

Abstract
We reported recently that the presenilin homologue signal peptide peptidase-like 2a (SPPL2a) is essential for B cell development by cleaving the N-terminal fragment (NTF) of the invariant chain (li, CD74). Based on this, we suggested that pharmacological modulation of SPPL2a may represent a novel approach to deplete B cells in autoimmune disorders. With regard to reported overlapping substrate spectra of SPPL2a and its close homologue, SPPL2b, we investigated the role of SPPL2b in CD74 NTF proteolysis and its impact on B and dendritic cell homeostasis. In heterologous expression experiments, SPPL2b was found to cleave CD74 NTF with an efficiency simliar to that of SPPL2a. For in vivo analysis, SPPL2b single-deficient and SPPL2a/SPPL2b double-deficient mice were generated and examined for CD74 NTF turnover/accumulation, B cell maturation and functionality, and dendritic cell homeostasis. We demonstrate that in vivo SPPL2b does not exhibit a physiologically relevant contribution to CD74 proteolysis in B and dendritic cells. Furthermore, we reveal that both proteases exhibit divergent subcellular localizations in B cells and different expression profiles in murine tissues. These findings suggest distinct functions of SPPL2a and SPPL2b and, based on a high abundance of SPPL2b in brain, a physiological role of this protease in the central nervous system.
INTRODUCTION
Transmembrane proteins can be substrates of a sequential proteolytic sequence referred to as regulated intramembrane proteolysis (RIP) (1). Usually, this involves the proteolytic release of the protein's ectodomain and the subsequent processing of the remaining membrane bound fragment by an intramembrane-cleaving protease (I-CLIP) (1). RIP can be actively involved in signal transduction by liberating intracellular domains that may trigger downstream signaling pathways and/or exert transcriptional control after nuclear translocation (2).
The signal peptide peptidase (SPP)/signal peptide peptidase-like (SPPL) intramembrane proteases, together with the presenilins, belong to the group of GxGD type aspartyl I-CLIPs (3). In mammals, the SPP/SPPL family includes five members: the ER protein SPP and the SPP-like proteins SPPL2a, SPPL2b, SPPL2c, and SPPL3, which were reported to exhibit diverse subcellular localizations within the biosynthetic pathway (SPPL2c and SPPL3), at the plasma membrane (SPPL2b), or in lysosomes/late endosomes (SPPL2a) (3). However, the subcellular localizations of the SPPL proteases demonstrated to date are based on overexpression studies, with the exception of SPPL2a, for which residence in lysosomes/late endosomes could also be shown at the endogenous level (17).
We and others recently identified the invariant chain (CD74) of major histocompatibility complex class II (MHC-II) as the first in vivo validated substrate of SPPL2a (4–6). In antigen-presenting cells, CD74 binds newly synthesized MHC-II dimers in the ER. It prevents premature acquisition of peptides by MHC-II in the biosynthetic pathways and mediates targeting of the complex to modified endosomal compartments. There, the luminal domain of CD74 is degraded by endosomal proteases, thereby releasing MHC-II, allowing the binding of antigenic peptides (7). Although RIP had been suggested earlier as a potential clearance mechanism for the remaining membrane-bound CD74 N-terminal fragment (NTF) (8), the responsible protease was unknown until recently (4). We could show that this CD74 NTF can be processed by coexpressed SPPL2a (4) in the standard overexpression-based experimental setup that had been used for the identification of previously reported substrates (9–13). More importantly, we demonstrated that significant amounts of this CD74 NTF accumulate in B cells of SPPL2a-deficient mice, indicating that under physiological conditions SPPL2a is required for the turnover of this fragment. Phenotypically, SPPL2a−/− mice exhibit a deficiency of B cells that is caused by a block of splenic B cell maturation at the transitional stage 1 (T1). Furthermore, the functionality of the residual B cells was found to be significantly impaired (4). Since these changes were significantly alleviated by additional ablation of CD74 in SPPL2a−/− CD74−/− mice, we could identify the accumulating CD74 NTF as the causative element behind this phenotype. Mechanistically, a disturbance of endosomal membrane traffic, as well as of central signaling pathways, caused by the CD74 NTF seemed to contribute to the B cell developmental arrest (4). Apparently, a major function of SPPL2a is to control the levels of this fragment. However, this proteolytic event also generates an inherently unstable cleavage product by liberating the CD74 intracellular domain into the cytosol. A putative role of this fragment in signal transduction has been suggested earlier (14). However, the molecular details remain poorly defined.
SPPL2a and SPPL2b are ca. 50% identical and 70% homologous to each other (15). Within the SPP/SPPL family, both proteases were suggested to form a distinct subgroup since they share a number of substrates that were not cleaved by SPPL3 and SPP (3). These include NTFs of tumor necrosis factor alpha (TNF-α) (9, 10), the Bri2 protein (12), and the transferrin receptor (13). Therefore, we considered that the SPPL2a substrate CD74 may also be cleaved by SPPL2b. Overexpressed SPPL2b has been reported to reside primarily at the plasma membrane (9, 17). However, the presence of minor but functionally relevant amounts of this protease in endosomes, where CD74 turnover takes place, is conceivable based on the continuous membrane exchange of these compartments with the plasma membrane. Furthermore, CD74 has been described to exhibit MHC-II-independent functions at the plasma membrane. There, CD74 binds the cytokine macrophage migration inhibitory factor (MIF) (18) and is critically involved in its signal transduction, together with CD44 (19) or CXCR4 (20) as coreceptors. MIF is a proinflammatory cytokine (21), which has also been implicated in B cell survival signaling (22, 23). It is currently unknown whether cell surface-localized CD74 is subjected to regulated proteolysis similar to other cell surface proteins (1). A role of SPPL2b in this context seems possible. In the present study, we sought to analyze the capability of SPPL2b to perform intramembrane cleavage of CD74 in a cell culture setup based on the coexpression of proteases and substrates, as it has been frequently used for the identification of SPPL substrates (9–13). In order to scrutinize these findings in vivo and precisely assess the individual contributions of SPPL2a and SPPL2b to CD74 proteolysis, we generated SPPL2b-deficient mice and bred these with our previously reported SPPL2a−/− mice (4) to obtain SPPL2a/SPPL2b (SPPL2a/b) double-deficient mice. We specifically analyzed these mice with respect to CD74 NTF turnover and accumulation, as well as downstream effects of this fragment on endosomal morphology, B cell maturation and functionality, and dendritic cell homeostasis. Furthermore, we have investigated the subcellular localization of endogenous SPPL2a and SPPL2b in B cells and demonstrate distinct expression profiles of both proteases in different murine tissues.
MATERIALS AND METHODS
Experimental animals.
Mice with a heterozygous gene trap insertion in the first intron of the SPPL2b gene [B6; CB-3110056O03RikGt(pU-21T)160Imeg] were generated at CARD Institute, Kumamoto University, Japan based on the embryonic stem (ES) cell clone Ayu21-T160. The exchangeable gene trap vector pU-21T (24), which is based on the pU-17 vector (25), contains an alternative splice acceptor sequence with stop codons in all three reading frames, followed by the coding sequence of the β-galactosidase gene and a polyadenylation signal. This leads to a fusion transcript of SPPL2b-exon 1 and the β-galactosidase gene, and expression of the β-galactosidase gene under the control of the SPPL2b promoter, thereby disrupting production of the SPPL2b wild-type transcript. The exact position of the gene trap insertion in the SPPL2b gene was determined by DNA-sequencing of PCR products, generated using primers binding in exon 1 of SPPL2b (forward [fw]) and the β-galactosidase gene sequence (reverse [rv]) and accordingly in the β-galactosidase gene sequence (fw) and exon 2 (rv) (data not shown). The position of the insertion was found to be ∼0.4 kb upstream of exon 2. Approximately ∼1.6 kb of the intronic sequence were not present in either analyzed PCR product, indicating that this part of the intron has been deleted upon insertion of the gene trap (data not shown). Heterozygous mice were interbred in order to generate SPPL2b−/− mice. A PCR amplifying a 408-bp fragment from the intronic sequence which has been deleted by the gene trap insertion and is therefore present only in the SPPL2b wild-type allele, was utilized to distinguish heterozygous from homozygous animals (fw, 5′-CAT GCC TAC CTC CTT ACT CTG-3′; rv, 5′-TGA GAT CTG ATA CCC TCT TCT G-3′). In addition, a PCR specifically detecting the β-galactosidase gene sequence (fw, 5′-TTA TCG ATG AGC GTG GTG GTT ATG C-3′, rv, 5′-GCG CGT ACA TCG GGC AAA TAA TAT C-3′) was performed. Absence of SPPL2b wild-type transcript was confirmed by reverse transcription-PCR (RT-PCR). Isolation of total RNA from murine embryonic fibroblasts (MEFs) and mouse tissues was achieved with the NucleoSpin RNA II kit (Macherey-Nagel, Düren, Germany). RT was conducted using the RevertAid first-strand cDNA synthesis kit (Fermentas) and random hexamer primers. Primers annealing in exon 1 and at the transition of exons 2 and 3 or exons 3 and 4, respectively, of the SPPL2b open reading frame (ORF; Ex1-fw, 5′-TTT GCT GCT ACT CGC GGC-3′; Ex2/3-rv, 5′-TCA GGA GAG ACA CTT TGC TG-3′; Ex3/4-rv, 5′-AGG GAC CAG CTT CTC CTT GC-3′) were used to amplify fragments of 153 or 331 bp of the SPPL2b wild-type transcript from the cDNA. In parallel, appropriate primers for fragments of β-actin (fw, 5′-GTT ACA ACT GGG ACG ACA TGG-3′; rv, 5′-GAT GGC TAC GTA CAT GGC TG-3′) or GAPDH (fw, 5′-CTG CAC CAC CAA CTG CTT AG-3′; rv, 5′-CAG TGA GCT TCC CGT TCA G-3′) were used as a control.
Mice deficient for SPPL2a have been described previously (4). SPPL2a−/− mice were bred with SPPL2b−/− mice for the generation of SPPL2a/b double-deficient mice. All mice were backcrossed for 10 generations in a C57BL/6N Crl background. Animal experimentation was performed in agreement with local guidelines for use of animals and their care.
cDNA constructs.
