duplicate compared to the invasion of WT to OmpA-untreated Caco-2 or INT-407 cells that were 4 to 7 days old. P values of <0.05 were considered significantly different, and bars with same lowercase letter are not significantly different from one another.
When Caco-2 cells 4 to 7 days old were pretreated with recombinant OmpA, the invasion rate of the WT was significantly reduced to 19.64%, compared to 100% for WT in Caco-2 cells without OmpA pretreatment. Furthermore, the rate of invasion for the Δinv mutant was more significantly attenuated in OmpA-pretreated Caco-2 cells (5.74%) than in cells without OmpA pretreatment (24.29%).
In contrast, no difference in invasion rate of the ΔompA mutant was observed between Caco-2 cells pretreated (18.77%) and not pretreated (17.72%) with purified OmpA. In addition, OmpA pretreatment of Caco-2 cells made only a marginal effect on the invasion rate of the double mutant compared to the effect seen with the Δinv mutant in OmpA-pretreated Caco-2 cells.
When 14- to 17-day-old Caco-2 cells were used, significantly increased WT invasiveness (1.7-fold) was observed compared to 4- to 7-day-old Caco-2 cells. Regarding the effect of pretreatment with purified OmpA, a similar pattern of invasion efficiency was observed for the 14- to 17-day-old OmpA-pretreated Caco-2 cells challenged with WT (18.23%), ΔompA (18.10%), Δinv (6.11%), and ΔompA Δinv (4.48%) (Fig. 3C) strains compared to a 100% rate for WT in 4- to 7-day-old Caco-2 cells without OmpA pretreatment.
In INT-407 cells, when 4- to 7-day-old cells were pretreated with purified OmpA, the invasion efficiency for the WT, ΔompA, Δinv, and ΔompA Δinv strains were 12.78, 13.11, 14.41, and 12.50%, respectively, compared to 100% for the WT in INT-407 without OmpA pretreatment. In addition, OmpA-pretreated 14- to 17-day-old INT-407 cells did not show any significant differences in invasion pattern compared to 4- to 7-day-old cells recovering 20.91, 19.02, 25.94, and 19.97% for WT, ΔompA, Δinv, and ΔompA Δinv strains, respectively, compared to 100% for WT in 4- to 7-day-old INT-407 cells at without OmpA pretreatment. These results favor the presence of an additive effect involving ompA and the inv gene products in C. sakazakii ATCC 29544 invasions into Caco-2 cells but not in INT-407 cells.
Introduction of the antisense inv gene further reduced the invasion of the ΔompA mutant into Caco-2 cells.
In order to further clarify the role of the inv gene in the invasion of C. sakazakii ATCC 29544 and its relationship with ompA, we introduced the reverse complement of the inv gene (r-inv) into the WT in which the resulting antisense mRNA would form a duplex with the mRNA of chromosomal inv to inhibit functional Inv production. The construct contained the reverse complement of the complete inv gene (without a stop codon) inserted into the pBAD-TOPO vector cloning site (723 to 727 bp), which would make a transcript fusion of upstream thioredoxin, r-inv, and the downstream his-tag sequence (see Fig. S1A in the supplemental material). The correct orientation of pBAD::r-inv construct was confirmed by sequencing (see Fig. S1B in the supplemental material). In addition, reverse transcriptase PCR targeting the region from bp 436 to 589 of the inv gene or inv reverse complement resulted in amplification of 0.15 kb in WT, WT(pBAD::r-inv), and Δinv(pBAD::r-inv) strains but no amplification in the Δinv mutant (Fig. 4A), confirming the formation of the antisense mRNA from pBAD::r-inv construct.
FIG 4.
