

Abstract
The mevalonate pathway is used by cells to produce sterol and nonsterol metabolites and is subject to tight metabolic regulation. We recently reported that squalene monooxygenase (SM), an enzyme controlling a rate-limiting step in cholesterol biosynthesis, is subject to cholesterol-dependent proteasomal degradation. However, the E3-ubiquitin (E3) ligase mediating this effect was not established. Using a candidate approach, we identify the E3 ligase membrane-associated RING finger 6 (MARCH6, also known as TEB4) as the ligase controlling degradation of SM. We find that MARCH6 and SM physically interact, and consistent with MARCH6 acting as an E3 ligase, its overexpression reduces SM abundance in a RING-dependent manner. Reciprocally, knockdown of MARCH6 increases the level of SM protein and prevents its cholesterol-regulated degradation. Additionally, this increases cell-associated SM activity but is unexpectedly accompanied by increased flux upstream of SM. Prompted by this observation, we found that knockdown of MARCH6 also controls the level of 3-hydroxy-3-methyl-glutaryl coenzyme A reductase (HMGCR) in hepatocytes and model cell lines. In conclusion, MARCH6 controls abundance of both SM and HMGCR, establishing it as a major regulator of flux through the cholesterol synthesis pathway.
INTRODUCTION
The mevalonate pathway leading to cholesterol synthesis is controlled transcriptionally and, for more rapid shutdown, posttranslationally (1). The third step in the pathway, catalyzed by 3-hydroxy-3-methyl-glutaryl coenzyme A reductase (HMGCR), is generally regarded as the rate-limiting step in cholesterol synthesis and has been intensively studied (2, 3). However, squalene monooxygenase (SM) is a neglected rate-limiting enzyme in cholesterol synthesis downstream of HMGCR. A flavin monooxygenase located in the endoplasmic reticulum (ER), SM catalyzes the conversion of squalene into monooxidosqualene (MOS), the step in the mevalonate pathway preceding cyclization to form the steroid backbone. It can also act on its product to yield dioxidosqualene (DOS), the precursor for the potent oxysterol regulator 24(S),25-epoxycholesterol, which fine-tunes acute cholesterol synthesis (4). SM resides after the isoprenoid branch of the mevalonate pathway, committing products to sterol synthesis. This may allow differential control of cholesterol synthesis from that of essential nonsterol products (5).
We recently reported that SM's activity is controlled at the posttranslational level via accelerated cholesterol-dependent ubiquitination and proteasomal degradation (5). This regulation requires the first 100 amino acids of the protein—a region that is highly conserved in vertebrates but lacking in lower organisms, such as yeast (Saccharomyces cerevisiae). Moreover, this sequence is sufficient to confer cholesterol-dependent turnover when fused to green fluorescent protein (GFP) (5). We have established that the process of degradation is distinct from the sterol-regulated ubiquitination and proteasomal degradation of HMGCR, as it does not require insulin-induced gene proteins (INSIGs) or side-chain oxysterols but rather responds to cholesterol itself (5). However, numerous details of the degradation of SM remain to be uncovered, including the identity of the E3 ubiquitin ligase involved.
The ubiquitin-proteasome system is the major nonlysosomal pathway for intracellular degradation of proteins and plays major roles in regulation of many cellular processes. Ubiquitin modification of substrate proteins is achieved by the activity of E1-activating, E2-conjugating, and E3 ligase enzymes. Ubiquitination occurs with exquisite spatial, temporal, and substrate specificity, much of which is dictated by the more than 600 E3 ligases encoded by the mammalian genome (6). The ubiquitin-proteasome system participates in diverse cellular processes. However, our understanding of the role this system plays in cholesterol metabolism is limited. Only a few E3 ligases have been identified in cholesterol-related processes, such as in the degradation of HMGCR (7, 8) and the low-density lipoprotein (LDL) receptor (9). Therefore, our aim in this study was to identify and characterize the E3 ligase responsible for mediating the cholesterol-dependent degradation of SM.
We report here that the E3, membrane-associated RING finger 6, MARCH6 (also known as TEB4 or, in yeast, Doa10), is the ligase required for this process. We provide gain- and loss-of function evidence for MARCH6's involvement in controlling basal and cholesterol-dependent degradation of SM. Interestingly, we demonstrate that silencing MARCH6 also stabilizes HMGCR. We propose therefore that MARCH6 defines a previously unrecognized point of control in cholesterol biosynthesis in mammalian cells.
MATERIALS AND METHODS
Materials.
V5 antibody, superscript III reverse transcriptase, Lipofectamine RNAiMAX, RPMI medium, newborn calf serum (NCS), Dulbecco's modified Eagle's medium (DMEM), DMEM–Ham's F-12 nutrient mixture (1:1) (DMEM–F-12), and NuPAGE Bis-Tris gels were obtained from Life Technologies (Carlsbad, CA). TransIT-2020 was from Mirus Bio (Madison, WI), and JetPrime was from Polyplus (Illkirch, France). α-Tubulin antibody, cycloheximide, compactin (mevastatin), small interfering RNA (siRNA), primers, mevalonate, MG132 (Z-Leu-Leu-Leu-al), protease inhibitor cocktail, and Tri reagent were from Sigma Aldrich (St. Louis, MO). SensiMix SYBR No-Rox was from Bioline (London, United Kingdom). Lanosterol synthase inhibitor, R048-8071, was from Sapphire Bioscience (Waterloo, NSW, Australia). Fetal calf serum (FCS) was from Bovogen (East Keilor, Victoria, Australia). Lipoprotein-deficient serum (LPDS) and lipoprotein-deficient fetal calf serum (FCLPDS) were prepared from heat-inactivated NCS and FCS, respectively, as previously described (10). Cholesterol was complexed to methyl-β-cyclodextrin (CD), as described previously (11). Myc antibody was from Abcam (Cambridge, United Kingdom). Endogenous SM antibody was from Protein Tech Group (Chicago, IL). The HMGCR and GFP antibodies were previously described (9). Protein A/G PLUS-agarose beads were from Santa Cruz Biotechnology (Dallas, TX). [14C]acetate (specific radioactivity, 55.3 mCi/mmol, 2.046 GBq/mmol) and [14C]mevalonate (specific radioactivity, 53.9 mCi/mmol, 1.994 GBq/mmol) were from PerkinElmer (Waltham, MA). Cholesterol and 25-hydroxycholesterol (25HC) were from Steraloids (Newport, RI). Hemagglutinin (HA) antibody was from Covance (Princeton, NJ).
