

ABSTRACT
The chemokine receptor CCR5 is essential for HIV infection and is thus a potential target for vaccine development. However, because CCR5 is a host protein, generation of anti-CCR5 antibodies requires the breaking of immune tolerance and thus carries the risk of autoimmune responses. In this study, performed in mice, we compared 3 different immunogens representing surface domains of murine CCR5, 4 different adjuvants, and 13 different immunization protocols, with the goal of eliciting HIV-blocking activity without inducing autoimmune dysfunction. In all cases the CCR5 sequences were presented as fusions to the Flock House virus (FHV) capsid precursor protein. We found that systemic immunization and mucosal boosting elicited CCR5-specific antibodies and achieved consistent priming in Peyer's patches, where most cells showed a phenotype corresponding to activated B cells and secreted high levels of IgA, representing up to one-third of the total HIV-blocking activity. Histopathological analysis revealed mild to moderate chronic inflammation in some tissues but failed in reporting signs of autoimmune dysfunction associated with immunizations. Antisera against immunogens representing the N terminus and extracellular loops 1 and 2 (Nter1 and ECL1 and ECL2) of CCR5 were generated. All showed specific anti-HIV activity, which was stronger in the anti-ECL1 and -ECL2 sera than in the anti-Nter sera. ECL1 and ECL2 antisera induced nearly complete long-lasting CCR5 downregulation of the receptor, and especially, their IgG-depleted fractions prevented HIV infection in neutralization and transcytosis assays. In conclusion, the ECL1 and ECL2 domains could offer a promising path to achieve significant anti-HIV activity in vivo.
IMPORTANCE The study was the first to adopt a systematic strategy to compare the immunogenicities of all extracellular domains of the CCR5 molecule and to set optimal conditions leading to generation of specific antibodies in the mouse model. There were several relevant findings, which could be translated into human trials. (i) Prime (systemic) and boost (mucosal) immunization is the best protocol to induce anti-self antibodies with the expected properties. (ii) Aluminum is the best adjuvant in mice and thus can be easily used in nonhuman primates (NHP) and humans. (iii) The Flock House virus (FHV) system represents a valid delivery system, as the structure is well known and is not pathogenic for humans, and it is possible to introduce constrained regions able to elicit antibodies that recognize conformational epitopes. (iv) The best CCR5 vaccine candidate should include either extracellular loop 1 or 2 (ECL1 or ECL2), but not N terminus domains.
INTRODUCTION
CCR5 plays a key role in HIV infection as the main coreceptor involved in primary infections occurring across genital and gut mucosa (1). Antibodies (Abs) to the molecule, either natural or elicited by virus exposure or by immunization, show HIV-blocking properties and may provide both systemic and local protection from virus (2–6). Anti-CCR5 antibodies have been isolated, not only from the sera of HIV-infected or HIV-exposed seronegative (ESN) individuals, but also from many different mucosal secretions, including saliva, breast milk, and genital secretions, a striking point in antiviral protection (2, 6–8).
Different types of antibodies to CCR5 have been isolated from HIV-infected and/or ESN subjects. Most anti-CCR5 antibodies recognized the N terminus (Nter), and especially the second extracellular loop (ECL2), of the receptor, the immunodominant region involved in chemokine and HIV binding (9, 10). According to studies employing anti-CCR5 monoclonal antibodies (MAbs), some of the immunoglobulins with these two specificities competed for chemokine binding, blocked HIV docking, or, more significantly, prevented cell fusion and virus entry (10, 11). A special subset of anti-CCR5 antibodies recognized the first external loop of the CCR5 receptor (ECL1), a domain not involved in ligand binding or in HIV docking. Anti-CCR5 antibodies to the ECL1 domain have been detected in serum and mucosal secretions from ESN individuals and in some HIV-positive subjects, both men and women, supporting the hypothesis that IgG and IgA with this specificity may be involved in HIV protection or in infection control (2, 4, 6, 12).
Various studies in animal models showed that anti-CCR5 antibodies could be elicited by suitable immunization protocols employing properly conformed antigens, thereby achieving significant breaking of immune tolerance, a key factor that can restrict the generation of antibodies to self-antigens (13). The resulting antibodies to CCR5 had desirable properties in the context of protection from HIV, such as blocking virus docking or the downregulation of endogenous CCR5 receptor from the surfaces of target cells (3, 5, 14–17).
In the study reported here, we compared various immunization procedures and formulations in order to optimize the production of systemic and mucosal anti-CCR5 antibodies. Four adjuvant molecules were also compared in terms of IgG and IgA profiles and tissue toxicity. Once we determined the most effective protocol, we characterized the anti-CCR5 sera in a variety of functional assays.
MATERIALS AND METHODS
Cloning and expression of a chimeric FHV CCR5 immunogen.
The FHV Epitope Presenting System, based on a modified capsid precursor protein of the Flock House virus (FHV) (Nodaviridae), was obtained through the courtesy of F. Baralle (International Center for Genetic Engineering and Biotechnology [ICGEB], Trieste, Italy) (18–21). This vector is based on the capsid precursor protein of the insect FHV (21). It has a number of sites suitable for epitope display without substantial changes in the whole structure (which would adversely affect antigenicity). In particular, the available insertion positions are L1 (amino acids [aa] 198 to 213), L2 (aa 252 to 278), L3 (aa 127 to 145), I2 (aa 153 to 159), and I3 (aa 305), corresponding to outer loops of the protein connecting beta-sheets of the eight-stranded beta-barrel structure (22). In this way, heterologous inserts are exposed at the surface of the protein. FHV-based antigens were extremely useful in studying the influence of the stereochemistry of the presenting molecule in the immune recognition of short heterologous loops (19, 20). The system is composed of a set of five modified capsid gene sequences (cloned in the expression vector pET-3 [Novagen, Madison, WI]), in each of which the nucleotide sequence corresponding to a peptide loop was replaced by a single Bsu36I restriction site to allow the oriented insertion of a synthetic polynucleotide coding for a foreign epitope in each of five distinct sites (L1, L2, L3, I2, and I3) suitable to expose and conform epitopes in their proper three-dimensional structure (19). In previous immunizations, we found position I2 was the most immunogenic for presenting CCR5 loops, and hence, we inserted all CCR5 loops in this placement (3, 5).
Pairs of synthetic oligonucleotides corresponding to CCR5 domains (ECL1, ECL2, Nter1, and Nter2; PRIMM, Milan, Italy) were denatured at 95°C and reannealed by letting the tubes return to ambient temperature. The resulting double-stranded molecules bore 5′ overhangs suitable for ligation into the Bsu36I restriction site, and all CCR5 domains were inserted in the I2 cloning site of the FHV antigen in the vector; previous experiments showed that the I2 cloning site ensured proper folding and presentation for ECL1 expression (3). Wild-type FHV antigen was expressed and purified to be used as an experimental negative control. Recombinant proteins were purified from transformed Escherichia coli BL21(DE3) bacteria (Novagen) by enzymatic and detergent-mediated lysis and subsequent electroelution from SDS-PAGE gels, as previously described (3). Recovered antigens were dialyzed in phosphate-buffered saline (PBS) to allow their refolding and quantified. In order to ensure complete lipopolysaccharide (LPS) purification, the eluted proteins were filtered on 0.45-μm membranes after folding. Residual LPS was quantified by a colorimetric commercial assay using Toll-like receptor 4 (TLR4) binding (HEK-Blue LPS detection kit; Invitrogen, San Diego, CA, USA).
Animal inoculation and antibody collection from sera and mucosal secretions.
Thirteen immunization protocols were compared; in each protocol, four recombinant antigens (FHV ECL1, FHV ECL2, FHV Nter1, and FHV Nter2) and the negative control (CTRL) (FHV without insertion of the CCR5 region) were assayed.
Four administration routes were tested (intraperitoneal [i.p.], intramuscular [i.m.], intrarectal [i.r.], and intranasal [i.n.]), either alone or in combination; all the immunization schemes are summarized in Table 1.
TABLE 1.
Scheme of the study, showing the immunization protocols used
| Protocol | Aga dose (μg) | Route for immunization: |
Adjuvantb | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| I | II | III | IV | V | VI | ||||||||
| A | 40 | I.r. | I.r. | I.r. | I.r. | I.r. | I.r. | No | No | No | No | No | No |
| B | 40 | I.p. | I.p. | I.p. | I.p. | I.p. | I.p. | CFA | IFA | IFA | No | No | No |
| C | 40 | I.m. | I.m. | I.m. | I.m. | I.m. | I.m. | CFA | IFA | IFA | No | No | No |
| D | 40 | I.n. | I.n. | I.n. | I.n. | I.n. | I.n. | No | No | No | No | No | No |
| E | 40 | I.r. | I.r. | I.r. | I.p. | I.p. | I.p. | No | CFA | IFA | IFA | No | No |
| F | 40 | I.p. | I.p. | I.p. | I.r. | I.r. | I.r. | CFA | IFA | IFA | No | No | No |
| G | 40 | I.r. | I.r. | I.r. | I.m. | I.m. | I.m. | No | No | No | CFA | IFA | IFA |
| H | 40 | I.m. | I.m. | I.m. | I.r. | I.r. | I.r. | CFA | IFA | IFA | No | No | No |
| I | 40 | I.n. | I.n. | I.n. | I.p. | I.p. | I.p. | No | No | No | CFA | IFA | IFA |
| L | 40 | I.p. | I.p. | I.p. | I.n. | I.n. | I.n. | CFA | IFA | IFA | No | No | No |
| M | 40 | I.p. | I.p. | I.p. | I.n. | I.n. | I.n. | Mo | Mo | Mo | No | No | No |
| N | 40 | I.p. | I.p. | I.p. | I.n. | I.n. | I.n. | Al | Al | Al | No | No | No |
| O | 40 | I.p. | I.p. | I.p. | I.n. | I.n. | I.n. | RB | RB | RB | No | No | No |
The antigens (Ag) used were ECL1, ECL2, Nter1, and Nter2.
