

Abstract
The shelterin protein TIN2 is required for the telomeric accumulation of TPP1/POT1 heterodimers and for the protection of telomeres by the POT1 proteins (POT1a and POT1b in the mouse). TIN2 also binds to TRF1 and TRF2, improving the telomeric localization of TRF2 and its function. Here, we ask whether TIN2 needs to interact with both TRF1 and TRF2 to mediate the telomere protection afforded by TRF2 and POT1a/b. Using a TIN2 allele deficient in TRF1 binding (TIN2-L247E), we demonstrate that TRF1 is required for optimal recruitment of TIN2 to telomeres and document phenotypes associated with the TIN2-L247E allele that are explained by insufficient TIN2 loading onto telomeres. To bypass the requirement for TRF1-dependent recruitment, we fused TIN2-L247E to the TRF2-interacting (RCT) domain of Rap1. The RCT-TIN2-L247E fusion showed improved telomeric localization and was fully functional in terms of chromosome end protection by TRF2, TPP1/POT1a, and TPP1/POT1b. These data indicate that when sufficient TIN2 is loaded onto telomeres, its interaction with TRF1 is not required to mediate the function of TRF2 and the TPP1/POT1 heterodimers. We therefore conclude that shelterin can protect chromosome ends as a TRF2-tethered TIN2/TPP1/POT1 complex that lacks a physical connection to TRF1.
INTRODUCTION
Shelterin represses DNA damage signaling by the ATM and ATR kinases and prevents double-stranded break (DSB) repair at chromosome ends (reviewed in reference 1). Conditional deletion of individual shelterin proteins has revealed considerable functional compartmentalization within shelterin. TRF2 is required for the repression of ATM-dependent signaling and classical nonhomologous end joining (c-NHEJ), whereas the closely related TRF1 protein prevents replication fork stalling in telomeric DNA and represses a fragile site phenotype at telomeres. POT1a prevents ATR kinase signaling at telomeres by blocking the binding of RPA to the single-stranded TTAGGG repeats. POT1b, on the other hand, is required for the correct formation of the 3′ telomeric overhang.
TIN2, the central component of shelterin, is critical for the function of POT1a and POT1b because it links TPP1/POT1 heterodimers to TRF1 and TRF2 (2–6). Consistent with this role, TIN2 deletion phenocopies the loss of TPP1 and/or POT1a/b, including the activation of ATR signaling, the deregulation of the amount of single-stranded telomeric DNA, and a low frequency of (postreplicative) sister telomere fusions (7–10). In addition, deletion of TIN2 elicits ATM kinase signaling and a moderate level of chromosome-type telomere fusions that are hallmarks of TRF2 loss (7, 10–12). The chromosome-type fusions associated with TIN2 deletion are due to decreased telomeric accumulation of TRF2, as overexpression of TRF2 in TIN2-null cells alleviates this phenotype (10). However, overexpression of TRF2 does not fully repress ATM kinase signaling in TIN2-knockout (KO) cells, and a TRF2 mutant that lacks the TIN2 binding domain is partially defective in preventing ATM activation, suggesting that TIN2 directly contributes to this TRF2 function. There is no indication for a role of TIN2 in the function of TRF1 at telomeres since TIN2-deleted cells do not display the fragile telomere phenotype typical of TRF1 loss (10, 13, 14).
Here, we ask whether TIN2 requires its interaction with both TRF1 and TRF2 in order to function as a tether for the TPP1/POT1 heterodimers. Deletion of TRF2 does not give rise to any of the phenotypes associated with POT1a/b deficiency (7, 11). Similarly, deletion of TRF1 does not appear to elicit the phenotypes typical of the loss of POT1a/b from telomeres (14). However, deletion of either TRF1 or TRF2 induces a prominent telomeric DNA damage response, which could mask (some of) the phenotypes of POT1a/b loss. Thus, to further elucidate the role of the TIN2-TRF1 and TIN2-TRF2 interactions in the protective function of shelterin, we generated a TIN2 allele defective in TRF1 association. We show that TRF1 is principally responsible for the recruitment of TIN2 to telomeres and describe a distinct set of telomere dysfunction phenotypes that arise from the severing of this interaction. By artificially tethering TIN2 to TRF2 via the fusion of the Rap1 TRF2-interacting domain (RCT) to TIN2, we provide direct evidence that TRF2-bound TIN2 is in principle sufficient to fulfill the TPP1/POT1 tethering function required to protect chromosome ends.
MATERIALS AND METHODS
Cell lines and expression plasmids.
SV40-LT-immortalized TIN2F/F, TIN2F/F ATM−/−, TIN2F/F ATRF/F (10), TRF2F/F, and TRF1F/F mouse embryonic fibroblasts (MEFs) (14) were grown in Dulbecco modified Eagle medium (DMEM) supplemented with l-glutamine, penicillin-streptomycin, nonessential amino acids, and 10% fetal bovine serum (Invitrogen). HT1080 and 293T cells were maintained in DMEM supplemented with l-glutamine, penicillin-streptomycin, nonessential amino acids, and 10% bovine calf serum (BCS) (HyClone). TIN2F/F, TIN2F/F ATM−/−, and TIN2F/F ATRF/F MEFs were infected with pWZL-hygromycin retroviruses expressing FLAG-hemagglutinin (HA)-HA (FLAG-HA2)-tagged mouse TIN2 alleles or an empty vector and selected with hygromycin. In some experiments, the hygromycin-selected MEFs were subsequently infected with pLPC-puromycin retroviruses expressing MYC-tagged TRF2, TPP1, or POT1b. In all cases, following three rounds of retroviral infection, MEFs were selected with hygromycin or puromycin for 2 to 4 days. The human HT1080 fibrosarcoma cells were first infected with pWZL-hygromycin retroviruses expressing short hairpin RNA (shRNA)-insensitive FH2-TIN2 alleles or an empty vector. TIN2i constructs possess silent mutations to generate shRNA resistance (AGACAATATGGTGTGGACAT to AGGCAGTACGGCGTGGACAT; underlining identifies which bases were changed to introduce silent mutations to confer shRNA resistance). Following selection with hygromycin, HT1080 cells were infected with a previously characterized shRNA (puromycin-resistant lentiviral vector) against human TIN2 and selected with puromycin (10). For immunoprecipitation studies, 293T cells were transiently cotransfected with MYC-tagged TRF1 or TRF2 plasmids (pLPC-based) and FLAG-HA2-tagged mouse TIN2 alleles (pLPC-based) using the calcium phosphate coprecipitation method. All introduced mutations and deletions in TIN2 were made by using a QuikChange II site-directed mutagenesis kit (Agilent Technologies) and confirmed by DNA sequencing. Deletion of floxed alleles in all MEF lines was induced by two infections with Hit&Run Cre retrovirus at 12-h intervals. Time points are defined in hours or days after the second infection.
Immunoblotting.
