

ABSTRACT
Serious permanent neurological or psychiatric dysfunction may result from virus infections in the central nervous system (CNS). Olfactory sensory neurons are in direct contact with the external environment, making them susceptible to infection by viruses that can enter the brain via the olfactory nerve. The rarity of full brain viral infections raises the important question of whether unique immune defense mechanisms protect the brain. Here we show that both RNA (vesicular stomatitis virus [VSV]) and DNA (cytomegalovirus [CMV]) virus inoculations of the nasal mucosa leading to olfactory bulb (OB) infection activate long-distance signaling that upregulates antiviral interferon (IFN)-stimulated gene (ISG) expression in uninfected remote regions of the brain. This signaling mechanism is dependent on IFN-α/β receptors deep within the brain, leading to the activation of a distant antiviral state that prevents infection of the caudal brain. In normal mice, VSV replication is limited to the OB, and these animals typically survive the infection. In contrast, mice lacking the IFN-α/β receptor succumbed to the infection, with VSV spreading throughout the brain. Chemical destruction of the olfactory sensory neurons blocked both virus trafficking into the OB and the IFN response in the caudal brain, indicating a direct signaling within the brain after intranasal infection. Most signaling within the brain occurs across the 20-nm synaptic cleft. The unique long-distance IFN signaling described here occurs across many millimeters within the brain and is critical for survival and normal brain function.
IMPORTANCE The olfactory mucosa can serve as a conduit for a number of viruses to enter the brain. Yet infections in the CNS rarely occur. The mechanism responsible for protecting the brain from viruses that successfully invade the OB, the first site of infection subsequent to infection of the nasal mucosa, remains elusive. Here we demonstrate that the protection is mediated by a long-distance interferon signaling, particularly IFN-β released by infected neurons in the OB. Strikingly, in the absence of neurotropic virus infection, ISGs are induced in the posterior regions of the brain, activating an antiviral state and preventing further virus invasion.
INTRODUCTION
The distance from the nasal mucosa and periphery to the brain, traversed by the olfactory nerve, is very small, and infection of the nasal mucosa is not uncommon (1), particularly in animals that use their sense of olfaction to constantly sample the external environment. A large number of viruses are capable of entering the brain through the olfactory mucosa. These viruses include rabies, Sindbis, pseudorabies, Borna, influenza, polio, herpes, and others (2–9). The brain is particularly susceptible to cytolytic agents, as the relative lack of neuronal replacement in most regions of the central nervous system (CNS) exacerbates the long-term ramifications of cytopathic viral infection. In some cases, viruses that presumably enter the brain through the olfactory system can lead to serious neurological disease, or death, both in laboratory rodents and in humans (5, 10).
Despite the presumptive high potential for viral infection leading to psychiatric or neurological consequences in the brain, these outcomes remain relatively uncommon, raising the question as to what protective mechanisms prevent virus spread to uninfected regions of the brain and the resultant neuron destruction. The fact that virus replication is limited within the olfactory bulb (OB) after intranasal infection has been well established, yet the viral and/or host factors that account for this restriction are poorly defined. One key factor that may limit virus spread in the brain is the antiviral response elicited by cytokines, including alpha/beta interferon (IFN-α/β), known as the type I IFNs. Cytokines induced by virus infection may act locally to attenuate cell-to-cell virus transmission, and they also potentiate the adaptive immune system, which ultimately eliminates pathogens.
We therefore characterized the IFN response in infected and uninfected regions of the mouse brain following intranasal infection with RNA (vesicular stomatitis virus [VSV]) and DNA (cytomegalovirus [CMV]) viruses. VSV is a rhabdovirus transmitted by insects to livestock, resulting in symptoms similar to those of foot-and-mouth disease, which generally resolve. VSV infection in humans is common in certain areas of Central America, with little adverse health consequence (11). Recombinant VSV vectors are currently being tested and used in humans as replication-competent immunogenic vaccines to evoke immunity against other pathogenic viruses such as human immunodeficiency virus (HIV) (clinical trial NCT01438606) and hepatitis B virus (HBV) (12). In addition, VSV is also being studied for its potential oncolytic application (clinical trial NCT01628640). CMV is a betaherpesvirus and is the most common infectious agent causing neurological dysfunction in the developing human brain (13–16). Developing neurons are more sensitive to the cytolytic actions of CMV, in part due to the lack of maturity of the innate and systemic immune response to CMV (17, 18). CMV is also a contributing factor in AIDS-related dementia (19).
The role of IFN-α/β in preventing virus spread throughout the brain following intranasal infection has been controversial. One recent study found that VSV did not generate an IFN-α/β response in the brain (20). In contrast, a second study suggested that localized IFN-α/β signaling within the glomerular layer of the olfactory bulb is critical for preventing spread of intranasally delivered VSV (21). More recently, a critical role was found for the IFN-stimulated gene (ISG) IFIT2 in preventing VSV spread in the brain but not in peripheral organs (22). In this study, we found that whereas virus infection was limited to the olfactory bulb after intranasal instillation, ISG induction could be detected at both the mRNA and protein levels throughout the uninfected brain in normal mice early after infection. In contrast, mice lacking the IFN-α/β receptor showed little ISG activation in the brain, and intranasal inoculation evoked infection throughout the brain and a fatal outcome. These results are consistent with a mechanism whereby IFN-α/β produced in the infected olfactory bulb induces a distant antiviral signaling that prevents virus spread to other regions of the brain.
MATERIALS AND METHODS
Viruses and reagents.
A recombinant VSV that carries a green fluorescent protein (GFP) reporter gene coupled to a second glycoprotein (G) gene in the 5th genome position was used (23, 24). A mouse CMV expressing GFP used in this study was described in detail elsewhere (18, 25, 26). For experiments involving IFN administration, cells were treated with a universal IFN-α(A/D) (PBL Interferon Source), which activates both the mouse and human IFN-α/β receptors.
Animal experiments.
VSV was applied to the external nares of Swiss-Webster adult mice (25 μl each side; 108 PFU/ml) during transient anesthesia. In other experiments, IFN-α was injected stereotactically by microsyringe directly into the lateral ventricle; control mice received sterile saline at the same site. To test the role of IFN-α/β signaling in virus spread, we used knockout (KO) mice lacking the IFN-α/β receptor (27), a gift of A. Iwasaki (Yale University). To destroy the olfactory receptor neurons, the selective toxin methimazole (50 mg/kg of body weight) was given intraperitoneally to 129Bl/SJ adult male mice (Jackson Laboratory), as described elsewhere (28, 29). Five days after olfactory neurons were eliminated, VSV was administered intranasally. Control 129Bl/SJ mice received saline instead of methimazole. Of the mice that received intranasal VSV, some were given an overdose of anesthetic and perfused transcardially with saline followed by 4% paraformaldehyde. Brains were sectioned on a cryostat and examined for GFP expression using an Olympus IX70 inverted fluorescence microscope fitted with a Spot digital camera. Contrast and gray scale were corrected with Adobe Photoshop CS5. All experiments using mice were approved by the Yale University Animal Care and Use Committee.