CFA, complete Freund adjuvant; IFA, incomplete Freund adjuvant; Mo, Montanide ISA 720; Al, aluminum hydroxide (alum); RB, RIBI; No, no adjuvant.
Each group of five female BALB/c mice (age, 6 weeks; 18 to 20 g) underwent immunization with 40 μg of immunogens resuspended in sterile PBS (volumes: i.p., 200 μl; i.m., 100 μl; i.n., 20 μl; i.r., 50 μl). The mice received systemic and/or mucosal inocula for six immunizations, carried out at weekly intervals from day 0 to day 42. An antigen dose of 40 μg was determined to be optimal in preliminary immunization experiments using the same routes chosen in the study.
Four different adjuvants were compared in the study; Freund's adjuvant, aluminum hydroxide, RIBI, and Montanide ISA 720 were added to immunogen preparations. All adjuvants but Montanide (Seppic, Valbonne, France) were provided by Sigma-Aldrich, Milan, Italy.
Blood samples, including those from mice immunized with FHV without CCR5 insertion (CTRL), were collected weekly from mouse tails, centrifuged at 1,500 rpm, pooled, heat inactivated at 56°C, and stored at −20°C before analysis. The antibody response upon immunization was determined by enzyme-linked immunosorbent assays (ELISAs), which quantified total immunoglobulins and IgG and IgA fractions in mouse sera (3).
Cervicovaginal wash (CVW) fluid for estrous staging and antibody determinations were collected daily by pipetting 50 μl of PBS in and out of the vagina six to eight times. The stage of the estrous cycle (estrus, metestrus, diestrus, or proestrus) for each mouse was based on smears from these washes. The washes corresponding to the estrous stage were collected twice a week. The cells were separated by centrifugation, while the supernatants were 10-fold concentrated on Ultrafree-15 Biomax 30 membranes with a cutoff of 30 kDa (Merck-Millipore, Milan, Italy). Vaginal secretions were pooled and stored at −20°C before analysis.
The institutional review board of the San Raffaele Scientific Institute, Milan, Italy, approved the investigations (IACUC no. 391).
Histopathological analysis.
Necropsies were performed on all immunized animals in protocols L to O. Skin, lymph nodes (iliac and cervical), spleen, stomach, gut, liver, kidney, lung, heart, female or male reproductive system, urinary bladder, and bone marrow were harvested, immediately fixed in 4% buffered formalin, and embedded in paraffin. Three-micrometer paraffin sections were stained with hematoxylin and eosin for histopathological examination.
Enrichment of IgA antibodies.
Agarose beads coupled with goat anti-mouse IgG antibodies (Sigma-Aldrich) were used to purify IgG from postimmune sera. The purified IgGs were obtained by elution with 0.2 M glycine-HCl buffer (pH 2), and the eluates were neutralized with 1 M Tris buffer, pH 11. The IgG-depleted fractions, which contained IgA, were concentrated on Ultrafree-15 Biomax 30 membranes (Merck-Millipore), dialyzed, and filtered through 0.22-μm filters. All IgG-depleted fractions that contained anti-CCR5-specific IgA were tested by IgG and IgA ELISAs to quantify both postimmune and specific immunoglobulins. IgA-enriched fractions, rather than purified IgA, from mouse sera and CVW samples were used, due to the small amounts of biologic fluids under examination.
Determination of total and CCR5-specific immunoglobulins. (i) Ig ELISA.
To quantify serum and mucosal immunoglobulins, microwell plates were coated with dilutions of sera or Ig-containing fractions (up to 1:128 by 2-fold dilutions) in 50 mM carbonate buffer, pH 9.5, for 1 h at 37°C. Commercial preparations of mouse Ig (total Ig, IgA, or IgG; Sigma-Aldrich) were used as standards at concentrations ranging from 4 to 0.06 μg/ml (2-fold serial dilutions) to generate a calibration curve. The plates were saturated for 1 h with 1% skim milk powder (Sigma-Aldrich) in PBS. Then, peroxidase-conjugated goat anti-mouse total Ig, IgA, or IgG (Sigma-Aldrich) was added and incubated for 30 min at 37°C. The enzymatic reaction was developed with the TMB Microwell Peroxidase Substrate System (KPL, Gaithersburg, MD, USA) and read at 492 nm.
(ii) CCR5 ELISA.
Anti-CCR5-specific responses were evaluated by CCR5 ELISA carried out on solid-phase synthetic peptides corresponding to the four CCR5 domains under study (3) (Table 2). The rest of the assay was exactly the same as for the Ig ELISA. The relative percentage of CCR5-specific Abs was calculated for each sample by comparison with Ig ELISA values.
TABLE 2.
Sequences of murine CCR5 domains expressed in the FHV system
| Domain | Amino acids | Sequencea |
|---|---|---|
| ECL1 | 89–103 | CYAANEWVFGNIMCK |
| ECL2 | 169–187 | CSPHFPHTQYHFWKSFQTLKC |
| Nter1 | 1–20 | CMDFQGSVPTYSYDIDYGMSAC |
| Nter2 | 16–35 | CYGMSAPCQKINVKQIAAQLLC |
The cysteine residues in boldface were added to maintain the three-dimensional conformation of the loop.
Isolation of Peyer's patches.
Peyer's patches (PPs) from mice immunized against ECL1 (protocol N; two immunizations at weekly intervals) and against FHV (as a negative control) were isolated from the intestinal ileum. PPs, were collected from immunized mice and processed so that a single-cell suspension could be obtained by dissociation with the syringe plunger in 15 ml RPMI plus 2% antibiotics on a Cell Strainer (BD Biosciences, Erembodegem, Belgium). The cells were washed three times and then cultured at 2 × 106/ml in 24-microwell plates in the presence of phorbol myristate acetate (PMA) (Sigma-Aldrich) at 10 μg/ml and ionomycin (Sigma-Aldrich) at 500 μg/ml to determine the extent of anti-CCR5 priming. One million cells were incubated for 4 h in B cell medium without antibiotics and then washed and incubated in complete medium supplemented with 100 μg/ml gentamicin for 24 h. The secreted immunoglobulins were quantified by ELISA, and the percentage of IgG- or IgA-expressing B cells in PPs was evaluated through cytometry. Total cells were stained with phycoerythrin (PE)-conjugated mouse anti-CD19, anti-CD138, and anti-CD40 (BD Biosciences) and fluorescein isothiocyanate (FITC)-conjugated anti-IgG (BD Biosciences) or anti-IgA (Serotec, Milan, Italy).
Cell lines.
U937; untransfected U87; and transfected U87-CD4-CXCR4, U87-CD4-CCR5 glioma, and CEM-NKR-CCR5 and CEM cell lines were obtained from the NIH AIDS Research and Reference Reagent Program (Germantown, MD, USA). HT29 cell lines used in transcytosis assays were obtained through ATCC.
Binding to CEM-NKR-CCR5 cells.
CEM-NKR-CCR5 cells (5 × 105) were incubated with the serum dilutions for 1 h at 4°C (the dilutions were made in RPMI-10% fetal calf serum [FCS]) and then washed with RPMI-10% FCS and incubated with FITC-conjugated goat anti-mouse antiserum for 30 min at 4°C.
Control of binding was done using 5 × 105 cells incubated with FITC-conjugated goat anti-mouse antibody for 30 min at 4°C. Binding competition was performed by preincubation of 10 μg MIP1β (R&D Systems, Minneapolis, MN) for 20 min at room temperature, and then serum samples were added at 1/600 dilution. The CEM cell line was used as a negative control. Ten thousand gated events were acquired using a FACSCalibur flow cytometer (Becton, Dickinson, San Jose, CA, USA), and data analysis was performed with Cytomix RXP software. Live cells initially gated by forward and side scatter were analyzed for FL1 expression. At least 10,000 events were counted. Positive control of binding was obtained with the anti-CCR5 antibody CD159 (BD Biosciences).
CCR5 downregulation assays.
CCR5 internalization was assayed using U937 cells stably expressing mouse CCR5 fused at its N terminus to the fluorogen-activating protein (FAP) HL4-MG and expressing a control receptor, B2AR, fused at its N terminus to a different fluorogen-activating protein, HL1.0.1-T01, as described previously (23, 24). Cells were grown at 37°C, 5% CO2 in RPMI 1640 plus 10% heat-inactivated fetal bovine serum (FBS), 1% penicillin-streptomycin. Receptor internalization was induced by exposing cells to ECL1, ECL2, Nter1, and FHV wild-type (WT) antisera (1/100 for 150 min to 48 h or when indicated at serial dilutions), to 1 μM RANTES for 40 min, or to 10 μM isoproterenol for 40 min. Membrane-impermeant fluorogens (MG-11p and TO1-2p) were added at a concentration of 100 nM and excited with HeNe 623-nm and argon 488-nm lasers, respectively. The cells were analyzed using a FACS Vantage SE flow cytometer with the FACS Diva option (Becton, Dickinson, Franklin Lakes, NJ).
Ex vivo CCR5 downregulation on mouse CD4+ lymphocytes was assayed on 50 μl of whole blood from immunized mice by immune detection with M20, a goat polyclonal IgG to mouse CCR5 (10 μg/ml; Santa Cruz Biotechnology, Santa Cruz, California), and a secondary anti-goat Ab conjugated with FITC (Sigma-Aldrich). All experiments were repeated three times, and the results were expressed as means with range values. CCR5-CD4 double labeling was also performed on mouse CD4+ lymphocytes. In brief, whole-blood samples (50 μl) were incubated first with goat IgG to mouse CCR5 (M20), then with FITC-conjugated rabbit anti-goat IgG, and finally with a phycoerythrin-conjugated rat MAb to mouse CD4 antigen (GK1.5; Santa Cruz Biotechnology, Santa Cruz, CA, USA). All antibodies were employed at 4 μg/assay for 30 min on ice. The relative CCR5 surface downregulation was calculated according to the following formula: 100 − [100 × (percentage of CCR5 expression stimulated − percentage of CCR5 expression by the negative control)/(percentage of CCR5 expression in the medium − percentage of CCR5 expression of the negative control)].
Block of virus infectivity. (i) SOS assay.