Expression constructs of murine SPPL2a and a catalytically inactive (D416A) mutant SPPL2a with C-terminally appended myc epitopes as well as constructs of the p31 isoform of murine CD74 (HA-mCD74p31-V5 and mCD74p31-HA) have been described previously (4, 17). For the generation of a SPPL2b expression construct, the murine SPPL2b-ORF was amplified from MEF cDNA and fused to a myc epitope at the 3′ end by PCR using mSPPL2b-KpnI-Fw (5′-ATT AGG TAC CGC CAC CAT GGC CGC GGC GCG GCT G-3′) and mSPPL2b-Myc-XbaI-Rv (5′-GTT ATC TAG ACT ACA GAT CCT CTT CTG AGA TGA GTT TTT GTT CGG CCG AAG TCT CTG GCT TCA CCA CAG G-3′) as forward and reverse primers, respectively. The PCR product was subcloned into pcDNA3.1/Hygro+ (Invitrogen, Carlsbad, CA). Based on this construct, a catalytically inactive mutant of SPPL2b (D414A) was produced by overlap-extension PCR using mSPPL2b-D414A-Fw (5′-GGC TTT GGA GCA ATA TTG GTG CCA GGG CTG CTG-3′) and mSPPL2b-D414A-Rv (5′-CAC CAA TAT TGC TCC AAA GCC CAG GAG GGA GAA-3′) as internal primers, as well as mSPPL2b-KpnI-Fw and mSPPL2b-Myc-XbaI-Rv as flanking primers. Triple Myc-tagged expression constructs of murine SPPL2a and SPPL2b (mSPPL2a-3xMyc and mSPPL2b-3xMyc) were generated based on the existing ORFs as described previously (26).
Cell culture and transfection.
HeLa cells (DSMZ, Braunschweig, Germany) were grown in Dulbecco modified Eagle medium (PAA, Cölbe, Germany) supplemented with 10% (vol/vol) fetal bovine serum (FBS; PAA), 100 U of penicillin (PAA)/ml, and 100 μg of streptomycin (PAA)/ml. The murine B lymphoma cell line Bal 17 (27) and the human B cell line Raji were maintained in RPMI 1640 with l-glutamine (Sigma) supplemented with 10% (vol/vol) FBS, 50 μM β-mercaptoethanol (Gibco/Invitrogen), and penicillin-streptomycin as specified above. The same medium without β-mercaptoethanol was used for culturing the human melanoma cell line MelJuso that was kindly provided by Jacques Neefjes, Netherlands Cancer Institute, Amsterdam, Netherlands. All cells were grown at 37°C in a humidified 5% CO2–95% air atmosphere. Treatment with the SPP/SPPL inhibitor (Z-LL)2-ketone (Peptanova, Sandhausen, Germany) was performed for 6 h at a final concentration of 10 μM. Transient transfections of HeLa and MelJuso cells were performed at semiconfluence utilizing Turbofect (Fermentas, St. Leon-Rot, Germany) according to the manufacturer's instructions. To reduce cytotoxicity, culture medium was replaced by fresh medium 6 h after transfection. Cells were harvested at 24 h posttransfection.
Protein extraction and immunoblotting.
Total lysates of cells or mouse tissues were prepared in 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1.0% (vol/vol) Triton X-100, 0.1% (wt/vol) sodium dodecyl sulfate (SDS), and 4 mM EDTA supplemented with protease inhibitors as described previously (28). Protein concentration of lysates was determined using a bicinchoninic acid (BCA) protein assay (Thermo). Electrophoretic separation of proteins prior to Western blotting was performed by SDS-PAGE either with a standard Tris-glycine (29) or a Tris-Tricine buffer system (30). Semidry transfer of proteins to nitrocellulose was conducted as described previously (28). For protein detection on Western blots, the primary antibodies anti-mSPPL2a (17), anti-CD74 (In-1; BD Biosciences, Heidelberg, Germany), and antiactin (Sigma-Aldrich, Taufkirchen, Germany) were used. For the detection of murine SPPL2b, a polyclonal antiserum was generated against the synthetic peptide NGDEAQPIPVVKPETSA (residues 562 to 578) in rabbits and affinity purified against the immobilized peptide (Pineda Antikörper-Service, Berlin, Germany). The hemagglutinin (HA) and Myc epitope tags were detected by using the monoclonal antibodies 3F10 and 9B11 from Roche and Cell Signaling, respectively. A monoclonal antibody against LAMP-1 (1D4B) was obtained from Developmental Studies Hybridoma Bank. Antibodies against transferrin receptor (clone H68.4), the α-1 subunit of the Na+/K+ ATPase (clone C464.6), and Rab5 (sc28570) were obtained from Invitrogen, Millipore, and Santa Cruz, respectively. To confirm equal protein loading, antibodies against actin (Sigma) or elongation factor 2 (EEF2; Abcam) were used. Horseradish peroxidase-labeled secondary antibodies were purchased from Dianova, Hamburg, Germany, and detection of chemiluminescence was performed with Amersham ECL Advance Western blotting detection reagent (GE Healthcare, Uppsala, Sweden).
Flow cytometric analysis.
Single cell suspensions of red bone marrow, spleen, thymus, and lymph nodes were prepared in cold fluorescence-activated cell sorting (FACS) buffer (phosphate-buffered saline [PBS] containing 2% [vol/vol] FBS, 0.1% [wt/vol] NaN3, and 2 mM EDTA [pH 7.4]) by flushing tibia and femur or cutting the organs into pieces, before passing them through a 100-μm-pore-size cell strainer. Peritoneal cells were isolated by flushing the peritoneal cavity with 7 ml of PBS. Cells were disseminated by gentle massage and collected afterward. For the lysis of erythrocytes, isolated cells were incubated in 155 mM NH4Cl, 10 mM KHCO3, and 0.1 mM EDTA for 10 min at room temperature. For staining, 5 × 105 cells were suspended in 100 μl of FACS buffer and incubated for 30 min at 4°C with the following fluorescein isothiocyanate-, phycoerythrin (PE)-, PE-Cy5-, PE-Cy7-, or allophycocyanin-conjugated monoclonal antibodies: anti-CD11c (N418), anti-CD21/CD35 (8D9), and anti-CD45R (RA3-6B2) from eBioscience (San Diego, CA); anti-CD19 (PeCa1) from Immunotools (Friesoythe, Germany); anti-IgM (R6-60.2) from BD Biosciences; and anti-CD23 (B3B4), anti-CD24 (M1/69), and anti-MHC-II (M5/114.15.2) from Biolegend (San Diego, CA). After washing, stained cells were resuspended in 0.5 μg of propidium iodide (PI)/ml for labeling dead cells (PI+). The data were recorded on a FACSCanto flow cytometer (BD Biosciences) and analyzed with FACSDiva or FlowJo (Tree Star, Inc.) software.
Isolation of B cells.
Single cell suspensions of murine spleens were obtained in MACS buffer (PBS supplemented with 2 mM EDTA and 0.5% [wt/vol] bovine serum albumin [BSA]) as described above. Depending on the devised experiments, B cells were ether isolated by positive selection (for Western blot analysis and electron microscopy) or negative depletion (immunofluorescence). Positive selection of IgM+ cells was performed by using anti-mouse IgM MicroBeads and LS columns of the MACS cell separation system (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer's instructions. Similarly, for immunofluorescent staining of B cells, total splenic B cells were recovered with the Pan B cell isolation kit (Miltenyi Biotec) and LD depletion columns. Isolated cells were analyzed by flow cytometry for the expression of CD45R (B220) and found to comprise between 80 and 90% CD45R+ cells.
Generation of BMDCs.
Bone marrow-derived dendritic cells (BMDCs) were generated as described previously (31) with slight modifications. Cells were isolated from red bone marrow as described above using PBS instead of FACS buffer and sedimented (210 × g, 10 min). After resuspension in BMDC medium (RPMI 1640 with l-glutamine [PAA] supplemented with 10% [vol/vol] FBS, 50 μM β-mercaptoethanol [Gibco/Invitrogen], 100 U of penicillin/ml, and 100 μg of streptomycin/ml), the cells were seeded at a density of 5 × 106 cells per 10 ml in the presence of 20 ng of murine granulocyte macrophage colony-stimulating factor (mGM-CSF; Immunotools, Friesoythe, Germany)/ml in a 10-cm culture dish. After 3 days, 10 ml of BMDC medium supplemented with 20 ng of mGM-CSF/ml was added to the cultures. At 6 days after cell isolation, 10 ml of the medium was collected, and the cells were sedimented (220 × g, 10 min), resuspended in 10 ml of fresh BMDC medium with 10 ng of mGM-CSF/ml, and replaced in the dish. BMDCs were used for further investigations on day 8 and were either stimulated for 24 h by the addition of 1 μg of Escherichia coli lipopolysaccharide (LPS; L8274; Sigma-Aldrich)/ml or left unstimulated. For all analyses, adherent cells were detached by scraping and then combined with the suspension cells.
Indirect immunofluorescence.
Cells were adhered to poly-l-lysine-coated coverslips as described previously (4) and fixed in 4% (wt/vol) paraformaldehyde in PBS for 20 min at room temperature. Immunocytochemical staining was performed as described previously (26). The primary antibodies included anti-mouse CD74 (In-1; BD Biosciences) and a polyclonal antiserum raised against the N terminus of murine CD74 (32) provided by N. Barois (Institut Pasteur de Lille, Lille, France) and W. Stoorvogel (Faculty of Veterinary Medicine, Utrecht University, Utrecht, Netherlands). Anti-human CD74 PIN1 (StressMarq Biosciences, Victoria, Canada), anti-mouse LAMP-2 (ABL93; Developmental Studies Hybridoma Bank), and anti-human LAMP-2 2D5 (33) were also used in combination with Alexa Fluor 488- or Alexa Fluor 594-conjugated secondary antibodies (MoBiTec). For the detection of human SPPL2a, a polyclonal rabbit antiserum against a C-terminal epitope of this protein (ATNEENPVISGEQIVQQ, residues 504 to 520) was generated and affinity purified against the immobilized immunogen (Pineda Antikörper-Service). For visualization of nuclei, DAPI (4′,6′-diamidino-2-phenylindole; Sigma-Aldrich) was added to the embedding medium at a final concentration of 1 μg/ml. Images were acquired using an FV1000 confocal laser scanning microscope (Olympus, Hamburg, Germany).