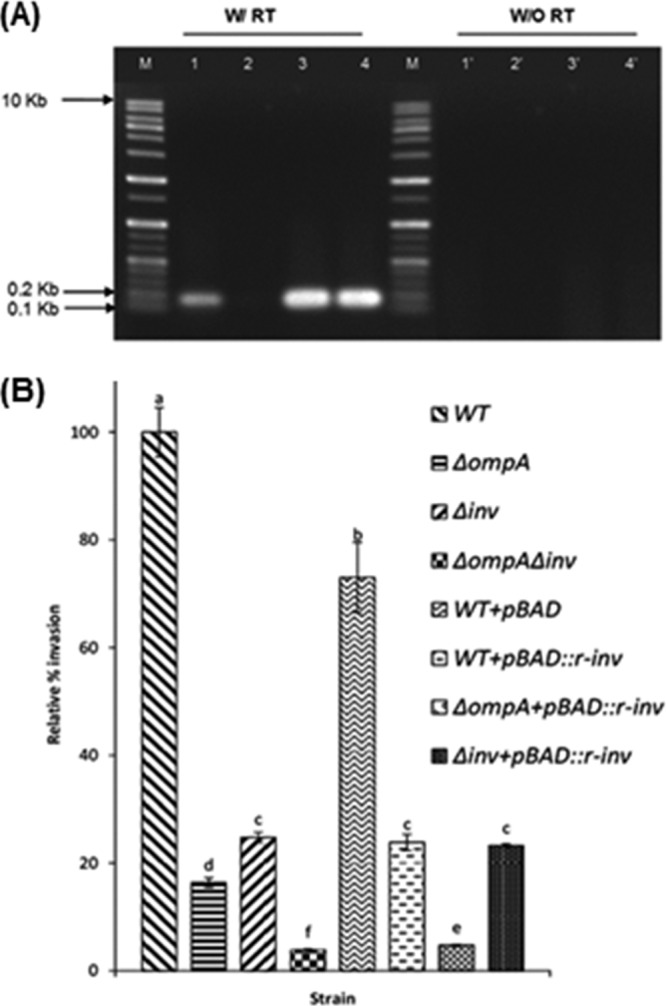
Antisense expression of C. sakazakii inv gene. (A) PCR-amplified cDNA from reverse transcription of inv or inv reverse complement (r-inv) genes. Total RNA from C. sakazakii and its isogenic strains were treated with DNase I, and cDNA was generated by reverse transcription using random hexamer primers. The inv gene (region from bp 436 to 589) or inv reverse complement were amplified by using Primer_2_F inV and Primer_2_R inV to give 153-bp PCR products. Lane M, DNA size marker; lane 1, C. sakazakii; lane 2, Δinv mutant; lane 3, C. sakazakii + pBAD::r-inv; lane 4, Δinv + pBAD::r-inv. (B) Invasion of C. sakazakii carrying pBAD-r-inv into Caco-2 cell monolayers (4 to 7 days old). The data represent the relative percentages of invasion ± the SD from at least three independent experiments performed in duplicate. P values of <0.05 were considered significantly different, and bars with same lowercase letter are not significantly different from one another.
When pBAD-TOPO without r-inv was introduced into WT as a control, the invasion rate decreased to 75% compared to WT (100%) without the plasmid. In contrast, when pBAD::r-inv was introduced into WT, the invasion ability decreased to 23.86% compared to WT without pBAD-TOPO, confirming the importance of functional Inv protein in C. sakazakii invasion. In addition, the presence of pBAD::r-inv further lowered the invasiveness of the ΔompA mutant, which was measured at 4.75%, compared to 100% for WT without pBAD-TOPO, whereas no significant difference was observed between WT(pBAD::r-inv) and Δinv (23.23%) strains. These data support an additive effect of putative Inv and OmpA protein in invasion of C. sakazakii ATCC 29544 in Caco-2 cells.
Additive effect of ompA and inv deletions is observed in tight-junction-disrupted Caco-2 cells.
To determine whether the opening of a tight junction and the age of the host cells have an effect on the invasion of C. sakazakii, we carried out invasion assays with EGTA-pretreated Caco-2 cells that were either 4 to 7 days old or 14 to 17 days old. In these experiments, we compared the invasion potential of each strain relative to that of WT (100%) in 4- to 7-day-old, EGTA-untreated Caco-2 cells (Fig. 5).
FIG 5.

Invasion of C. sakazakii into Caco-2 cells (4 to 7 days old or 14 to 17 days old) after pretreatment with EGTA. Cell monolayers were pretreated with 5 mM EGTA (or not pretreated) and used for an invasion assay. The data represent the relative percent invasion ± the SD from at least three independent experiments performed in duplicate compared to the invasion of WT to EGTA-untreated Caco-2 cells that were 4 to 7 days old. P values of <0.05 were considered significantly different, and bars with same lowercase letter are not significantly different from one another.
When the tight junctions of Caco-2 cells (4 to 7 days old) were disrupted by EGTA pretreatment (Fig. 5B), the invasion efficiency of WT (265.77%) increased ∼2.6-fold compared to EGTA-untreated Caco-2 cells that were 4 to 7 days old (100%) (Fig. 5A). For EGTA-treated Caco-2 cells that were 14 to 17 days old, the WT invasiveness (762.13%) (Fig. 5B) increased by 7.6-fold compared to untreated Caco-2 cells that were 4 to 7 days old.
When Caco-2 cells (4 to 7 days old) were pretreated with EGTA, the invasion efficiencies of the ΔompA and Δinv mutants were 26.62% ± 1.03% and 28.81% ± 5.08%, respectively (Fig. 5B). Furthermore, the invasiveness of the ΔompA Δinv double mutant (8.54% ± 1.24%) was significantly lower compared to the ΔompA and Δinv single mutants (Fig. 5B). A similar pattern was found when EGTA-pretreated 14- to 17-day-old Caco-2 cells were used; we recovered 33.15% ± 4.89%, 58.98% ± 11.31%, and 9.29% ± 1.84% for the ΔompA, Δinv, and ΔompA Δinv mutants, respectively (Fig. 5B), compared to EGTA-untreated Caco-2 cells that were 4 to 7 days old (100%) (Fig. 5A).