Cell extracts were made from MEFs using 450 mM NaCl lysis buffer (50 mM Tris-HCl [pH 7.4], 1% Triton X-100, 0.1% SDS, 450 mM NaCl, 1 mM EDTA, 1 mM dithiothreitol [DTT]) and protease inhibitors. Lysates were centrifuged at 13,200 rpm for 10 min. Protein concentrations from supernatants were determined using the Bradford protein assay (Bio-Rad). Samples were suspended in 2× sample buffer (75 mM Tris-HCl [pH 6.8], 10% glycerol, 2% SDS, 0.05% bromophenol blue, 2.5% β-mercaptoethanol) prior to being loaded on 8% or 10% SDS-PAGE gels and then transferred to nitrocellulose membranes in transfer buffer (25 mM Tris, 0.192 M glycine, 20% methanol). After being blocked with 5% nonfat dry milk in phosphate-buffered saline (PBS)–0.1% Tween 20 (PBST) at room temperature for 20 min, membranes were incubated in PBST for 1 h with the following primary antibodies: rabbit polyclonal anti-mTRF1 antibody (1449), rabbit polyclonal anti-mTRF2 antibody (1254), rabbit polyclonal anti-mRap1 antibody (1252), rabbit polyclonal anti-hTRF1 antibody (370), mouse monoclonal anti-α-tubulin (Sigma-Aldrich), mouse monoclonal anti-MYC antibody (9E10 [Calbiochem]) and mouse monoclonal anti-HA antibody (12CA5 [Roche]). Membranes were developed with enhanced chemiluminescence (ECL; Amersham).
Coimmunoprecipitation.
Cell extracts were made from MEFs or 293T cells using 150 mM NaCl lysis buffer (50 mM Tris-HCl [pH 7.4], 1% Triton X-100, 0.1% SDS, 150 mM NaCl, 1 mM EDTA, 1 mM DTT) and protease inhibitors. Lysates were centrifuged at 13,200 rpm for 10 min. One percent of the supernatant was taken to serve as the input. Following preclearing with protein G beads (GE Healthcare) for 30 min at 4°C, supernatants were incubated with anti-HA affinity matrix (Roche) overnight at 4°C on a rotator wheel. Following three 10-min washes with lysis buffer, beads were pelleted by centrifugation and resuspended in 2× sample buffer (75 mM Tris-HCl [pH 6.8], 10% glycerol, 2% SDS, 0.05% bromophenol blue, 2.5% β-mercaptoethanol) prior to being loaded on 8% or 10% SDS-PAGE gels and then transferred to nitrocellulose membranes in transfer buffer (25 mM Tris, 0.192 M glycine, 20% methanol). Membranes were immunoblotted as described above.
Indirect immunofluorescence.
Cells were grown on coverslips and fixed with 2% paraformaldehyde (PFA) in PBS for 10 min. Telomeric DNA fluorescence in situ hybridization (FISH) was combined with immunofluorescence (IF) as described previously (7) by using the following antibodies: polyclonal rabbit anti-mTIN2 (1447), polyclonal rabbit anti-mRap1 (1252), polyclonal rabbit anti-human 53BP1 antibody NB 100-304 (Novus), mouse monoclonal anti-MYC antibody 9E10 (Sigma), and anti-HA antibody (12CA5 [Roche]). Secondary antibodies were Alexa Fluor 555 goat anti-rabbit IgG (Molecular Probe) or RRX-conjugated donkey anti-mouse IgG (Jackson Laboratory). A fluorescein isothiocyanate (FITC)-TelC (FITC-OO-CCCTAACCCTAACCCTAA; Applied Biosystems) probe was used to detect telomeric DNA using the protocol developed by Herbig and colleagues (15). DNA was stained with 4,6-diamino-2-phenylindole (DAPI). Where noted, nucleoplasmic proteins were removed using Triton X-100 buffer (0.5% Triton X-100, 20 mM HEPES-KOH [pH 7.9], 50 mM NaCl, 3 mM MgCl2, 300 mM sucrose) on ice for 2 min prior to fixation with 2% paraformaldehyde (PFA) in PBS. Digital images were captured with a Zeiss Axioplan II microscope with a Hamamatsu C4742-95 camera using Improvision OpenLab software.
ChIP.
Chromatin immunoprecipitation (ChIP) was performed as described previously (16) with minor modifications. Cells were fixed in medium with 1% paraformaldehyde for 60 min at room temperature. Glycine was added to 0.2 M to stop the cross-linking. Cells were pelleted by centrifugation, washed once with cold PBS, followed by a final wash in PBS-1 mM phenylmethylsulfonyl fluoride (PMSF). The cells were resuspended in cell lysis buffer [5 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES) at pH 8.0, 85 mM KCl, 0.5% NP-40, 1 mM PMSF, Complete protease inhibitor cocktail (Roche)] and incubated on ice for 15 min. After sonication, the lysates were centrifuged at 13,200 rpm for 10 min. The supernatants were incubated with crude rabbit antibody serum raised against mTRF1 (1448), mTRF2 (1254), mRap1 (1252), or mTIN2 (1447) or with commercial purified antibodies against MYC (9B11 Cell Signaling) or HA (Abcam). Samples were incubated at 4°C overnight and for 45 min with ChIP-grade protein G magnetic beads (Invitrogen Dynal). The immunoprecipitated DNA was collected and washed using a 16-tube magnetic separation rack (Cell Signaling) and precipitated with ethanol after reversal of the cross-links. The DNA samples were dissolved in Tris-EDTA (TE), blotted using a slot blotter, and hybridized with an 800-bp probe labeled with Klenow and a primer for the C-rich telomeric repeat strand. The signal intensity measured by ImageQuant software was normalized to the signals of the input DNA on the same blot.
Telomere analysis.
Telomeric overhang signals and telomeric restriction fragment patterns were analyzed by in-gel analysis as previously described (11). Briefly, a (CCCTAA)4 oligonucleotide was hybridized to native MboI-digested genomic DNA fractionated on contour-clamped homogeneous electric field (CHEF) gels to determine the overhang signal. After capture of the signal, the DNA was denatured in situ, neutralized, and then rehybridized with the same probe to determine the total telomeric DNA signals. The overhang signal in each lane is normalized to the duplex telomeric signal so that comparisons of these ratios reveal changes in the overhang status. The procedures for telomeric FISH on metaphase spreads were as described previously (14). Briefly, cells at the indicated time points and treatments were incubated for 2 h with 0.2 μg/ml Colcemid. The cells were harvested, swollen in 75 mM KCl, fixed in methanol-acetic acid (3:1), and dropped onto glass slides. After aging overnight, the slides were washed in 1× PBS for 5 min followed by consecutive incubation with 75%, 95%, and 100% ethanol. The slides were allowed to air dry before applying hybridizing solution (70% formamide, 1 mg/ml blocking reagent [Roche], 10 mM Tris-HCl [pH 7.2]) containing FITC-OO-(CCCTAA)3 PNA probe (Applied Biosystems). The spreads were denatured for 3 min at 80°C on a heat block and hybridized at room temperature for 2 h. The slides were washed twice for 15 min with 70% formamide-10 mM Tris-HCl (pH 7.0), followed by three 5-min washes in 0.1 M Tris-HCl (pH 7.0)-0.15 M NaCl-0.08% Tween 20. The chromosomal DNA was counterstained with DAPI added to the second wash. Slides were mounted in antifade reagent (ProLong Gold; Invitrogen), and digital images were captured with a Zeiss Axioplan II microscope with a Hamamatsu C4742-95 camera using Improvision OpenLab software.