RT-qPCR.
One microgram of total RNA prepared from cells or snap-frozen mouse brain tissue was reverse transcribed by random hexamer priming using the TaqMan reverse transcription kit (Applied Biosystems, Foster City, CA). Quantitative PCR (qPCR) was performed using an Applied Biosystems 7500 real-time PCR system as previously described (18). Gene expression was normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression and quantified by the threshold cycle (ΔΔCT) method using the 7500 system sequence detection software (Applied Biosystems). The primer sequences used for PCR were as follows: GAPDH, 5′-TCT GGA AAG CTG TGC CGT G-3′ (sense) and 5′-CCA GTG AGC TTC CCG TTC AG-3′ (antisense); alpha interferon 2 (IFN-α2), 5′-TAC TCA GCA GAC CTT GAA CCT-3′ (sense) and 5′-CAG TCT TGG CAG CAA GTT GAC-3′ (antisense); IFN-α4, 5′-TGA TGA GCT ACT ACT GGT CAG C-3′ (sense) and 5′-GAT CTC TTA GCA CAA GGA TGG C-3′ (antisense); IFN-α5, 5′-CCT GTG TGA TGC AAC AGG TC-3′ (sense) and 5′-TCA CTC CTC CTT GCT CAA TC-3′ (antisense); IFN-β, 5′-CAG CTC CAA GAA AGG ACG AAC-3′ (sense) and 5′-GGC AGT GTA ACT CTT CTG CAT-3′ (antisense); IFN-λ2/3, 5′-AGC TGC AGG CCT TCA AAA AG-3′ (sense) and 5′-TGG GAG TGA ATG TGG CTC AG-3′ (antisense); 2′-5′ oligoadenylate synthetase 1A (OAS), 5′-GAT GTC AAA TCA GCC GTC AA-3′ (sense) and 5′-AGT GTG GTG CCT TTG CCT GA-3′ (antisense); IFN-induced protein with tetratricopeptide repeats 3 (IFIT3), 5′-GGG AAA CTA CGC CTG GAT CTA CT-3′ (sense) and 5′-CAT GCT GTA AGG ATT CGC AAAC-3′ (antisense); IFIT2, 5′-AGT ACA ACG AGT AAG GAG TCA CT-3′ (sense) and 5′-AGG CCA GTA TGT TGC ACA TGG-3′ (antisense); GFP, 5′-GAG CGC ACC ATC TTC TTC AAG-3′ (sense) and 5′-TGT CGC CCT CGA ACT TCA C-3′ (antisense); and VSV-N, 5′-GAT AGT ACC GGA GGA TTG ACG ACT A-3′ (sense) and 5′-TCA AAC CAT CCG AGC CAT TC-3′ (antisense).
Brain cultures.
Mouse brain cultures were generated from embryonic day 18 mice. Brains were removed and incubated in papain to disaggregate cells. Cells were then plated on poly-l-lysine-coated plastic 35-mm dishes. Cultures were maintained at 37°C in a 5% CO2 incubator. Human brain cells were cultured from adult human temporal lobe removed during surgery for intractable epilepsy. Human tissue use was approved by the Yale Human Investigations Committee.
Statistical analysis.
Statistical significance was determined by Student's t test. P values are indicated on figures.
RESULTS
Intranasal virus infection activates ISG expression in the brain distant from the site of virus replication.
Following intranasal virus inoculation, we used a sensitive RT-qPCR-based assay to measure VSV-encoded GFP RNA, IFN production, and ISG expression in four regions of the brain, including the olfactory bulb (OB), and respective rostral to caudal coronal slices of the whole mouse brain containing the frontal cortex (FC), hypothalamus and thalamus (HT), and cerebellum and medulla (CB). VSV-encoded RNA was exclusively limited to the OB 1 day after intranasal infection (Fig. 1A). In contrast to the case with the OB, no VSV RNA was detected in the hypothalamic and cerebellar regions of the brain of any mouse. Similarly, VSV RNA was also typically absent from the FC, with the exception of a single mouse that showed a barely detectable level of VSV RNA that was 1,000-fold lower than that found in the OB. In addition, IFN production corresponded to the site of virus infection (Fig. 1B). High mRNA levels of IFNs were detected in the OB but not in the CB. Among the multiple type I and type III IFNs we tested, IFN-β and IFN-λ were the predominant IFNs activated in response to intranasal VSV infection. Interestingly, the multiple isoforms of IFN-α that we examined were not significantly produced in the OB. Importantly, in contrast to IFN mRNA, which was limited to the OB, we found an induction of IFN-dependent antiviral response in all regions of the brain, as measured by the expression of the canonical ISGs such as the moderately inducible IFIT3 and the strongly inducible OAS (Fig. 1C and E). ISG expression in the HT and CB was particularly striking given the complete absence of detectable VSV RNA in these brain regions. Another important ISG, IFIT2, critical in mediating protection against VSV and West Nile virus infections in the CNS (22, 30), was also significantly induced in all parts of the brain (data not shown). Collectively, these data suggest that considerable amounts of ISGs are activated even in the absence of virus and IFN in the caudal region of the brain.
FIG 1.

ISG expression is induced throughout the brain following intranasal VSV and CMV infections. (A) VSV-encoded GFP RNA 1 day after intranasal administration of saline (n = 4) (−) or 5 × 106 PFU of VSV-GFP (n = 4). The RNA levels are displayed as relative to infection in the OB, which is assigned an arbitrary value of 100. (B) IFN induction 1 day after intranasal administration of saline (n = 4) (−) or 5 × 106 PFU of VSV-GFP (n = 4). The expression levels are displayed relative to that of one of the uninfected mice. (C) ISG induction 1 day after intranasal administration of saline (−) or 5 × 106 PFU of VSV-GFP. Values represent fold changes relative to saline administration. (D) ISG expression in mice 1 day after administration of intranasal saline (n = 3) (−) or CMV-GFP (n = 3). Values represent fold changes relative to saline administration. All RNA levels in panels A to D are normalized to GAPDH. Error bars represent standard errors of the means, and P values show results of Student's t test for VSV RNA (A) or OAS (C and D). (E) Representative raw RT-qPCR data showing IFIT3 and OAS expression increase in the CB between 3 h and 1 day after intranasal VSV infection. Single asterisk, IFIT3 1 day after VSV infection; double asterisks, IFIT3 3 h after VSV infection; single dagger, OAS 1 day after VSV infection; double daggers, OAS 3 h after VSV infection.