For EGTA-untreated Caco-2 cells that were 14 to 17 days old, a slight but significant increase in invasiveness was observed for all four strains: 194.41% ± 1.49%, 19.10% ± 5.74%, 33.88% ± 1.94%, and 5.72% ± 3.66% for the WT, ΔompA, Δinv, and ΔompA Δinv strains, respectively, compared to Caco-2 cells that were 4 to 7 days old with intact tight junctions (Fig. 5A).
Use of a transwell system confirmed the opening of tight junctions and the additive effect of ompA and inv in tight-junction-disrupted Caco-2 cells.
In order to verify the importance of the inv gene in the basolateral invasion of C. sakazakii, we carried out the invasion assay in a transwell system. First, disruption of the tight junction by EGTA treatment was confirmed by measuring the TEER values. When 14-day-old Caco-2 cells were treated with PBS, EGTA, and LPS, the TEER values were 506.5, 131.5, and 143.0 Ω/cm2, respectively, indicating disruption of the tight junction (see Fig. S2A in the supplemental material). Second, when PBS-treated 14-day-old Caco-2 cells were infected with C. sakazakii, the invasiveness values of the mutants were 13.38, 29.88, and 1.51% for the ΔompA, Δinv, and ΔompA Δinv mutants compared to the WT (100%). A similar decrease in invasion rates was observed in EGTA-pretreated Caco-2 cells, showing 25.09, 111.15, and 3.66% for the ΔompA, Δinv, and ΔompA Δinv mutants compared to the WT (509.69%). Comparable results were also observed in LPS-treated Caco-2 cells (see Fig. S2B in the supplemental material).
Complementation of either ompA or inv partially recovered the invasion efficiency of the ΔompA Δinv double mutant.
When complement strains were used, partial recovery of the invasion ability was found in the double mutant (Fig. 5). The invasion efficiencies of the ΔompA Δinv(pompA) and ΔompA Δinv(pinv) strains were 9.80% ± 0.20% and 9.29% ± 0.84%, respectively, for EGTA-untreated Caco-2 cells that were 4 to 7 days old and 13.66% ± 3.54% and 11.67% ± 2.52%, respectively, for untreated cells that were 14 to 17 days old.
In addition, when EGTA-pretreated Caco-2 cells were used, the invasion efficiencies of the ΔompA Δinv(pompA) and ΔompA Δinv(pinv) mutant strains were 12.64% ± 2.89% and 13.08% ± 1.40%, respectively, for cells that were 4 to 7 days old and 35.56 ± 6.08 and 21.36% ± 5.20%, respectively, for cells that were 14 to 17 days old.
inv is essential and exhibits an additive effect with ompA for the translocation of C. sakazakii to deeper organs in a rat pup model.
We applied a CI assay to examine the ability of C. sakazakii and its isogenic strains to translocate into deeper organs such as the liver and spleen. We first compared the translocation efficiencies of Δinv or ΔompA mutants relative to the WT using WT–Δinv::Kanr or WT–ΔompA::Kanr strain combinations (1:1 CFU ratios) to infect a single host. At 24 h postinfection, the calculated average CI values for recovered C. sakazakii administered in a WT–Δinv::Kanr (or WT–ΔompA::Kanr) strain combination inoculum from the liver (Fig. 6A) and spleen (Fig. 6B) were 4.65 (5.8) and 4.64 (6.1), respectively. Next, we tested the potential additive effect of inv and ompA mutants in vivo using ΔompA Δinv–ompA::Kanr and ΔompA Δinv–Δinv::Kanr strain combinations. The average CI values in the liver (Fig. 6A) were 0.18 and 0.19, respectively, and those in the spleen (Fig. 6B) were 0.17 and 0.35, respectively.
FIG 6.

In vivo animal study. C sakazakii WT and mutants (ΔompA::Kanr, Δinv::Kanr, and ΔompA Δinv) were grown to exponential phase and mixed in a 1:1 ratio (reference strain to test strain) to give four different mixed cultures that were fed (107 CFU) to each group of five (n = 5) rat pups (4 days old) by oral gavage. The pups were then sacrificed at 24 h postinfection. The livers and spleens were removed, homogenized, and plated on BHI1/2 agar and BHI1/2 agar containing 50 μg of kanamycin/ml. The CI score per organ [(the CFU of the reference strain – the CFU of the test strain)/(the CFU of the test strain)] was plotted with the mean CI ± the SD, where a CI of 1.0 indicates no difference in virulence between the reference and test strains. P values of <0.05 were considered significantly different, and bars with same lowercase letter are not significantly different from one another.