SOS pseudoviruses (a kind gift of D. Burton and J. Binley) were used to infect the CCR5-transfected U87 cell line, as described previously (6, 25). Briefly, the plasmid pCAGGS was used to express membrane-bound Env of the R5 isolate JR-FL. Env proteins, either as full-length gp160 or as a mutant truncated at residue 708, leaving 3 amino acids of the gp41 cytoplasmic tail, were expressed. Mutations were made to introduce cysteine at residues 501 and 605 (the SOS mutant). A mutation was also generated to replace the gp120-gp41 cleavage site REKR with the inefficiently cleaved GEKR. The plasmid pNL4-3.Luc.R-E-, expressing an HIV-1 genome fragment with frameshifts in Env and Vpr and a luciferase reporter gene in place of Nef34, was also used. Pseudoviruses were produced by transfection of 293T cells with pNL4-3.Luc.R-E- and Env-expressing pCAGGS-based plasmids. As a negative control, vesicular stomatitis virus (VSV)-G pseudovirus was used. U87 cells (2 × 104/well) were incubated with different concentrations of Abs or, when indicated, undiluted PP supernatant for 48 h; thus, SOS pseudoviruses (HIV-R5 and VSV-G) were incubated with U87 cells for 2 h, after which the cultures were washed and treated with 5 mM dithiothreitol (DTT) for 10 min, and the medium was replaced. The cells were then cultured for an additional 48 h, and luciferase activity was measured. The reaction was read with the use of a Top Count apparatus (Packard, Meriden, CT).
(ii) Transcytosis assay.
HIV-1 transcytosis was assayed using HT-29 intestinal epithelial tumor cells as previously described (26). Briefly, cells were grown as a tight, polarized monolayer (1 × 106 cells/12-mm-diameter filter unit in Dulbecco's modified Eagle's medium [DMEM] GlutaMax, 20% FCS) for 7 to 10 days on a permeable filter support (0.45-μm pore size) forming the interface between two independent chambers, the upper one bathing the apical surface of the epithelial monolayer, and the lower one bathing the basolateral surface. Several dilutions of mouse antisera were preincubated with the apical pole of the epithelial monolayer for 1 h at 37°C. A pool of preimmune sera was used as a negative control, and 2F5-IgG at 15 μg/ml was used as a positive control. HIV-1-infected human T lymphocytes were inoculated into the apical chamber (20 ng/ml; strain 93Br029). After 2 h, the extent of transcytosis was determined by quantifying p24 in the basolateral medium by ELISA (Coulter, Villepinte, France).
C-C chemokine assay.
Mouse serum chemokine concentrations (RANTES, Mip1α, and Mip1β) were determined with commercial ELISA kits (R&D Systems).
Statistical analyses.
Differences between variables were calculated using parametric tests (Student's t test for comparisons between antisera to wild-type FHV [CTRL] and to FHV containing the different CCR5 regions and analysis of variance[ANOVA] followed by the Student-Newman-Keuls posttest for multiple comparisons). Calculations were conducted using the software GraphPad Prism 5.00.288 (GraphPad Software, Inc., San Diego, CA, USA).
RESULTS
Immunogenicity, efficacy, and safety of immunization protocols.
The aim of the study was to elicit and characterize anti-CCR5 antibodies recognizing four specific domains of the homologous murine CCR5 receptor. For this purpose (Table 2), peptides corresponding to four CCR5 extracellular domains were used as immunogens: ECL1 (aa 89 to 103), ECL2 (aa 169 to 187), Nter1 (aa 1 to 20), and Nter2 (aa 16 to 35). Amino acids 1 to 20 (which contain a known CCR5 epitope [9, 16]) and 16 to 35 were assayed separately and defined as Nter1 and Nter2. The ECL3 domain was not included in the analysis because it has no direct interaction with endogenous chemokines or with HIV, and previous studies did not find any specific immune reactivity to it (10, 11).
With the goal of finding a protocol that would break immune tolerance for murine CCR5, a variety of different administration routes, adjuvants, and formulations were compared in 13 protocols, from A to O, as shown in Table 1.
Doses for inocula were determined on the basis of observations made during previous immunization experiments with ECL1 (3, 5). According to previous findings for ECL1, all CCR5 loops were inserted at the I2 position within the FHV protein (3, 5).
Protocols A to D were aimed at comparing different routes of immunization for generating specific CCR5 antibodies against the four peptides. Protocols G to L were aimed at evaluating whether combinations of immunization routes would elicit higher-titer antibodies than the single routes. Protocols M to O were aimed at defining the best adjuvant.
The four routes in protocols A to D were i.m., i.p., i.r., and i.n. I.r. and i.m. protocols failed to elicit antibodies with the desired properties; in particular, there were no significant differences with antisera to FHV alone, whereas i.p. and i.n. inoculations elicited IgA and IgG of interest, except for Nter1 and Nter2, which generated low-level CCR5-specific Igs. I.p. and i.n. immunizations showed increased antibody titers to ECL1 and ECL2 compared to antisera to CTRL (FHV), although the difference was not statistically significant (Table 3, protocols B and D).
TABLE 3.
Efficacy of immunizations evaluated at 7 days after the sixth immunization obtained with protocols B, D, I, and L
| Ag | Protocol | Mean titera |
Downregulation |
||
|---|---|---|---|---|---|
| IgA | IgG | Ex vivob | In vitroc | ||
| FHV-ECL1 | B | 20 | 100 | 39 | ND |
| D | 60 | 300 | 32.5 | ND | |
| I | 60 | 900 | 53 | 69 | |
| L | 250 | 1,300 | 69.6 | 75 | |
| FHV-ECL2 | B | 20 | 100 | 33.3 | ND |
| D | 66 | 300 | 49 | ND | |
| I | 10 | 433 | 67 | 53 | |
| L | 300 | 1,235 | 67 | 62 | |
| FHV-Nter1 | B | 0 | 30 | 22 | ND |
| D | 0 | 200 | 25.7 | ND | |
| I | 30 | 1,500 | 39 | 37 | |
| L | 100 | 750 | 43 | 43 | |
| FHV-Nter2 | B | 0 | 0 | 0 | ND |
| D | 0 | 200 | 0 | ND | |
| I | 0 | 50 | 0 | ND | |
| L | 0 | 150 | 0 | ND | |
| FHV (CTRL) | B | 0 | 0 | 0 | ND |
| D | 0 | 0 | 0 | ND | |
| I | 0 | 0 | 0 | ND | |
| L | 0 | 0 | 0 | ND | |
The values are mean titers of binding antibodies to the different CCR5 domains, expressed as a serum dilution of 1/n (where n is the number shown).
Ex vivo CCR5 downregulation is expressed as percent reduction of CCR5 molecules on the surfaces of murine PBMC from immunized animals.
In vitro CCR5 downregulation was induced in the U937 cell line by murine antisera, and it is expressed as percent receptor internalization obtained at a serum dilution of 1/40. ND, not detectable.
Based on these results, combined systemic and mucosal routes were chosen for further tests; Freund's adjuvants (one complete [CFA] and two incomplete [IFA]) were added to systemic formulations to potentiate immunogenicity. The other five protocols (from E to I) examined whether combined systemic-mucosal routes could enhance antibody generation. Antigens were administered by systemic i.m. and i.p. routes, with CFA or IFA. The four combinations of systemic and mucosal routes tested in protocols E to H gave poor results compared with B and D immunizations (data not shown). In mucosal immunizations, no adjuvants were added, because nasal immunization requires only a few microliters and addition of adjuvants would have required increased volumes.
The combination of i.n. and i.p. routes appeared to provide better results (protocols I and L), as shown in Table 3; comparison of protocols I and L showed that combined routes, and especially the i.p. plus i.n. combination (protocol L), provided the highest IgA and IgG titers, especially with ECL1 and ECL2 antigens (P ≤ 0.03 and P ≤ 0.04, respectively) (Table 3). The L protocol achieved high titers of IgG (Fig. 1A) and IgA (Fig. 1B) antibodies with three out of four antigens assayed (ECL2, ECL1, and Nter1), although the highest values were reached with ECL1 and ECL2, as shown in Fig. 1. Antisera to CTRL did not induce antibodies to any domain of CCR5. Antibody titers achieved a plateau after five immunizations, but antibodies to Nter2 were poorly elicited (IgG and IgA responses reached titers of 1/100 and 1/25 at the last immunization, respectively).
FIG 1.
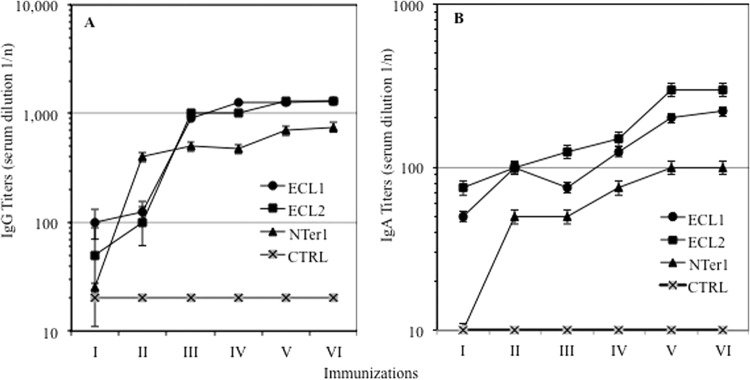
Immunogenicities of antigens after six immunizations using protocol N (three i.p. administrations followed by three i.n. administrations). Antigen Nter2 elicited no immunoglobulins (not shown). (A) IgG antibodies. (B) IgA antibodies. Mean Ig titers and standard deviations are shown.