Cytomegalovirus elicits long-range IFN signaling in the brain.
Although the majority of experiments in this study were performed using VSV, we also tested whether other neurotropic viruses might induce similar protective ISG activation in the brain. Mice were intranasally inoculated with mouse CMV (25 μl/naris of 108 PFU/ml), and ISG expression was measured afterwards in a similar manner. In parallel to VSV, CMV stimulated an upregulation of ISGs 1 day postinfection (dpi) in all brain regions (Fig. 1D) despite the absence of detectable virus in the brain at this time point, as measured by histological analysis of GFP reporter gene expression (data not shown), implying that long-distance IFN signaling is universal to both RNA and DNA virus infections in the brain.
Intravenous virus infection also induces long-distance IFN signaling in the CNS.
To further explore whether the long-range IFN protection is specific to intranasal infection, we also tested other routes of infection, including intravenous VSV administration. Consistent with our hypothesis, by systemic infection we found viral RNA in the peripheral tissues such as lung and liver, and also in the olfactory mucosa (OM) and OB, but not in the CB (Fig. 2A). Importantly, ISGs, represented by OAS and IFIT2 expression, were induced in all tissues, including the CB (Fig. 2B). Collectively, these data are consistent with the protective role of long-range IFN signaling within the brain that prevents the spread of viruses arising in the olfactory system.
FIG 2.

ISG expression is activated in the cerebellum following intravenous VSV infection. (A) The RNA level of VSV-encoded nucleocapsid (N) 1 day after intravenous administration of saline (n = 4) (−) or 5 × 106 PFU of VSV (n = 4). The expression levels are displayed relative to those in mice receiving the saline control. (B) RNA level of ISG induction 1 day after intravenous administration of saline (n = 4) (−) or 5 × 106 PFU of VSV (n = 4). The expression levels are displayed relative to those in mice receiving the saline control.
Histological verification of lack of VSV spread.
After VSV inoculation of the external nares, we also examined the brain histologically for the presence of VSV by tracking expression of the viral GFP reporter. By 2 days postinoculation, VSV had entered the olfactory bulb via the olfactory nerve and was found in the periglomerular cells of the olfactory bulb, cells which the olfactory nerve terminals contact (Fig. 3A to C); GFP was also found in dendrites in the external plexiform layer (EPL). The fluorescence was associated with the plasma membrane, typical of the expression of the GFP reporter when fused to the VSV G protein. Strong virus labeling was found in cells of the granule cell layer and their dendrites that reached into the EPL. Immunocytochemical verification of neuronal labeling was done with antisera against the neuronal antigen NeuN. Granule cells infected with VSV (Fig. 3D to G) showed NeuN in the cell nucleus (Fig. 3F). Interestingly, little or no infection of mitral cells was found, based on the lack of cell body infection in the mitral cell layer and the lack of GFP reporter in mitral dendrites that extend through the EPL into the periglomerular layer. No virus was found histologically in frontal cortex or other brain regions (Fig. 3H). By 4 days postinoculation, infected granule cells in the center of the bulb were detected. Little or no virus was found caudal to the olfactory bulb at any time between inoculation and day 4 postinfection. Control mice not infected with VSV showed no fluorescence.
FIG 3.

VSV is limited to the OB, whereas IFN-induced proteins are widely expressed following intranasal infection. (A) A few periglomerular cells (arrows) show VSV-GFP, and strong expression is found in dendrites in the external plexiform layer (EPL). (B) A 4′,6-diamidino-2-phenylindole (DAPI) counterstain shows cell nuclei in the olfactory bulb. (C) Merged photomicrograph showing the images in panels A and B. Scale bar, 34 μm. (D) VSV-GFP is found in the granule cell somata (GC), indicated by three arrows, and their dendrites in the GCL. (E) NeuN immunostaining shows the nuclei of neurons. (F) Merged photomicrograph shows that VSV-GFP-labeled cell bodies contain red NeuN-labeled nuclei, indicating the infected cells are neurons. Scale bar, 22 μm. (G) Higher magnification of granule cell dendrites in the EPL. Scale bar, 9 μm. (H) Despite strong infection of neurons in the olfactory bulb, no virus was detected in cortex. Scale bar, 30 μm. GL, glomerular layer. (I) Western blot analysis shows upregulation of IFIT3 and actin (ACT) protein expression in mice administered intranasal VSV compared with saline (−) controls.
In addition to RT-qPCR analysis and fluorescence imaging, we also performed plaque assays using different regions of the brain tissues harvested from mice receiving intranasal VSV infection. As expected, tissues from HT and CB harvested at 6 h postinfection (hpi) did not generate plaques after 48 h postincubation (data not shown). In contrast, tissues harvested from OB after intranasal VSV inoculation showed strong significant infectivity of resident cultured cells within 6 h. These data support the view that VSV enters the olfactory system relatively fast but does not spread to the more caudal parts of the CNS.
To corroborate that the increased RNA levels of ISG translated to protein expression, we performed Western blot analysis of the different brain regions to examine the relative expression of IFIT3. IFIT3 protein showed the strongest upregulation in the olfactory bulb; in addition, the other three more caudal brain regions showed increased expression of IFIT3 compared with noninfected control mice, confirming the ISG induction in all parts of the brain (Fig. 3I).
Time course of ISG upregulation.
We next examined the temporal dynamics of VSV and ISG RNA expression in the different brain regions over a 4-day time course. VSV RNA was found only in the OB over a 4-day period and peaked at 2 dpi (Fig. 4A). ISG expression showed a strong increase in the OB over the 4-day time course. Importantly, despite the lack of VSV detection in other brain regions, we again found an increase in IFIT3 and OAS expression in all brain regions examined, including the FC, HT, and CB. At both 1 and 2 days postinfection, noninfected regions of the brain showed an increase of 30- to >120-fold over uninfected controls. This decreased by 3 days postinfection and returned to near saline control levels by 4 days postinfection (Fig. 4B). Therefore, intranasal VSV infection induces a substantial ISG response throughout the brain, even though the viral RNA is limited to the olfactory bulb.
FIG 4.