RESULTS
TIN2 is dependent on TRF1 for optimal telomeric association.
Human TIN2 uses an FXLXP motif around position 260 to interact with the TRFH domain of TRF1, and substitution of the leucine at position 260 of TIN2 with glutamate (L260E) abolishes its binding to TRF1 but not to TRF2 (17). A mouse TIN2 allele harboring the analogous leucine-to-glutamate substitution at residue 247 was introduced as a FLAG-HA2-tagged version into previously characterized SV40-LT-immortalized TIN2F/F mouse embryonic fibroblasts (MEFs) (Fig. 1A) (10). As expected, coimmunoprecipitation (co-IP) analysis showed that TIN2-L247E failed to interact with TRF1, whereas the mutated allele interacted normally with TRF2 (Fig. 1B and C). Similar to the effect of complete deletion of TIN2 (10), cells expressing only the TIN2-L247E allele showed diminished TRF1 protein levels compared to those of TRF2 or Rap1 (Fig. 1D).
FIG 1.

TIN2 allele lacking TRF1 binding is lost at telomeres. (A) Schematic of the mouse TIN2 protein indicating regions of shelterin binding and location of mutations identified in dyskeratosis congenita (DC) patients. Homology between human and mouse TIN2 is shown surrounding the FXLXP TRF1 binding motif. Amino acids in red indicate mutations introduced to generate TRF1 binding mutants. (B) Cell extracts from TIN2F/F MEFs exogenously expressing the indicated FLAG-HA2 TIN2 alleles were immunoprecipitated (IP) with anti-HA resin, and immunocomplexes were probed for the indicated proteins. (C) Cell extracts from 293T cells coexpressing the indicated FLAG-HA2 TIN2 alleles and TRF2 were immunoprecipitated with anti-HA resin, and immunocomplexes were probed for the indicated proteins. (D) Immunoblotting of cell extracts from TIN2F/F MEFs expressing TIN2 alleles (as in panel B) or empty vector (EV) with or without Cre treatment (72 h). (E) Left, loss of TIN2 as observed by IF-FISH from TIN2F/F MEFs with or without Cre treatment (72 h). Right, IF of FLAG-HA2 TIN2 alleles (anti-HA antibody, red) and telomeric FISH (green) following Triton X-100 extraction of soluble proteins in TIN2−/− MEFs (72 h post-Cre). (F) Left, IF of TRF2 (anti-TRF1 antibody, red) in cells expressing FLAG-HA2 TIN2 alleles and telomeric FISH (green) following Triton X-100 extraction of soluble proteins in TIN2−/− MEFs (72 h post-Cre). Middle, IF of TRF2 (anti-TRF2 antibody, red) in cells expressing FLAG-HA2 TIN2 alleles and telomeric FISH (green) following Triton X-100 extraction of soluble proteins in TIN2−/− MEFs (72 h post-Cre). Right, IF of Rap1 (anti-Rap1 antibody, red) in cells expressing FLAG-HA2 TIN2 alleles and telomeric FISH (green) following Triton X-100 extraction of soluble proteins in TIN2−/− MEFs (72 h post-Cre). DNA is stained with DAPI (blue). At least 200 cells were used for quantification of TIN2 and Rap1 foci. Bars, 1.5 μm.
Indirect immunofluorescence (IF) showed diminished accumulation of TIN2-L247E at telomeres, resulting in a dispersed nucleoplasmic staining pattern (Fig. 1E). This IF pattern was clearly distinct from FLAG-HA2-tagged wild-type (WT) TIN2 assayed in parallel (Fig. 1E) and from endogenous TRF1, TRF2, or Rap1 in these cells (Fig. 1F).
Based on chromatin immunoprecipitation (ChIP), the amount of TIN2-L247E at mouse telomeres was ∼5-fold lower than that of wild-type TIN2 and comparable to the background levels observed in cells infected with the empty vector (Fig. 2A and B). The amounts of TRF1 at telomeres in TIN2-L247E-expressing cells were also reduced (Fig. 2A and B). Although the IF signals for TRF2 or Rap1 did not show a strong reduction in cells expressing TIN2-L247E (Fig. 1F), TRF2 and Rap1 ChIP suggested that the TIN2-L247E mutant also had a small effect on the accumulation of TRF2 and Rap1 at telomeres (Fig. 2B and D). However, this effect was not statistically significant when analyzed based on three independent experiments.
FIG 2.

TIN2-L247E protein is absent from telomeres by ChIP. (A) Telomeric DNA ChIP for FLAG-HA2 TIN2 alleles (anti-HA), TRF1, or Rap1 in TIN2F/F MEFs with or without Cre treatment (72 h). (B) Quantification of the ChIP signals from three identical experiments, as described for panel A. ChIP signals were normalized to the input, and the background (PI) was subtracted. **, P < 0.05 (paired Student's t test). (C) Telomeric DNA ChIP for TRF2 in TIN2F/F MEFs expressing FLAG-HA2 TIN2 alleles (anti-HA) with or without Cre treatment (72 h). (D) Quantification of the ChIP signals from three identical experiments, as described for panel C. ChIP signals were normalized to the input, and the background (PI) was subtracted. **, P < 0.05 (paired Student's t test).
Similar to the effect of the TIN2-L247E allele in mouse cells, a reduction in human TRF1 protein was also observed in HT1080 cells treated with a TIN2 shRNA and expressing an shRNA-resistant version of the TIN2-L260E allele (TIN2-L260Ei) that binds TRF2 but does not interact with TRF1 (Fig. 3A to C). Human TIN2-L260Ei also did not accumulate efficiently at telomeres in HT1080 cells after the endogenous TIN2 was knocked down with an shRNA (Fig. 3D and E). The effect of TIN2-L260E on TRF1 abundance in human cells is consistent with a previously published role of TIN2 in protecting TRF1 from tankyrase-mediated PARsylation and subsequent degradation (18, 19). The reason TIN2 binding is required for optimal expression of mouse TRF1, which does not interact with tankyrase (20), is unclear.
FIG 3.