Plasmids.
SM plasmids encoding either full-length SM or the first 100 amino acids (SM-N100) have been described previously (5). Full-length SM was also cloned into pDEST40 using gateway-based recombination and was used in some experiments. The human MARCH6 gene was amplified from human cDNA and inserted into pcDNA4/mycHisB to obtain pcDNA4/MARCH6-myc. To create a nonfunctional MARCH6, a C9A mutation (12) was introduced using megaprimed PCR site-directed mutagenesis (13). An independent set of corresponding MARCH6 expression plasmids (12), which were kind gifts from Emmanuel Wiertz (University of Utrecht, The Netherlands), were also used to confirm some of the key findings. pTK-HMGCR-V5 (human) was prepared by replacing the coding sequence of squalene monooxygenase in pTK SM-V5 (5) with that of amino acids 1 to 887 from pRed-227 (14), a kind gift from Michael Brown and Joseph Goldstein (UT Southwestern Medical Center, Dallas, TX). The sequence encoding the first 350 amino acids of hamster HMGCR was amplified from pIRESneo-HMG350-3XHA (15), a kind gift from Joseph Roitelman (Sheba Medical Center, Israel) and subcloned into pENTR1A-GFP (Addgene no. 19364) to create pENTR1A-HMGCR1–350-GFP. Subsequently, pLenti6.3-HMGCR1–350-GFP was obtained by LR gateway recombination with pLenti6.3-DEST (Invitrogen). The resulting construct was used to produce viral particles for infection of A431 cells. Plasmids were transfected into cells using TransIT-2020 or JetPrime reagent according to standard protocols.
Cell lines.
Flp-In T-Rex human embryonic kidney (HEK293) cells were obtained from Life Technologies and stably transfected with a plasmid encoding the first 100 amino acids of SM fused to GFP and V5 (SM-N100-GFP-V5) and selected by growth in hygromycin B. CHO-7 cells stably overexpressing full-length SM-V5 or HMGCR-V5 were created using a similar Flp-In system. All other cell lines were gifts as follows: HeLaT cells from Noel Whitaker (University of New South Wales [UNSW], Sydney, Australia), Be(2)C cells from Louise Lutze-Mann (UNSW, Sydney, Australia), SRD-1 cells from Michael Brown and Joseph Goldstein (UT Southwestern Medical Center, Dallas, TX), HepG2 cells from the Centre for Vascular Research (UNSW, Sydney, Australia), and A431 cells from Vilja Pietiainen and Elina Ikonen (University of Helsinki, Helsinki, Finland).
HEK293, A431, and Be(2)C cells were grown in 10% (vol/vol) FCS–high-glucose DMEM and pretreated and treated in 10% (vol/vol) FCLPDS–high-glucose DMEM. CHO-7 cells were grown, pretreated and treated in 5% (vol/vol) LPDS–DMEM–F-12. HeLaT cells were grown in 10% (vol/vol) FCS-RPMI, and pretreated and treated in 10% (vol/vol) FCLPDS-RPMI. HepG2 cells were grown in 10% (vol/vol) FCS–low-glucose DMEM and pretreated and treated in 10% (vol/vol) FCLPDS–low-glucose DMEM. SRD-1 cells were grown in 5% (vol/vol) LPDS–DMEM–F-12 with 1 μg/ml 25-hydroxycholesterol and pretreated and treated in 5% (vol/vol) LPDS–DMEM–F-12.
siRNA transfection.
To knock down specific E3 ubiquitin ligase genes, cells were transfected with 25 nM siRNA using Lipofectamine RNAiMAX or PolyPlus JetPrime for 24 h, followed by overnight statin pretreatment with compactin and mevalonate as described previously (5) and then treated as indicated in the figure legends.
qRT-PCR.
Following siRNA transfection and pretreatment, total RNA was harvested from cells using Tri reagent and then reverse transcribed to cDNA using Superscript III reverse transcriptase. Gene expression levels of gp78, TRC8, HRD1, MARCH6, and WSB1 were determined and normalized to the housekeeping gene coding for porphobilinogen deaminase (PBGD). The primer sequences for quantitative reverse transcription-PCR (qRT-PCR) are listed in Table S1 in the supplemental material.
Western blotting.
To probe for SM, HMGCR, GFP, and α-tubulin, protein lysates were prepared in 10% (wt/vol) SDS with protease inhibitors as previously described (5). When probing for MARCH6, protein lysates were prepared using a modified radioimmunoprecipitation assay (RIPA) buffer, passed through a 22-gauge needle 40 times, and then rotated at 4°C for 30 min as described previously (16). Protein lysates were separated by SDS-PAGE and probed with the following antibodies: V5 (SM or HMGCR), endogenous SM, endogenous HMGCR, GFP [HMGCR(1–350)], α-tubulin, HA (ubiquitin), and myc (MARCH6) as required. Densitometric analysis was performed with the ImageJ software package (NIH).
Immunoprecipitation.
HEK293 cells were transiently transfected with SM-V5 and wild-type (WT) or C9A MARCH6-myc expression plasmids and treated with or without 25 μM MG132 for 6 h. To control for transfection efficiencies, pEGFP-N3 was cotransfected. Lysates were prepared in RIPA buffer with protease inhibitors and precleared by centrifugation at 20,000 × g for 10 min at 4°C. MARCH6-myc was immunoprecipitated by incubation with myc antibody (1:1,000) for 2 h followed by addition of protein A/G PLUS-agarose beads for 16 h. SM-V5 was immunoprecipitated as described above with V5 antibody. Beads were then washed three times with RIPA buffer. All steps were performed at 4°C. Proteins were eluted from the beads by being heated in loading buffer for 15 min at 65°C. Samples were separated on NuPAGE Bis-Tris gels and immunoblotted as indicated.
Nonsaponifiable lipid synthesis and SM activity.
To measure neutral lipid synthesis following siRNA transfection and pretreatment, cells were incubated for 2 h with 1 μCi [14C]acetate/well in a 6-well plate. For the SM activity assay, cells were incubated for 2 h with 8 μCi [14C]mevalonate/well and 10 μM oxidosqualene cyclase inhibitor in a 6-well plate. For both assays, cells were then lysed in 0.1 M NaOH and saponified as previously described (5). The nonsaponifiable lipids were subjected to either scintillation counting, and dpm values per mg protein were determined, or thin-layer chromatography and phosphorimaging as described previously (5). Densitometry was performed using ScienceLab ImageGuage (v4.0).