Time course of ISG expression after intranasal VSV-GFP infection reveals a transient widespread IFN-α/β response throughout the brain. (A) Relative GFP RNA levels in the OB, FC, HT, and CB 1 to 4 days after intranasal infection with 5 × 106 PFU of VSV-GFP (n = 2 at each time point; total n = 8). RNA levels are displayed as relative to day 1 postinfection in the OB, which is given an arbitrary value of 100. The dotted line indicates the background of the assay. (B) ISG expression in the OB, FC, HT, and CB 1 to 4 days after intranasal infection with 5 × 106 PFU of VSV-GFP. Values represent fold change between 3 h postinfection and the indicated time point. All RNA levels in panels A and B are normalized to GAPDH. Error bars represent standard errors of the means.
VSV spreads throughout the brain in the absence of an IFN-α/β response.
If induction of an IFN response throughout the brain is an important mechanism that prevents virus spread, we hypothesized that in the absence of this response, VSV would more efficiently spread caudally to the hypothalamus and cerebellum. To test this hypothesis, we infected wild-type 129 control or IFN-α/β receptor (IFNAR) knockout mice intranasally with VSV and measured VSV and ISG RNA throughout the brain. By 3 dpi, VSV RNA levels were 650- and 970-fold higher in the IFN-α/β receptor KO mice than in normal animals in the HT and CB, respectively (Fig. 5A). Unlike wild-type mice which survived infection without any adverse symptoms, intranasal administration of VSV to the IFN-α/β receptor KO mice led to CNS dysfunction characterized by reduced mobility, reduction of food intake and grooming, and, in some cases, hind limb paralysis; the outcome was lethal by 4 days postinfection (n = 4). Whereas robust ISG (OAS and IFIT3) activation was detected throughout the brain in normal mice, OAS and IFIT3 inductions were both minimal in the IFN-α/β receptor-deficient animals (Fig. 5B). Combined with the predominant induction of IFN-β and IFN-λ over IFN-α in the normal brain in response to VSV infection (Fig. 1B), these data support the view that a functional IFN-β response plays a critical role in limiting virus spread within the brain.
FIG 5.

Efficient spread of VSV throughout the brain in mice lacking the IFN-α/β receptors. (A) Relative GFP RNA in the brain 1 and 3 days postinfection in IFNAR−/− or IFNAR+/+ mice following intranasal infection with 5 × 106 PFU of VSV-GFP. RNA levels are displayed as relative to day 1 postinfection in the IFNAR+/+ OB, which is given an arbitrary value of 100. The dotted line indicates the background of the assay. Three mice were infected in each group, except in IFNAR−/− day 3, where n = 2. (B) ISG expression in the brain 1 day postinfection in IFNAR−/− or IFNAR+/+ mice following intranasal infection with 5 × 106 PFU of VSV-GFP. IFNAR+/+ mice were standardized to 100% for IFIT3 and OAS for each brain region, allowing a straightforward comparison of the relative decreased expression in IFNAR−/− mice. All RNA levels in panels A and B are normalized to GAPDH. Error bars represent standard errors of the means, and P values show results of Student's t test for VSV RNA (A) or OAS (B).
The magnitude of ISG expression in the brain is sufficient to inhibit VSV replication.
The peak magnitude of ISG expression observed in HT and CB approached 100-fold by 1 to 2 days postinoculation (Fig. 1 and 4). As this was lower than the magnitude of induction observed in the OB, we next determined whether this 100-fold-increased level of ISG induction was sufficient to inhibit virus replication in brain cells. Cultured mouse brain cells were isolated and treated with different concentrations of IFN-α for 18 h, after which ISG mRNA expression was measured. As expected, IFIT3 and OAS expression was induced in a dose-dependent manner (Fig. 6A). To further examine the ability of different amounts of IFN-α to protect brain cells from virus infection, we cultured mouse or human brain cells and infected them with VSV expressing GFP in the presence of different concentrations of IFN-α. After VSV inoculation, we compared infectivity in brain cultures with no IFN-α and in cultures with 1, 10, or 100 U/ml of IFN-α added 18 h before infection. In the absence of IFN-α, 93% and 84% of mouse and human brain cell cultures were efficiently infected with VSV at 18 h postinfection, respectively. Importantly, even at the lowest concentration, 1 U/ml of IFN-α reduced the percentage of infection to 30% and 50% of mouse and human cells, respectively, and an increased amount of 10 U/ml IFN-α almost completely obliterated virus infection in both mouse and human brain cell cultures (Fig. 6B and C). No infection was found with 100 U/ml of IFN-α in either mouse or human brain cells. These data suggest that minimal levels of IFN-α induction and ISG expression can significantly impact the outcome of VSV infection in the brain by generating a potent antiviral state.
FIG 6.
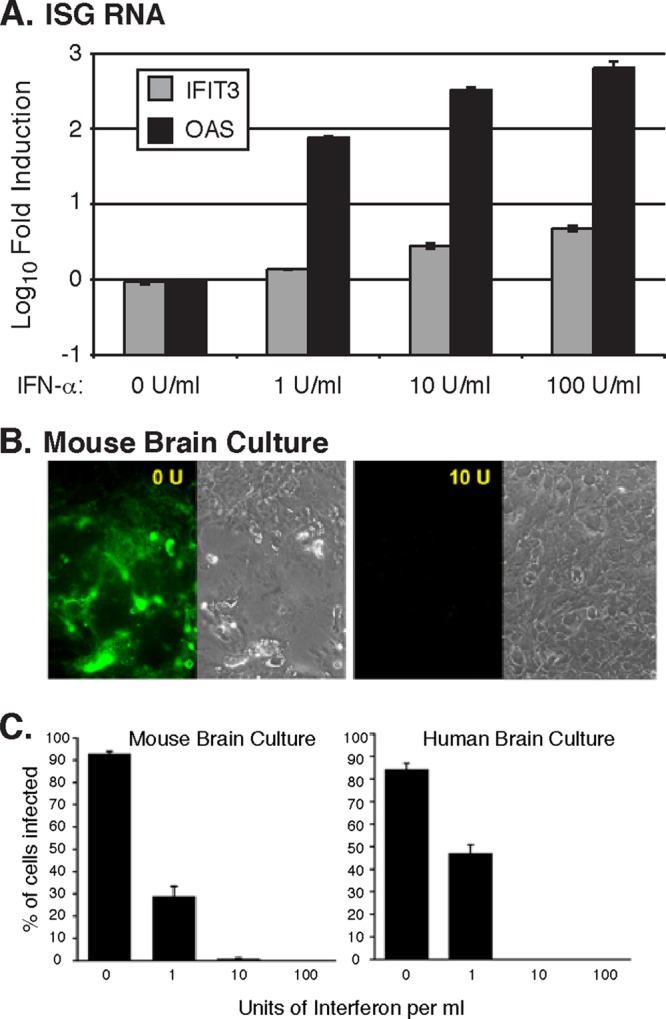
IFN-α induces ISG expression and protects against VSV infection in mouse and human brain cells. (A) IFIT3 and OAS expression in cultured mouse brain cells was measured following an 18-h stimulation with the indicated concentrations of IFN-α. Values represent fold changes relative to untreated cells. RNA levels are normalized to GAPDH, and error bars represent standard errors of the means. (B) Photomicrographs showing GFP reporter indicating VSV infection in control untreated mouse brain cultures and in cultures treated with 10 U/ml of IFN-α, which blocked infection. (C) Bar graph showing the percentage of mouse and human brain cell cultures treated with indicated concentrations of IFN-α that were infected with VSV at the multiplicity of infection of 1.