It is of interest that the ECL2, ECL1, and Nter1 antigens, administered by combined routes (Table 3, protocols I and L), showed a marked reduction of CCR5 in CD4+ T lymphocytes from immunized mice (ex vivo) and induced CCR5 internalization on the cell membranes of U937 cells in vitro, as shown in Table 3. Moreover, the L protocol was superior to the I protocol in terms of receptor internalization; ECL1 L immunization achieved levels of ex vivo CCR5 internalization comparable to those observed with RANTES (75% versus 78% for RANTES) (Table 3). In summary, ECL1 and ECL2 were more immunogenic than the N terminus domains (either Nter1 or Nter2), and antibodies to both ECL1 and ECL2 were more effective in internalizing CCR5 than antibodies specific to Nter1. The differences among ECL1, ECL2, and Nter1 were statistically significant. In particular, ECL1 and ECL2 showed P values of ≤0.001 for all parameters considered, whereas Nter1 reached significance (P ≤ 0.04) for immunoglobulin elicitation only, and not for downregulation. Therefore, the i.p. plus i.n. scheme from protocol L was further applied to different adjuvant formulations (protocols M to O).
Mucosal antibodies were scarcely elicited by all protocols. IgGs were more abundant in CVW fluid from ECL2-L mice (range, 1:20 to 1:150), and IgA responses were found in ECL2 L and in ECL1 I and Nter1-L groups (range, 0 to 1:30). Nter2 antigen was unable to provide any response in mucosal (CVW) fluids (<1:2). Due to the low responses at mucosal sites, mucosal antibodies were not further tested.
Four different adjuvants were compared in i.p. inocula to assess their efficacies and safety profiles: Freund's adjuvant, aluminum hydroxide, RIBI, and Montanide ISA 720. Freund's adjuvant is known to elicit systemic humoral and cellular immunity, but it can often cause chronic inflammation and tissue necrosis at the injection site. Aluminum gel generates mild inflammatory responses and achieves immune memory, and therefore, it is considered a very safe adjuvant, and it is widely used in human vaccines. RIBI favors antigen presentation of hydrophobic epitopes and can address humoral responses to native epitopes rather than denatured domains. Montanide ISA 720 is a stable oil-water emulsion, similar to but less aggressive than incomplete Freund's adjuvant (27, 28). Adjuvants were chosen with the aim of eliciting high-titer antibodies to a self-antigen; other adjuvants, such as CpG and other TLR ligands, were excluded in preliminary experiments due to their poor efficacy in the mouse model and the present experimental goal.
Montanide induced a high yield in specific IgA (Fig. 2A), while RIBI obtained the largest amount of anti-CCR5 IgG (Fig. 2B); however, both adjuvants caused a nonspecific depletion of CCR5 in an ex vivo assay (Fig. 2C).
FIG 2.

Comparative efficacies of adjuvants on induction of immunoglobulins (protocols L to O). (A) Total and CCR5-specific serum IgA concentrations obtained with different adjuvants. (B) Total and CCR5-specific serum IgG concentrations obtained with different adjuvants. The mean values and standard deviations for 3 different experiments are shown. (C) Nonspecific depletion of CCR5 receptors on PBMC upon adjuvant administration. The CCR5 density on PBMC was determined by cytofluorimetric analysis before and after immunization with the individual adjuvants to determine whether each adjuvant molecule per se affects the CCR5 expression profile. The proportions express reductions in CCR5 expression on PBMC observed with each adjuvant versus saline immunization. The mean values and standard deviations for 2 different experiments are shown. (D) Immune priming in Peyer's patches with CCR5-specific IgA, total IgA, CCR5-specific IgG, and total IgG profiles. (E) IgG and IgA levels in Peyer's patches from mice immunized with FHV alone. The mean values and standard deviations for 2 independent experiments are shown. (F) HIV-blocking activities of Peyer's patch supernatants from mice immunized with FHV-ECL1-CCR5 (containing IgA specific for CCR5) and FHV alone (containing IgA not specific for CCR5).
We then performed histopathological analysis to evaluate the effects of each adjuvant on the different tissues, as described in Materials and Methods. Necropsies were performed on all immunized animals from protocols L to O. As shown in Table 4, we established the histopathological effect by evaluating the inflammatory level and the presence of granulomatous reactions; in particular, we defined three different levels: 0 when we did not observe any alterations, 1 if we observed a low level of inflammation, 2 when moderate inflammation was shown, and 3 when severe inflammation with granulomatous reaction was observed (Table 4). As determined by histopathological analyses, the inflammatory effects, where observed, did not increase in the presence of CCR5 antigen. No histopathological abnormalities were observed in brain and heart. As expected, Freund's adjuvant induced severe chronic inflammation in all organs, but not the heart and brain, while Montanide-treated animals showed moderate chronic inflammation in various organs hosting key immune regions, such as the lymph nodes, liver, kidney, stomach, gut, and urogenital tract. Aluminum and RIBI showed very low chronic inflammation levels, although only aluminum caused slight interference with the generation of specific antibodies. However, all animals were in good health, and no histopathology indicative of autoimmune responses was observed in most organs and tissues (Table 4). Representative micrographs showing the effects of RIBI, Montanide, and aluminum with and without CCR5 antigen are shown in Fig. 3. Of note, immunization with aluminum and three out of four antigens under study achieved significant receptor downregulation (Fig. 4C and D); downregulation activities achieved by Montanide and RIBI formulations were not assayed, because immunization with adjuvants alone was found to cause a nonspecific depletion of CCR5 receptors on peripheral blood mononuclear cells (PBMC) from immunized mice (Fig. 2C). In conclusion, aluminum emerged from the comparative assays as the safest and most effective adjuvant among the four that were evaluated in the study.
TABLE 4.
Histopathology scores
| Antigen | Adjuvant | Histopathology scorea |
||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Spleen and thymus |
Liver |
Kidney and lung |
Gut and pancreas |
Brain, heart, urogenital tract | Pelvis |
Stomach | ||||||||||||
| Adipose | Spleen | Thymus | Lymph | Parenchyma | Adipose | Adipose | Kidney | Lung | Lymph | Adipose | Parenchyma | Lymph | Adipose | Lymph | ||||
| CCR5 | Montanide | 1.5 | 0 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1.5 | 0 | 2 |
| Aluminum | 0.5 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0.5 | 0 | 0 | 0 | 0.5 | 0 | 1 | |
| RIBI | 0.5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Freund's | 1.5 | 2 | 0 | 3 | 1 | 3 | 3 | 0 | 1 | 2 | 3 | 2 | 2 | 0 | 3 | 1 | 3 | |
| Ctrl | Montanide | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 2 | 0 | 2 |
| Aluminum | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 1.5 | |
| RIBI | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Freund's | 3 | 3 | 0 | 3 | 3 | 3 | 3 | 0 | 2 | 3 | 3 | 2 | 3 | 0 | 3 | 2 | 3 | |
| No adjuvant | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
0, normal; 1, mild inflammation; 2, moderate inflammation; 3, severe inflammation with granulomatous reaction.
FIG 3.
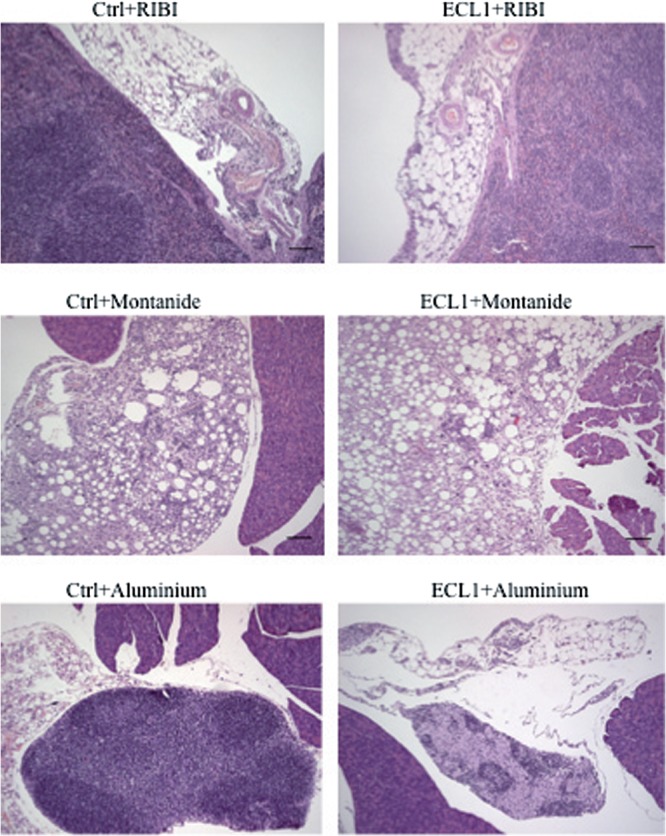
Representative histopathology analysis performed on perisplenic adipose tissues (top row), peripancreatic adipose tissues (middle row), and lymph node tissues (bottom row) from mice receiving ECL1 antigen in complete formulation or as the sole immunization adjuvant. All the images show moderate to severe chronic inflammation of the adipose tissue. RIBI plus ECL1, protocol O; Montanide plus ECL1, protocol M; aluminum plus ECL1, protocol N.
FIG 4.

CCR5 binding and downregulation in in vitro assays. (A) Binding to CCR5-expressing CEM-NKR cells by all antisera (protocol N) at dilutions ranging from 1:300 to 1:900. All antisera diluted 1:600 were also used when MIP1β was preincubated on CEM-NKR cells; 2D7, MAb to the ECL2 domain, was used as a positive control. The values indicate the percentages of mean fluorescence intensity per antiserum compared to that obtained with 2D7, which represents 100% binding. (B) A representative experiment showing binding to CCR5-CEM cells. Thick line, ECL1 antiserum (protocol N); thin line, mouse antiserum to CTRL (FHV wild type); dashed line, secondary antibody to mouse Igs. (C and D) CCR5 downregulation in CCR5-expressing U937 cells, measured with FAP technology. Shown are ECL1, ECL2, Nter1, and CTRL (FHV) antisera (protocol N) at 150 min (C) and at 48 h (D). Unstained cells and cells stained with fluorogen only (MG-11p) are also shown.
Immune priming of B cells within Peyer's patches.