Confocal imaging.
A431 cells stably expressing HMGCR(1–350)-GFP were fixed and stained with DAPI (4′,6-diamidino-2-phenylindole) before examination. A Leica TCS SP8 confocal microscope equipped with a 405-nm laser (for DAPI) and 488-nm laser (for GFP) and a 63× objective (Leica Microsystems, Mannheim, Germany) was used. The GFP images were taken with identical laser power, gain, and offset in order to compare localizations and intensities between the different samples. The GFP fluorescence is depicted in the “glow-over-under” mode.
RESULTS
Knockdown of MARCH6 stabilizes SM-N100 and endogenous SM.
To identify the E3 ligase involved in degradation of SM, we used a candidate approach in a human embryonic kidney 293 cell line (HEK293) that stably overexpresses SM-N100-GFP-V5 (the first 100 amino acids of the N terminus, known to be the regulatory region [5], fused to GFP, with a V5 epitope tag). Degradation of SM-N100-GFP-V5 showed robust cholesterol regulation in these cells both with and without the protein synthesis inhibitor cycloheximide (Fig. 1A). We then used siRNA to knock down expression of a number of candidate E3 ligases (Fig. 1B). The siRNA oligonucleotides decreased target gene expression by between ∼60 and 90%.
FIG 1.

Effect of various E3 ligases on the cholesterol-dependent degradation of SM-N100. (A) HEK-SM-N100-GFP-V5 cells were statin pretreated overnight, and then treated with or without 20 μg/ml cholesterol-cyclodextrin (Chol/CD) and 10 μg/ml cycloheximide (CHX) for 8 h. Protein lysates were subjected to SDS-PAGE and immunoblotted as indicated. (B) HEK-SM-N100-GFP-V5 cells were transfected with the indicated siRNA for 24 h and statin pretreated overnight, RNA was isolated, and expression of the indicated genes was analyzed by qRT-PCR. Control siRNA has been set to 1 for each gene. Data are presented as means ± standard errors of the means (SEM). (C to E) HEK-SM-N100-GFP-V5 cells were transfected with the indicated siRNA for 24 h, statin pretreated overnight, and then treated with or without 20 μg/ml cholesterol-cyclodextrin for 8 h. Total cell lysates were subjected to SDS-PAGE and immunoblotted as indicated. Immunoblots are representative of at least 2 independent experiments with similar results.
gp78 and TRC8 (also known as AMFR and RNF139, respectively) are two E3 ligases implicated in sterol-regulated HMGCR degradation (7, 8), although this has been recently questioned (17). siRNA targeting gp78 or TRC8 did not affect degradation of SM-N100-GFP-V5 (Fig. 1C). HRD1 (also known as SYVN1) has been shown to play a role in basal degradation of HMGCR (18), but siRNA against this E3 ligase also had no effect on SM-N100-GFP-V5 levels (Fig. 1D).
In yeast, the only other E3 ligase, besides HRD1, known to mediate ubiquitination of ER-associated degradation (ERAD) substrates is membrane-bound Doa10 (3). Therefore, we knocked down expression of MARCH6, the mammalian homolog of Doa10. We achieved an ∼60% knockdown of MARCH6 mRNA (Fig. 1B), and this abolished the robust cholesterol-dependent turnover of ectopic SM-N100 (Fig. 1D), as well as increasing basal levels of the reporter construct. The best-characterized substrate of MARCH6 (besides itself [12]) is the ER resident thyroid hormone-activating type 2 deiodinase (D2), which is also inactivated by ubiquitination via the hedgehog-inducible WSB1 (19). We therefore tested if WSB1 also degrades SM, but found no effect, contrary to the marked effect observed with MARCH6 siRNA (Fig. 1E). We also employed a second, independent, siRNA targeting MARCH6 and found a similar stabilizing effect on both SM-N100 and, importantly, endogenous SM (Fig. 2A). Furthermore, MARCH6 mRNA was not affected by changing sterol conditions (data not shown). Importantly, we found that the effect of MARCH6 on endogenous SM levels could be generalized, as it was observed in several relevant cell types, including those of neuronal [Be(2)C] and hepatic (HepG2) origins, with or without cycloheximide treatment (Fig. 2B), or when cholesterol was introduced by addition of lipoprotein-containing full serum (data not shown). Collectively, basal levels of ectopic and endogenous SM protein were increased markedly when MARCH6 was silenced, suggesting that cholesterol-dependent degradation may involve acceleration of the same mechanism used for basal degradation.
FIG 2.
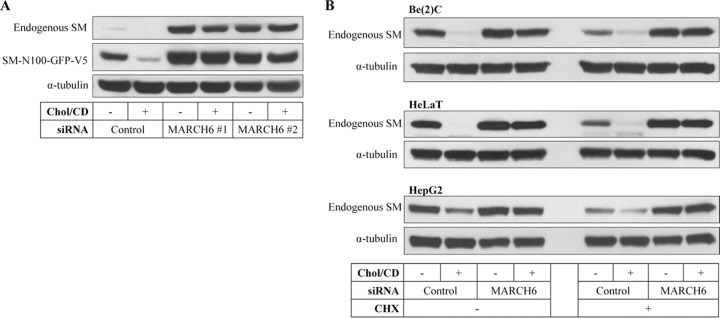
Knockdown of MARCH6 abolishes cholesterol-regulated degradation of SM in a range of cell types. (A) HEK-SM-N100-GFP-V5 cells were transfected with two independent MARCH6 siRNAs for 24 h, statin pretreated overnight, and then treated with or without 20 μg/ml cholesterol-cyclodextrin (Chol/CD) for 8 h. (B) Be(2)C, HeLaT, and HepG2 cells were transfected with control or MARCH6 siRNA for 24 h, pretreated overnight, and then treated with or without 20 μg/ml cholesterol-cyclodextrin. Where indicated, 10 μg/ml cycloheximide (CHX) was added for 8 h. Total cell lysates were subjected to SDS-PAGE and immunoblotted as indicated. Immunoblots are representative of at least 2 independent experiments with similar results.
MARCH6 interacts with and degrades SM.