Destruction of the olfactory epithelium blocks ISG induction.
To test the hypothesis that the olfactory receptor neurons are critical for the induction of an ISG response to virus infection, we used methimazole to eliminate the olfactory neurons, as described previously (28, 29). Prior to the primary experiment, we performed pilot experiments by testing different methimazole concentrations and exposures; the parameters described here resulted in the absence of detectable VSV in the olfactory bulb after intranasal inoculation, suggesting good ablation of olfactory receptor neurons. Mice were administered methimazole (50 mg/kg of body weight intraperitoneally) plus intranasal VSV, intraperitoneal methimazole plus intranasal saline, intraperitoneal saline plus intranasal VSV, or intraperitoneal saline plus intranasal saline. Intranasal VSV (25 μl/naris of 108 PFU/ml) was administered 5 days after methimazole treatment. In all regions of the brain studied at 1 day postinoculation, VSV-infected mice lacking the olfactory receptor neurons showed a marked reduction in the ISG response to the virus infection of the nasal cavity (Fig. 7). In control mice, methimazole alone in the absence of VSV infection had little effect on ISG stimulation. This supports the view that the olfactory neuron is critical for the ISG response, probably as a conduit for the virus to enter the olfactory bulb and initiate the CNS infection, as VSV RNA was also substantially reduced in the olfactory bulb in methimazole-treated animals (data not shown). This result also corroborates the view that signaling is not dependent on a vascular or alternate pathway outside the CNS to increase ISG expression within the brain.
FIG 7.

Methimazole prevents ISG expression throughout the brain. ISG expression in the OB, FC, HT, and CB was measured 1 day after intranasal infection with 5 × 106 PFU of VSV-GFP (n = 3 per group; total n = 12). Mice treated with the olfactory receptor neuron toxin methimazole (+) and untreated mice (−) are compared. Values represent fold changes relative to control uninfected, untreated mice. All RNA levels are normalized to GAPDH, and error bars represent standard errors of the means. P values show results of Student's t test for OAS comparing VSV alone to VSV plus methimazole.
IFN in the lateral ventricle stimulates ISG expression throughout the brain.
To provide mechanistic insights into how the distant type I IFN signaling is transmitted, we tested whether IFN-α/β can spread to the back of the brain through the ventricular system. Consistent with this hypothesis, we found that when directly injected into the lateral ventricle, IFN-α (2 μl containing 200 U of IFN-α) potently stimulated ISG expression throughout the brain (Fig. 8A).
FIG 8.

IFN in the lateral ventricle induces ISG expression throughout the brain. (A) ISG expression in the OB, FC, HT, and CB 24 h after administration of 4 × 105 units of IFN-α in the lateral ventricle (n = 3). Values represent fold changes relative to saline administration. (B) IFN activity was measured via MX1 expression induced by CSF collected from uninfected mice (−) (n = 3) and mice receiving intranasal infection (VSV) (n = 3), compared to the positive control (10 U/ml of universal type I IFN). RNA levels are normalized to GAPDH, and error bars represent standard errors of the means. P values show results of Student's t test for OAS comparing saline to IFN.
Importantly, when cerebrospinal fluid (CSF) collected from mice intranasally infected with VSV was used to stimulate cells in culture, we found an induction of MX1 expression comparable to that activated by the universal type I IFN, which served as the positive control (Fig. 8B). In contrast, this IFN-like activity was not observed in CSF extracted from the mice in the control group. These data are in line with our hypothesis that the possibility of IFN transmission via CSF to the posterior brain regions at least partially accounts for one of the mechanisms that mediate long-distance brain protection. In addition, we did not detect infectious viral particles in the CSF 1 dpi as determined by plaque assay. These data collectively demonstrate that neurotropic viruses infecting the olfactory mucosa and olfactory bulb via different routes potently activate type I IFNs, in particular IFN-β, which induces ISG expression and protects the brain from infection.
DISCUSSION
A broad spectrum of viruses can traverse the short distance from the olfactory mucosa to the rostral regions of the CNS and continue with a productive infection that results in serious neurological or psychiatric dysfunction. We show here that the presence of virus in the olfactory bulb leads to long-distance type I IFN (primarily IFN-β) signaling in distant regions of the brain, resulting in an enhanced antiviral immunity which protects against virus spread to the central parts of the brain. In the absence of the IFN-α/β receptor, cells in the brain show an increased vulnerability to virus infection due to a lack of ISG induction, leading to neurological dysfunction and mortality. Not only can the olfactory system be infected from environmental incursions on the nasal mucosa but also a number of blood-borne neurotropic viruses invade the brain by transfer of the virus from the vascular system to the olfactory mucosa, with subsequent spread into the brain along the olfactory nerve (31, 32). Thus, the long-distance IFN signaling that we describe within the brain may protect the brain from infections arising from multiple origins, including blood-borne infections and those from the external environment. Consistent with this idea, intravenous infection of VSV also resulted in virus efficiently infecting the olfactory mucosa and olfactory bulb, and the long-distance IFN signaling successfully protected the caudal region of the brain from infection (Fig. 2).