In order to verify the occurrence of immune priming specific to CCR5, a small set of mice immunized by the ECL1 N protocol (ECL1 as the antigen and aluminum as the adjuvant in an i.p. plus i.n. immunization scheme) were sacrificed, and the Peyer's patches were isolated and cultured for 24 h. The cultured cells displayed a CD19+ (25%) CD138+ (36%) CD40+ (67%) phenotype, corresponding to activated B lymphocytes. Cell supernatants from such B cell populations were found to contain high titers of CCR5-specific IgA, confirming that mucosal priming of B cells had taken place (Fig. 2D). As expected, IgG was not elicited in PP supernatants. In addition, mice immunized with FHV alone showed similar levels of total IgA but were completely negative for CCR5-specific IgA, as shown in Fig. 2E. To evaluate if such PP supernatants could block HIV infection, the supernatants obtained from mice immunized with FHV-ECL1-CCR5 and FHV alone were tested in neutralization assays. As shown in Fig. 2F, the PP supernatant positive for IgA to CCR5 and not the negative supernatant specifically blocked SOS virus.
Biological characterization of antibodies to CCR5.
Once protocol N was selected as the optimal immunization method, the resulting anti-CCR5 antibodies and cells were used in a variety of functional bioassays, including CCR5 receptor binding and downregulation in vitro and ex vivo, blocking of HIV infection, and inhibition of virus transcytosis. Due to poor immunogenicity, antisera to the Nter2 antigen were not tested. IgA antibodies were measured directly in serum IgG-depleted fractions, due to their relative paucity of IgG. For the same reason, antibodies from the CVW fluids were not tested in bioassays.
Binding to CCR5 target cells.
Antisera obtained from ECL1 N, ECL2 N, and Nter1 N immunizations were tested for the presence of antibodies that could bind to CCR5-CEM-NKR cells. As shown in Fig. 4A, all three antisera were positive in the test and showed equivalent binding at 1:300, 1:600, and 1:900 dilutions. Anti-CCR5 antibody 2D7, recognizing the ECL2 domain, was used as a positive control; a pool of antisera to FHV, the carrier antigen, was used as a negative control (CTRL) (Fig. 4A). To verify the specificity of binding, CEM-NKR cells were preincubated with MIP1β before performing flow cytometry assays; as expected, no competition with antisera to Nter1 N was observed, while specific and strong competition was shown with ECL2 N antisera (P ≤ 0.001). Decreased binding was observed with antisera to ECL1 N, as well, although it was not statistically significant, probably due to steric hindrance, as previously shown with natural human antibodies (4) (Fig. 4A). As negative controls, CEM cells, which are negative for CCR5, were used (data not shown). Representative results obtained with antisera to ECL1 N are shown in Fig. 4B.
In vitro downregulation of CCR5 receptor.
Antisera to ECL1 N, ECL2 N, and Nter1 N also induced CCR5 receptor downregulation over short and long incubation times (150 min and 48 h), as measured using FAP technology (23, 24). All sera were tested at a 1:100 dilution. The assay used U937 cells that stably expressed a recombinant form of CCR5 receptor carrying a FAP domain at its N terminus and a control receptor, B2AR, carrying a different FAP at its N terminus. Experiments were performed in the presence of thiazole orange and malachite derivatives emitting either yellow-green (530-nm) or red-orange (650-nm) fluorescence when bound to their cognate FAPs.
When exposed to its natural ligand, RANTES, CCR5 was promptly internalized (70%). After 150 min of incubation, ECL1 antisera also caused receptor internalization (40%), while ECL2 and Nter1 achieved 30% and 18% downregulation, respectively (Fig. 4C). The incubation time was chosen because natural antibodies to the ECL1 domain of CCR5 begin receptor internalization at 150 min (L. Lopalco, unpublished data). Mouse antisera to FHV (CTRL) did not induce receptor downregulation (Fig. 4C).
CCR5 internalization persisted after 48 h of incubation, although ECL1 and ECL2 antibodies achieved complete CCR5 internalization, while Nter1 achieved 93%, as indicated by fluorescence peak shifts observed at 685 nm (Fig. 4D). Mouse antisera to FHV (CTRL) induced a very low level of receptor downregulation (4%), as shown in Fig. 4D. In addition, ECL1 antibodies induced maximal internalization at up to 1:350 dilution, ECL2 at 1:300 dilution, and Nter1 at 1:150 dilution. The specificity of CCR5 internalization was previously demonstrated and recently published (29). Briefly, the control B2AR receptor did not respond to RANTES but was promptly internalized in response to isoproterenol (29).
Ex vivo downregulation of CCR5 receptor.
In ex vivo internalization, a significant reduction of CCR5 expression on the cell membranes of CD4+ T lymphocytes was obtained by sera from protocol L after the last immunization. As summarized in Table 3, antibodies to ECL1 and ECL2 achieved the highest reductions (69% and 67%, respectively), while Nter2 did not produce any downregulation. Similar CCR5 downregulation was also observed when IgG- and IgA-enriched fractions (N protocol; immunization VI) were incubated with PBMC from nonimmunized control mice (at concentrations corresponding to a 1:10 serum dilution), as a confirmation of previous results obtained with anti-ECL1 antibodies (3, 5).
Blocking of HIV infection.
Mouse antisera elicited by all antigens (ECL1, ECL2, Nter1, and CTRL) were assayed in neutralization assays to assess their HIV-blocking properties. Antisera were assayed at 2-fold dilutions ranging from 1:20 to 1:160. ECL2 and ECL1 showed almost complete inhibition with high levels of statistical significance, while Nter1 reduced HIV infectivity no more than 60% and was not statistically significant in IgG-depleted fractions (Fig. 5A). In ECL1 and ECL2 antisera, both IgG and IgA fractions contributed to inhibition, because IgG-depleted fractions retained more than half of the total activity for both antigens; conversely, IgA activity accounted for less than a third of the total inhibition in Nter1 antiserum (Fig. 5B). All antisera did not block VSV-G control virus (Fig. 5C). Maraviroc was used as a control, as shown in Fig. 5D. As previously demonstrated, mouse anti-CCR5 antibodies specifically block HIV-1 R5 but not HIV-1 R5-X4 viruses (3, 5). To exclude the possibility that the observed inhibition may have been due to high serum β-chemokine concentrations, we measured the concentrations of RANTES, Mip1α, and Mip1β in all antisera, including those elicited to FHV (CTRL). The concentrations of all three chemokines were comparable among all sera (data not shown).
FIG 5.

HIV-blocking activities of all antisera. (A) HIV-JRFL-SOS-R5 pseudovirus-blocking activities of total antisera. (B) HIV-JRFL-SOS-R5 pseudovirus-blocking activities of IgG-depleted fractions. (C) VSV-G pseudovirus-blocking activities of all antisera. CTRL, antiserum to FHV carrier protein. Statistically significant differences between CTRL and the other antisera are shown. ns, not significant. (D) HIV-JRFL-SOS-R5 pseudovirus-blocking curve for maraviroc. (E) Blocking of HIV transcytosis of 93Br029 (B-R5) or of 91US054 (B-R5-X4) HIV strains by anti-CCR5 antisera. FHV antiserum was used as a negative control (CTRL); 2F5, an anti-gp41 MAb IgG, was used as a positive control for both viruses. The error bars indicate standard deviations.
Inhibition of HIV transcytosis.
A further biological activity of anti-CCR5 antibodies, inhibition of virus transcytosis across mucosae, was assayed in a model of the human mucosa (26). As shown in Fig. 5E, all three antisera nearly abolished HIV transcytosis of the R5-B virus strain 93Br029 and achieved inhibition comparable or even superior to that of the control antibody (2F5 MAb). Of note, ECL1 maintained the same inhibition activity at 1:75 dilution and showed the highest level of statistical significance (P ≤ 0.007) compared to the control (CTRL), while ECL2 and Nter1 had P values of ≤0.02 and ≤0.03, respectively. All sera were unable to block the X4-B strain 91US054, confirming their specificity for CCR5 receptor (Fig. 5E). Antisera to CTRL (wild-type FHV) did not affect viral infectivity either in classical neutralization or in transcytosis, as shown in Fig. 5A, B, C, and E.
DISCUSSION
Despite worldwide efforts, no effective AIDS vaccine has yet been formulated. In view of the difficulties encountered using traditional vaccine development approaches, nontraditional strategies must be explored.
CCR5 is required for initial virus entry in most primary infections, making CCR5 an attractive target for vaccine development (11, 30, 31).
The study reported here was inspired by our observation that some exposed, uninfected sexual partners of HIV-positive individuals generated anti-CCR5 antibodies (2, 4). Our previous studies, performed in small-animal models, showed the feasibility of anti-CCR5 vaccination (3, 5, 15, 17). In particular, we demonstrated that it is possible to induce anti-CCR5 antibodies in mice at levels in serum 300-fold greater than those found in humans and that the amount of mouse CCR5-specific immunoglobulins reached 30% of total serum IgG (3). Here, we optimized the immunization protocols, and thus, CCR5-specific serum IgG reached 50% of total IgG.
In our previous works on the mouse model (3, 5), we verified the specificity of CCR5 modulation, which was not evident after 1 h of anti-CCR5 antibody incubation, thus demonstrating that the receptor was not simply occluded from detection by the presence of the bound mouse antibodies, and in addition, we demonstrated that the internalization was dependent on clathrin-coated pits. Our present goal was to go two steps further: on one hand, we sought full breaking of immune tolerance to autologous CCR5 domains and the induction of protective antibodies to the endogenous CCR5 protein; on the other hand, we compared biological properties and protection conferred by immunoglobulins recognizing four different CCR5 domains to assess whether features previously observed for anti-ECL1 antibodies (i.e., long-lasting receptor downregulation and recycling) could extend to other anti-CCR5 antibodies. Moreover, we accomplished our goals through the first (to our knowledge) extensive evaluation of peptide targets, routes of vaccine delivery, and adjuvants, although the reproduction of similar responses in other animal (or human) models requires the use of suitable adjuvants and a different schedule to be properly set.