Ultrastructural analysis of IgM+ B cells.
IgM+ B cells were isolated by magnetic cell sorting from spleens as described above. Sample preparation and transmission electron microscopy were performed as described previously (4). In brief, cells were fixed in 2.5% glutaraldehyde in suspension and incorporated into blocks of BSA for further processing. Samples were postfixed with 2% OsO4 and embedded in araldite. Ultrathin sections were stained with uranyl acetate and lead citrate. Fifty B cells of each specimen were photographed at a magnification of ×7,000. Vacuoles with a diameter of ≥250 nm were counted on the computer screen, and the mean number of vacuoles per cell profile was calculated for each mouse.
Determination of basal immunoglobulin levels.
The Clonotyping System-HRP (5300-05; Southern Biotech, Birmingham, AL) was utilized for the isotype specific detection of immunoglobulins in mouse serum by enzyme-linked immunosorbent assay (ELISA) according to the manufacturer's instructions. Purified mouse immunoglobulins (BD Biosciences) were used for the determination of immunoglobulin concentrations. BM Blue POD substrate (Roche, Mannheim, Germany) was applied as a substrate for the colorimetric detection of POD activity. After the addition of 1 M H2SO4, the absorbance was measured at 450 nm in a microplate reader (Powerwave 340; BioTek Instruments, Winooski, VT).
Subcellular fractionation by Percoll density gradient centrifugation.
Bal17 B cells were harvested by centrifugation (210 × g, 5 min) and washed twice with PBS and homogenization buffer containing 250 mM sucrose, 10 mM HEPES-NaOH (pH 7.4), and 1 mM EDTA. Cells were resuspended in homogenization buffer supplemented with Complete protease inhibitor (Roche) and disrupted mechanically by 5 passages through a 25G cannula. A postnuclear supernatant was obtained by centrifugation for 5 min at 500 × g. In a 10-ml ultracentrifuge tube, 8.7 ml of a 20% Percoll solution in homogenization buffer (GE Healthcare) was carefully underlayered with 0.5 ml of 65% (wt/vol) sucrose in 10 mM HEPES-NaOH using a glass capillary fitted to a peristaltic pump. Finally, 1 ml of the postnuclear supernatant was layered on top. Centrifugation was performed in a 70.1 Ti fixed-angle rotor (Beckman Coulter) at 40,500 × gmax for 60 min at 4°C. Fractions of 500 μl were collected from the bottom (fraction 20) to the top (fraction 1) with a capillary and pump, assayed for β-hexosaminidase activity (34), and analyzed by Western blotting.
Histology and β-galactosidase staining.
Mice were anesthetized by intraperitoneal injection of xylazine/ketamine and perfused transcardially with 0.1 M phosphate buffer, pH 7.4, followed by fixative (4% paraformaldehyde in 0.1 M phosphate buffer). Brains were dissected and postfixed for 6 h in the same fixative. Thereafter, tissue was incubated in 30% sucrose–0.1 M phosphate buffer overnight before cutting 45-μm free-floating cryosections. Visualization of the β-galactosidase activity was carried out according to common laboratory protocols. Briefly, a 5-bromo-4-chloro-3-indolyl-β-d-galactoside (Sigma-Aldrich, Steinheim, Germany) stock solution, dissolved in dimethylformamide, was diluted in staining buffer (phosphate buffer supplemented with 2 mM MgCl2, 5 mM potassium ferricyanide, 5 mM potassium ferricyanide, 0.02% Nonidet P-40, and 0.01% sodium deoxycholate) to a final concentration of 1 mg/ml. Free-floating sections were incubated in staining solution at 37°C for 5 h, followed by washing (three times) in fresh phosphate buffer. Sections were costained with nuclear red (Chroma, Münster, Germany) and dehydrated in an ascending alcohol series before mounting in Eukitt (Sigma-Aldrich) and visualized using a BX50 microscope (Olympus).
Statistics.
Data are shown as means ± the standard deviations (SD). For statistical analyses, an unpaired two-tailed t test or one-way analysis of variance (ANOVA), followed by Bonferroni post hoc testing, was used as indicated. Significance levels (P) of <0.05, <0.01, and <0.001 were applied.
RESULTS
SPPL2b cleaves CD74 in vitro.
After the delivery of MHC-II to endosomal compartments, CD74 is degraded by stepwise proteolysis (Fig. 1a). This involves different endosomal proteases cleaving sequentially within the ectodomain and, as we showed recently, the intramembrane protease SPPL2a for the turnover of the final membrane-bound NTF (4). Based on the high degree of sequence homology between SPPL2a and SPPL2b and the reported overlap in their substrate spectrum (9, 12, 13), we sought to evaluate the capability of SPPL2b to process CD74. We initially assessed this in vitro by coexpressing the epitope-tagged substrate (Fig. 1b) and protease in HeLa cells (Fig. 1c). As shown previously (4), in this setup SPPL2a was able to reduce the steady-state levels of the CD74 NTF (Fig. 1c), which was not observed with an active-site mutant of SPPL2a (D416A). Interestingly, also upon cotransfection with SPPL2b, a decrease in CD74 NTF levels could be detected. This effect was absent when the catalytically inactive SPPL2b D414A mutant was coexpressed, demonstrating that overexpressed SPPL2b is capable of processing the NTF of CD74 in a manner similar to that of SPPL2a. Since these experiments were performed with expression constructs for murine CD74 and SPPL proteases, we checked for any putative differences in the human system. Thus, we coexpressed the epitope-tagged p33 isoform of human CD74 with human SPPL2a or SPPL2b in HeLa cells. Also in this setup, both proteases were capable of reducing CD74 NTF levels upon coexpression (not shown). Thus, the general ability of SPPL2b to cleave the CD74 NTF was not limited to the murine system.
FIG 1.

SPPL2b is capable of cleaving N-terminal fragments of CD74 in vitro. (a) Scheme of CD74-proteolysis in MHC-II compartments. The luminal domain of CD74 is sequentially degraded by several endosomal proteases. Finally, the proteolytic cleavage by cathepsin S releases the MHC-II dimer. Thereby, a small fragment (class II-associated li chain peptide [CLIP]) remains inside the MHC-II binding groove until it is replaced by an antigenic peptide. The N-terminal fragment of CD74 (NTF; 82 amino acids) is then further processed by the intramembrane protease SPPL2a. Colored asterisks indicate the catalytic motifs YD and GxGD within SPPL2a. (b) Layout of the utilized expression construct of the p31 isoform of murine CD74 with N- and C-terminally fused HA and V5 epitopes, respectively. TM, transmembrane segment. (c) HeLa cells were transiently transfected with CD74 alone or in combination with SPPL2a or SPPL2b or their catalytically inactive D416A or D414A mutant forms, respectively. Detection of CD74 was performed with an antibody against the N-terminally appended HA epitope tag after Western blotting and protein separation by SDS-PAGE using a Tris-Tricine buffer system with improved resolution in the low-molecular-weight range. Equal protein loading was confirmed by the detection of actin. Full-length CD74 and the CD74 NTF remaining after degradation of the luminal domain by endosomal proteases are marked with closed and open arrowheads, respectively.
CD74 cleavage and B cell development in SPPL2b−/− mice.
In light of the general capability of SPPL2b to cleave CD74, we sought to analyze the contribution of SPPL2b to CD74 proteolysis under endogenous conditions in vivo. Therefore, we generated SPPL2b-deficient mice based on ES cells with a gene trap insertion in the first intron of the SPPL2b gene (Fig. 2a and b). The gene trap vector harbors a splice acceptor sequence in order to induce fusion of the first SPPL2b exon to the β-galactosidase reporter upon splicing of the SPPL2b primary transcript, thereby disrupting the SPPL2b open reading frame (ORF). We sought to verify functionality of this splice acceptor sequence by RT-PCR in MEF cells (Fig. 2c), as well as in brain, spleen, and liver cells from homozygous mice (Fig. 2d). For this purpose, we amplified fragments between exon 1 and borders of exon 2/3 or exon 3/4 of the SPPL2b wild-type ORF, respectively. Under the conditions used, we observed quantitative usage of the splice acceptor and full disruption of the wild-type transcript in these cells or tissues. In agreement, absence of SPPL2b protein was confirmed by Western blotting in MEF cells (not shown), B cells (Fig. 2e), BMDCs (Fig. 2f), and other tissues (see Fig. 8b) of SPPL2b−/− mice. These mice were viable and without any overt impairment.
FIG 2.

CD74 proteolysis, as well as B cell development and function, is not impaired in SPPL2b-deficient mice. (a) Exon-intron structure of the murine SPPL2b gene and the insertion of the gene trap in the first intron. Positions of primers used for genotyping by PCR and for detection of SPPL2b transcript by RT-PCR are indicated. (b) PCRs performed for genotyping of SPPL2b−/− mice. Isolated DNA from SPPL2b+/+ and SPPL2b−/− tail biopsy specimens was used for amplification with either gene-specific (SPPL2b, red in panel a) or β-galactosidase gene-specific primers (green in panel a). (c) Wild-type transcript of SPPL2b was not detected in SPPL2b−/− MEFs. After total RNA isolation from SPPL2b+/+ and SPPL2b−/− MEF cells, brain, spleen, or liver RT-PCR was performed using primers annealing in exon 1 and between exons 3 and 4 (c and d) or exons 2 and 3 (d) of the SPPL2b ORF as indicated in panel a. Fragments of 331 bp (c and d) or 153 bp (d) of the SPPL2b wild-type ORF were amplified from the cDNA. Appropriate primers for β-actin or GAPDH fragments were used as control. (e and f) Steady-state levels of CD74 and its degradation intermediates were analyzed in total lysates of IgM+ B cells (e) and LPS-activated or unstimulated BMDCs (f) from wild-type or SPPL2b−/− mice by Western blotting with an antibody directed against an N-terminal epitope of CD74. In SPPL2a-deficient B cells, large amounts of CD74 NTF with an apparent molecular mass of ∼10 kDa were observed that were not present in wild-type controls and SPPL2b-deficient cells (e). Similarly, processing of CD74 NTF in SPPL2b-deficient BMDCs was indistinguishable from wild-type controls (f). Full-length CD74, which is present as p31 and p41 isoforms in mice, and the CD74 NTF, remaining after degradation of the luminal domain by endosomal proteases in SPPL2a−/− cells, are marked by filled and open arrowheads, respectively. Electrophoretic separation prior to detection of CD74 was performed using a Tris-Tricine buffer system (e) or a standard Tris-glycine SDS-PAGE (f). Actin levels were determined to confirm equal protein loading. (g) The frequency of B cells in different lymphatic tissues of SPPL2b−/− mice in comparison to wild-type mice was quantified by flow cytometry. No reduction of B cells (B220+, % of PI− cells) was detected in bone marrow (BM), spleen, lymph nodes (LN), and peritoneal cells (PC) of SPPL2b-deficient mice (n = 6). (h) Basal immunoglobulin concentrations in the plasma of SPPL2b−/− mice and wild-type mice were measured by enzyme-linked immunosorbent assay (n = 6). An unpaired, two-tailed Student t test was performed (g and h).