The strong upregulation of ISGs we observed raises the question of the cellular source of IFN production. We and others have previously shown that glia, microglia, and neurons can participate in IFN signaling (18, 33, 34), indicating that IFN can be produced locally within the brain. Another possibility is that cells outside the brain may produce IFN-α/β after nasal infection and then signal the brain to upregulate ISGs; however, that seems unlikely as an explanation for the present results, as intraperitoneal inoculation with VSV evoked no upregulation of ISGs in the CNS (data not shown). Furthermore, intranasal inoculation in mice in which infection of the olfactory bulb had been blocked by chemically eliminating the olfactory receptor neurons induced no ISG expression in response to nasal virus. These data strongly suggest that the signaling after intranasal inoculation was not due to peripheral mechanisms but rather due to direct signaling of cells within the CNS. Within the brain, VSV has been reported to selectively infect neurons over glia (22), suggesting that IFN release may be neuronal in origin; our data using combined VSV-reporter together with immunostaining against the neuronal antigen NeuN corroborate this perspective. Many types of neurons have been reported to respond to and release IFN (33, 34). Interestingly, we detected strong induction of IFN-β as well as IFN-λ, but not multiple IFN-α isoforms, in response to VSV infection. These data, combined with those from the IFN-α/β receptor knockout mouse experiment, suggest that distant IFN signaling is most likely mediated by IFN-β.
Another critical question is how the long-distance signaling is transmitted to the back of the brain. One potential route through which IFN-α/β may spread to the back of the brain is the ventricular system. Consistent with this mechanism, IFN activity but no virus was detected in the cerebrospinal fluid, at least partially accounting for the protection. We did not exclude other possibilities, for instance, the trafficking of exosome-containing antiviral molecules which can potentially mediate antiviral activity without the local induction of IFNs (35).
As shown in the classic work of Sabin and Olitsky (36), VSV entering the adult mouse brain by nasal mucosa inoculation is blocked and does not yield symptoms, whereas the same virus injected directly into the brain is fatal. However, there is controversy over the role of IFN in protection of the brain against VSV. Our data support the view that IFN responses are critical for preventing virus spread throughout the brain. Local IFN-α/β receptor signaling within the glomerular layer of the olfactory bulb has been shown to be critical for preventing the spread of intranasally delivered VSV to the rest of the brain (21, 37). However, in previous studies, IFN-α/β-mediated tyrosine-phosphorylated STAT-1 was found exclusively in the olfactory bulb but not in other brain regions, implying a local but not widespread IFN response (21). On the basis of a more sensitive RT-qPCR-based assay for elevated ISG expression, our results suggest a previously unappreciated long-distance global IFN response that serves to protect the uninfected brain caudal to the olfactory bulb. ISG upregulation has been found in uninfected cells near VSV-infected cells in the olfactory bulb (22). The notion that IFN signaling operates within the brain and is not part of a systemic immune response is supported by our experiments showing the absence of a CNS effect with intraperitoneal VSV inoculation. In contrast to these findings, however, another study demonstrated that wild-type VSV delivered intranasally to BALB/c mice did not trigger an IFN-α/β response in the brain (20). Nevertheless, the same study found that a VSV mutant (matrix protein M51R, which is less capable of suppressing IFN-α/β induction) hyperactivates the IFN-α/β response in the brain, and this activation is associated with reduced virus spread and pathogenesis. Therefore, notwithstanding virus dose and virus and mouse strain differences among the studies, these results are also broadly consistent with the view that IFN-α/β production and signaling within the brain can prevent virus spread in the CNS.
In addition to the IFN-α/β response, IFN-γ released primarily by NK and T cells may also protect tissue from infection by activating the IFN-γ receptor; however, IFN-γ has been suggested to show minimal efficacy against VSV in the brain (38). Outside the brain, the type III IFN-λ response is important for protection of various cell types against virus infection, including cells of the lungs, gut, and liver, where the IFN-λ receptor is highly expressed (39, 40). In contrast, mouse brain cells express low levels of the IFN-λ receptor and do not respond to type III IFN signaling (41). Human brain cells express relatively low levels of IFNLR1 mRNA and, compared to IFN-α, respond only weakly to IFN-λ (42). Although we observed robust induction of IFN-λ in the OB of infected mice (Fig. 1B), the fact that IFN-α/β receptor knockout mice display robust virus replication in the brain and rapidly succumb to intranasal VSV infection argues that the roles of IFN-γ and IFN-λ in blocking virus spread in the brain are likely minor compared to that of IFN-α/β.
Although most of our experiments were based on the negative-strand RNA virus VSV, we also tested CMV, a DNA virus in the betaherpesvirus family. CMV in the olfactory mucosa also evoked an increase in ISG expression in caudal brain regions, even in the absence of any detectable CMV in those brain sites. Furthermore, the slow replication of CMV (43) would further make it improbable that this virus had spread into the caudal brain a day after inoculation. Together, these data based on two very different viruses, one a negative-strand RNA virus and the other a double-strand DNA virus, suggest that our findings may generalize to other different potentially neurotropic opportunistic viruses that can infect the nasal mucosa and then the brain.
The level of neuronal replacement in the olfactory system is much greater than in other regions of the brain or body. Olfactory receptor neurons can be replaced in as little as 6 weeks (44, 45), and the periglomerular cells within the olfactory bulb are also constantly undergoing replacement from the rostral migratory stream originating from the subventricular zone in the brain (46). Although a number of viable theories have suggested that the ongoing olfactory neuron replacement underlies some functional attribute of olfaction, an alternate but not mutually exclusive view is that this replacement relates to the ongoing attacks on olfactory neurons by viral or other pathogens.
Although VSV has been suggested to potentially show axonal transport, no axonal transport was found within the brain for the VSV G protein and GFP used in the present study (24). Similarly, no axonal transport was found for the murine CMV used in this study. Furthermore, neither infected cells expressing the virus GFP reporter protein nor viral RNA was detected outside the olfactory bulb. This was despite the fact that the limit of detection of the RT-qPCR assay used was very sensitive (105-fold less than the viral RNA levels detected in the olfactory bulb). Thus, an explanation for our observation of distant IFN signaling based on axonal transport of the virus, followed by IFN-α/β activation, seems unlikely.
Many viruses, including CMV, lymphocytic choriomeningitis virus, and rubella virus, are particularly problematic in the developing brain (1). CMV, for instance, is the most common viral pathogen causing neurological dysfunction in the developing human brain (13–16). Although the majority of adults have been infected by CMV, there are relatively few associated CNS problems despite the lifelong presence of the active or latent virus. Both intranasal VSV and CMV can be lethal in early development, but they evoke only mild symptoms in adult mice (17, 36, 47). Relevant to our findings here, one mechanism that may contribute to the developmental susceptibility of the brain to viral infections is the attenuated nature of the intrinsic immune response to infection. Interestingly, immature brain cells respond to IFN-α/β, but upregulation of IFN-α/β itself appears to be blunted in developing brain cells (18). Therefore, the long-distance IFN-mediated signaling may be a critical mediator that prevents infection of adult, but not young, brains.
ACKNOWLEDGMENTS
Grant support was provided by NIH grants NS48454, CA175577, and CA124737 (A.N.V.D.P.) and the Dana Foundation Neuroimmunology Program (M.D.R.).