DISCUSSION
In an effort to identify genes related to C. sakazakii pathogenesis, inv was recognized as an outer-membrane-localized potential virulence factor in in silico genomic sequence analysis. It contains a number of bacterial Ig-like domains belonging to the Big_1 superfamily. Bacterial Ig-like domains are found in bacterial surface proteins such as intimin/invasin, which have been reported to be involved in bacterial pathogenesis (15). Previously, Invs of Yersinia or Salmonella spp. exhibiting Ig domains similar to those of the C. sakazakii inv gene product have been reported to be involved in the penetration of various host epithelial cells (Hep-2 and M cells) (22–27). Putative Inv of C. sakazakii BAA-894 exhibits homologies to those of Yersinia (50% similarity) and Salmonella (68% similarity) spp. at the protein level. This correspondence implies the possibility of a role for the putative inv gene product in C. sakazakii pathogenesis.
Based on the inv sequence of C. sakazakii BAA-894 (GenBank accession number NC_009778.1), we amplified and sequenced the inv homolog (GenBank accession number KC602378) in C. sakazakii ATCC 29544. Interestingly, when the DNA sequences are compared, the inv of C. sakazakii ATCC 29544 is significantly different from those of C. sakazakii BAA-894, ES15 (GenBank accession number NC_017933.1), and SP291 (GenBank accession number NC_020260.1). The differences were due to the deletion of a nucleotide sequence from nucleotides 1951 to 2239 that is present in C. sakazakii BAA-894, which encodes 96 amino acids (651- to 746-amino-acid region). This missing region contains an Ig-like domain (3 found in ATCC 29544 and 4 found in other sequenced C. sakazakii strains, according to the pfam database) (Fig. 1C). However, other previously reported outer membrane proteins, such as OmpA and OmpX, exhibit no difference among C. sakazakii isolates (BAA-894, ES15, SP291, and ATCC 29544), showing 100% identity at the amino acid level.
Phenotypic variation, including virulence properties among the different C. sakazakii isolates, has been well described in previous studies (12, 20, 33, 34, 35), and BAA-894 and ATCC 29544 also differ in their growth (data not shown). However, strain heterogeneity has not been elucidated at the molecular level in C. sakazakii. Currently, it is not clear whether the differences in C. sakazakii inv genes that exist among the isolates result in any functional differences. Previously, studies on inv of Y. pseudotuberculosis and Y. enterocolitica showed that, despite their high sequence homology (71% identity at the DNA level and 77% identity at the protein level), E. coli harboring Y. enterocolitica inv (92-kDa protein) was noninvasive to MDCK cells, whereas inv of Y. pseudotuberculosis (103-kDa protein) conferred the invasive property to E. coli (27).Thus, it would be interesting to determine whether the absence of the extra Ig domain found in C. sakazakii ATCC 29544 might be able to explain the observed phenotypic variation, such as the invasive property of various C. sakazakii isolates.
When the Δinv mutant was tested for invasion, a significant reduction was observed in three epithelial cell lines (Caco-2, INT-407, and Hep-2) compared to the WT strain (Fig. 2). The blocking assay (Fig. 3B and C) and antisense approach (Fig. 4B, see below) also clearly showed reduced invasion by C. sakazakii due to an absence of functional inv gene product. Furthermore, complete restoration of invasion phenotype by inv complementation strain in all three cell lines confirms the importance of the inv gene in C. sakazakii invasion. Inv is the third putative outer membrane protein, after OmpA and OmpX (17), to be identified in C. sakazakii.
Repeated trials to construct an inv-overexpressing E. coli strain using several expression vector systems with tightly controlled promoters have failed, suggesting a possible toxicity of the putative inv gene product in host E. coli. The toxicity of certain overexpressed transmembrane proteins is well described. For example, E. coli unc, which encodes the membrane-spanning subunits a (36) and b and c (37) of F-ATPase were shown to be extremely toxic, causing total growth inhibition of host E. coli [BL21(DE3)] upon IPTG (isopropyl-β-d-thiogalactopyranoside) induction (36, 37). The toxicity of overexpressed membrane proteins was suggested to be due to Sec translocan saturation (38), increased protonophoric activity, unbalanced synthesis of membrane proteins and lipids, and misinsertion of the protein into the membrane (36, 37, 38). In this regard, it is worth mentioning that predicted three-dimensional structure of putative Inv (using Phyre 2.0) mimics the beta-barrel membrane protein structure that is found in proteins of the outer membranes of Gram-negative bacteria and often functions as aqueous pores that allow the passage of small molecules (39). Interestingly, C. sakazakii outer membrane proteins, OmpA and OmpX, when successfully overexpressed in E. coli (17), turned out to be toxic to the host once they were induced (data not shown). Currently, the toxic mechanism of C. sakazakii Inv in E. coli is not clear, but the capacity to act as a porin/ion channel might be possible.