FIG 8.

Distinct tissue expression profiles of SPPL2a and SPPL2b in mice. (a and b) Total lysates from the indicated tissues were prepared from wild type, SPPL2a−/−, and SPPL2b−/− mice. Aliquots of 50 μg of protein were subjected to Western blotting in order to determine the abundance of SPPL2a (a) and SPPL2b (b) in lysates from wild-type mice (+/+). Tissue lysates were analyzed on two separate gels. The two membranes were assembled prior to chemiluminescent detection. To control for the specificity of the SPPL2a and SPPL2b antibodies, equivalent aliquots of lysates from SPPL2a and SPPL2b knockout animals (−/−) were analyzed in parallel. To demonstrate equal protein loading between the respective +/+ and −/− samples, membranes were stained with Ponceau S. Bands representing SPPL2a and SPPL2b are marked by arrowheads. (c) Representative sagittal brain sections from heterozygous (SPPL2b+/−) and wild-type mice as a negative control were stained for β-galactosidase activity as a reporter for the expression of SPPL2b. Sections were counterstained with nuclear red (cblm, cerebellum; hip, hippocampus; p. cx, piriform cortex; g, granular cell layer, P, Purkinje cell layer; m, molecular cell layer).
We analyzed processing of endogenous CD74 in splenic IgM+ B cells of these mice by Western blotting with an antibody directed against an N-terminal epitope. In contrast to SPPL2a-deficient B cells, which accumulated large amounts of the CD74 NTF as described before (4), no accumulation of this fragment was observed in SPPL2b−/− IgM+ cells (Fig. 2e). We scrutinized this observation in dendritic cells that had been differentiated from bone marrow cells (i.e., BMDCs) isolated from wild-type or SPPL2b−/− mice. Again, no increase of full-length CD74 or N-terminal fragments in SPPL2b-deficient compared to wild-type BMDCs was seen (Fig. 2f) irrespective of the maturation status (±LPS). Thus, in both cell types no indication for a major alteration of CD74 NTF turnover in the absence of SPPL2b was obtained. Conspicuously, a minor reduction of an ∼15-kDa CD74 degradation intermediate of CD74 was observed in nonstimulated (−LPS) SPPL2b−/− versus wild-type BMDCs. Furthermore, SPPL2b seemed to exhibit individual band patterns upon detection in wild-type BMDCs and B cells (Fig. 2e and f). Since modification with N-glycans has been reported for this protein (35), this may reflect differential glycosylation in these cell types.
Subtle reductions in NTF turnover may not be reflected in a significant increase of steady-state NTF levels but could still account for a reduced production of the inherently unstable CD74 intracellular domain (ICD) with putative phenotypic consequences. Therefore, we assessed B cell maturation and functionality in these SPPL2b−/− mice. We determined the frequency of B cells (B220+) in bone marrow, spleen, lymph nodes, and the peritoneal cavity of SPPL2b-deficient mice in comparison to wild-type mice (Fig. 2g). No significant differences were found. Thus, the loss of SPPL2b did not lead to a depletion of B cells. Furthermore, the basal immunoglobulin concentrations in the plasma of SPPL2b−/− mice were comparable to those of wild-type animals (Fig. 2h). Therefore, we could not obtain any indication of a functional impairment of SPPL2b-deficient B cells. These findings strongly argue against a major indispensable role of SPPL2b for the intramembrane proteolysis of CD74 in vivo. This is in contrast to the described significant effects caused by the loss of SPPL2a (4). Thus, this corroborates the role of SPPL2a as the predominant intramembrane protease of CD74. However, based on these results a minor contribution of SPPL2b to this process could not be excluded, since SPPL2a, which is still present in the SPPL2b−/− mice, may have a compensatory effect.
Characterization of SPPL2a/b double-deficient mice.
In support of a functional cross talk between SPPL2a and SPPL2b, we noticed an increased abundance of SPPL2b in SPPL2a-deficient BMDCs compared to wild-type cells by Western blotting (Fig. 3). SPPL2b protein levels in SPPL2a−/− splenic IgM+ B cells were found to be enhanced to a similar degree, albeit with a higher variability (not shown). Although the mechanisms behind this have not yet been analyzed in detail, the observed upregulation of SPPL2b in the absence of SPPL2a was strongly suggestive of a cellular compensatory mechanism. Based on this, we sought to determine whether the severity of the phenotypic effects of SPPL2a deficiency on CD74 proteolysis and B cell homeostasis would be enhanced after disabling this putative compensatory mechanism by additional genetic ablation of SPPL2b.
FIG 3.

Upregulation of SPPL2b in SPPL2a-deficient dendritic cells. (a) Expression of SPPL2b was analyzed in SPPL2a-deficient BMDCs by Western blotting. BMDCs were either stimulated with LPS or left unstimulated. Electrophoretic separation was performed by SDS-PAGE with a standard Tris-glycine buffer system. Actin levels were determined in parallel to control for differences in protein loading. (b) Densitometric quantification of the Western blot depicted in panel a based on n = 3 biological replicates. SPPL2b levels were determined in relation to actin (SPPL2b/actin) and normalized to those of unstimulated (−LPS) wild-type (wt) BMDCs. Means ± the SD are shown. *, P < 0.05; **, P < 0.01 (unpaired, two-tailed Student t test).
Therefore, we generated SPPL2a/b double-deficient mice and specifically assessed the development and maturation of B cells in these mice compared to SPPL2a or SPPL2b single-deficient and wild-type mice. The total B cells (B220+), as well as the respective subpopulations, were quantified in the bone marrow, spleens, lymph nodes, and peritoneal cavities in mice of all four genotypes (Fig. 4 and Table 1). As expected, SPPL2a/b double-deficient mice exhibited a significant reduction of total B cells in the spleen, lymph nodes and peritoneal cavity reflecting the absence of SPPL2a (Fig. 4a and Table 1). However, no significant further reduction of B cells in the double-deficient versus the SPPL2a single-deficient mice could be detected. We demonstrated previously that the loss of B cells in SPPL2a−/− mice is caused by an arrest of splenic maturation beyond the transitional stage 1 (T1) B cells (4), which manifests as a reduction of transitional stage 2 (T2) and mature (M) B cells (Fig. 4b and Table 1). The abundance of these splenic subpopulations in SPPL2a/b double-deficient mice was determined, but we could not ascertain any significant differences compared to SPPL2a−/− mice (Fig. 4b to d and Table 1). This applies to the depletion of T2 and mature B cells, as well as to the preservation of T1 cells. No obvious shift to an earlier stage was detected for the onset of the B cell loss in the SPPL2a/b double-deficient mice. In addition, we measured the plasma immunoglobulin levels in SPPL2a/b single or double-deficient and wild-type mice. However, due to the reported severe functional impairment of B cells in SPPL2a−/− mice (4), basal immunoglobulin concentrations in the plasma of the SPPL2a single-deficient mice were already in the range of the detection limit of the ELISA performed (not shown). As expected, the same applied to the SPPL2a/b double-deficient mice, allowing no definite conclusion if the severely compromised functionality of SPPL2a-deficient B cells is further impaired by the ablation of SPPL2b. In conclusion, we did not obtain any indication that the additional loss of SPPL2b leads to a deterioration of the B cell deficiency and maturation arrest associated with the absence of SPPL2a.
FIG 4.

The B cell phenotype of SPPL2a−/− mice is not significantly exacerbated by additional ablation of SPPL2b. (a) The frequency (%) of B220+ viable B cells (B220+, PI−) was determined in bone marrow (BM), spleen, lymph nodes (LN), and peritoneal cells (PC) from wild type, SPPL2a−/−, SPPL2b−/−, and SPPL2a/b double-deficient (SPPL2a−/− SPPL2b−/− [Do-ko]) mice (n = 6) by flow cytometry. The data show the means ± the SD. (b to d) Splenic B cell subsets (transitional stage 1 [T1], transitional stage 2 [T2], and mature [M]) were determined by flow cytometry with antibodies against B220, CD21, and CD24. (b) Dot plots depict the viable B220-positive splenocytes (PI−, B220+) from a representative experiment. Numbers in the plots indicate the frequency (%) of the respective populations among viable splenocytes (PI−). (c and d) Graphs summarize the results from 6 mice per genotype regarding the abundance of total (B220+), transitional stage 1 (B220+ CD21low CD24high), transitional stage 2 (B220+ CD21high CD24high), or mature B cells (B220+ CD21low CD24low) in the spleens of wild type, SPPL2a−/−, SPPL2b−/−, and SPPL2a/b double-deficient mice. The data are depicted as the means ± the SD and display the percentages of all viable cells (PI−) (c) or the absolute number of cells (d) for the different populations. ***, P < 0.001; **, P < 0.01; *, P < 0.05 (one-way ANOVA with Bonferroni post hoc testing).
TABLE 1.