TIN2-L260E affects TRF1 accumulation at human telomeres. (A) Cell extracts from 293T cells exogenously expressing the indicated human FLAG-HA2 TIN2 alleles (WT and L260E) and MYC-tagged TRF1 and MYC-tagged TRF2 were immunoprecipitated (IP) with anti-HA resin, and immunocomplexes were probed for the indicated proteins. (B) FLAG-HA2-tagged TIN2 alleles were introduced into HT1080 human fibrosarcoma cells. Immunoblotting for these proteins was conducted from whole-cell extracts. Endogenous and exogenous TIN2 proteins were identified using an anti-TIN2 antibody. (C) Protein levels of FLAG-HA2-tagged shRNA-insensitive proteins (indicated as TIN2i and L247Ei) and TRF1 in HT1080 human fibroblasts transduced with a TIN2 shRNA. Immunoblotting for these proteins was conducted in whole-cell extracts and detected by anti-HA and anti-TIN2 antibodies. (D) Loss of TIN2 as observed by IF-FISH from HT1080 cells transduced with a TIN2 shRNA, as described for panel C. DNA is stained with DAPI (blue). (E) IF of FLAG-HA2 TIN2i alleles (anti-HA antibody, red) and telomeric FISH (green) following Triton X-100 extraction of soluble proteins in HT1080 cells, as described for panel C. DNA is stained with DAPI (blue). At least 200 cells were used for quantification of TIN2 foci. Bars, 1.5 μm.
Consistent with the role of TRF1 in promoting the accumulation of TIN2 at telomeres, there was also a reduction in the telomeric TIN2 signals after deletion of TRF1 from immortalized TRF1F/F MEFs, whereas the telomeric Rap1 foci were unaltered (Fig. 4A and B). Importantly, this reduction of TIN2 or changes in the levels of TRF1 at telomeres was not observed following deletion of TRF2 from TRF2F/F cells (Fig. 4C and D). Together, these data demonstrate the significance of the TIN2-TRF1 association for optimal accumulation of shelterin at telomeres.
FIG 4.

Accumulation of TIN2 at telomeres is reduced in TRF1-deficient MEFs. (A) An anti-TRF1 antibody was used to detect TRF1 protein in immunoblots in TRF1F/F MEFs with or without Cre treatment (72 h). (B) Endogenous TIN2 (left) and endogenous Rap1 (right) were detected by IF in combination with telomeric FISH (green) with or without Cre treatment (72 h). Triton X-100 was used to extract soluble proteins. DNA is stained with DAPI (blue). Bars, 1.5 μm. (C) An anti-TRF2 antibody was used to detect TRF2 protein in immunoblots in TRF2F/F MEFs with or without Cre treatment (72 h). *, unspecific bands. (D) Endogenous TIN2 (left) and endogenous TRF1 (right) were detected by IF in combination with telomeric FISH (green) with or without Cre treatment (72 h). Triton X-100 was used to extract soluble proteins. DNA is stained with DAPI (blue). Bars, 1.5 μm.
Phenotypes of TIN2-L247E suggest impaired function of TRF2 but not TRF1.
TIN2 deletion results in a telomeric DNA damage response mediated by both the ATM and ATR kinase pathways (10). This DNA damage response, detected based on 53BP1 telomere dysfunction-induced foci (TIFs) (21), was fully repressed by expression of wild-type TIN2 but not by TIN2-L247E (Fig. 5A). However, TIN2-L247E-expressing cells had significantly fewer TIFs than TIN2-null cells, suggesting that this allele is partially competent in repressing the DNA damage response despite its diminished association with telomeres (Fig. 5B). Exogenous expression of TRF2 does not blunt activation of the DNA damage response following TIN2 deletion (10), and overexpression of TRF2 also failed to improve the ability of TIN2-L247E to fully repress the DNA damage response in TIN2-deleted cells (Fig. 5C and D). To determine whether TIN2-L247E differentially affected ATM and ATR signaling, the effect of this allele was monitored in ATM- or ATR-deficient cells. The expression of TIN2-L247E in TIN2F/F ATM−/− cells, where the TIFs are induced by ATR signaling, showed a significantly reduced number of TIFs per nucleus (Fig. 5E), indicating that TIN2-L247E is partially competent for repression of the ATR-dependent DNA damage response. The deficiency of TIN2-L247E in repression of the ATM-dependent DNA damage response was more pronounced, resulting in the same level of TIF formation in TIN2F/F ATRF/F cells treated with Cre, regardless of whether they expressed TIN2-L247E or contained no TIN2 (Fig. 5E). In agreement with these results, ATR activation, as monitored by Chk1 phosphorylation, was repressed in TIN2-deleted MEFs expressing wild-type TIN2 and TIN2-L247E, while the ATM response, as monitored by Chk2 phosphorylation, was readily observed in cells expressing TIN2-L247E (Fig. 5F).
FIG 5.

DNA damage response in TIN2-L247E-expressing cells. (A) IF of 53BP1 (red) and telomeric FISH (green) in TIN2−/− MEFs (72 h post-Cre) expressing empty vector (EV) or the indicated FLAG-HA2 TIN2 alleles. DNA is stained with DAPI (blue). Bars, 1.5 μm. (B) Quantification of TIFs in TIN2F/F MEFs transduced with an empty vector (top), wild-type TIN2 (middle), or L247E (bottom), as scored for 53BP1 TIFs per nucleus (n > 200) after Cre (72 h). Averages and median values are from three independent experiments and standard deviations (SDs). **, P < 0.05 (paired Student's t test). (C) An anti-TRF2 antibody and an anti-MYC antibody were used to detect exogenously expressed MYC-tagged TRF2 proteins in immunoblots in TIN2F/F MEFs expressing FLAG-HA2 TIN2 alleles (anti-HA) and MYC-TRF2. (D) Quantification of a median number of TIFs per nucleus in TIN2-deleted cells expressing an EV or TIN2-L247E with or without exogenously expressed TRF2. Median values are from three independent experiments and SDs. (E) Left, quantification of TIFs in TIN2F/F ATM−/− MEFs transduced with an empty vector (top), wild-type TIN2 (middle), or L247E (bottom), as scored for 53BP1 TIFs per nucleus (n > 100) after Cre (72 h post-Cre). Averages and median values are from three independent experiments and SDs. **, P < 0.05 (paired Student's t test). Right, quantification of TIFs in TIN2F/F ATRF/F MEFs transduced with an empty vector (top), wild-type TIN2 (middle), or L247E (bottom), as scored for 53BP1 TIFs per nucleus (n > 100) after Cre (72 h post-Cre). Averages and median values are from three independent experiments and SDs. **, P < 0.05 (paired Student's t test). Endoreduplicated (tetraploid) cells were excluded from the analysis. (F) Immunoblots for Chk1 and Chk2 phosphorylation in TIN2F/F MEFs expressing the indicated FLAG-HA2 TIN2 alleles and treated with Cre (72 h) as indicated.