CCR5 structure includes an external N-terminal domain and three external loops connecting the seven membrane-spanning helices, any of which could provide vaccine immunogens. Many attempts to elicit anti-CCR5 vaccines have failed, however, possibly because the tested CCR5 domains were not immunogenic enough or because the antigens were not presented in an adequate conformation (14). Indeed, the use of scaffold proteins able to present the CCR5 ECL2 domain in a constrained conformation, as occurred in virus-like particles (VLPs), succeeded in overcoming tolerance and induced the desired response (32). As for ECL2, immunogens presenting the CCR5 ECL1 domain in a linear conformation were previously shown to be unable to elicit immunoglobulins recognizing either the native receptor or the domain itself in a conformed, circular structure (3). Most importantly, structural studies showed that modifications to the flanking regions of the ECL1 sequences could alter the epitope conformation and consequently affected the biological properties of the resulting antibodies (5). Therefore, the adoption of a vector with a well-determined three-dimensional conformation and different positions to accept foreign peptides and protein domains, as is the case for the FHV protein, offered us the chance of presenting epitope conformations needed to elicit the desired immune responses. Indeed, the FHV capsid protein has a well-characterized structure, where external loops of the protein connecting beta-sheets of the eight-stranded beta-barrel structure allow optimal display of conformed epitopes and enhance their immunogenicity (21, 22). As a confirmation of the native conformation of antigens presented through the FHV scaffold, antibodies to ECL1 were found to bind the CCR5 receptor both on murine and on human PBMC (3, 5). In our previous studies with VLPs, we achieved optimal presentation of gp140 and gp41 and the generation of neutralizing antibodies recognizing conformed epitopes within trimeric, full-length structures that were very similar to the natural spikes observed on HIV particles (33, 34).
The present study was aimed at achieving a complete break in immune tolerance for endogenous CCR5 using the mouse model to set and compare immunization conditions. In our previous studies, ECL1 immunogens based on the human CCR5 sequence proved to be very immunogenic in murine and chicken hosts (3, 5). Conversely, in the present study, mice were immunized to four different CCR5 sequences that were fully murine, always presented in a suitable conformation through the Flock House virus vector (3, 18). Due to exogenous CCR5 sequences, these self-immunogens required five immunizations to achieve their response plateaus, whereas the previous immunizations reached their plateaus at the third immunization (3, 5).
The use of combined immunization routes, i.e., systemic plus mucosal, achieved generation of circulating antibodies and, most importantly, of mucosal antibodies. The i.m. and i.r. routes failed in eliciting significant amounts of antibodies, either individually or combined (Tables 2 and 3), whereas the combination of both i.p. and i.n. routes in a unique scheme (protocols I and L) elicited large amounts of IgG and IgA to three antigens out of four, especially when systemic i.p. immunizations preceded mucosal i.n. administrations (protocol L) (Fig. 1). Nter2 was poorly immunogenic in relation to other antigens, probably due to the lack of strong immunogenic epitopes (9, 16, 35). These results agreed with findings from immunizations performed in other animal models, also confirmed in recent clinical trials, where mucosal boosting after intramuscular immunization was superior to intramuscular immunization alone in providing full protective mucosal responses (36, 37). In human immunization, mucosal boosting following systemic immunization was considered safe and was found effective, even in the absence of mucosal adjuvants (38, 39).
Binding of endogenous chemokines has been suggested to play a role in protection from HIV infection (40); however, our previous studies found that chemokine levels in natural carriers of anti-CCR5 antibodies are comparable to those observed in healthy controls, thus excluding ligand overexpression as the cause of CCR5 downregulation in vivo (4). In the present study, we show that enriched IgG and IgA fractions are able to downregulate CCR5 receptors in mouse PBMC, showing a receptor reduction similar to that observed in ex vivo experiments (Table 3). Moreover, we did not observe any difference in chemokine levels in all immunized mice; this finding agrees with previous results achieved with anti-ECL1 antibodies and supports the role of immunoglobulins, but not of chemokines, in receptor downregulation (3, 5).
As is known, ECL2, Nter1, and ECL1, but not ECL3, are directly involved in HIV binding and/or receptor downregulation and can be highly immunogenic; indeed, some antibodies to these sequences could block HIV-1 binding, while others might induce inhibition of the infection via receptor internalization (3, 4, 9–11, 35). Antibodies of some classes and subclasses can provide protection by fixing complement; on the other hand, such activities could also promote pathogenic antiself effects and/or inhibit the effectiveness of activated cell traffic and immune responses. In order to address all such questions, CCR5 immunization was tested in a methodical way that examined systematically the responses to single, constrained epitopes of the molecule with different routes of immunization and adjuvants. To our knowledge, this is the first study in which the immunogenicities of all significant extracellular domains of the CCR5 receptor were examined systematically, employing different protocols and comparing different routes and formulations.
Due to the small size of the animal model we used, it was mandatory to keep nasal instillations within a tiny volume (20 μl); therefore, the effects of mucosal adjuvants on immunization efficacy could not be evaluated. Similarly, mucosal immunity could not be evaluated because CVW fluid amounts were too exiguous to proceed with further assays; the size limitations connected to the animal model under study also prevented the evaluation of vaginal immunization as a possible route.
Comparison of four adjuvants showed that strong immunogenicity could be achieved without inducing severe tissue toxicity (Table 4); on the other hand, two good adjuvants, RIBI and Montanide, were found to exert immunogen-independent CCR5 depletion effects that led us to exclude them (Fig. 2C). This effect deserves attention and could even be exploited to enhance immune protection conferred by anti-CCR5 antibodies; however, in the context of the present study, we felt that it would obscure antibody-specific effects that we wished to measure.
The optimal immunization scheme, coupling systemic and mucosal antigen presentation with a suitable adjuvant, achieved strong mucosal immune priming in Peyer's patches, as shown by the high proportion of cells with the phenotype of activated B cells and by the consistent production of CCR5-specific IgA, a finding particularly evident in blocking assays (Fig. 2F and 5B). Although Fig. 5B shows an IgG-depleted fraction, it contains predominantly IgA. IgA induction takes place in mucosal immune nodes, i.e., in Peyer's patches, and especially in isolated lymphoid follicles (ILFs), where B cells can be activated by dendritic cells (DCs) with or without the intervention of T cells (41, 42). The widespread distribution of Peyer's patches and ILFs in the murine gut and the consistent B1 cell population in adult mice may account for the strong induction of specific IgA that was observed in the study and might have facilitated the breaking of immune tolerance that we observed. Strong activity observed in bioassays, namely, receptor binding and downregulation, blocking of infectivity, and transcytosis inhibition, may mean that antibody affinity maturation took place in gut-associated lymphoid tissue (GALT) upon immunization, although this possibility was not investigated in the study (42).
IgA antibodies act in blood, as well as in local regions, for example, in the gut and the genital mucosae. Although IgAs are usually found in concentrations at least 10-fold lower than those of IgG, their presence in mucosal fluids could block most pathogens through various mechanisms, such as blocking of viral transcytosis and ADCC, even in the absence of systemic responses (37). Even when their concentration in mucosal fluids is measured at a few nanograms per milliliter, or even less, tens of billions of IgA molecules are ready to catch viral pathogens and can rely on stoichiometric relationships very favorable to host defense (38).
Different antibody isotypes, such as IgG and IgA, may provide immune protection by exerting different but synergistic functions (43); interestingly, IgA can bind antigens with higher affinity and provide stronger neutralizing activity than IgG sharing the same specificity, which implies different roles of the respective CH domains (44). Recent human clinical trials showed that mucosal IgG and IgA can be promptly elicited with suitable immunogens able to present conformed antigens in the presence of safe and effective adjuvants (38). Boosting was shown to increase IgG, but not IgA, levels; however, the possibility that boosting may increase the affinity of antibodies for their molecular targets and/or their antiviral function even in the absence of detectable quantitative effects could not be excluded (38).
The notion of anti-CCR5 vaccination has naturally raised concerns that the breaking of immune tolerance might lead to autoimmune dysfunction (45, 46) or affect T cell function. In the present study, no signs of histopathological alterations in the tissues and organs or immune dysfunction in T cell responses (as demonstrated in a previous work) due to the induction or the presence of anti-CCR5 antibodies were observed. The possibility of long-term toxicity and any functional impact of anti-CCR5 antibodies deserve further evaluation; nonetheless, our findings are supported by previous studies, where no adverse events were reported in CCR5-immunized macaques after 3 years of follow-up (15).
In a previous study (3), we performed a splenic cell proliferation assay after activation by several mitogens (concanavalin A, staphylococcal enterotoxin B, and phytohemagglutinin [PHA] in the presence of mouse interleukin 12 [IL-12]). DNA incorporation did not show significant differences between preimmunized and postimmunized animals (3). Thus, we demonstrated that the antibody response obtained with a protocol similar to L does not interfere with T cell function.
ECL1, ECL2, and Nter1 antibodies, elicited by a systemic plus mucosal immunization protocol, achieved total ex vivo downregulation of CCR5 receptor (Fig. 4C). In in vitro assays, ECL1 antiserum produced downregulation comparable to or even greater than that produced by RANTES, an endogenous CCR5 agonist (Fig. 4C), thus affirming observations in previous studies that antibodies recognizing the domain cause receptor internalization (3, 5). ECL1 and ECL2 antibodies were the most effective, showing complete downregulation at 48 h (Fig. 4D); this is the first study reporting that long-lasting CCR5 downregulation was also achieved by antibodies to the ECL2 domain and, to a lesser extent, by those to Nter1. We did not examine CCR5 recycling in this study; according to our previous results, receptor downregulation lasts up to 2 to 4 weeks in mice and can be promptly refreshed upon boosting (3).
Both ECL1 and ECL2 antisera confirmed their remarkable functional properties, showing nearly complete HIV-blocking activity in neutralization. This blocking activity was especially associated with IgG-depleted fractions, with high statistical significance (P ≤ 0.0001 and P ≤ 0.0003 for ECL1 and ECL2, respectively), suggesting that it resided mostly in specific IgA antibodies elicited through immune priming (Fig. 5A and B). Immunoglobulins to the Nter1 domain showed lower antiviral activity; this could be explained by the high flexibility of the CCR5 N terminus, which might have taken a nonnative conformation within the FHV vector and therefore could have elicited antibodies presenting consistent binding properties but lower functional activity.