B cell deficiency and impairment of B cell maturation are comparable in SPPL2a−/− and SPPL2a/b double-deficient micea
| Cell type | Phenotype | Mean ± SD |
P (SPPL2a−/− [Do-ko]) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Wild type |
SPPL2a−/− |
SPPL2b−/− |
SPPL2a−/− SPPL2b−/− |
|||||||
| Mean | SD | Mean | SD | Mean | SD | Mean | SD | |||
| Bone marrow (% viable cells) | ||||||||||
| B cells | B220+ | 17.7 | 5.1 | 19.6 | 5.4 | 19.4 | 5.5 | 16.0 | 7.1 | >0.05 |
| Pro-/pre-B cells | B220+ IgM− | 9.1 | 3.5 | 14.1 | 4.5 | 10.8 | 3.3 | 11.4 | 4.4 | >0.05 |
| Immature B cells | B220+ IgM+ | 3.0 | 0.9 | 4.8 | 1.5 | 3.2 | 1.1 | 4.2 | 2.8 | >0.05 |
| Recirculating B cells | B220high | 5.6 | 2.2 | 0.6 | 0.3 | 5.3 | 2.3 | 0.4 | 0.2 | >0.05 |
| Spleen (% viable cells) | ||||||||||
| B cells | B220+ | 54.4 | 1.9 | 23.5 | 2.8 | 53.3 | 4.3 | 22.3 | 4.6 | >0.05 |
| T1 transitional | B220+ CD21low CD24high | 8.8 | 2.8 | 14.5 | 1.4 | 8.4 | 2.0 | 14.2 | 3.3 | >0.05 |
| T2 transitional | B220+ CD21high CD24high | 9.3 | 2.5 | 0.4 | 0.3 | 10.9 | 2.5 | 0.2 | 0.2 | >0.05 |
| Mature B cells | B220+ CD21low CD24low | 32.1 | 2.6 | 3.3 | 2.3 | 30.0 | 2.7 | 2.6 | 1.0 | >0.05 |
| Spleen (106 splenocytes) | ||||||||||
| B cells | B220+ | 55.7 | 7.7 | 8.5 | 4.5 | 47.3 | 19.6 | 5.3 | 1.4 | >0.05 |
| T1 transitional | B220+ CD21low CD24high | 9.1 | 3.0 | 5.2 | 2.8 | 7.6 | 3.9 | 3.4 | 1.0 | >0.05 |
| T2 transitional | B220+ CD21high CD24high | 9.5 | 2.2 | 0.2 | 0.1 | 9.5 | 4.1 | 0.05 | 0.03 | >0.05 |
| Mature B cells | B220+ CD21low CD24low | 32.9 | 5.4 | 1.3 | 1.2 | 2.7 | 1.1 | 0.6 | 0.3 | >0.05 |
| Lymph nodes (% viable cells) | ||||||||||
| B cells | B220+ | 20.5 | 5.3 | 6.1 | 5.1 | 23.9 | 6.5 | 3.0 | 1.0 | >0.05 |
| Lymph nodes (106 cells) | ||||||||||
| B cells | B220+ | 9.1 | 5.7 | 0.5 | 0.4 | 5.1 | 2.0 | 0.2 | 0.2 | >0.05 |
| Peritoneal cells (% viable cells) | ||||||||||
| B cells | B220+ | 37.6 | 12.9 | 7.5 | 4.2 | 38.2 | 9.1 | 5.5 | 1.8 | >0.05 |
| B1 | B220neg/low CD19high | 22.3 | 8.3 | 7.1 | 4.2 | 24.1 | 2.8 | 5.1 | 1.5 | >0.05 |
| B2 | B220high CD19+/low | 15.3 | 10.3 | 0.4 | 0.3 | 14.0 | 8.6 | 0.4 | 0.4 | >0.05 |
B cell subsets were analyzed by flow cytometry in bone marrow, spleen, lymph node, and peritoneal lavage samples of wild-type, SPPL2a−/−, SPPL2b−/−, and SPPL2a/b double-deficient (SPPL2a−/− SPPL2b−/−/do-ko) mice. The proportions of cell populations are indicated as the percentages of viable cells (PI−) or absolute cell numbers, respectively. Results represent means from 6 mice per genotype. Data were analyzed by one-way ANOVA with Bonferroni post hoc testing.
We aimed to assess whether this also applies to the phenotypic effects of SPPL2a deficiency at the level of the individual cell. We have shown previously that in SPPL2a−/− B cells undegraded CD74 NTF disturbs the membrane trafficking of the endocytic system (4), resulting in an accumulation of endosomal vacuoles. Hence, we compared the ultrastructure of splenic IgM+ B cells from wild-type, SPPL2a−/−, SPPL2b−/−, and SPPL2a/b double-deficient mice by electron microscopy (Fig. 5a). Whereas wild-type and SPPL2b-deficient cells were morphologically indistinguishable, SPPL2a−/− and the SPPL2a/b double-deficient cells exhibited numerous endosomal vacuoles. We determined the mean numbers of these vacuoles per cellular profile (Fig. 5b) and observed no significant difference between SPPL2a−/− and SPPL2a/b double-deficient B cells. Finally, we compared the CD74 NTF accumulation under steady-state conditions between SPPL2a−/− and SPPL2a/b double-deficient IgM+ B cells (Fig. 5c). However, we did not find the abundance of this fragment to be obviously increased in B cells lacking both proteases compared to cells only deficient in SPPL2a. In addition, also the localization of the accumulating CD74 NTF in compartments positive for LAMP-2 (Fig. 5d) and early endosome antigen 1 (EEA1; data not shown) was similar in SPPL2a−/− and SPPL2a/b double-deficient B cells.
FIG 5.
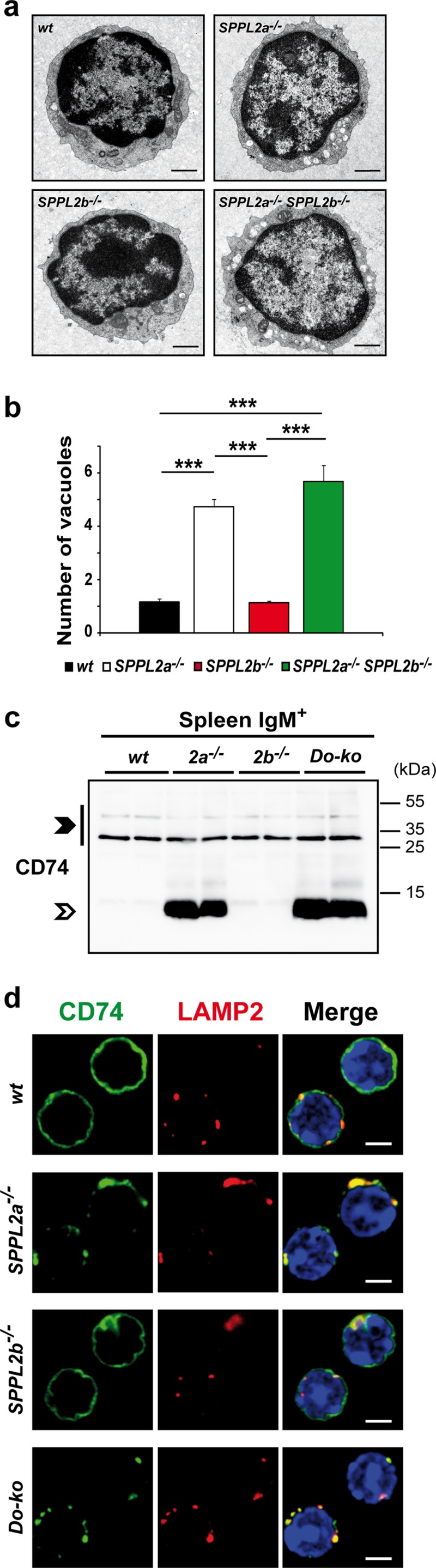
Evaluation of CD74 NTF accumulation and effects on the endocytic system in SPPL2b−/− and SPPL2a/b double-deficient B cells. (a) IgM+ splenic B cells isolated from wild-type (wt), SPPL2a−/−, SPPL2b−/−, and SPPL2a/b double-deficient (SPPL2a−/− SPPL2b−/−) mice were examined by transmission electron microscopy. Bars, 500 nm. (b) Quantification of accumulating endosomal vacuoles in B cells as shown in panel a. The number of vesicles with a diameter ≥250 nm was determined in 50 cellular profiles per mouse from three mice per genotype analyzed. The mean number of vacuoles per cell profile is depicted for each genotype ± the SD. ***, P < 0.001 (one-way ANOVA with Bonferroni post hoc testing). (c) The levels of full-length CD74 and CD74 NTF were determined in splenic IgM+ B cells from wild-type, SPPL2a−/−, SPPL2b−/−, and SPPL2a/b double-deficient (Do-ko) mice by Western blotting. Electrophoretic separation was performed by SDS-PAGE with a standard Tris-glycine buffer system. Isoforms of full-length CD74 and the CD74 NTF accumulating in SPPL2a−/− and SPPL2a/b double-deficient cells are marked with closed and open arrowheads, respectively. Full-length CD74 (p31) served as a control for equal protein loading. (d) CD74 was visualized in splenic B cells isolated from wild-type, SPPL2a−/−, SPPL2b−/−, and SPPL2a/b double-deficient (Do-ko) mice by indirect immunofluorescence. The antibody used was directed against an N-terminal epitope of CD74 and therefore detected NTF and full-length protein. In parallel, costaining for LAMP-2 as marker for lysosomes/late endosomes was performed. Scale bar, 2 μm.
DCs in SPPL2a−/− and SPPL2a/b double-deficient mice.
The SPPL2a dependence of the CD74 NTF turnover is not restricted to B cells but also applies to dendritic cells, as documented by the accumulation of this fragment in SPPL2a-deficient BMDCs (Fig. 6a), as well as primary splenic CD11c+ DCs (data not shown). In relation to this, mouse strains harboring mutations in the SPPL2a gene that had been derived from ENU mutagenesis screens were reported to exhibit a depletion of distinct DC subsets (5, 6). We aimed to recapitulate this observation in our SPPL2a-deficient mice and to assess a possible contribution of SPPL2b to CD74 proteolysis in this cell type. Therefore, we examined CD74 processing in BMDCs from wild-type, SPPL2a−/−, SPPL2b−/−, and SPPL2a/b double-deficient mice by Western blotting (Fig. 6a). Similar to the situation in B cells, a prominent accumulation of the CD74 NTF was detected in SPPL2a single- and SPPL2a/b double-deficient BMDCs; however, no consistent increase caused by the additional ablation of SPPL2b was evident. To assess the phenotypic consequences, we analyzed the frequency of CD11c+ MHC-II+ cells in spleens and lymph nodes from mice of all four genotypes by flow cytometry (Fig. 6b and c). In absolute cell numbers, spleens and lymph nodes of SPPL2a single-deficient mice were found to contain significantly less CD11c+ MHC-II+ cells than wild-type mice, which correlates well with observations in the SPPL2a mutant mouse strains (5, 6). In agreement with biochemical findings, no reduction of these cells was seen in SPPL2b−/− mice. The depletion of CD11c+ MHC-II+ cells seen in SPPL2a/b double-deficient mice was very similar to that observed in SPPL2a−/− mice, reflecting the absence of SPPL2a. Therefore, no evidence for an additive effect of SPPL2b ablation on this phenotype was obtained.