In order to assess the effect of inv gene expression on invasion, an antisense mRNA technique was used (Fig. 4). The expression of antisense mRNA from a multicopy plasmid has been successfully used in previous studies (40, 41) that were based on the silencing of the gene expression by preventing the synthesis of the protein (42, 43). We introduced here a reverse complement of the complete inv gene (r-inv) into the pBAD-TOPO vector, resulting in the production of a fused transcript that was flanked by thioredoxin- and His tag-coding sequences. We also confirmed the synthesis of antisense mRNA using reverse transcriptase PCR in an Δinv mutant transformed with pBAD-TOPO::r-inv (Fig. 4A), and the expression of r-inv in the WT strain resulted in a significant reduction in invasion efficiency and further supports the importance of functional Inv protein for the invasion of C. sakazakii into Caco-2 cells.
Tight junctions act as a physical barrier preventing the invasion of pathogens through the basolateral side of host cells (17, 18, 35). Previously, Kim et al. (17, 18) had reported a critical role for ompA and ompX in mediating the basolateral invasion of C. sakazakii. In the present study and consistent with previous data (18), the disruption of tight junctions enhanced the penetration of C. sakazakii WT into Caco-2 cells (with 2.6- and 7.6-fold increases in EGTA-treated Caco-2 cells that were 4 to 7 days old and 14 to 17 days old, respectively, compared to EGTA-untreated Caco-2 cells that were 4 to 7 days old) (Fig. 5). In contrast, the invasion potentials of the Δinv mutant (29% in EGTA-untreated Caco-2 cells that were 4 to 7 days old) did not increase as significantly as those of the WT, showing 29 and 58% invasion potentials in EGTA-treated Caco-2 cells that were 4 to 7 days old and 14 to 17 days old, respectively. If the putative inv gene product mediates C. sakazakii invasion through the apical side only in Caco-2 cells, the invasion potential of the Δinv mutant should have increased as significantly as or close to that of the WT. Comparable data were obtained when 14-day-old Caco-2 cells were pretreated with EGTA or LPS and used for invasion assay in a transwell system (see Fig. S2B in the supplemental material). Therefore, this information suggests that the putative inv gene product is essential for the basolateral invasion of C. sakazakii in Caco-2 cells.
Previously, it was reported that the C. sakazakii invasion rate increased over the age of the Caco-2 cells used (17, 18). In the present study, we showed that the Inv-dependent invasion potential also increased as the Caco-2 cells advanced in age. When the invasion potentials of the WT were compared between EGTA-untreated Caco-2 cells that were 4 to 7 days old and those that were 14 to 17 days old, a 1.9-fold increase (6.8 × 104 CFU/well and 1.3 × 105 CFU/well, respectively) was observed, whereas a significantly lower fold increase (1.1) was found with the Δinv mutant (Fig. 5A). A similar pattern was observed in EGTA-treated Caco-2 cells (2.9- and 2.1-fold increases, respectively) (Fig. 5B). These data indicate that the inv-dependent invasion potential increased as the age of the Caco-2 cells increased.
We examined the effect of the inv deletion on the invasion phenotype in the ΔompA background by using a ΔompA Δinv double mutant (Fig. 2). Interestingly, a severely attenuated invasion rate was detected for the ΔompA Δinv double mutant relative to that of either single mutant (ΔompA or Δinv), indicating an additive effect in Caco-2 invasion by C. sakazakii in the absence of both inv and ompA (Fig. 2A). This phenomenon was further supported by the results of the blocking assay using the Δinv mutant with recombinant OmpA-pretreated Caco-2 cells (Fig. 3) and antisense expression of the inv gene in the ΔompA mutant (Fig. 4). In addition, the additive effect of OmpA and Inv was also observed in tight-junction-disrupted Caco-2 cells (Fig. 5), as well as in the in vivo rat pup model (Fig. 6) (see below). The present study is the first to demonstrate that two outer membrane proteins work additively in C. sakazakii's invasion of Caco-2 cells and in an in vivo animal model.
The additive effect of the two invasin molecules, suggested in comparing the invasion potentials of single and double mutants, may imply that OmpA and Inv exhibit independent modes of action. However, the complementation study hints at the presence of different routes to penetrate Caco-2 cells by OmpA and Inv. When inv or ompA complementation in a respective single mutant background was studied, complete recovery of the invasion potential was observed (Fig. 2) (17). On the other hand, when it was performed in a double mutant, a significantly lower level of complementation was observed (Fig. 5). These data indicate a possible invasion mechanism in which OmpA and Inv are dependent upon each other.