Since MARCH6 has been implicated in ERAD (12), we reasoned that stabilization of SM when MARCH6 is silenced is due to decreased ubiquitination and subsequent proteasomal degradation of SM. In line with this, we found that stabilization of SM with the proteasomal inhibitor MG132 was similar to that observed with MARCH6 siRNA (Fig. 3A).
FIG 3.

MARCH6 interacts with SM and targets SM-N100 for proteasomal degradation. (A) HEK-SM-N100-GFP-V5 cells were transfected with control or MARCH6 siRNA for 24 h, statin pretreated overnight, and then treated with or without 20 μg/ml cholesterol-cyclodextrin (Chol/CD) and 10 μM MG132 for 8 h. (B) HEK293 cells were cotransfected with expression plasmids for SM-N100-GFP-V5 and either wild-type (WT) or mutant (C9A) MARCH6-myc for 24 h, statin pretreated overnight, and then treated with or without 20 μg/ml cholesterol-cyclodextrin for 8 h. (C) HEK293 cells were transfected with expression plasmids for SM-V5 and either wild-type or mutant (C9A) MARCH6-myc, as well as a GFP construct to control for transfection efficiency. Cells were treated with or without 25 μM MG132 for 6 h prior to harvest, and MARCH6 was immunoprecipitated (IP). Samples were subjected to SDS-PAGE and immunoblotted as indicated. Immunoblots are representative of at least 2 independent experiments with similar results. *, nonspecific band.
Conversely, cotransfection of wild-type MARCH6 with SM-N100 resulted in lower levels of the cotransfected SM-N100, which was nevertheless still sterol responsive (Fig. 3B). SM-N100 levels were restored when a nonfunctional MARCH6 with an inactivating mutation (C9A) in the RING finger domain was used (12). Note that the level of wild-type MARCH6 protein following transfection is substantially lower than that detected with mutant MARCH6 (Fig. 3B and C). This most likely reflects, at least in part, autoubiquitination and subsequent degradation by the wild-type form, a common feature of E3 ligases, also previously reported for MARCH6 (12).
To further demonstrate the interaction between SM and MARCH6, we used an immunoprecipitation approach. Here, we similarly observed that cotransfection of full-length SM with MARCH6 resulted in lower expression of SM, which was restored by the C9A mutation or the proteasomal inhibitor MG132 (Fig. 3C, lower panel). Following immunoprecipitation of MARCH6, we could detect coimmunoprecipitating SM, suggesting that the two proteins interact (Fig. 3C, upper panel).
To directly confirm that MARCH6 ubiquitinates SM, we employed a ubiquitination assay with epitope-tagged ubiquitin cotransfected with SM and the MARCH6 wild type or mutant. This showed that MARCH6 promotes ubiquitination of SM (Fig. 4).
FIG 4.
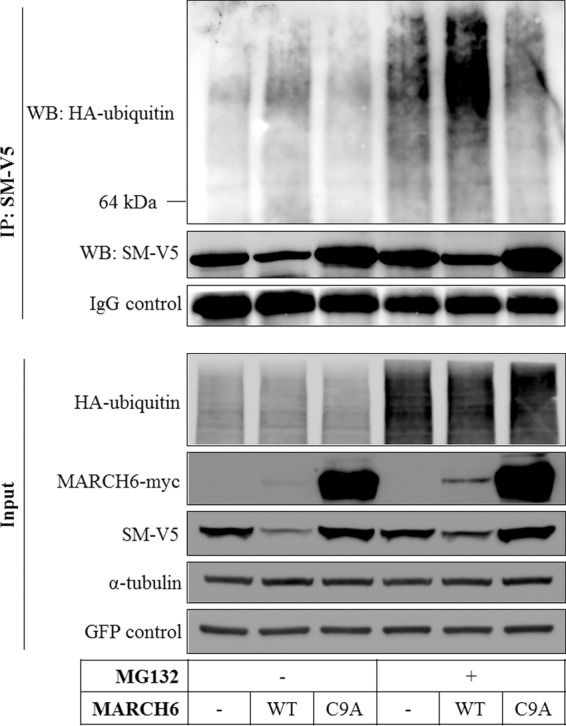
MARCH6 ubiquitinates SM. HEK293 cells were transfected with expression plasmids for SM-V5, HA-ubiquitin, and either wild-type (WT) or mutant (C9A) MARCH6-myc as indicated, as well as a GFP construct to control for transfection efficiency. Cells were treated with or without 50 μM MG132 for 5 h prior to harvest, and SM was immunoprecipitated (IP). Samples were subjected to SDS-PAGE and immunoblotted (Western blotting [WB]) as indicated. The 64-kDa marker indicates the position of SM. Immunoblots are representative of at least 2 independent experiments with similar results.
Knockdown of MARCH6 increases flux through the mevalonate pathway and SM activity.
To examine functional consequences of MARCH6 on SM, we utilized an SRD-1 cell line that has an overactive sterol regulatory element binding protein (SREBP) pathway (20), and thus transcriptional effects are excluded. First, we confirmed that MARCH6 knockdown stabilizes SM in this cell line (Fig. 5A).
FIG 5.
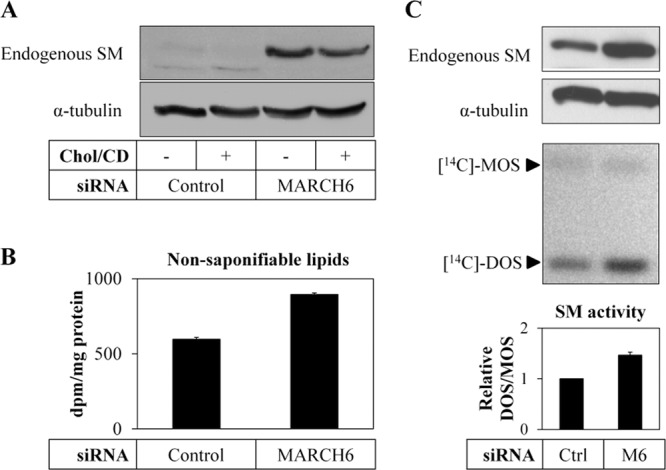
Knockdown of MARCH6 increases SM activity. (A) SRD-1 cells were transfected with control or MARCH6 siRNA for 24 h, statin pretreated overnight, and then treated with or without 20 μg/ml cholesterol-cyclodextrin (Chol/CD) for 8 h. Total cell lysates were subjected to SDS-PAGE and immunoblotted as indicated. (B) SRD-1 cells were transfected with control or MARCH6 siRNA for 24 h, statin pretreated overnight, and subsequently incubated with [14C]acetate for 2 h. A statin-treated control was included to determine the background. Nonsaponifiable lipids were isolated and measured by scintillation counting. Data are means ± SEM from 3 independent experiments. (C) SRD-1 cells were transfected with control (Ctrl) or MARCH6 (M6) siRNA for 24 h and statin pretreated overnight, and total cell lysates were subjected to SDS-PAGE and immunoblotted as indicated (top panels) or incubated with [14C]mevalonate for 2 h in the presence of 10 μM lanosterol synthase inhibitor (bottom panels). Nonsaponifiable lipids were separated by thin-layer chromatography and visualized using phosphorimaging. MOS, monooxidosqualene; DOS, dioxidosqualene; Ctrl, control. SM activity is expressed as the DOS/MOS ratio with means ± SEM from 3 independent experiments. The control siRNA condition has been set to 1.