FIG 6.

Dendritic cells in SPPL2a−/− and SPPL2a/b double-deficient mice. (a) The levels of full-length CD74 and the CD74 NTF were determined in LPS-stimulated BMDCs from wild type, SPPL2a−/−, SPPL2b−/−, and SPPL2a/b double-deficient (Do-ko) mice by Western blotting. SDS-PAGE with a standard Tris-glycine buffer system was used for electrophoretic separation. Isoforms (p31 and p41) of full-length CD74 and the CD74 NTF accumulating in SPPL2a−/− and SPPL2a/b double-deficient cells are indicated by filled and open arrowheads, respectively. (b and c) CD11c and MHC-II double-positive cells were quantified by flow cytometry in spleens (b) or lymph nodes (c) of wild-type, SPPL2a−/−, SPPL2b−/−, and SPPL2a/b double-deficient mice. Graphs show the mean absolute number of these cells (CD11c+ MHC-II+) in the spleen (b) or in lymph nodes (c) quantified as the means ± the SD in 6 mice per genotype. ***, P < 0.001; **, P < 0.01; *, P < 0.05 (one-way ANOVA with Bonferroni post hoc testing).
Distinct subcellular localization and tissue distribution of SPPL2a and SPPL2b.
Based on the described different roles of SPPL2a and SPPL2b in B and dendritic cells, we considered divergent subcellular localizations of both proteases as a possible explanation and aimed to analyze these in B cells. Therefore, we performed subcellular fractionation of Bal 17 murine B lymphoma cells using Percoll density gradient centrifugation (Fig. 7a). The distributions of SPPL2a and SPPL2b within this gradient were distinctly different. The bulk of SPPL2a was recovered in the bottom fractions cosedimenting with the lysosomal markers β-hexosaminidase and LAMP-1. In contrast, the profile of SPPL2b showed a maximum in fractions 8 and 9 similar to the plasma membrane protein Na+/K+ ATPase. The fractions enriched in lysosomal compartments (18/19) did not contain significant amounts of SPPL2b. However, the presence of SPPL2b in early endosomes could not be excluded based on the overlapping profiles of the membrane-bound pool of Rab5 and Na+/K+ ATPase. In addition to the major peak of SPPL2a in the bottom fractions, a second less intense maximum was detected in fractions 8/9, thus cosedimenting with the bulk of SPPL2b. This indicates a partial overlap between the subcellular distributions of SPPL2a and SPPL2b and argues for presence of a minor fraction of SPPL2a in early endosomes or at the cell surface. However, we found that the major pools of both proteases were differentially localized in B cells which could explain the observed functional differences. In the human Raji B cell line available antibodies allowed visualization of endogenous SPPL2a by immunocytochemistry (Fig. 7b and c). In agreement with the Bal 17 fractionation, we observed significant colocalization between SPPL2a and the lysosomal/late endosomal protein LAMP-2 (Fig. 7b). Under steady-state conditions, mainly full-length CD74 presumably localized within the biosynthetic pathway was detected in Bal 17 (Fig. 7a) and human B cell lines (not shown) with an antibody against an N-terminal epitope of CD74. However, treatment with the SPP/SPPL inhibitor (Z-LL)2-ketone induced the accumulation of the CD74 NTF in vesicular compartments that were positive for SPPL2a (Fig. 7c). In summary, these findings support the view that the CD74 NTF intramembrane cleavage takes place in intracellular compartments with characteristics of late endosomes/lysosomes that, according to the subcellular fractionation shown in Fig. 7a, do not exhibit significant amounts of SPPL2b.
FIG 7.

Distinct subcellular localizations of SPPL2a and SPPL2b. (a) Subcellular fractionation of murine B cells. A postnuclear supernatant generated from the murine B lymphoma cell line Bal 17 was subjected to centrifugation on a self-forming Percoll density gradient. Fractions were analyzed spectrophotometrically for activity of the lysosomal marker enzyme β-hexosaminidase. Furthermore, the distribution of SPPL2a, SPPL2b, LAMP-1 (lysosomes/late endosomes), transferrin receptor (TfR; recycling endosomes and plasma membrane), Na+/K+-ATPase (plasma membrane), and CD74 and Rab5 (early endosomes) was analyzed by immunoblotting. Fractions 1 to 10 and fractions 11 to 20 were analyzed on two separate gels. The two membranes were assembled prior to chemiluminescent detection. (b and c) Immunocytochemical visualization of endogenous SPPL2a and LAMP-2 (b) or CD74 (c) in the human B cell line Raji. (c) Cells were incubated for 6 h in the presence of the SPP/SPPL inhibitor (Z-LL)2-ketone (10 μM final concentration) or dimethyl sulfoxide as a control prior to fixation. CD74 was detected with a primary antibody directed against an N-terminal epitope detecting full-length CD74, as well as the CD74 NTF accumulating in the inhibitor-treated cells. (d and e) HeLa cells (d) and MelJuso cells (e) were transiently transfected with murine CD74 (HA-CD74p31-V5). (d) Increasing amounts (0.125 to 1 μg) of murine SPPL2a or SPPL2b expression constructs with a C-terminally fused triple Myc epitope (mSPPL2a-3×Myc, m-SPPL2b-3×Myc) were cotransfected. (e) Coexpression of the catalytically inactive SPPl2a D416A and SPPL2b D414A mutant forms was included in the experiment as a control in addition to the wild-type proteases. CD74 was detected via the N-terminally appended HA epitope tag following Western blotting and electrophoretic separation using Tris-Tricine buffer system. Equal protein loading was confirmed by detection of actin and EEF2, respectively. Full-length CD74 and the CD74 NTF are marked with closed and open arrowheads, respectively.
Based on these findings, it seems likely that the diverging subcellular localizations of SPPL2a and SPPL2b determine their in vivo substrate spectrum. It is possible that the subcellular localization of overexpressed substrates and/or proteases does not entirely match that of the endogenous proteins, thus explaining why coexpressed SPPL2b cleaved CD74 NTF in HeLa cells (Fig. 1c). We assessed different modifications of this in vitro system in order to approximate the physiological conditions. First of all, we reduced and titrated the amount of coexpressed SPPL2a and SPPL2b in the HeLa cell setup (Fig. 7d). Furthermore, we repeated the coexpression experiment in the MHC-II+ MelJuso melanoma cell line (36) since the absence of MHC-II may alter the trafficking of CD74 (Fig. 7e). In both modified setups, the effect of overexpressed SPPL2b on CD74 NTF levels appeared to be less pronounced than that of SPPL2a, thus more closely reflecting the in vivo findings than the experiment shown in Fig. 1c. However, a significant reduction of CD74 NTF by SPPL2b was still observed, indicating that, even with the described modifications, the endogenous in vivo situation cannot be fully recapitulated in cell-based assays.
To provide initial insights into the physiologic function of SPPL2b, we have analyzed the expression of SPPL2b (Fig. 8b) in different murine tissues and compared it with that of SPPL2a (Fig. 8a). By far the highest level of SPPL2b expression was observed in brain. In addition, significantly smaller amounts of SPPL2b were detected in lymphatic tissues, specifically bone marrow, thymus, and spleen in decreasing order. In contrast, SPPL2a showed a rather ubiquitous expression in all tissues analyzed, conspicuously with the lowest abundance in brain. Based on the high level of SPPL2b in the total brain lysate (Fig. 8b), we sought to analyze the distribution of SPPL2b expression in the central nervous system. Therefore, we made use of the β-galactosidase reporter, which is part of the SPPL2b gene trap allele. The activity of β-galactosidase was visualized in sagittal sections from SPPL2b+/− mice. Wild-type mice were utilized as a negative control. Staining for β-galactosidase, as an indicator of SPPL2b promoter activity, was particularly pronounced throughout the hippocampus, but was also observed in the piriform cortex, as well as the Purkinje cell layer of the cerebellum (Fig. 8c). In addition, faint staining was detected in the striatum, thalamus, midbrain, amygdala, and inner cortical layers. Based on these findings, it seems likely that SPPL2b fulfills a major physiological function in the central nervous system.
DISCUSSION
We and others have previously documented the essential role of the intramembrane protease SPPL2a in B cells in cleaving the NTF of CD74 (4–6). In the present study, we have analyzed the contribution of the homologous protease SPPL2b to this process relating to previous studies that suggested overlapping functions of both proteases (9, 12). We demonstrated that SPPL2b has the intrinsic ability to process CD74 similar to SPPL2a, which was evident upon coexpression of CD74 and the respective proteases. To evaluate the in vivo relevance of this, we generated SPPL2b−/− and SPPL2a/b double-deficient mice and compared them to SPPL2a−/− and wild-type mice. We specifically assessed a possible impairment of CD74 NTF turnover as well as possible cellular or immunological consequences of this regarding endosomal morphology and B and dendritic cell homeostasis. We observed no changes of these parameters in SPPL2b−/− mice and furthermore demonstrated that by additional ablation of SPPL2b in SPPL2a−/− mice the phenotypic changes were not significantly exacerbated. Therefore, we conclude that SPPL2b does not play a physiologically relevant role for the intramembrane proteolysis of CD74 in vivo.