Consistent with the inability of TIN2-L247E to function like wild-type TIN2, expression of this allele did not improve the growth of TIN2F/F cells treated with Cre (Fig. 6A). Furthermore, TIN2-L247E was partially defective in protecting telomeres from c-NHEJ, resulting in a substantial telomere fusion phenotype after the deletion of TIN2 (Fig. 6B and C). Exogenous expression of TRF2 was previously shown to reduce the amount of chromosome-type fusions resulting from TIN2 deletion, suggesting that TIN2 is not required for the ability of TRF2 to repress c-NHEJ (10), and exogenous expression of TRF2 in TIN2-deleted cells expressing TIN2-L247E similarly reduced the amount of chromosome-type fusions (Fig. 6D). Thus, the L247E allele of TIN2, deficient in TRF1 binding, acts as if it has a diminished capacity to support TRF2 function at telomeres.
FIG 6.

Growth arrest and telomere fusions in cells expressing TIN2-L247E. (A) Growth curve of SV40-LT-immortalized TIN2−/− MEFs expressing an empty vector (EV) or the indicated FLAG-HA2 TIN2 alleles. (B) Examples of telomere fusions in metaphases of TIN2-deficient cells expressing the indicated TIN2 alleles or no TIN2 (EV) (72 h post-Cre). Green, TelC PNA probe; red, DAPI DNA stain. (C) Summary of telomere phenotypes in TIN2-deficient cells expressing TIN2 alleles, as determined by FISH, as shown in panel B. Values are averages and SDs from 3 independent experiments. Chromosome-type and chromatid-type telomere fusions were scored on 3,000 to 5,000 telomeres/experiment. Long-arm sister telomere fusions and fragile telomeres were scored on 2,000 to 3,000 telomeres/experiment. (D) Quantification of chromosome-type fusions in TIN2-deleted cells expressing an EV or TIN2-L247E with or without exogenously expressed TRF2. Averages and SDs are from three independent experiments (2,000 to 3,000 telomeres/experiment). **, P < 0.05 (paired Student t test).
On the other hand, TIN2-L247E-expressing cells did not show one of the hallmarks of TRF1 deficiency, the formation of fragile telomeres (Fig. 6B and C), arguing that the TIN2-TRF1 interaction is not essential for the replication function of TRF1, despite the fact that the overall expression level of TRF1 is lowered in these cells. This conclusion is consistent with the absence of a fragile telomere phenotype in TIN2-KO cells (10). The occurrence of telomere fusions does not mask our ability to detect fragile telomeres since they were readily detected after the complete removal of shelterin in a setting where ∼30% of the telomeres undergo fusions (22).
TIN2-L247E interacts with TPP1.
The partial deficiency of TIN2-L247E for repression of ATR signaling in ATM−/− cells could be explained if the L247E mutation affected the ability of TIN2 to adequately recruit TPP1 to telomeres. Although immunoprecipitation (IP) analysis for TPP1 binding showed the L247E protein was fully capable of interacting with TPP1 (Fig. 7A), ChIP for exogenous MYC-tagged TPP1 indicated that the accumulation of TPP1 at telomeres was compromised in cells expressing the TIN2-L247E mutant (Fig. 7B and C). Furthermore, IF for MYC-tagged TPP1 and POT1b showed that little TPP1 and POT1b associated with telomeres when the TIN2-L247E mutant was the only TIN2 protein in cells (Fig. 7D). However, cells expressing L247E did not display Chk1 phosphorylation (as observed following TPP1 or POT1a deletion) (Fig. 5D) or a statistically significant increase in the single-stranded TTAGGG repeats compared to TIN2-deficient MEFs (Fig. 7E and F), as would have been expected if TPP1 or the POT1 proteins were completely absent. This result would suggest that despite the obvious reduction in telomeric TPP1/POT1, there is sufficient TPP1/POT1a and TPP1/POT1b heterodimers at telomeres in the TIN2-L247E cells for the POT1 proteins to fulfill most of their functions. These results are consistent with a prior analysis of MEFs bearing the TPP1 ACD allele, which also show severely reduced TPP1 accumulation at telomeres yet do not have an increase in the telomeric overhang or a high level of TIFs (23). Moreover, despite a substantial reduction of endogenous TIN2 at telomeres (Fig. 4B), TRF1-deficient MEFs also do not display an increase in the single-stranded telomeric overhang (14). Nevertheless, given that TIN2-L247E does not accumulate at telomeres efficiently, our results are difficult to interpret; thus, we sought to improve the loading of the TIN2-L247E mutant on telomeres.
FIG 7.

Unaltered telomeric overhangs despite diminished TPP1 and POT1b at telomeres. (A) TIN2F/F MEFs exogenously expressing MYC-TPP1 were infected with empty vector (EV) or the indicated FLAG-HA2 TIN2 alleles. Cell extracts were immunoprecipitated (IP) with anti-HA resin, and immunocomplexes were probed for the indicated proteins. (B) Telomeric DNA ChIP for FLAG-HA2 TIN2 alleles (anti-HA) and MYC-TPP1 (anti-MYC) in TIN2F/F MEFs with or without Cre treatment (72 h). (C) Quantification of the ChIP signals from two identical experiments, as described for panel B. ChIP signals were normalized to the input, and the background (PI) was subtracted. Error bars represent standard errors of the means. (D) TIN2F/F MEFs exogenously expressing MYC-tagged TPP1 (left) or MYC-tagged POT1b (right) were infected with EV or the indicated FLAG-HA2 TIN2 alleles. Indirect immunofluorescence (IF) of MYC-tagged proteins (anti-MYC antibody, red) and telomeric FISH (green) was conducted following Triton X-100 extraction of soluble proteins in TIN2−/− MEFs (72 h post-Cre). DNA is stained with DAPI (blue). Bars, 1.5 μm. (E) In-gel hybridization assay for single-stranded (ss) telomeric DNA after TIN2 deletion in cells expressing EV, TIN2, or L247E-TIN2. Left, TelC signals under the native condition; right, same gel rehybridized after in situ denaturation of the DNA. (F) Quantification of overhang signals from three independent experiments, as described for panel E, were normalized to the total telomeric signals and compared to TIN2F/F MEFs without Cre. **, P < 0.05 (paired Student's t test).
TRF2-tethered TIN2-L247E restores telomere end protection.
In order to test whether TIN2-L247E could protect telomeres like wild-type TIN2 when its localization to telomeres was restored, TIN2-L247E was fused at its N terminus to the TRF2 binding (RCT) domain of Rap1 (Fig. 8A). Immunoblotting of whole-cell extracts from TIN2F/F MEFs expressing TIN2 or the RCT fusion derivatives confirmed expression of the higher-molecular-weight fusion proteins (Fig. 8B). Similar to TIN2-L247E, RCT-TIN2-L247E resulted in reduced TRF1 protein levels in TIN2-deleted cells, whereas the RCT-fused wild-type TIN2 did not have this effect (Fig. 8B). Interestingly, Rap1 protein levels were reduced in cells expressing the RCT-TIN2 alleles (Fig. 8B), suggesting that the RCT-TIN2 alleles compete with Rap1 for binding to TRF2 and result in destabilization of the displaced Rap1. As expected, RCT-TIN2 retained its ability to bind TRF1 in co-IP experiments, whereas the RCT-TIN2-L247E allele failed to interact with TRF1 (Fig. 8C). RCT-TIN2 and RCT-TIN2-L247E were equally efficient in binding to TRF2 (Fig. 8D).