We then investigated whether CCR5 Igs could block in vitro mucosal transmission of HIV-1. Remarkably, all three antisera also inhibited HIV infectivity in transcytosis assays (Fig. 5E); the highest significance was reached for ECL1 and ECL2, although some antibodies to ECL2, such as 2D7 monoclonal antibody, do not work in this assay (26; L. Lopalco, unpublished data). It is possible that activity in transcytosis is mostly due to IgA; indeed, IgA was shown to be more effective in antiviral functional assays than the corresponding IgG sharing the same specificity (44). Isotype switching could offer a way to overcome poor reactivity of the 2D7 MAb; the present results suggest that selection of new anti-ECL2 antibodies, endowed with HIV-protective functional properties, could be accomplished through suitable immunization.
Of note, CCR5 antibodies to ECL1, ECL2, and Nter1 inhibit transcytosis of the B clade HIV-1 R5 primary isolate BR9309; in contrast, transcytosis of a CXCR4-tropic strain, the B clade HIV-1 primary isolate 91US054, remained insensitive to the presence of the same Igs. This suggested that the CCR5 Igs that have a transcytosis-blocking activity were specific for CCR5. Nonspecific Igs were also unable to block viral transcytosis of both the CCR5 and CXCR4 viruses, confirming the specificity of inhibition. Furthermore, virus produced at the apical pole of the monolayer during coculture in the absence or presence of antibodies was routinely measured and found to be constant (not shown). Additionally, we also found that production of virus by the infected cells only (without epithelial cells) after 2 h of incubation was not modified by the presence of anti-CCR5 antibodies (not shown). These results rule out the possibility that the anti-CCR5 antibodies inhibited virus release or reinfection within the producer cells and, in turn, virus transcytosis in the basolateral medium as a result of the presence of fewer viruses at the apical surface.
We also observed in our previous study that preincubation for 1 h or 24 h increased transcytosis blockade activity and that it did not modify CCR5 surface expression by epithelial cells (26). This indicates that in the 1-h preincubation the antibodies reach an intracellular pool of CCR5. Clearly, the role of CCR5 in HIV-1 transcytosis is different from that of its coreceptor in cell infection. Hence, agrin and galactosyl ceramide (GalCer) serve as epithelial receptors for HIV-1. As HIV transcytosis does not change GalCer apical polarity, it suggests that HIV detaches from GalCer in the endosome and is free to bind a second transporter to achieve the second leg of transcytosis to the basal pole of the cell. Our data suggest that CCR5 interaction with the virus most likely occurs at a later time point in transcytosis, once the virus has been endocytosed in the endosome. Whether this interaction between HIV and CCR5 in the acidic endosome depends on pH remains to be determined.
This study is the first to adopt a systematic strategy to compare the immunogenicities of all extracellular domains of the CCR5 molecule and to set optimal conditions leading to generation of specific antibodies in the mouse model. Indeed, the successful breaking of immune tolerance for this self-molecule was achieved, as confirmed by the wide population of activated B lymphocytes that were isolated from Peyer's patches and were found to secrete specific IgA. These immunoglobulins, and to a lesser extent IgG antibodies, generated to the ECL1, ECL2, and Nter1 domains were found to almost completely block HIV infectivity in cells and in mucosal layers. Most importantly, anti-CCR5 antibodies to different extracellular domains were also found to share similar functional and protective properties.
In other words, the immunization protocol set and tested in the mouse produced anti-CCR5 antibodies with different specificities but endowed with the expected properties, i.e., the ability to downregulate the host receptor and prevent virus docking to it nearly completely. Notably, this immune response was accomplished without signs of tissue toxicity. The lack of signs of autoimmunity, at least from a short-term point of view, suggests that generation of anti-self antibodies by vaccination could be exploited for therapeutic or prophylactic purposes without necessarily causing severe adverse events.
Due to their peculiar and unique properties, immunization using the ECL1 and ECL2 domains could offer a promising path to achieving significant anti-HIV activity in vivo and warrants further investigation in a primate host.
ACKNOWLEDGMENTS
We thank Maria Rescigno for help in PP isolation and culturing, Silvia Russo for her editorial help, and Arianna Vino and Martina Rocchi for their technical help in histopathological analysis.
Footnotes
Published ahead of print 8 January 2014
REFERENCES
- 1.Klasse PJ, Shattock R, Moore JP. 2008. Antiretroviral drug-based microbicides to prevent HIV-1 sexual transmission. Annu. Rev. Med. 59:455–471. 10.1146/annurev.med.59.061206.112737 [DOI] [PubMed] [Google Scholar]
- 2.Barassi C, Lazzarin A, Lopalco L. 2004. CCR5-specific mucosal IgA in saliva and genital fluids of HIV-exposed seronegative subjects. Blood 104:2205–2206. 10.1182/blood-2004-06-2134 [DOI] [PubMed] [Google Scholar]
- 3.Barassi C, Soprana E, Pastori C, Longhi R, Buratti E, Lillo F, Marenzi C, Lazzarin A, Siccardi AG, Lopalco L. 2005. Induction of murine mucosal CCR5-reactive antibodies as an anti-human immunodeficiency virus strategy. J. Virol. 79:6848–6858. 10.1128/JVI.79.11.6848-6858.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lopalco L, Barassi C, Pastori C, Longhi R, Burastero SE, Tambussi G, Mazzotta F, Lazzarin A, Clerici M, Siccardi AG. 2000. CCR5-reactive antibodies in seronegative partners of HIV-seropositive individuals down-modulate surface CCR5 in vivo and neutralize the infectivity of R5 strains of HIV-1 in vitro. J. Immunol. 164:3426–3433 [DOI] [PubMed] [Google Scholar]
- 5.Pastori C, Clivio A, Diomede L, Consonni R, De Mori GM, Longhi R, Colombo G, Lopalco L. 2008. Two amino acid substitutions within the first external loop of CCR5 induce human immunodeficiency virus-blocking antibodies in mice and chickens. J. Virol. 82:4125–4134. 10.1128/JVI.02232-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pastori C, Weiser B, Barassi C, Uberti-Foppa C, Ghezzi S, Longhi R, Calori G, Burger H, Kemal K, Poli G, Lazzarin A, Lopalco L. 2006. Long-lasting CCR5 internalization by antibodies in a subset of long-term nonprogressors: a possible protective effect against disease progression. Blood 107:4825–4833. 10.1182/blood-2005-06-2463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bouhlal H, Latry V, Requena M, Aubry S, Kaveri SV, Kazatchkine MD, Belec L, Hocini H. 2005. Natural antibodies to CCR5 from breast milk block infection of macrophages and dendritic cells with primary R5-tropic HIV-1. J. Immunol. 174:7202–7209 [DOI] [PubMed] [Google Scholar]
- 8.Eslahpazir J, Jenabian MA, Bouhlal H, Hocini H, Carbonneil C, Gresenguet G, Keou FX, LeGoff J, Saidi H, Requena M, Nasreddine N, de Dieu Longo J, Kaveri SV, Belec L. 2008. Infection of macrophages and dendritic cells with primary R5-tropic human immunodeficiency virus type 1 inhibited by natural polyreactive anti-CCR5 antibodies purified from cervicovaginal secretions. Clin. Vaccine Immunol. 15:872–884. 10.1128/CVI.00463-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blanpain C, Doranz BJ, Vakili J, Rucker J, Govaerts C, Baik SS, Lorthioir O, Migeotte I, Libert F, Baleux F, Vassart G, Doms RW, Parmentier M. 1999. Multiple charged and aromatic residues in CCR5 amino-terminal domain are involved in high affinity binding of both chemokines and HIV-1 Env protein. J. Biol. Chem. 274:34719–34727. 10.1074/jbc.274.49.34719 [DOI] [PubMed] [Google Scholar]
- 10.Blanpain C, Vanderwinden JM, Cihak J, Wittamer V, Le Poul E, Issafras H, Stangassinger M, Vassart G, Marullo S, Schlndorff D, Parmentier M, Mack M. 2002. Multiple active states and oligomerization of CCR5 revealed by functional properties of monoclonal antibodies. Mol. Biol. Cell 13:723–737. 10.1091/mbc.01-03-0129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Olson WC, Jacobson JM. 2009. CCR5 monoclonal antibodies for HIV-1 therapy. Curr. Opin. HIV AIDS 4:104–111. 10.1097/COH.0b013e3283224015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grene E, Pinto LA, Landay AL, Kessler HA, Anderson SA, Dolan MJ, Shearer GM. 2001. Anti-CCR5 antibodies in sera of HIV-positive individuals. Hum. Immunol. 62:143–145. 10.1016/S0198-8859(00)00243-3 [DOI] [PubMed] [Google Scholar]
- 13.Haynes BF, Moody MA, Verkoczy L, Kelsoe G, Alam SM. 2005. Antibody polyspecificity and neutralization of HIV-1: a hypothesis. Hum. Antibodies 14:59–67 [PMC free article] [PubMed] [Google Scholar]