This clearly indicates a discrepancy between the substrate spectrum of SPPL2b that is only defined by its intrinsic properties and the substrates actually cleaved by this protease in vivo under endogenous conditions. The observation that CD74 was cleaved by SPPL2a as well as SPPL2b upon coexpression relates well to similar findings for TNF-α (9) and Bri2 (12). Thus, it may be assumed that the substrate recognition properties of SPPL2a and SPPL2b are equivalent or at least very similar. Subcellular trafficking is considered a key regulatory mechanism controlling encounters of intramembrane proteases and their substrates (37). Therefore, we have analyzed the subcellular localization of SPPL2a/b in B cells and observed distinct, although partially overlapping distributions of both proteases. This analysis represents the first report to date on the subcellular residence of SPPL2b at the endogenous level. In general, our findings were in agreement with the reported localizations of overexpressed SPPL2a and SPPL2b in standard cell lines (9, 17). In conclusion, it seems likely that the divergent subcellular localizations of SPPL2a/b determine, to a significant degree, the in vivo substrate spectra of both proteases. These may not always be precisely reflected in overexpression systems. We could demonstrate that carefully titrating the amount of coexpressed SPPL proteases can help to increase the specificity of such analyses. Certainly, the findings on CD74 strongly advocate that the individual protease-substrate relationships between SPPL2a/b and TNF-α, Bri2, and the TFR, which are thus far largely based on in vitro setups, should also be scrutinized under endogenous conditions in vivo. For this purpose, the mouse strains deficient for SPPL2a and/or SPPL2b that we have generated will be valuable tools to dissect these questions. Apparently, a final definition of the substrate spectrum of individual SPPL proteases will require such a thorough in vivo analysis.
Our finding that SPPL2b appears not to be involved in CD74 proteolysis in vivo may have a second implication. We previously suggested that pharmacological inhibition of SPPL2a may represent a putative therapeutic strategy for depleting B cells and controlling their function in autoimmunity (4). Inhibition of SPPL2a, at least in the murine model system, seems to be sufficient to block CD74 proteolysis and to deplete B cells. Therefore, for this effect a compound targeting SPPL2a but sparing the activity of SPPL2b would be effective and minimize the risk of unwanted side effects. In light of the high degree of homology between SPPL2a and SPPL2b, it is certainly speculative, if inhibitors exhibiting such a high specificity could be achieved. However, since the physiological functions of SPPL2b in vivo have not yet been fully characterized as discussed above, it may be reasonable to restrict the specificity of putative therapeutic compounds as far as possible.
If SPPL2b is not significantly involved in CD74 NTF proteolysis although it is expressed in B cells and dendritic cells at well detectable levels, its actual physiological substrates in these and other cell types remain elusive. TNF-α was suggested to be a substrate of SPPL2a and/or SPPL2b in DCs (9, 10) with possible implications for TNF-α reverse signaling (38). Together with the observed minor reduction of a CD74 15-kDa degradation intermediate in SPPL2b−/− BMDCs (Fig. 2f), this may justify the study of the activation properties and signaling responses of these cells in more detail. Conspicuously, we observed pronounced expression of SPPL2b in mouse brain, which was significantly higher than in the other tissues tested. In contrast to a previous microarray analysis of human tissues, SPPL2b transcript levels in brain were not found to be higher than those of other major tissues (35). This may reflect differences between both species or a poor correlation between transcript and protein abundance. Among the currently known substrates, Bri2 is particularly highly expressed in the central nervous system (39). Beyond that, the existence of additional not yet identified substrates cannot be excluded at this stage. In general, the loss of SPPL2b and even the combined deficiency of SPPL2a and SPPL2b was well tolerated in mice since the double-deficient mice were viable and without any overt impairment.
Prior to the described mouse models, the in vivo functions of SPPL proteases have been analyzed by knockdown in zebrafish (40). The zebrafish genome harbors a single orthologue (SPPL2) for the three related proteases SPPL2a, SPPL2b, and SPPL2c that are found in the human and murine genomes. Interestingly, among the three mammalian proteins, SPPL2b exhibits the highest sequence homology toward the zebrafish SPPL2 (40). Upon knockdown of SPPL2, zebrafish embryos displayed a distinct phenotype characterized by an accumulation of erythrocytes in an enlarged caudal vein (40). In contrast to these severe impairments of embryonic development in zebrafish, it may appear surprising that SPPL2a/b double-deficient mice are so mildly affected. This could be related to a compensatory or independent role of the yet poorly characterized third mammalian SPPL2 orthologue: SPPL2c. Based on its reported residence in the endoplasmic reticulum (9), a functional overlap with SPPL2a and SPPL2b localized in lysosomes/late endosomes and the plasma membrane, respectively, is not very obvious. In agreement with this, the SPPL2a/b substrate TNF-α was not cleaved by coexpressed SPPL2c (9). To date, no substrates of SPPL2c or any nonproteolytic functions are known.
In summary, the results presented here strongly suggest distinct functions of the homologous proteases SPPL2a and SPPL2b in vivo, which is supported by the described distinct subcellular localization and tissue distribution profiles of both proteases. Further studies will be needed to further evaluate this with regard to other substrates and cell types. The described findings are reminiscent of the relationship between presenilin 1 and 2 (PS1/2), the catalytic subunits of the γ-secretase complex. Both proteins exhibit significant homology, comparable to that between SPPL2a and SPPL2b, and were reported to have overlapping functional properties in vitro (41, 42). However, whereas PS1 knockout mice exhibited severe developmental defects (43), PS2-deficient mice were only mildly affected and did not show detectable alterations of amyloid precursor protein processing (44). Altogether, this strongly emphasizes the requirement of suitable in vivo models as we have provided for SPPL2a/b to assess a functional redundancy under physiological conditions.
ACKNOWLEDGMENTS
We thank Marlies Rusch, Sebastian Held, Dagmar Niemeier, Rafael Kurz, Leslie Elsner, Gabi Sonntag, Katrin Westphal, and Katrin Streeck for excellent technical assistance and Zane Orinska, Department of Immunology and Cell Biology, Research Center Borstel, for help with initial flow cytometric analyses. We are also grateful to Willem Stoorvogel, Utrecht University, and Nicolas Barois for sharing a polyclonal antibody against CD74 and to Jacques Neefjes, Netherlands Cancer Institute, Amsterdam, for providing MelJuso cells. We thank Guido Looft and Tobias Obser, Pediatric Hematology, UKE Hamburg, for the generous gift of Raji cells.
This study was supported by the Deutsche Forschungsgemeinschaft as part of the SFB 877 and the Center of Excellence Inflammation at Interfaces.
Footnotes
Published ahead of print 3 February 2014
REFERENCES
- 1.Lichtenthaler SF, Haass C, Steiner H. 2011. Regulated intramembrane proteolysis: lessons from amyloid precursor protein processing. J. Neurochem. 117:779–796. 10.1111/j.1471-4159.2011.07248.x [DOI] [PubMed] [Google Scholar]
- 2.Urban S, Freeman M. 2002. Intramembrane proteolysis controls diverse signalling pathways throughout evolution. Curr. Opin. Genet. Dev. 12:512–518. 10.1016/S0959-437X(02)00334-9 [DOI] [PubMed] [Google Scholar]
- 3.Voss M, Schröder B, Fluhrer R. 2013. Mechanism, specificity, and physiology of signal peptide peptidase (SPP) and SPP-like proteases. Biochim. Biophys. Acta 1828:2828–2839. 10.1016/j.bbamem.2013.03.033 [DOI] [PubMed] [Google Scholar]
- 4.Schneppenheim J, Dressel R, Hüttl S, Lüllmann-Rauch R, Engelke M, Dittmann K, Wienands J, Eskelinen EL, Hermans-Borgmeyer I, Fluhrer R, Saftig P, Schröder B. 2013. The intramembrane protease SPPL2a promotes B cell development and controls endosomal traffic by cleavage of the invariant chain. J. Exp. Med. 210:41–58. 10.1084/jem.20121069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beisner DR, Langerak P, Parker AE, Dahlberg C, Otero FJ, Sutton SE, Poirot L, Barnes W, Young MA, Niessen S, Wiltshire T, Bodendorf U, Martoglio B, Cravatt B, Cooke MP. 2013. The intramembrane protease Sppl2a is required for B cell and DC development and survival via cleavage of the invariant chain. J. Exp. Med. 210:23–30. 10.1084/jem.20121072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bergmann H, Yabas M, Short A, Miosge L, Barthel N, Teh CE, Roots CM, Bull-Jeelall KRY, Horikawa K, Whittle B, Balakishnan B, Sjollema G, Bertram EM, MacKay F, Rimmer AJ, Cornall RJ, Field MA, Andrews TD, Goodnow CC, Enders A. 2013. B cell survival, surface BCR and BAFFR expression, CD74 metabolism, and CD8− dendritic cells require the intramembrane endopeptidase SPPL2A. J. Exp. Med. 210:31–40. 10.1084/jem.20121076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neefjes J, Jongsma ML, Paul P, Bakke O. 2011. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 11:823–836. 10.1038/nri3084 [DOI] [PubMed] [Google Scholar]
- 8.Becker-Herman S, Arie G, Medvedovsky H, Kerem A, Shachar I. 2005. CD74 is a member of the regulated intramembrane proteolysis-processed protein family. Mol. Biol. Cell 16:5061–5069. 10.1091/mbc.E05-04-0327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Friedmann E, Hauben E, Maylandt K, Schleeger S, Vreugde S, Lichtenthaler SF, Kuhn PH, Stauffer D, Rovelli G, Martoglio B. 2006. SPPL2a and SPPL2b promote intramembrane proteolysis of TNFα in activated dendritic cells to trigger IL-12 production. Nat. Cell Biol. 8:843–848. 10.1038/ncb1440 [DOI] [PubMed] [Google Scholar]
- 10.Fluhrer R, Grammer G, Israel L, Condron MM, Haffner C, Friedmann E, Bohland C, Imhof A, Martoglio B, Teplow DB, Haass C. 2006. A gamma-secretase-like intramembrane cleavage of TNFα by the GxGD aspartyl protease SPPL2b. Nat. Cell Biol. 8:894–896. 10.1038/ncb1450 [DOI] [PubMed] [Google Scholar]
- 11.Kirkin V, Cahuzac N, Guardiola-Serrano F, Huault S, Luckerath K, Friedmann E, Novac N, Wels WS, Martoglio B, Hueber AO, Zornig M. 2007. The Fas ligand intracellular domain is released by ADAM10 and SPPL2a cleavage in T cells. Cell Death. Differ. 14:1678–1687. 10.1038/sj.cdd.4402175 [DOI] [PubMed] [Google Scholar]
- 12.Martin L, Fluhrer R, Reiss K, Kremmer E, Saftig P, Haass C. 2008. Regulated intramembrane proteolysis of Bri2 (Itm2b) by ADAM10 and SPPL2a/SPPL2b. J. Biol. Chem. 283:1644–1652. 10.1074/jbc.M706661200 [DOI] [PubMed] [Google Scholar]
- 13.Zahn C, Kaup M, Fluhrer R, Fuchs H. 2013. The transferrin receptor-1 membrane stub undergoes intramembrane proteolysis by signal peptide peptidase-like 2b. FEBS J. 280:1653–1663. 10.1111/febs.12176 [DOI] [PubMed] [Google Scholar]
- 14.Matza D, Kerem A, Shachar I. 2003. Invariant chain, a chain of command. Trends Immunol. 24:264–268. 10.1016/S1471-4906(03)00073-5 [DOI] [PubMed] [Google Scholar]
- 15.Golde TE, Wolfe MS, Greenbaum DC. 2009. Signal peptide peptidases: a family of intramembrane-cleaving proteases that cleave type 2 transmembrane proteins. Semin. Cell Dev. Biol. 20:225–230. 10.1016/j.semcdb.2009.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Reference deleted.