Next we determined flux through the mevalonate pathway by measuring the synthesis of nonsaponifiable lipids from [14C]acetate (Fig. 5B) and found that MARCH6 siRNA increased these by 50% over the control. To more directly test SM activity, we acutely labeled cells with [14C]mevalonate, which enabled us to measure monooxidosqualene (MOS) and dioxidosqualene (DOS), a substrate and product of SM, respectively. We treated cells with a lanosterol synthase inhibitor to prevent further flux beyond SM (4). Under these conditions, MARCH6 siRNA stabilized SM and accordingly increased its activity by ∼50% as evident from the greater DOS/MOS ratio (Fig. 5C).
Knockdown of MARCH6 stabilizes HMGCR.
Paradoxically, MARCH6 gene silencing increased [14C]squalene accumulation (but not sterol production) when cells were acutely labeled with [14C]acetate (Fig. 6A, left panel). The [14C]squalene accumulation observed with [14C]acetate labeling represents increased flux through the pathway, as there was no corresponding detectable increase in cell squalene levels as assessed by gas chromatography-mass spectrometry (GC-MS) (data not shown). This suggests that MARCH6 may be affecting a target upstream of SM. We therefore considered the possibility that MARCH6 is affecting the stability of HMGCR, the other major control point in cholesterol synthesis. To test this, we labeled cells with [14C]mevalonate, thus bypassing the HMGCR control point. Importantly, MARCH6 knockdown still increased flux through the pathway (total nonsaponifiable lipids, 2,000 ± 370 dpm/mg for control siRNA, versus 4,000 ± 560 dpm/mg for MARCH6 siRNA) but did not lead to an accumulation of squalene (Fig. 6A, right panel). One plausible scenario that could resolve these apparently conflicting results is that MARCH6 knockdown increases HMGCR protein and activity, and with acute [14C]acetate labeling, flux through the beginning of the pathway is accelerated, accumulating at the SM control point. By eliminating confounding effects from HMGCR, acute [14C]mevalonate labeling allows synthesis to proceed through the SM control point, with negligible squalene accumulation but increased downstream sterol production (notably lanosterol) (Fig. 6A, right panel).
FIG 6.

Knockdown of MARCH6 increases ectopic HMGCR expression. (A) SRD-1 cells were transfected with control (Ctrl) or MARCH6 (M6) siRNA for 24 h, statin pretreated overnight, and subsequently incubated with [14C]acetate (left) or [14C]mevalonate (right) for 2 h. Nonsaponifiable lipids were separated by thin-layer chromatography and visualized using phosphorimaging. (B and C) A431 cells stably overexpressing the sterol-sensing domain of HMGCR fused to GFP (SSD-GFP) were transfected with control or MARCH6 siRNA for 24 h, grown in low-sterol medium overnight, and then treated with or without 2.5 μM 25-hydroxycholesterol (25HC) for 2 h. Subsequently, cells were fixed and imaged by confocal fluorescence microscopy (B), or total cell lysates were subjected to SDS-PAGE and immunoblotted as indicated (C). (D) CHO-7 cells stably overexpressing HMGCR-V5 were transfected with control or MARCH6 siRNA for 24 h, statin pretreated overnight, and then statin treated with or without 10 μM 25HC for 4 h. Total cell lysates were subjected to SDS-PAGE and immunoblotted as indicated.
To confirm that HMGCR is indeed stabilized by MARCH6 siRNA, we next engineered cells that stably overexpress the sterol-sensing-domain (SSD) of HMGCR fused to GFP. Since the SSD is a major determinant of HMGCR-regulated degradation (21), the abundance of this reporter construct reflects its posttranslational stability. In line with the reported regulation of HMGCR, we found that sterol depletion increases, while 25-hydroxycholesterol (25HC) decreases, abundance of the HMGCR-SSD-GFP protein (Fig. 6B and C). Under these conditions, silencing of MARCH6 enhanced the level of the reporter protein (Fig. 6B and C). MARCH6 knockdown was associated with a slight blunting of sterol-mediated degradation (reduction by 44% versus 61% for control; P < 0.05). We observed similar stabilization following silencing of MARCH6 in a cell line that stably overexpresses a full-length HMGCR-V5 construct, with and without added 25HC (Fig. 6D). The effect of MARCH6 on HMGCR was not limited to ectopic systems. Silencing of MARCH6 in HepG2 cells increased the abundance of endogenous HMGCR (Fig. 7) without changing its mRNA levels (data not shown). It should be noted, however, that the effects of 25HC here are likely to be both transcriptional (via SREBP suppression) as well as posttranslational (accelerating proteasomal degradation) (3). Collectively, these results point to MARCH6 being a regulator of HMGCR abundance, although this may occur through indirect mechanisms.
FIG 7.

Knockdown of MARCH6 increases endogenous HMGCR expression. HepG2 cells were transfected with control (Ctrl) or MARCH6 (M6) siRNA for 24 h and grown in full serum or low-sterol medium overnight. Subsequently, cells were treated with or without 2.5 μM 25-hydroxycholesterol (25HC) for 0, 2, 4, or 8 h, and total cell lysates were immunoblotted as indicated. Immunoblots are representative of at least 3 independent experiments with similar results. Densitometry is shown relative to the control siRNA condition at 0 h, which has been set to 1. Data are presented as means ± SEM.