FIG 8.

Fusion of RCT domain of Rap1 to TIN2 alleles. (A) Schematic of the Rap1 RCT-domain TIN2 fusion protein indicating regions of shelterin binding (TRF1, TRF2, and TPP1) and location of mutations identified in DC patients. The RCT domain of mouse Rap1 (amino acids [aa] 295 to 393) was fused to the N terminus (aa 2) of TIN2 and L247E to generate RCT-TIN2 alleles. All alleles have an N-terminal FLAG-HA2 tag. The residue in red indicates the mutation introduced to generate the TRF1 binding mutant. (B) The indicated FLAG-HA2-tagged proteins were introduced into TIN2F/F MEFs. Immunoblotting for the indicated proteins was conducted in cell extracts from MEFs with or without Cre treatment (72 h). (C) Extracts from TIN2F/F MEFs infected with empty vector (EV) or the indicated FLAG-HA2 TIN2 alleles following Cre treatment (72 h) were immunoprecipitated (IP) with anti-HA resin, and immunocomplexes were probed for the indicated proteins. (D) Extracts from TIN2F/F MEFs infected with EV or the indicated FLAG-HA2 RCT-TIN2 alleles following Cre treatment (72 h) were immunoprecipitated with anti-HA resin, and immunocomplexes were probed for the indicated proteins.
The addition of the RCT domain to TIN2-L247E restored its association with telomeres as evaluated by IF analysis (Fig. 9A). However, ChIP analysis indicated that the RCT-TIN2 fusion proteins were significantly impaired in their telomeric association compared to that of wild-type TIN2 (Fig. 9B and C). Nonetheless, the RCT domain improved the accumulation of TIN2-L247E at telomeres, as intended (Fig. 7E and F).
FIG 9.

Fusion of the Rap1 RCT domain to TIN2-L247E restores telomere association. (A) Indirect immunofluorescence (IF) for the indicated FLAG-HA2 TIN2 alleles (anti-HA antibody, red) and telomeric FISH (green) in TIN2−/− MEFs (72 h post-Cre). Soluble proteins were extracted by Triton X-100 treatment prior to fixation. DNA is stained with DAPI (blue). Bars, 1.5 μm. (B) Telomeric DNA ChIP for the FLAG-HA2 TIN2 alleles (anti-HA), TRF1, and Rap1 in TIN2F/F MEFs with or without Cre treatment (72 h). (C) Quantification of the ChIP signals from three identical experiments, as performed for panel B. ChIP signals were normalized to the input, and the background (PI) was subtracted. **, P < 0.05 (paired Student's t test).
We also observed that Rap1 levels at the telomere were considerably reduced in cells expressing the RCT-L247E allele, which is likely due to both competition for TRF2 binding and a reduction in Rap1 protein levels (Fig. 9B and C). Interestingly, the RCT-TIN2 (WT) fusion did not completely outcompete Rap1 compared to RCT-TIN2-L247E, perhaps because it can interact with TRF1. ChIP analysis for TRF2 indicated that, as expected, the loss of Rap1 from telomeres did not affect TRF2 at telomeres (Fig. 9B and C). The reduction in Rap1 at telomeres is not anticipated to interfere with the functional analysis of TIN2 since Rap1 is not involved in the repression of ATM/ATR signaling or c-NHEJ at telomeres (24, 25).
Despite its suboptimal telomeric accumulation, RCT-TIN2-L247E showed a restored ability to promote telomere protection by TRF2. Expression of RCT-TIN2-L247E in TIN2-null cells rescued the growth arrest phenotype of MEFs as effectively as wild-type TIN2 and repressed the occurrence of telomere fusions (Fig. 10A, B, and C). Furthermore, the DNA damage response observed in TIN2-deleted cells expressing TIN2-L247E was no longer observed in cells expressing RCT-TIN2-L247E or RCT-TIN2 (Fig. 10D, E, and F). As expected, RCT-TIN2-L247E was also fully competent in repressing the increased overhang phenotype associated with TIN2 deletion (Fig. 11A and B). Thus, despite the lack of connection to TRF1, RCT-TIN2-L247E is proficient in protecting telomeres in conjunction with TRF2 and the TPP1/POT1 heterodimers.
FIG 10.

RCT-L247E rescues telomere deprotection phenotypes of TIN2 deletion. (A) Growth curve of SV40-LT-immortalized TIN2−/− MEFs expressing an empty vector (EV) or the indicated FLAG-HA2 RCT-TIN2 alleles. (B) Examples of metaphases in TIN2-deficient cells expressing RCT-TIN2 alleles by telomeric FISH. Green, TelC PNA probe; red, DAPI DNA stain. (C) Summary of telomere phenotype TIN2-deficient cells expressing RCT-TIN2 alleles, as determined by FISH, as shown in panel B. Values are averages and SDs from 3 independent experiments. Chromosome-type and chromatid-type telomere fusions were scored on 3,000 to 5,000 telomeres/experiment. Long-arm sister telomere fusions and fragile telomeres were scored on 2,000 to 3,000 telomeres/experiment. (D) IF for 53BP1 (red) and telomeric FISH (green) in TIN2−/− MEFs (72 h post-Cre) transduced with RCT-TIN2 or RCT-L247E. DNA is stained with DAPI (blue). Bars, 1.5 μm. (E) Quantification of 53BP1 TIFs, as determined for panel D (n > 100 nuclei) after Cre (72 h). Averages and median values are from three independent experiments and SDs. **, P < 0.05 (paired Student's t test). (F) Immunoblots for Chk1 and Chk2 phosphorylation in TIN2F/F MEFs expressing the indicated FLAG-HA2 TIN2 alleles and treated with Cre (72 h) as indicated.
FIG 11.
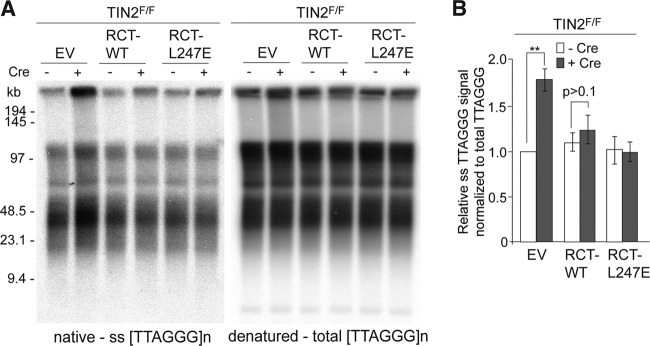
RCT-TIN2-L427E expressing cells have telomeres with normal 3′ overhangs. (A) In-gel hybridization assay for ss telomeric DNA after TIN2 deletion in cells expressing empty vector, RCT-TIN2, or RCT-L247E fusion proteins. Left, TelC signals under the native condition; right, same gel rehybridized after in situ denaturation of the DNA. (B) Quantification of overhang signals from three identical experiments, as described for panel A, were normalized to the total telomeric signals and compared to TIN2F/F MEFs without Cre. **, P < 0.05 (paired Student's t test).