- 17.Behnke J, Schneppenheim J, Koch-Nolte F, Haag F, Saftig P, Schröder B. 2011. Signal-peptide-peptidase-like 2a (SPPL2a) is targeted to lysosomes/late endosomes by a tyrosine motif in its C-terminal tail. FEBS Lett. 585:2951–2957. 10.1016/j.febslet.2011.08.043 [DOI] [PubMed] [Google Scholar]
- 18.Leng L, Metz CN, Fang Y, Xu J, Donnelly S, Baugh J, Delohery T, Chen Y, Mitchell RA, Bucala R. 2003. MIF signal transduction initiated by binding to CD74. J. Exp. Med. 197:1467–1476. 10.1084/jem.20030286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi X, Leng L, Wang T, Wang W, Du X, Li J, McDonald C, Chen Z, Murphy JW, Lolis E, Noble P, Knudson W, Bucala R. 2006. CD44 is the signaling component of the macrophage migration inhibitory factor-CD74 receptor complex. Immunity 25:595–606. 10.1016/j.immuni.2006.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwartz V, Lue H, Kraemer S, Korbiel J, Krohn R, Ohl K, Bucala R, Weber C, Bernhagen J. 2009. A functional heteromeric MIF receptor formed by CD74 and CXCR4. FEBS Lett. 583:2749–2757. 10.1016/j.febslet.2009.07.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Calandra T, Roger T. 2003. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat. Rev. Immunol. 3:791–800. 10.1038/nri1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gore Y, Starlets D, Maharshak N, Becker-Herman S, Kaneyuki U, Leng L, Bucala R, Shachar I. 2008. Macrophage migration inhibitory factor induces B cell survival by activation of a CD74-CD44 receptor complex. J. Biol. Chem. 283:2784–2792. 10.1074/jbc.M703265200 [DOI] [PubMed] [Google Scholar]
- 23.Sapoznikov A, Pewzner-Jung Y, Kalchenko V, Krauthgamer R, Shachar I, Jung S. 2008. Perivascular clusters of dendritic cells provide critical survival signals to B cells in bone marrow niches. Nat. Immunol. 9:388–395. 10.1038/ni1571 [DOI] [PubMed] [Google Scholar]
- 24.Araki K, Imaizumi T, Sekimoto T, Yoshinobu K, Yoshimuta J, Akizuki M, Miura K, Araki M, Yamamura K. 1999. Exchangeable gene trap using the Cre/mutated lox system. Cell Mol. Biol. 45:737–750 [PubMed] [Google Scholar]
- 25.Taniwaki T, Haruna K, Nakamura H, Sekimoto T, Oike Y, Imaizumi T, Saito F, Muta M, Soejima Y, Utoh A, Nakagata N, Araki M, Yamamura K, Araki K. 2005. Characterization of an exchangeable gene trap using pU-17 carrying a stop codon-beta geo cassette. Dev. Growth Differ. 47:163–172. 10.1111/j.1440-169X.2005.00792.x [DOI] [PubMed] [Google Scholar]
- 26.Savalas LR, Gasnier B, Damme M, Lubke T, Wrocklage C, Debacker C, Jezegou A, Reinheckel T, Hasilik A, Saftig P, Schröder B. 2011. Disrupted in renal carcinoma 2 (DIRC2), a novel transporter of the lysosomal membrane, is proteolytically processed by cathepsin L. Biochem. J. 439:113–128. 10.1042/BJ20110166 [DOI] [PubMed] [Google Scholar]
- 27.Kim KJ, Kanellopoulos-Langevin C, Merwin RM, Sachs DH, Asofsky R. 1979. Establishment and characterization of BALB/c lymphoma lines with B cell properties. J. Immunol. 122:549–554 [PubMed] [Google Scholar]
- 28.Schröder B, Wrocklage C, Hasilik A, Saftig P. 2010. Molecular characterisation of “transmembrane protein 192” (TMEM192), a novel protein of the lysosomal membrane. Biol. Chem. 391:695–704. 10.1515/BC.2010.062 [DOI] [PubMed] [Google Scholar]
- 29.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. 10.1038/227680a0 [DOI] [PubMed] [Google Scholar]
- 30.Schagger H. 2006. Tricine-SDS-PAGE. Nat. Protoc. 1:16–22. 10.1038/nprot.2006.4 [DOI] [PubMed] [Google Scholar]
- 31.Lutz MB, Kukutsch N, Ogilvie AL, Rossner S, Koch F, Romani N, Schuler G. 1999. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J. Immunol. Methods 223:77–92. 10.1016/S0022-1759(98)00204-X [DOI] [PubMed] [Google Scholar]
- 32.Barois N, Forquet F, Davoust J. 1997. Selective modulation of the major histocompatibility complex class II antigen presentation pathway following B cell receptor ligation and protein kinase C activation. J. Biol. Chem. 272:3641–3647. 10.1074/jbc.272.6.3641 [DOI] [PubMed] [Google Scholar]
- 33.Radons J, Faber V, Buhrmester H, Volker W, Horejsi V, Hasilik A. 1992. Stimulation of the biosynthesis of lactosamine repeats in glycoproteins in differentiating U937 cells and its suppression in the presence of NH4Cl. Eur. J. Cell Biol. 57:184–192 [PubMed] [Google Scholar]
- 34.von Figura K. 1977. Human α-N-acetylglucosaminidase. 1. Purification and properties. Eur. J. Biochem. 80:523–533 [PubMed] [Google Scholar]
- 35.Friedmann E, Lemberg MK, Weihofen A, Dev Dengler KKU, Rovelli G, Martoglio B. 2004. Consensus analysis of signal peptide peptidase and homologous human aspartic proteases reveals opposite topology of catalytic domains compared with presenilins. J. Biol. Chem. 279:50790–50798. 10.1074/jbc.M407898200 [DOI] [PubMed] [Google Scholar]
- 36.Wubbolts R, Fernandez-Borja M, Oomen L, Verwoerd D, Janssen H, Calafat J, Tulp A, Dusseljee S, Neefjes J. 1996. Direct vesicular transport of MHC class II molecules from lysosomal structures to the cell surface. J. Cell Biol. 135:611–622. 10.1083/jcb.135.3.611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sannerud R, Annaert W. 2009. Trafficking, a key player in regulated intramembrane proteolysis. Semin. Cell Dev. Biol. 20:183–190. 10.1016/j.semcdb.2008.11.004 [DOI] [PubMed] [Google Scholar]
- 38.Horiuchi T, Mitoma H, Harashima S, Tsukamoto H, Shimoda T. 2010. Transmembrane TNF-alpha: structure, function and interaction with anti-TNF agents. Rheumatology (Oxford) 49:1215–1228. 10.1093/rheumatology/keq031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsachaki M, Ghiso J, Efthimiopoulos S. 2008. BRI2 as a central protein involved in neurodegeneration. Biotechnol. J. 3:1548–1554. 10.1002/biot.200800247 [DOI] [PubMed] [Google Scholar]
- 40.Krawitz P, Haffner C, Fluhrer R, Steiner H, Schmid B, Haass C. 2005. Differential localization and identification of a critical aspartate suggest non-redundant proteolytic functions of the presenilin homologues SPPL2b and SPPL3. J. Biol. Chem. 280:39515–39523. 10.1074/jbc.M501645200 [DOI] [PubMed] [Google Scholar]
- 41.Bentahir M, Nyabi O, Verhamme J, Tolia A, Horre K, Wiltfang J, Esselmann H, De SB. 2006. Presenilin clinical mutations can affect gamma-secretase activity by different mechanisms. J. Neurochem. 96:732–742. 10.1111/j.1471-4159.2005.03578.x [DOI] [PubMed] [Google Scholar]
- 42.Lai MT, Chen E, Crouthamel MC, Muzio-Mower J, Xu M, Huang Q, Price E, Register RB, Shi XP, Donoviel DB, Bernstein A, Hazuda D, Gardell SJ, Li YM. 2003. Presenilin-1 and presenilin-2 exhibit distinct yet overlapping gamma-secretase activities. J. Biol. Chem. 278:22475–22481. 10.1074/jbc.M300974200 [DOI] [PubMed] [Google Scholar]
- 43.Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, Tonegawa S. 1997. Skeletal and CNS defects in presenilin-1-deficient mice. Cell 89:629–639. 10.1016/S0092-8674(00)80244-5 [DOI] [PubMed] [Google Scholar]
- 44.Herreman A, Hartmann D, Annaert W, Saftig P, Craessaerts K, Serneels L, Umans L, Schrijvers V, Checler F, Vanderstichele H, Baekelandt V, Dressel R, Cupers P, Huylebroeck D, Zwijsen A, Van Leuven F, De Strooper B. 1999. Presenilin 2 deficiency causes a mild pulmonary phenotype and no changes in amyloid precursor protein processing but enhances the embryonic lethal phenotype of presenilin 1 deficiency. Proc. Natl. Acad. Sci. U. S. A. 96:11872–11877. 10.1073/pnas.96.21.11872 [DOI] [PMC free article] [PubMed] [Google Scholar]