We thank Yang Yang, Vitaliy Rogulin, and Prasanthi Bandi for technical assistance.
Footnotes
Published ahead of print 15 January 2014
REFERENCES
- 1.Johnson RT. 1998. Viral infections of the nervous system. Lippincott-Raven, Philadelphia, PA [Google Scholar]
- 2.Aronsson F, Robertson B, Ljunggren HG, Kristensson K. 2003. Invasion and persistence of the neuroadapted influenza virus A/WSN/33 in the mouse olfactory system. Viral Immunol. 16:415–423. 10.1089/088282403322396208 [DOI] [PubMed] [Google Scholar]
- 3.Babic N, Mettenleiter TC, Ugolini G, Flamand A, Coulon P. 1994. Propagation of pseudorabies virus in the nervous system of the mouse after intranasal inoculation. Virology 204:616–625. 10.1006/viro.1994.1576 [DOI] [PubMed] [Google Scholar]
- 4.Boggian I, Buzzacaro E, Calistri A, Calvi P, Cavaggioni A, Mucignat-Caretta C, Palu G. 2000. Asymptomatic herpes simplex type 1 virus infection of the mouse brain. J. Neurovirol. 6:303–313. 10.3109/13550280009030756 [DOI] [PubMed] [Google Scholar]
- 5.Morales JA, Herzog S, Kompter C, Frese K, Rott R. 1988. Axonal transport of Borna disease virus along olfactory pathways in spontaneously and experimentally infected rats. Med. Microbiol. Immunol. 177:51–68 [DOI] [PubMed] [Google Scholar]
- 6.Lafay F, Coulon P, Astic L, Saucier D, Riche D, Holley A, Flamand A. 1991. Spread of the CVS strain of rabies virus and of the avirulent mutant AvO1 along the olfactory pathways of the mouse after intranasal inoculation. Virology 183:320–330. 10.1016/0042-6822(91)90145-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Conomy JP, Leibovitz A, McCombs W, Stinson J. 1977. Airborne rabies encephalitis: demonstration of rabies virus in the human central nervous system. Neurology 27:67–69. 10.1212/WNL.27.1.67 [DOI] [PubMed] [Google Scholar]
- 8.Harberts E, Yao K, Wohler JE, Maric D, Ohayon J, Henkin R, Jacobson S. 2011. Human herpesvirus-6 entry into the central nervous system through the olfactory pathway. Proc. Natl. Acad. Sci. U. S. A. 108:13734–13739. 10.1073/pnas.1105143108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mori I, Nishiyama Y, Yokochi T, Kimura Y. 2005. Olfactory transmission of neurotropic viruses. J. Neurovirol. 11:129–137. 10.1080/13550280590922793 [DOI] [PubMed] [Google Scholar]
- 10.Messenger SL, Smith JS, Rupprecht CE. 2002. Emerging epidemiology of bat-associated cryptic cases of rabies in humans in the United States. Clin. Infect. Dis. 35:738–747. 10.1086/342387 [DOI] [PubMed] [Google Scholar]
- 11.Letchworth GJ, Rodriguez LL, Del Cbarrera J. 1999. Vesicular stomatitis. Vet. J. 157:239–260. 10.1053/tvjl.1998.0303 [DOI] [PubMed] [Google Scholar]
- 12.Cobleigh MA, Wei X, Robek MD. 2013. A vesicular stomatitis virus-based therapeutic vaccine generates a functional CD8 T cell response to hepatitis B virus in transgenic mice. J. Virol. 87:2969–2973. 10.1128/JVI.02111-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hanshaw JB, Scheiner AP, Moxley AW, Gaev L, Abel V. 1975. CNS sequelae of congenital cytomegalovirus infection, p 47–54 In Krugman S, Gershoen AA. (ed), Infections of the fetus and newborn infant. A. R. Liss, New York, NY [Google Scholar]
- 14.Hicks T, Fowler K, Richardson M, Dahle A, Adams L, Pass R. 1993. Congenital cytomegalovirus infection and neonatal auditory screening. J. Pediatr. 123:779–782. 10.1016/S0022-3476(05)80859-5 [DOI] [PubMed] [Google Scholar]
- 15.Ho M. 1991. Cytomegalovirus. Biology and infection. Plenum, New York, NY [Google Scholar]
- 16.Luo MH, Hannemann H, Kulkarni AS, Schwartz PH, O'Dowd JM, Fortunato EA. 2010. Human cytomegalovirus infection causes premature and abnormal differentiation of human neural progenitor cells. J. Virol. 84:3528–3541. 10.1128/JVI.02161-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van den Pol AN, Reuter J, Santarelli J. 2002. Enhanced cytomegalovirus infection of the developing brain independent of the adaptive immune system. J. Virol. 76:8842–8854. 10.1128/JVI.76.17.8842-8854.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van den Pol AN, Robek MD, Ghosh PK, Ozduman K, Bandi P, Whim MD, Wollmann G. 2007. Cytomegalovirus induces interferon-stimulated gene expression and is attenuated by interferon in the developing brain. J. Virol. 81:332–348. 10.1128/JVI.01592-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fiala M, Singer EJ, Graves MC, Tourtellotte WW, Stewart JA, Schable CA, Rhodes RH, Vinters HV. 1993. AIDS dementia complex complicated by cytomegalovirus encephalopathy. J. Neurol. 240:223–231. 10.1007/BF00818709 [DOI] [PubMed] [Google Scholar]
- 20.Trottier MD, Lyles DS, Reiss CS. 2007. Peripheral, but not central nervous system, type I interferon expression in mice in response to intranasal vesicular stomatitis virus infection. J. Neurovirol. 13:433–445. 10.1080/13550280701460565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Detje CN, Meyer T, Schmidt H, Kreuz D, Rose JK, Bechmann I, Prinz M, Kalinke U. 2009. Local type I IFN receptor signaling protects against virus spread within the central nervous system. J. Immunol. 182:2297–2304. 10.4049/jimmunol.0800596 [DOI] [PubMed] [Google Scholar]
- 22.Fensterl V, Wetzel JL, Ramachandran S, Ogino T, Stohlman SA, Bergmann CC, Diamond MS, Virgin HW, Sen GC. 2012. Interferon-induced Ifit2/ISG54 protects mice from lethal VSV neuropathogenesis. PLoS Pathog. 8(5):e1002712. 10.1371/journal.ppat.1002712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dalton KP, Rose JK. 2001. Vesicular stomatitis virus glycoprotein containing the entire green fluorescent protein on its cytoplasmic domain is incorporated efficiently into virus particles. Virology 279:414–421. 10.1006/viro.2000.0736 [DOI] [PubMed] [Google Scholar]