DISCUSSION
In this study, we elucidate the role of the TIN2-TRF1 interaction in shelterin. Our results establish that a subcomplex of shelterin, composed of a modified TIN2 fusion protein (RCT-TIN2-L247E), TRF2, Rap1, TPP1, and POT1a/b, can protect chromosome ends from ATM and ATR signaling, prevent c-NHEJ, and ensure appropriate processing of the telomere ends. Thus, the TIN2-TRF1 link is not required for the tethering of TPP1/POT1a and TPP1/POT1b to telomeres, as observed by the lack of Chk1 phosphorylation and maintenance of the single-stranded telomeric overhang. Furthermore, while loss of TIN2 compromises the ability of TRF2 to repress ATM signaling and c-NHEJ, TRF2-linked TIN2 does not need to associate with TRF1 in order for it to promote these functions. Finally, these data demonstrate that TRF1 can function in the repression of replication problems at telomeres even when it is not connected to the rest of shelterin. However, the TRF1-TIN2 link is clearly important for the shelterin complex since it improves the expression level and telomeric accumulation of TRF1.
Consistent with earlier data on human TIN2, the TIN2-L247E mutant used in this study has no ability to interact with TRF1, while its interactions with TPP1 and TRF2 are unaltered. The use of this mutant revealed the dependency of TIN2 on TRF1 for its normal accumulation at telomeres in vivo. It is not clear why TIN2 is so reliant on TRF1 compared to TRF2 for this purpose. It is possible that the binding of TIN2 to TRF2 detected via co-IP experiments and far-Western analysis is based on a low-affinity interaction and does not reflect how poorly TIN2 interacts with TRF2 in vivo (2, 3, 5, 17). Therefore, it will be of interest to determine the structure and biochemical characteristics of the TRF2-TIN2 interface. Importantly, the interaction of TIN2 with TRF1 not only is critical for the accumulation of TIN2 at telomeres but also improves the expression and telomeric accumulation of TRF1. This effect is most likely due to the previously described tethering role of TIN2, where its ability to link TRF1 and TRF2 was found to increase the telomeric occupancy of both proteins (3, 5, 26). However, in the case of TRF1, its binding to TIN2 does not appear to play an additional role other than ensuring optimal loading of TRF1 on telomeres. As shown here, TRF1 function is not obviously impaired in cells expressing the TIN2-L247E mutant since the fragile telomere phenotype typical of TRF1 deficiency is not observed. This finding is consistent with the prior analysis of TIN2-knockout MEFs in which there was also no indication of impaired TRF1 function (10). Thus, the severely reduced levels of TRF1 that are observed when the protein cannot bind TIN2 are nonetheless sufficient to ensure efficient replication of the duplex telomeric repeat array.
The data obtained with these TIN2 binding mutants further underscore the importance of TIN2 for the proper functioning of TRF2. Previous work using TIN2 KO cells and TRF2 KO cells complemented with a TRF2 mutant that does not bind TIN2 showed that TRF2 requires TIN2 for its optimal accumulation at telomeres (10). Furthermore, the prior work had shown that even when sufficient TRF2 is localized to telomeres, its ability to repress ATM signaling to its full extent requires that TRF2 interacts with TIN2. In contrast, full repression of c-NHEJ by TRF2 does not require TIN2 as long as sufficient TRF2 is present at telomeres, and exogenous expression of TRF2 in TIN2-deleted cells expressing TIN2-L247E fulfilled the same function. The current data provide further insight into how TIN2 modulates the control of ATM signaling by TRF2, showing that TIN2 itself and not its ability to connect TRF2 to TRF1 is important. It will be of obvious interest to understand what aspect of TIN2 is responsible for this attribute and how the binding of TIN2 affects the way TRF2 executes its protective functions.
These results also shed light on the quantities of shelterin required for telomere protection. Remarkably, cells expressing TIN2-L247E have a severely diminished ability to load TPP1 and thus recruit less POT1a and POT1b to telomeres. However, the residual POT1b at telomeres is sufficient to ensure the correct processing at telomere termini, resulting in a wild-type-length 3′ overhang of the telomeres. Previous estimates indicated that mouse telomeres contain in the order of only 20 to 70 copies of POT1b per telomere (10). Since the loading of TPP1 on telomeres in cells expressing TIN2-L247E is diminished by ∼5-fold, the number of POT1b molecules per telomere that can sustain the processing of the telomere terminus may be as low as 4 to 14. Similarly, a dozen POT1a proteins per telomere may be sufficient to repress ATR signaling.
TIN2 has emerged as a remarkably versatile component of shelterin, not only affecting the accumulation of TRF1 and the accumulation and function of TRF2 but also being required for the association of TPP1 and POT1 with telomeres. TIN2 is mutated in a subset of dyskeratosis congenita (DC) patients with severe disease manifestation (27, 28). Although the DC mutations in TIN2 are close to the TRF1 binding site, most do not affect TRF1 binding. It therefore remains unclear which aspect of TIN2 is compromised in these patients. One suggestion is that the TIN2 residues mutated in DC contribute to the telomeric recruitment of HP1γ, which has been implicated in telomere cohesion (29). These distinct functions of TIN2 within shelterin suggest the need for further analysis on how the DC TIN2 mutations may affect the function of shelterin, with regard to both telomere protection and telomerase recruitment.
ACKNOWLEDGMENTS
We thank the members of the de Lange lab for discussion of this work.
This research was supported by grants from the NIH (5R37GM49046 and 5RO1AG16642) and an American Cancer Society postdoctoral fellowship to D.F. T.D.L. is an American Cancer Society Research Professor.