The importance of the putative inv gene product and its additive effect with OmpA was also shown in an in vivo murine model (Fig. 6). CI values for WT versus the Δinv::Kanr strain were 4.65 and 4.64 in the liver (Fig. 6A) and spleen (Fig. 6B), respectively, while those for double mutant versus the single mutant were 0.12 (0.21 for ΔompA::Kanr) and 0.39 (0.25 for ΔompA::Kanr) in the liver (Fig. 6A) and spleen (Fig. 6B), respectively. The presence of the kanamycin gene did not compromise the virulence of the ΔompA and Δinv mutants, as determined by an in vitro invasion assay (see Fig. S3 in the supplemental material), and we also confirmed the maintenance of the kanamycin gene during infection.
Previously, it was reported that C. sakazakii OmpA and OmpX showed an additive effect in invasion using an in vitro cell culture model—INT-407 cells—but not in Caco-2 cells (17). On the other hand, the same study described no additive effect for those factors in an in vivo rat pup model (17). In the present study, an additive effect was found in the in vitro Caco-2 cell model (but not in INT-407 cells) and in an in vivo animal model. At present, there is no clear explanation as to why invasion-related proteins behave differently in different cell lines, which obviously would be an interesting subject for further study.
In conclusion, we showed here that putative Inv is essential for invasion of C. sakazakii ATCC 29544 to various mammalian epithelial cells (Caco-2, INT-407, and Hep-2) and for penetration through the basolateral side of Caco-2 cells and to deeper organs in a rat pup model. In addition, an additive effect of ompA and inv was observed in Caco-2 cells and in an animal model. We have also suggested a possible dual mode of invasion involving OmpA and Inv. This report is the first to relate the findings for two proteins working additively in vitro as well as in vivo in C. sakazakii pathogenesis.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by a National Research Foundation of Korea grant funded by the Korean government (MEST no. 2010-0026030) and the Basic Sciences Research Program through the National Research Foundation of Korea funded by the Ministry of Science, ICT & Future Planning (NRF-2012R1A1A1010596).
Footnotes
Published ahead of print 18 February 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01397-13.
REFERENCES
- 1.Iversen CM, Waddington SL, Forsythe S. 2004. Identification and phylogeny of Enterobacter sakazakii relative to Enterobacter and Citrobacter species. J. Clin. Microbiol. 42:5368–5370. 10.1128/JCM.42.11.5368-5370.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Health Organization. 2004. Enterobacter sakazakii and other microorganisms in powdered infant formula: meeting report. MRA series 6. World Health Organization, Geneva, Switzerland [Google Scholar]
- 3.Caubilla-Barron J, Hurrell ES, Townsend P, Cheetham Loc-Carrillo CO, Fayet MF, Prere Forsythe SJ. 2007. Genotypic and phenotypic analysis of Enterobacter sakazakii strains from an outbreak resulting in fatalities in a neonatal intensive care unit in France. J. Clin. Microbiol. 45:3979–3985. 10.1128/JCM.01075-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nazarowec-White M, Farber JM. 1997. Enterobacter sakazakii: a review. Int. J. Food Microbiol. 34:103–113. 10.1016/S0168-1605(96)01172-5 [DOI] [PubMed] [Google Scholar]
- 5.Van Acker J, de Smet F, Muyldermans G, Bougatef A, Naessens A, Lauwers S. 2001. Outbreak of necrotizing enterocolitis associated with Enterobacter sakazakii in powdered milk formula. J. Clin. Microbiol. 39:293–297. 10.1128/JCM.39.1.293-297.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hunter CJ, Petrosyan M, Ford HR, Prasadarao NV. 2008. Enterobacter sakazakii: an emerging pathogen in infants and neonates. Surg. Infect. 9:533–539. 10.1089/sur.2008.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mullane NR, Iversen C, Healy B, Walsh C, Whyte P, Wall PG, Quinn T, Fanning S. 2007. Enterobacter sakazakii an emerging bacterial pathogen with implications for infant health. Minerva Pediatr. 59:137–148 [PubMed] [Google Scholar]
- 8.Skovgaard N. 2007. New trends in emerging pathogens. Int. J. Food Microbiol. 120:217–224. 10.1016/j.ijfoodmicro.2007.07.046 [DOI] [PubMed] [Google Scholar]
- 9.Willis J, Robinson JE. 1988. Enterobacter sakazakii meningitis in neonates. Pediatr. Infect. Dis. J. 7:196–199. 10.1097/00006454-198803000-00012 [DOI] [PubMed] [Google Scholar]