DISCUSSION
The mevalonate pathway is subject to tight transcriptional and posttranscriptional regulation, the latter allowing rapid adaptation of the metabolic flux to cellular demand (1). With this in mind, the most important finding of our study is the identification of the E3 ubiquitin ligase MARCH6 as a central component of this posttranslational regulatory network. Using gain- and loss-of-function approaches, we provide evidence indicating that MARCH6 controls degradation of SM and HMGCR in mammalian cells and, as a consequence, cholesterol synthesis.
MARCH6 is one of 11 members of the MARCH family of membrane-associated E3 ligases (22). Members of this family are reported to modulate intracellular trafficking and turnover of membrane proteins following ubiquitination of their substrate. Among these, the ER resident MARCH6 is a relatively unstudied member of this family (12), and before this work, its only other reported substrate was type 2 iodothyronine deiodinase (19). We report here that MARCH6 promotes cholesterol-dependent degradation of SM. As none of the other ER-resident E3 ligases implicated in ERAD and cholesterol metabolism that were tested could account for this activity, we suggest that MARCH6 provides the molecular basis for our earlier report describing SM degradation (5). Interestingly, anecdotal evidence suggests that regulation of SM activity via proteasomal degradation may be an evolutionarily conserved process. Doblas et al. recently reported that Arabidopsis strains carrying a hypomorphic SM allele display massive accumulation of squalene in their roots (23). Importantly, when a mutant allele of the plant homolog of MARCH6, SUD1, was crossed into this background, squalene accumulation was dramatically relieved. This observation is consistent with increased SM activity in SUD1 plants, plausibly due to decreased degradation of the hypomorphic SM allele in the absence of SUD1. Although the authors did not raise the possibility that plant SM is a substrate of SUD1, their work contributes to the concept that an E3 ligase closely related to MARCH6 degrades SM across eukaryotic life.
In addition to promoting SM degradation, we find that silencing MARCH6 increases the basal level of HMGCR protein and flux through the cholesterol synthesis pathway. Intriguingly, in the mutant SUD1 allele described above, HMGCR activity is also elevated but apparently not via stabilization of the plant HMGCR protein (23). However, the contribution of MARCH6 to sterol-stimulated degradation of HMGCR is less clear. Despite a slight decrease in the rate of HMGCR degradation, sterols are still able to promote HMGCR degradation when MARCH6 is silenced. Similarly, silencing of the Drosophila homolog of MARCH6, dTeb4, in a heterologous system does not affect sterol-regulated degradation of hamster HMGCR (16).
The E3 ligases gp78 (8, 24) and TRC8 (7) are implicated in controlling the sterol-regulated degradation of HMGCR. In support of gp78's role, hepatic-specific loss of gp78 in mice increased Hmgcr protein and rendered isolated gp78−/− hepatocytes resistant to sterol-induced degradation of Hmgcr (25). However, regulated degradation of INSIGs by both gp78 (25–27) and TRC8 (28, 29) is a confounding factor, as INSIGs are also used to regulate the SREBP pathway and are required for HMGCR degradation (21, 30, 31). Accordingly, a recent report questions the involvement of these two ligases in sterol-regulated degradation of HMGCR (17). Tsai et al. report that absence of gp78 in MEFs derived from gp78−/− mice or following siRNA-mediated silencing in the same cells used in references 7 and 26 does not abrogate the sterol-stimulated turnover of HMGCR, even though this dramatically increases INSIG-1 (17). Silencing of TRC8 alone or in cells devoid of gp78 activity also did not prevent sterol-stimulated degradation of HMGCR. The reason for the discrepancies between the aforementioned studies is unclear and may be attributed to methodological differences (e.g., the concentration of siRNAs used [17] or cell- and tissue-specific factors [7, 25]). However, it is also possible that another, yet unidentified, E3 ligase accounts for regulated degradation of HMGCR, as suggested by Tsai et al. (17). Based on our results showing that silencing of MARCH6 increases the abundance of endogenous HMGCR in HepG2 cells and of ectopic HMGCR in A431 and CHO cells, we propose that MARCH6 represents an E3 candidate for HMGCR. Although our results have confirmed that MARCH6 affects basal levels of HMGCR (comparable to what has been shown for HRD1 [18]), its contribution to sterol-stimulated degradation of HMGCR needs to be investigated further.
While many of the mechanistic details underlying how MARCH6 controls the level of HMGCR remain to be investigated, it is important to point out that degradation of SM by this E3 does not require INSIGs (5), and it remains to be seen whether this holds for HMGCR as well. It should also be noted that while 25HC promotes rapid proteasomal degradation of HMGCR, SM is degraded in response to cholesterol itself. Collectively, this points to MARCH6 using distinct mechanisms to control SM and HMGCR degradation that may allow cells to selectively shut down cholesterol synthesis, while sparing the isoprenoid branch of the mevalonate pathway (Fig. 8). Our work may inform new strategies that selectively target cholesterol synthesis rather than the statin class of drugs that also inhibit nonsterols.
FIG 8.

A model for how MARCH6 affects the cholesterol synthesis pathway. MARCH6 appears to degrade SM through a cholesterol-dependent mechanism and to be involved in the basal degradation of HMGCR. The contribution of MARCH6 to sterol-dependent degradation of HMGCR is less clear.
During the preparation of this article, Foresti et al. (32) reported that yeast Doa10 degrades Erg1, the yeast homolog of SM. Doa10-dependent degradation of Erg1 was proposed to be triggered in response to lanosterol accumulation and occurred via a single lysine residue that is not conserved in mammalian SM. Foresti et al. then went on to show that the genetic depletion of the Doa10 homolog MARCH6 stabilizes endogenous SM in HEK293 cells. In the present study, we have considerably extended the investigation of the degradation of SM by MARCH6. We narrowed down the domain of SM targeted for destruction by MARCH6 to the first 100 amino acids (representing 17% of the protein), which is notably absent in yeast. Furthermore, we demonstrate that MARCH6 interacts with SM and promotes its ubiquitination and that silencing MARCH6 has functional consequences, increasing flux through the cholesterol synthesis pathway, including SM activity. Importantly, we also implicate MARCH6 in controlling the abundance of HMGCR, although this effect may be indirect.