Footnotes
Published ahead of print 27 January 2014
REFERENCES
- 1.Palm W, de Lange T. 2008. How shelterin protects mammalian telomeres. Annu. Rev. Genet. 42:301–334. 10.1146/annurev.genet.41.110306.130350 [DOI] [PubMed] [Google Scholar]
- 2.Houghtaling BR, Cuttonaro L, Chang W, Smith S. 2004. A dynamic molecular link between the telomere length regulator TRF1 and the chromosome end protector TRF2. Curr. Biol. 14:1621–1631. 10.1016/j.cub.2004.08.052 [DOI] [PubMed] [Google Scholar]
- 3.Liu D, O'Connor MS, Qin J, Songyang Z. 2004. Telosome, a mammalian telomere-associated complex formed by multiple telomeric proteins. J. Biol. Chem. 279:51338–51342. 10.1074/jbc.M409293200 [DOI] [PubMed] [Google Scholar]
- 4.Liu D, Safari A, O'Connor MS, Chan DW, Laegeler A, Qin J, Songyang Z. 2004. PTOP interacts with POT1 and regulates its localization to telomeres. Nat. Cell Biol. 6:673–680. 10.1038/ncb1142 [DOI] [PubMed] [Google Scholar]
- 5.Ye JZ, Donigian JR, Van Overbeek M, Loayza D, Luo Y, Krutchinsky AN, Chait BT, de Lange T. 2004. TIN2 binds TRF1 and TRF2 simultaneously and stabilizes the TRF2 complex on telomeres. J. Biol. Chem. 279:47264–47271. 10.1074/jbc.M409047200 [DOI] [PubMed] [Google Scholar]
- 6.Ye JZ, Hockemeyer D, Krutchinsky AN, Loayza D, Hooper SM, Chait BT, de Lange T. 2004. POT1-interacting protein PIP1: a telomere length regulator that recruits POT1 to the TIN2/TRF1 complex. Genes Dev. 18:1649–1654. 10.1101/gad.1215404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Denchi EL, de Lange T. 2007. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature 448:1068–1071. 10.1038/nature06065 [DOI] [PubMed] [Google Scholar]
- 8.Hockemeyer D, Daniels JP, Takai H, de Lange T. 2006. Recent expansion of the telomeric complex in rodents: two distinct POT1 proteins protect mouse telomeres. Cell 126:63–77. 10.1016/j.cell.2006.04.044 [DOI] [PubMed] [Google Scholar]
- 9.Kibe T, Osawa GA, Keegan CE, de Lange T. 2010. Telomere protection by TPP1 is mediated by POT1a and POT1b. Mol. Cell. Biol. 30:1059–1066. 10.1128/MCB.01498-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takai KK, Kibe T, Donigian JR, Frescas D, de Lange T. 2011. Telomere protection by TPP1/POT1 requires tethering to TIN2. Mol. Cell 44:647–659. 10.1016/j.molcel.2011.08.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Celli GB, de Lange T. 2005. DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat. Cell Biol. 7:712–718. 10.1038/ncb1275 [DOI] [PubMed] [Google Scholar]
- 12.van Steensel B, Smogorzewska A, de Lange T. 1998. TRF2 protects human telomeres from end-to-end fusions. Cell 92:401–413. 10.1016/S0092-8674(00)80932-0 [DOI] [PubMed] [Google Scholar]
- 13.Martinez P, Thanasoula M, Munoz P, Liao C, Tejera A, McNees C, Flores JM, Fernandez-Capetillo O, Tarsounas M, Blasco MA. 2009. Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes Dev. 23:2060–2075. 10.1101/gad.543509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, de Lange T. 2009. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 138:90–103. 10.1016/j.cell.2009.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herbig U, Jobling WA, Chen BP, Chen DJ, Sedivy JM. 2004. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol. Cell 14:501–513. 10.1016/S1097-2765(04)00256-4 [DOI] [PubMed] [Google Scholar]
- 16.Loayza D, de Lange T. 2003. POT1 as a terminal transducer of TRF1 telomere length control. Nature 424:1013–1018. 10.1038/4241013a [DOI] [PubMed] [Google Scholar]
- 17.Chen Y, Yang Y, van Overbeek M, Donigian JR, Baciu P, de Lange T, Lei M. 2008. A shared docking motif in TRF1 and TRF2 used for differential recruitment of telomeric proteins. Science 319:1092–1096. 10.1126/science.1151804 [DOI] [PubMed] [Google Scholar]
- 18.Chang W, Dynek JN, Smith S. 2003. TRF1 is degraded by ubiquitin-mediated proteolysis after release from telomeres. Genes Dev. 17:1328–1333. 10.1101/gad.1077103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ye JZ, de Lange T. 2004. TIN2 is a tankyrase 1 PARP modulator in the TRF1 telomere length control complex. Nat. Genet. 36:618–623. 10.1038/ng1360 [DOI] [PubMed] [Google Scholar]
- 20.Donigian JR, de Lange T. 2007. The role of the poly(ADP-ribose) polymerase tankyrase1 in telomere length control by the TRF1 component of the shelterin complex. J. Biol. Chem. 282:22662–22667. 10.1074/jbc.M702620200 [DOI] [PubMed] [Google Scholar]
- 21.Takai H, Smogorzewska A, de Lange T. 2003. DNA damage foci at dysfunctional telomeres. Curr. Biol. 13:1549–1556. 10.1016/S0960-9822(03)00542-6 [DOI] [PubMed] [Google Scholar]
- 22.Sfeir A, de Lange T. 2012. Removal of shelterin reveals the telomere end-protection problem. Science 336:593–597. 10.1126/science.1218498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hockemeyer D, Palm W, Else T, Daniels JP, Takai KK, Ye JZ, Keegan CE, de Lange T, Hammer GD. 2007. Telomere protection by mammalian POT1 requires interaction with TPP1. Nat. Struct. Mol. Biol. 14:754–761. 10.1038/nsmb1270 [DOI] [PubMed] [Google Scholar]
- 24.Martinez P, Thanasoula M, Carlos AR, Gomez-Lopez G, Tejera AM, Schoeftner S, Dominguez O, Pisano DG, Tarsounas M, Blasco MA. 2010. Mammalian Rap1 controls telomere function and gene expression through binding to telomeric and extratelomeric sites. Nat. Cell Biol. 12:768–780. 10.1038/ncb2081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sfeir A, Kabir S, van Overbeek M, Celli GB, de Lange T. 2010. Loss of Rap1 induces telomere recombination in the absence of NHEJ or a DNA damage signal. Science 327:1657–1661. 10.1126/science.1185100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim SH, Beausejour C, Davalos AR, Kaminker P, Heo SJ, Campisi J. 2004. TIN2 mediates functions of TRF2 at human telomeres. J. Biol. Chem. 279:43799–43804. 10.1074/jbc.M408650200 [DOI] [PubMed] [Google Scholar]
- 27.Savage SA, Bertuch AA. 2010. The genetics and clinical manifestations of telomere biology disorders. Genet. Med. 12:753–764. 10.1097/GIM.0b013e3181f415b5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Savage SA, Giri N, Baerlocher GM, Orr N, Lansdorp PM, Alter BP. 2008. TINF2, a component of the shelterin telomere protection complex, is mutated in dyskeratosis congenita. Am. J. Hum. Genet. 82:501–509. 10.1016/j.ajhg.2007.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Canudas S, Houghtaling BR, Bhanot M, Sasa G, Savage SA, Bertuch AA, Smith S. 2011. A role for heterochromatin protein 1gamma at human telomeres. Genes Dev. 25:1807–1819. 10.1101/gad.17325211 [DOI] [PMC free article] [PubMed] [Google Scholar]