- 14.Bogers WM, Bergmeier LA, Oostermeijer H, ten Haaft P, Wang Y, Kelly CG, Singh M, Heeney JL, Lehner T. 2004. CCR5 targeted SIV vaccination strategy preventing or inhibiting SIV infection. Vaccine 22:2974–2984. 10.1016/j.vaccine.2004.02.050 [DOI] [PubMed] [Google Scholar]
- 15.Chackerian B, Briglio L, Albert PS, Lowy DR, Schiller JT. 2004. Induction of autoantibodies to CCR5 in macaques and subsequent effects upon challenge with an R5-tropic simian/human immunodeficiency virus. J. Virol. 78:4037–4047. 10.1128/JVI.78.8.4037-4047.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chain BM, Noursadeghi M, Gardener M, Tsang J, Wright E. 2008. HIV blocking antibodies following immunisation with chimaeric peptides coding a short N-terminal sequence of the CCR5 receptor. Vaccine 26:5752–5759. 10.1016/j.vaccine.2008.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Misumi S, Nakayama D, Kusaba M, Iiboshi T, Mukai R, Tachibana K, Nakasone T, Umeda M, Shibata H, Endo M, Takamune N, Shoji S. 2006. Effects of immunization with CCR5-based cycloimmunogen on simian/HIVSF162P3 challenge. J. Immunol. 176:463–471 [DOI] [PubMed] [Google Scholar]
- 18.Buratti E, McLain L, Tisminetzky S, Cleveland SM, Dimmock NJ, Baralle FE. 1998. The neutralizing antibody response against a conserved region of human immunodeficiency virus type 1 gp41 (amino acid residues 731–752) is uniquely directed against a conformational epitope. J. Gen. Virol. 79:2709–2716 [DOI] [PubMed] [Google Scholar]
- 19.Buratti E, Tisminetzky SG, Scodeller ES, Baralle FE. 1996. Conformational display of two neutralizing epitopes of HIV-1 gp41 on the Flock House virus capsid protein. J. Immunol. Methods 197:7–18. 10.1016/0022-1759(96)00097-X [DOI] [PubMed] [Google Scholar]
- 20.Schiappacassi M, Buratti E, D'Agaro P, Ciani L, Scodeller ES, Tisminetzky SG, Baralle FE. 1997. V3 loop core region serotyping of HIV-1 infected patients using the FHV epitope presenting system. J. Virol. Methods 63:121–127. 10.1016/S0166-0934(96)02120-9 [DOI] [PubMed] [Google Scholar]
- 21.Scodeller EA, Tisminetzky SG, Porro F, Schiappacassi M, De Rossi A, Chiecco-Bianchi L, Baralle FE. 1995. A new epitope presenting system displays a HIV-1 V3 loop sequence and induces neutralizing antibodies. Vaccine 13:1233–1239. 10.1016/0264-410X(95)00058-9 [DOI] [PubMed] [Google Scholar]
- 22.Speir J, Chen Z, Reddy VS, Johnson JE. 2012. Flock House Virus capsid structure Protein Data Bank (PDB at EMBL-EBI) 4fsj record (X-ray diffraction, resolution 2.5A). http://www.ebi.ac.uk/pdbe-srv/view/entry/4fsj/summary_details.html
- 23.Fisher GW, Adler SA, Fuhrman MH, Waggoner AS, Bruchez MP, Jarvik JW. 2010. Detection and quantification of beta2AR internalization in living cells using FAP-based biosensor technology. J. Biomol. Screen. 15:703–709. 10.1177/1087057110370892 [DOI] [PubMed] [Google Scholar]
- 24.Wu Y, Tapia PH, Fisher GW, Waggoner AS, Jarvik J, Sklar LA. 2013. High-throughput flow cytometry compatible biosensor based on fluorogen activating protein technology. Cytometry A 83:220–226. 10.1002/cyto.a.22242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Binley JM, Cayanan CS, Wiley C, Schulke N, Olson WC, Burton DR. 2003. Redox-triggered infection by disulfide-shackled human immunodeficiency virus type 1 pseudovirions. J. Virol. 77:5678–5684. 10.1128/JVI.77.10.5678-5684.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bomsel M, Pastori C, Tudor D, Alberti C, Garcia S, Ferrari D, Lazzarin A, Lopalco L. 2007. Natural mucosal antibodies reactive with first extracellular loop of CCR5 inhibit HIV-1 transport across human epithelial cells. AIDS 21:13–22. 10.1097/QAD.0b013e328011049b [DOI] [PubMed] [Google Scholar]
- 27.Hanly W, Bennet BT, Artwohl JE. 1994. Overview of adjuvants. U.S. Department of Agriculture, Washington, DC: http://www.nal.usda.gov/awic/pubs/antibody/overview.htm [Google Scholar]
- 28.Vogel FR, Powell MF. 1995. A compendium of vaccine adjuvants and excipients. Pharm. Biotechnol. 6:141–228. 10.1007/978-1-4615-1823-5_7 [DOI] [PubMed] [Google Scholar]
- 29.Joshi A, Nyakeriga AM, Ravi R, Garg H. 2011. HIV ENV glycoprotein-mediated bystander apoptosis depends on expression of the CCR5 co-receptor at the cell surface and ENV fusogenic activity. J. Biol. Chem. 286:36404–36413. 10.1074/jbc.M111.281659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lopalco L. 2011. Natural anti-CCR5 antibodies in HIV-infection and -exposure. J. Transl. Med. 9(Suppl 1):S4. 10.1186/1479-5876-9-S1-S4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lopalco L, Bomsel M. 2011. Protecting the initial site of viral entry: an alternative HIV vaccine target. Expert Rev. Vaccines 10:1253–1256. 10.1586/erv.11.98 [DOI] [PubMed] [Google Scholar]
- 32.Peabody DS, Manifold-Wheeler B, Medford A, Jordan SK, do Carmo Caldeira J, Chackerian B. 2008. Immunogenic display of diverse peptides on virus-like particles of RNA phage MS2. J. Mol. Biol. 380:252–263. 10.1016/j.jmb.2008.04.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pastori C, Tudor D, Diomede L, Drillet AS, Jegerlehner A, Rohn TA, Bomsel M, Lopalco L. 2012. Virus like particle based strategy to elicit HIV-protective antibodies to the alpha-helic regions of gp41. Virology 431:1–11. 10.1016/j.virol.2012.05.005 [DOI] [PubMed] [Google Scholar]
- 34.Visciano ML, Diomede L, Tagliamonte M, Tornesello ML, Asti V, Bomsel M, Buonaguro FM, Lopalco L, Buonaguro L. 2011. Generation of HIV-1 virus-like particles expressing different HIV-1 glycoproteins. Vaccine 29:4903–4912. 10.1016/j.vaccine.2011.05.005 [DOI] [PubMed] [Google Scholar]
- 35.Lee B, Sharron M, Blanpain C, Doranz BJ, Vakili J, Setoh P, Berg E, Liu G, Guy HR, Durell SR, Parmentier M, Chang CN, Price K, Tsang M, Doms RW. 1999. Epitope mapping of CCR5 reveals multiple conformational states and distinct but overlapping structures involved in chemokine and coreceptor function. J. Biol. Chem. 274:9617–9626. 10.1074/jbc.274.14.9617 [DOI] [PubMed] [Google Scholar]
- 36.Barnett SW, Srivastava IK, Kan E, Zhou F, Goodsell A, Cristillo AD, Ferrai MG, Weiss DE, Letvin NL, Montefiori D, Pal R, Vajdy M. 2008. Protection of macaques against vaginal SHIV challenge by systemic or mucosal and systemic vaccinations with HIV-envelope. AIDS 22:339–348. 10.1097/QAD.0b013e3282f3ca57 [DOI] [PubMed] [Google Scholar]
- 37.Bomsel M, Tudor D, Drillet AS, Alfsen A, Ganor Y, Roger MG, Mouz N, Amacker M, Chalifour A, Diomede L, Devillier G, Cong Z, Wei Q, Gao H, Qin C, Yang GB, Zurbriggen R, Lopalco L, Fleury S. 2011. Immunization with HIV-1 gp41 subunit virosomes induces mucosal antibodies protecting nonhuman primates against vaginal SHIV challenges. Immunity 34:269–280. 10.1016/j.immuni.2011.01.015 [DOI] [PubMed] [Google Scholar]
- 38.Leroux-Roels G, Maes C, Clement F, van Engelenburg F, van den Dobbelsteen M, Adler M, Amacker M, Lopalco L, Bomsel M, Chalifour A, Fleury S. 2013. Randomized phase I: safety, immunogenicity and mucosal antiviral activity in young healthy women vaccinated with HIV-1 Gp41 P1 peptide on virosomes. PLoS One 8:e55438. 10.1371/journal.pone.0055438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vajdy M, Singh M, Kazzaz J, Soenawan E, Ugozzoli M, Zhou F, Srivastava I, Bin Q, Barnett S, Donnelly J, Luciw P, Adamson L, Montefiori D, O'Hagan DT. 2004. Mucosal and systemic anti-HIV responses in rhesus macaques following combinations of intranasal and parenteral immunizations. AIDS Res. Hum. Retroviruses 20:1269–1281. 10.1089/aid.2004.20.1269 [DOI] [PubMed] [Google Scholar]
- 40.Cocchi F, DeVico AL, Garzino-Demo A, Arya SK, Gallo RC, Lusso P. 1995. Identification of RANTES, MIP-1 alpha, and MIP-1 beta as the major HIV-suppressive factors produced by CD8+ T cells. Science 270:1811–1815. 10.1126/science.270.5243.1811 [DOI] [PubMed] [Google Scholar]
- 41.Macpherson AJ, McCoy KD, Johansen FE, Brandtzaeg P. 2008. The immune geography of IgA induction and function. Mucosal Immunol. 1:11–22. 10.1038/mi.2007.6 [DOI] [PubMed] [Google Scholar]
- 42.Pabst O. 2012. New concepts in the generation and functions of IgA. Nat. Rev. Immunol. 12:821–832. 10.1038/nri3322 [DOI] [PubMed] [Google Scholar]
- 43.Plotkin SA. 2010. Correlates of protection induced by vaccination. Clin. Vaccine Immunol. 17:1055–1065. 10.1128/CVI.00131-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tudor D, Yu H, Maupetit J, Drillet AS, Bouceba T, Schwartz-Cornil I, Lopalco L, Tuffery P, Bomsel M. 2012. Isotype modulates epitope specificity, affinity, and antiviral activities of anti-HIV-1 human broadly neutralizing 2F5 antibody. Proc. Natl. Acad. Sci. U. S. A. 109:12680–12685. 10.1073/pnas.1200024109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Corbeau P, Reynes J. 2009. CCR5 antagonism in HIV infection: ways, effects, and side effects. AIDS 23:1931–1943. 10.1097/QAD.0b013e32832e71cd [DOI] [PubMed] [Google Scholar]
- 46.Telenti A. 2009. Safety concerns about CCR5 as an antiviral target. Curr. Opin. HIV AIDS 4:131–135. 10.1097/COH.0b013e3283223d76 [DOI] [PubMed] [Google Scholar]