In conclusion, we show in a variety of relevant cell types that MARCH6 controls the abundance of SM and HMGCR and propose that this E3 ligase is a central regulator of cholesterol synthesis. Ongoing studies addressing the in vivo role of MARCH6 in cholesterol metabolism will critically test this assertion. Although MARCH6 was not identified as a lipid-modifying gene in a large genome-wide-association study (33), the contribution of uncommon MARCH6 variants to lipid levels warrants further investigation. The present study marks the first step in the characterization of MARCH6-dependent degradation of HMGCR and SM. Future studies will contribute to our understanding of MARCH6's role in this process and in cholesterol metabolism.
Supplementary Material
ACKNOWLEDGMENTS
We thank James Krycer, Sandhya Shrestha, and Vincenzo Sorrentino for developing various cell lines and Eric Reits for assistance with imaging. The microscopy images were taken at the core facility Cellular Imaging/LCAM-AMC.
A.J.B.'s lab is supported by grants from the National Health and Medical Research Council (1008081) and the National Heart Foundation of Australia (G11S5757), and N.Z.'s lab is supported by a Career Development Award from the Human Frontier Science Program Organization (HFSPO) and a VIDI grant (17.106.355) from the Netherlands Organization of Scientific Research (NWO).
Footnotes
Published ahead of print 21 January 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01140-13.
REFERENCES
- 1.Sharpe LJ, Brown AJ. 2013. Controlling cholesterol synthesis beyond 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR). J. Biol. Chem. 288:18707–18715. 10.1074/jbc.R113.479808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burg JS, Espenshade PJ. 2011. Regulation of HMG-CoA reductase in mammals and yeast. Prog. Lipid Res. 50:403–410. 10.1016/j.plipres.2011.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jo Y, Debose-Boyd RA. 2010. Control of cholesterol synthesis through regulated ER-associated degradation of HMG CoA reductase. Crit. Rev. Biochem. Mol. Biol. 45:185–198. 10.3109/10409238.2010.485605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wong J, Quinn CM, Gelissen IC, Brown AJ. 2008. Endogenous 24(S),25-epoxycholesterol fine-tunes acute control of cellular cholesterol homeostasis. J. Biol. Chem. 283:700–707. 10.1074/jbc.M706416200 [DOI] [PubMed] [Google Scholar]
- 5.Gill S, Stevenson J, Kristiana I, Brown AJ. 2011. Cholesterol-dependent degradation of squalene monooxygenase, a control point in cholesterol synthesis beyond HMG-CoA reductase. Cell Metab. 13:260–273. 10.1016/j.cmet.2011.01.015 [DOI] [PubMed] [Google Scholar]
- 6.Metzger MB, Hristova VA, Weissman AM. 2012. HECT and RING finger families of E3 ubiquitin ligases at a glance. J. Cell Sci. 125:531–537. 10.1242/jcs.091777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jo Y, Lee PC, Sguigna PV, DeBose-Boyd RA. 2011. Sterol-induced degradation of HMG CoA reductase depends on interplay of two Insigs and two ubiquitin ligases, gp78 and Trc8. Proc. Natl. Acad. Sci. U. S. A. 108:20503–20508. 10.1073/pnas.1112831108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Song BL, Sever N, DeBose-Boyd RA. 2005. Gp78, a membrane-anchored ubiquitin ligase, associates with Insig-1 and couples sterol-regulated ubiquitination to degradation of HMG CoA reductase. Mol. Cell 19:829–840. 10.1016/j.molcel.2005.08.009 [DOI] [PubMed] [Google Scholar]
- 9.Zelcer N, Hong C, Boyadjian R, Tontonoz P. 2009. LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science 325:100–104. 10.1126/science.1168974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goldstein JL, Basu SK, Brown MS. 1983. Receptor-mediated endocytosis of low-density lipoprotein in cultured cells. Methods Enzymol. 98:241–260. 10.1016/0076-6879(83)98152-1 [DOI] [PubMed] [Google Scholar]
- 11.Brown AJ, Sun L, Feramisco JD, Brown MS, Goldstein JL. 2002. Cholesterol addition to ER membranes alters conformation of SCAP, the SREBP escort protein that regulates cholesterol metabolism. Mol. Cell 10:237–245. 10.1016/S1097-2765(02)00591-9 [DOI] [PubMed] [Google Scholar]
- 12.Hassink G, Kikkert M, van Voorden S, Lee SJ, Spaapen R, van Laar T, Coleman CS, Bartee E, Fruh K, Chau V, Wiertz E. 2005. TEB4 is a C4HC3 RING finger-containing ubiquitin ligase of the endoplasmic reticulum. Biochem. J. 388:647–655. 10.1042/BJ20041241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tseng WC, Lin JW, Wei TY, Fang TY. 2008. A novel megaprimed and ligase-free, PCR-based, site-directed mutagenesis method. Anal. Biochem. 375:376–378. 10.1016/j.ab.2007.12.013 [DOI] [PubMed] [Google Scholar]
- 14.Chin DJ, Gil G, Russell DW, Liscum L, Luskey KL, Basu SK, Okayama H, Berg P, Goldstein JL, Brown MS. 1984. Nucleotide sequence of 3-hydroxy-3-methyl-glutaryl coenzyme A reductase, a glycoprotein of endoplasmic reticulum. Nature 308:613–617. 10.1038/308613a0 [DOI] [PubMed] [Google Scholar]
- 15.Doolman R, Leichner GS, Avner R, Roitelman J. 2004. Ubiquitin is conjugated by membrane ubiquitin ligase to three sites, including the N terminus, in transmembrane region of mammalian 3-hydroxy-3-methylglutaryl coenzyme A reductase: implications for sterol-regulated enzyme degradation. J. Biol. Chem. 279:38184–38193. 10.1074/jbc.M405935200 [DOI] [PubMed] [Google Scholar]