- 24.van den Pol AN, Dalton K, Rose J. 2002. Relative neurotropism of a recombinant rhabdovirus expressing a green fluorescent envelope glycoprotein. J. Virol. 76:1309–1327. 10.1128/JVI.76.3.1309-1327.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van den Pol AN, Mocarski E, Saederup N, Vieira J, Meier TJ. 1999. Cytomegalovirus cell tropism, replication, and gene transfer in brain. J. Neurosci. 19:10948–10965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van den Pol AN, Vieira J, Spencer DD, Santarelli JG. 2000. Mouse cytomegalovirus in developing brain tissue: analysis of 11 species with GFP-expressing recombinant virus. J. Comp. Neurol. 427:559–580. [DOI] [PubMed] [Google Scholar]
- 27.Müller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. 1994. Functional role of type I and type II interferons in antiviral defense. Science 264:1918–1921. 10.1126/science.8009221 [DOI] [PubMed] [Google Scholar]
- 28.Sammeta N, McClintock TS. 2010. Chemical stress induces the unfolded protein response in olfactory sensory neurons. J. Comp. Neurol. 518:1825–1836. 10.1002/cne.22305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Booker-Dwyer Y, Hirsh S, Zhao H. 2008. A unique cell population in the mouse olfactory bulb displays nuclear beta-catenin signaling during development and olfactory sensory neuron regeneration. Dev. Neurobiol. 68:859–869. 10.1002/dneu.20606 [DOI] [PubMed] [Google Scholar]
- 30.Cho H, Shrestha B, Sen GC, Diamond MS. 2013. A role for Ifit2 in restricting West Nile virus infection in the brain. J. Virol. 87:8363–8371. 10.1128/JVI.01097-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Charles PC, Walters E, Margolis F, Johnston RE. 1995. Mechanism of neuroinvasion of Venezuelan equine encephalitis virus in the mouse. Virology 208:662–671. 10.1006/viro.1995.1197 [DOI] [PubMed] [Google Scholar]
- 32.Cook SH, Griffin DE. 2003. Luciferase imaging of a neurotropic viral infection in intact animals. J. Virol. 77:5333–5338. 10.1128/JVI.77.9.5333-5338.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Delhaye S, Paul S, Blakqori G, Minet M, Weber F, Staeheli P, Michiels T. 2006. Neurons produce type I interferon during viral encephalitis. Proc. Natl. Acad. Sci. U. S. A. 103:7835–7840. 10.1073/pnas.0602460103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang J, Campbell IL, Zhang H. 2008. Systemic interferon-alpha regulates interferon-stimulated genes in the central nervous system. Mol. Psychiatry 13:293–301. 10.1038/sj.mp.4002013 [DOI] [PubMed] [Google Scholar]
- 35.Li J, Liu K, Liu Y, Xu Y, Zhang F, Yang H, Liu J, Pan T, Chen J, Wu M, Zhou X, Yuan Z. 2013. Exosomes mediate the cell-to-cell transmission of IFN-α-induced antiviral activity. Nat. Immunol. 14:793–803. 10.1038/ni.2647 [DOI] [PubMed] [Google Scholar]
- 36.Sabin AB, Olitsky PK. 1937. Influence of host factors on neuroinvasiveness of vesicular stomatitis virus. 1. Effect of age on the invasion of the brain by virus instilled in the nose. J. Exp. Med. 66:15–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kalinke U, Bechmann I, Detje CN. 2011. Host strategies against virus entry via the olfactory system. Virulence 2:367–370. 10.4161/viru.2.4.16138 [DOI] [PubMed] [Google Scholar]
- 38.van den Broek MF, Muller U, Huang S, Zinkernagel RM, Aguet M. 1995. Immune defense in mice lacking type I and/or type II interferon receptors. Immunol. Rev. 148:5–18. 10.1111/j.1600-065X.1995.tb00090.x [DOI] [PubMed] [Google Scholar]
- 39.Mordstein M, Neugebauer E, Ditt V, Jessen B, Rieger T, Falcone V, Sorgeloos F, Ehl S, Mayer D, Kochs G, Schwemmle M, Gunther S, Drosten C, Michiels T, Staeheli P. 2010. Lambda interferon renders epithelial cells of the respiratory and gastrointestinal tracts resistant to viral infections. J. Virol. 84:5670–5677. 10.1128/JVI.00272-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pott J, Mahlakoiv T, Mordstein M, Duerr CU, Michiels T, Stockinger S, Staeheli P, Hornef MW. 2011. IFN-lambda determines the intestinal epithelial antiviral host defense. Proc. Natl. Acad. Sci. U. S. A. 108:7944–7949. 10.1073/pnas.1100552108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sommereyns C, Paul S, Staeheli P, Michiels T. 2008. IFN-lambda (IFN-lambda) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. 4:e1000017. 10.1371/journal.ppat.1000017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pagliaccetti NE, Robek MD. 2010. Interferon-lambda in innate immunity to HBV and HCV. J. Interferon Cytokine Res. 30:585–590. 10.1089/jir.2010.0060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mocarski ES, Courcelle CT. 2001. Cytomegaloviruses and their replication, p 2629–2674 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 4th ed. Lippincott Williams & Wilkins, New York, NY [Google Scholar]
- 44.Graziadei PC, Monti-Graziadei GA. 1979. Neurogenesis and neuron regeneration in the olfactory system of mammals. I. Morphological aspects of differentiation and structural organization of the olfactory sensory neurons. J. Neurocytol. 8:1–18 [DOI] [PubMed] [Google Scholar]
- 45.Mackay-Sim A, Kittel P. 1991. Cell dynamics in the adult mouse olfactory epithelium: a quantitative autoradiographic study. J. Neurosci. 11:979–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Curtis MA, Kam M, Nannmark U, Anderson MF, Axell MZ, Wikkelso C, Holtås S, van Roon-Mom WM, Björk-Eriksson T, Nordborg C, Frisén J, Dragunow M, Faull RL, Eriksson PS. 2007. Human neuroblasts migrate to the olfactory bulb via a lateral ventricular extension. Science 315:1243–1249. 10.1126/science.1136281 [DOI] [PubMed] [Google Scholar]
- 47.van den Pol AN, Davis JN. 2013. Highly attenuated recombinant vesicular stomatitis virus VSV-12′GFP displays immunogenic and oncolytic activity. J. Virol. 87:1019–1034. 10.1128/JVI.01106-12 [DOI] [PMC free article] [PubMed] [Google Scholar]