- 10.Gurtler JB, Kornacki JL, Beuchat LR. 2005. Enterobacter sakazakii: a coliform of increased concern to infant health. Int. J. Food Microbiol. 104:1–34. 10.1016/j.ijfoodmicro.2005.02.013 [DOI] [PubMed] [Google Scholar]
- 11.Pagotto FJ, Nazarowec-White M, Bidawid S, Farber JM. 2003. Enterobacter sakazakii: infectivity and enterotoxin production in vitro and in vivo. J. Food Prot. 66:370–375 [DOI] [PubMed] [Google Scholar]
- 12.Kothary MH, McCardell BA, Frazar CD, Deer D, Tall BD. 2007. Characterization of the zinc-containing metalloprotease encoded by zpx and development of a species-specific detection method for Enterobacter sakazakii. Appl. Environ. Microbiol. 73:4142–4151. 10.1128/AEM.02729-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Townsend SM, Hurrell E, Gonzalez-Gomez I, Lowe J, Frye JG, Forsythe S, Badger JL. 2007. Enterobacter sakazakii invades brain capillary endothelial cells, persists in human macrophages influencing cytokine secretion and induces severe brain pathology in the neonatal rat. Microbiology 153:3538–3547. 10.1099/mic.0.2007/009316-0 [DOI] [PubMed] [Google Scholar]
- 14.Franco AA, Kothary MH, Gopinath G, Jarvis KG, Grim CJ, Hu L, Datta AR, McCardell BA, Tall BD. 2011. Cpa, outer membrane protease of Cronobacter sakazakii activates plasminogen and mediates resistance to serum bactericidal activity. Infect. Immun. 79:1578–1587. 10.1128/IAI.01165-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marchler-Bauer A, Zheng C, Chitsaz F, Derbyshire MK, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Lanczycki CJ, Lu F, Lu S, Marchler GH, Song JS, Thanki N, Yamashita RA, Zhang D, Bryant SH. 2013. CDD: conserved domains and protein three-dimensional structure. Nucleic Acids Res. 41:D384–D352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choi Y, Kim KP, Kim K, Choi J, Shin H, Kang DH, Ryu S. 2012. Possible roles of LysR-type transcriptional regulator (LTTR) homolog as a global regulator in Cronobacter sakazakii ATCC 29544. Int. J. Med. Microbiol. 302:270–275. 10.1016/j.ijmm.2012.06.001 [DOI] [PubMed] [Google Scholar]
- 17.Kim K, Kim KP, Choi J, Lim JA, Lee J, Hwang S, Ryu S. 2010. Outer membrane protein A (OmpA) and X (OmpX) are essential for basolateral invasion of Cronobacter sakazakii. Appl. Environ. Microbiol. 76:5188–5198. 10.1128/AEM.02498-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim KP, Loessner MJ. 2008. Enterobacter sakazakii invasion in human intestinal Caco-2 cells requires the host cell cytoskeleton and is enhanced by disruption of tight junction. Infect. Immun. 76:562–570. 10.1128/IAI.00937-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mohan Nair MK, Venkitanarayanan K. 2007. Role of bacterial OmpA and host cytoskeleton in the invasion of human intestinal epithelial cells by Enterobacter sakazakii. Pediatr. Res. 62:664–669. 10.1203/PDR.0b013e3181587864 [DOI] [PubMed] [Google Scholar]
- 20.Giri CP, Shima K, Tall BD, Sathyamoorthy V, Hanisch B, Kim KS, Kopecko DJ. 2012. Cronobacter spp. (previously Enterobacter sakazakii) invade and translocate across both cultured human intestinal epithelial cells and human brain microvascular endothelial cells. Microb. Pathol. 52:140–147. 10.1016/j.micpath.2011.10.003 [DOI] [PubMed] [Google Scholar]
- 21.Leong JM, Fournier RS, Isberg RR. 1990. Identification of the integrin binding domain of the Yersinia pseudotuberculosis invasin protein. EMBO J. 9:1979–1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Isberg RR, Voorhis DL, Falkow S. 1987. Identification of invasin: a protein that allows enteric bacteria to penetrate cultured mammalian cells. Cell 50:769–778 [DOI] [PubMed] [Google Scholar]
- 23.Amini K, Salehi TZ, Nikbakht G, Ranjbar R, Amini J, Ashrafganjooei SB. 2010. Molecular detection of invA and spv virulence genes in Salmonella enteritidis isolated from human and animals in Iran. Afr. J. Microbiol. Res. 4:2202–2210 [Google Scholar]
- 24.Galan JE, Iii RC. 1989. Cloning and molecular characterization of genes whose products allow Salmonella typhimurium to penetrate tissue culture cells. Proc. Natl. Acad. Sci. U. S. A. Microbiology 86:6383–6387. 10.1073/pnas.86.16.6383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Isberg RR, Bernes P, Wong KW. 2003. Uptake of Yersinia pseudotuberculosis into cultured cells and integrin receptor signaling, p 37–51 In O'Connor CD, Smith DGE. (ed), Microbial subversion of host cells. Cambridge University Press, Cambridge, United Kingdom [Google Scholar]