- 16.Faulkner RA, Nguyen AD, Jo Y, DeBose-Boyd RA. 2013. Lipid-regulated degradation of HMG-CoA reductase and Insig-1 through distinct mechanisms in insect cells. J. Lipid Res. 54:1011–1022. 10.1194/jlr.M033639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsai YC, Leichner GS, Pearce MM, Wilson GL, Wojcikiewicz RJ, Roitelman J, Weissman AM. 2012. Differential regulation of HMG-CoA reductase and Insig-1 by enzymes of the ubiquitin-proteasome system. Mol. Biol. Cell 23:4484–4494. 10.1091/mbc.E12-08-0631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kikkert M, Doolman R, Dai M, Avner R, Hassink G, van Voorden S, Thanedar S, Roitelman J, Chau V, Wiertz E. 2004. Human HRD1 is an E3 ubiquitin ligase involved in degradation of proteins from the endoplasmic reticulum. J. Biol. Chem. 279:3525–3534. 10.1074/jbc.M307453200 [DOI] [PubMed] [Google Scholar]
- 19.Zavacki AM, Arrojo EDR, Freitas BC, Chung M, Harney JW, Egri P, Wittmann G, Fekete C, Gereben B, Bianco AC. 2009. The E3 ubiquitin ligase TEB4 mediates degradation of type 2 iodothyronine deiodinase. Mol. Cell. Biol. 29:5339–5347. 10.1128/MCB.01498-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang J, Sato R, Goldstein JL, Brown MS. 1994. Sterol-resistant transcription in CHO cells caused by gene rearrangement that truncates SREBP-2. Genes Dev. 8:1910–1919. 10.1101/gad.8.16.1910 [DOI] [PubMed] [Google Scholar]
- 21.Sever N, Yang T, Brown MS, Goldstein JL, DeBose-Boyd RA. 2003. Accelerated degradation of HMG CoA reductase mediated by binding of insig-1 to its sterol-sensing domain. Mol. Cell 11:25–33. 10.1016/S1097-2765(02)00822-5 [DOI] [PubMed] [Google Scholar]
- 22.Ohmura-Hoshino M, Goto E, Matsuki Y, Aoki M, Mito M, Uematsu M, Hotta H, Ishido S. 2006. A novel family of membrane-bound E3 ubiquitin ligases. J. Biochem. 140:147–154. 10.1093/jb/mvj160 [DOI] [PubMed] [Google Scholar]
- 23.Doblas VG, Amorim-Silva V, Pose D, Rosado A, Esteban A, Arro M, Azevedo H, Bombarely A, Borsani O, Valpuesta V, Ferrer A, Tavares RM, Botella MA. 2013. The SUD1 gene encodes a putative E3 ubiquitin ligase and is a positive regulator of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity in Arabidopsis. Plant Cell 25:728–743. 10.1105/tpc.112.108696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cao J, Wang J, Qi W, Miao HH, Wang J, Ge L, DeBose-Boyd RA, Tang JJ, Li BL, Song BL. 2007. Ufd1 is a cofactor of gp78 and plays a key role in cholesterol metabolism by regulating the stability of HMG-CoA reductase. Cell Metab. 6:115–128. 10.1016/j.cmet.2007.07.002 [DOI] [PubMed] [Google Scholar]
- 25.Liu TF, Tang JJ, Li PS, Shen Y, Li JG, Miao HH, Li BL, Song BL. 2012. Ablation of gp78 in liver improves hyperlipidemia and insulin resistance by inhibiting SREBP to decrease lipid biosynthesis. Cell Metab. 16:213–225. 10.1016/j.cmet.2012.06.014 [DOI] [PubMed] [Google Scholar]
- 26.Lee JN, Song B, DeBose-Boyd RA, Ye J. 2006. Sterol-regulated degradation of Insig-1 mediated by the membrane-bound ubiquitin ligase gp78. J. Biol. Chem. 281:39308–39315. 10.1074/jbc.M608999200 [DOI] [PubMed] [Google Scholar]
- 27.Shmueli A, Tsai YC, Yang M, Braun MA, Weissman AM. 2009. Targeting of gp78 for ubiquitin-mediated proteasomal degradation by Hrd1: cross-talk between E3s in the endoplasmic reticulum. Biochem. Biophys. Res. Commun. 390:758–762. 10.1016/j.bbrc.2009.10.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Irisawa M, Inoue J, Ozawa N, Mori K, Sato R. 2009. The sterol-sensing endoplasmic reticulum (ER) membrane protein TRC8 hampers ER to Golgi transport of sterol regulatory element-binding protein-2 (SREBP-2)/SREBP cleavage-activated protein and reduces SREBP-2 cleavage. J. Biol. Chem. 284:28995–29004. 10.1074/jbc.M109.041376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee JP, Brauweiler A, Rudolph M, Hooper JE, Drabkin HA, Gemmill RM. 2010. The TRC8 ubiquitin ligase is sterol regulated and interacts with lipid and protein biosynthetic pathways. Mol. Cancer Res. 8:93–106. 10.1158/1541-7786.MCR-08-0491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gong Y, Lee JN, Lee PC, Goldstein JL, Brown MS, Ye J. 2006. Sterol-regulated ubiquitination and degradation of Insig-1 creates a convergent mechanism for feedback control of cholesterol synthesis and uptake. Cell Metab. 3:15–24. 10.1016/j.cmet.2005.11.014 [DOI] [PubMed] [Google Scholar]
- 31.Sever N, Song BL, Yabe D, Goldstein JL, Brown MS, DeBose-Boyd RA. 2003. Insig-dependent ubiquitination and degradation of mammalian 3-hydroxy-3-methylglutaryl-CoA reductase stimulated by sterols and geranylgeraniol. J. Biol. Chem. 278:52479–52490. 10.1074/jbc.M310053200 [DOI] [PubMed] [Google Scholar]
- 32.Foresti O, Ruggiano A, Hannibal-Bach HK, Ejsing CS, Carvalho P. 2013. Sterol homeostasis requires regulated degradation of squalene monooxygenase by the ubiquitin ligase Doa10/Teb4. eLife 2:e00953. 10.7554/eLife.00953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, Ripatti S, Chasman DI, Willer CJ, Johansen CT, Fouchier SW, Isaacs A, Peloso GM, Barbalic M, Ricketts SL, Bis JC, Aulchenko YS, Thorleifsson G, Feitosa MF, Chambers J, Orho-Melander M, Melander O, Johnson T, Li X, Guo X, Li M, Shin Cho Y, Jin Go M, Jin Kim Y, Lee JY, Park T, Kim K, Sim X, Twee-Hee Ong R, Croteau-Chonka DC, Lange LA, Smith JD, Song K, Hua Zhao J, Yuan X, Luan J, Lamina C, Ziegler A, Zhang W, Zee RY, Wright AF, Witteman JC, Wilson JF, Willemsen G, et al. 2010. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466:707–713. 10.1038/nature09270 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.