- 26.Isberg RR, Leong JM. 1990. Multiple β1 chain integrins are receptors for invasin, a protein that promotes bacterial penetration into mammalian cells. Cell 60:861–871. 10.1016/0092-8674(90)90099-Z [DOI] [PubMed] [Google Scholar]
- 27.Pepe JC, Miller VL. 1990. The Yersinia enterocolitica inv gene product is an outer membrane protein that shares epitopes with Yersinia pseudotuberculosis invasion. J. Bacteriol. 172:3780–3789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hofmann K, Stoffel W. 1993. TMbase: a database of membrane spanning proteins segments. Biol. Chem. Hoppe-Seylers 374:166 [Google Scholar]
- 29.Tusnády GE, Simon I. 2001. The HMMTOP transmembrane topology prediction server. Bioinformatics 17:849–850. 10.1093/bioinformatics/17.9.849 [DOI] [PubMed] [Google Scholar]
- 30.Chang AC, Cohen SN. 1978. Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J. Bacteriol. 134:1141–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yeung CY, Chiau JSC, Chan WT, Jiang CB, Cheng ML, Liu HL, Lee HC. 2013. In vitro prevention of Salmonella lipopolysaccharide-induced damages in epithelial barrier function by various Lactobacillus strains. Gastroenterol. Res. Pract. 2013:973209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jerse AE, Yu J, Tall BD, Kaper JB. 1990. A genetic locus of enteropathogenic Escherichia coli necessary for the production of attaching and effacing lesions on tissue culture cells. Proc. Natl. Acad. Sci. U. S. A. 87:7839–7843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mange JP, Stephan R, Borel N, Wild P, Kim KS, Pospischil A, Lehner A. 2006. Adhesive properties of Enterobacter sakazakii to human epithelial and brain microvascular endothelial cells. BMC Microbiol. 6:58–67. 10.1186/1471-2180-6-58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mittal R, Wang Y, Hunter CJ, Gonzalez-Gomez I, Prasadarao NV. 2009. Brain damage in newborn rat model of meningitis by Enterobacter sakazakii: a role for outer membrane protein A. Lab. Invest. 89:263–277. 10.1038/labinvest.2008.164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Townsend S, Hurrell E, Forsythe S. 2008. Virulence studies of Enterobacter sakazakii isolates associated with a neonatal intensive care unit outbreak. BMC Microbiol. 8:64. 10.1186/1471-2180-8-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arechaga I, Miroux B, Runswick MJ, Walker JE. 2003. Over-expression of Escherichia coli F1Fo[ens]ATPase subunit a is inhibited by instability of the uncB gene transcript. FEBS Lett. 547:97–100. 10.1016/S0014-5793(03)00677-X [DOI] [PubMed] [Google Scholar]
- 37.Miroux B, Walker JE. 1996. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 260:289–298. 10.1006/jmbi.1996.0399 [DOI] [PubMed] [Google Scholar]
- 38.Narayanan A, Ridilla M, Yernool DA. 2011. Restrained expression, a method to overproduce toxic membrane proteins by exploiting operator-repressor interactions. Protein Sci. 20:51–61. 10.1002/pro.535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sugawaras E, Nikaido H. 1994. OmpA protein of Escherichia coli outer membrane occurs in open and closed channel forms. J. Biochem. Chem. 269:17981–17987 [PubMed] [Google Scholar]
- 40.Chen G, Patten CL, Schellhorn HE. 2003. Controlled expression of an rpoS antisense RNA can inhibit RpoS function in Escherichia coli. Antimicrob. Agents Chemother. 47:3485–3493. 10.1128/AAC.47.11.3485-3493.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yinduo JI, Marra A, Rosenberg M, Woodnutt G. 1999. Regulated antisense RNA eliminates alpha-toxin virulence in Staphylococcus aureus infection. J. Bacteriol. 181:6585–6590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Louise CVR, Hans USP, Kim KM. 2007. Hitting bacteria at the heart of the central dogma: sequence-specific inhibition. Microb. Cell Fact. 6:24. 10.1186/1475-2859-6-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao RC, McIvor RS, Griffin JD, Verfaillie CM. 1997. Gene therapy for chronic myelogenous leukemia (CML): a retroviral vector that renders hematopoietic progenitors methotrexate-resistant and CML progenitors functionally normal and nontumorigenic in vivo. Blood 90:4687–4698 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.