

Abstract
Errors during DNA replication are one likely cause of gross chromosomal rearrangements (GCRs). Here, we analyze the role of RNase H2, which functions to process Okazaki fragments, degrade transcription intermediates, and repair misincorporated ribonucleotides, in preventing genome instability. The results demonstrate that rnh203 mutations result in a weak mutator phenotype and cause growth defects and synergistic increases in GCR rates when combined with mutations affecting other DNA metabolism pathways, including homologous recombination (HR), sister chromatid HR, resolution of branched HR intermediates, postreplication repair, sumoylation in response to DNA damage, and chromatin assembly. In some cases, a mutation in RAD51 or TOP1 suppressed the increased GCR rates and/or the growth defects of rnh203Δ double mutants. This analysis suggests that cells with RNase H2 defects have increased levels of DNA damage and depend on other pathways of DNA metabolism to overcome the deleterious effects of this DNA damage.
INTRODUCTION
DNA replication, homologous recombination (HR), and repair are central pathways of DNA metabolism responsible for duplicating and maintaining the integrity of the genome. Each process relies on the coordination of many different proteins, and in addition, the three processes interact intimately with each other. Many DNA repair pathways, including multiple excision repair and HR pathways, rely on DNA replication proteins for the synthesis of new DNA. Similarly, normal DNA replication requires processing of Okazaki fragments, which are the discontinuously synthesized products of lagging-strand DNA replication, and this Okazaki fragment processing involves the action of what are essentially DNA repair pathways.
Okazaki fragments consist of 7 to 14 nucleotides of RNA primer covalently linked to the 5′ end of 150 to 200 nucleotides of DNA (1, 2). Several nucleases, including RNase H2, Rad27/FEN1, and Dna2, have been implicated in the processing of the 5′ ends of Okazaki fragments (2–4). RNase H2 is an endonuclease that degrades the RNA strand of RNA/DNA hybrids by cleaving the 5′ end of RNA phosphodiester bonds (5–8). RNase H2 is comprised of three subunits that are encoded by the genes RNH201 (formerly known as RNH35), RNH202, and RNH203 in Saccharomyces cerevisiae (9) and the genes RNASEH2A, RNASEH2B, and RNASEH2C in humans (10). Initial studies found that sequential actions of RNase H2 and FEN1, coupled to strand displacement synthesis and followed by the action of DNA ligase, were sufficient to complete Okazaki fragment processing (8, 11–13). In contrast, more recent studies have indicated that the major in vitro mechanism for RNA primer removal is flap processing by Rad27/FEN1 and Dna2 (14–16), with Exo1 apparently being able to substitute for Rad27/FEN1 in S. cerevisiae (17, 18). Because RNase H2 is not essential in S. cerevisiae (19), it is possible that RNase H2 plays either a redundant or a stimulatory role in Okazaki fragment processing, which could underlie the substantial growth defects caused by combining mutations affecting RNase H2 with mutations affecting Rad27/Fen1 (7, 20–23).
RNase H2 also links DNA replication and DNA repair through ribonucleotide excision repair (RER), which removes the estimated 10,000 ribonucleotides misincorporated into the S. cerevisiae genome DNA during replication (24–26). RNase H2, not RNase H1, appears to be the major RNase used to initiate RER in reconstituted reactions with S. cerevisiae proteins in vitro (26). This is consistent with the expression of these genes, as only RNase H2 has increased expression during S phase and G2/M (27). In addition, RNase H1 is most active on RNA/DNA hybrids containing several consecutive ribonucleotides (28), which are predicted to be rare due to the stochastic nature of ribonucleotide misincorporation (26), whereas RNase H2 shows prominent activity on single ribonucleotides embedded within double-stranded DNA (9). RER by RNase H2 is likely achieved in concert with other key proteins, including Rad27/FEN1, Pol δ, RFC, PCNA, and DNA ligase, by flap cleavage mechanisms similar to those that occur during the processing of Okazaki fragments and base excision repair (14, 26, 29). In the absence of RNase H2, Top1 can nick DNA at sites of ribonucleotide misincorporation (30, 31). Interestingly, Top1-initiated RER appears to lead to the formation of small deletions in short tandem repeats in DNA (30).
Defects in DNA repair mechanisms can lead to genome instability, including the accumulation of mutations and gross chromosomal rearrangements (GCRs) (32). However, little is known about the role of RNase H2 in suppressing genome instability. This is despite the fact that replication-generated Okazaki fragments, whose processing involves RNase H2, may be the largest source of DNA damage in the cell (29). Consequently, the observation that mutations in RAD27 and DNA2 cause increased genome instability, including GCRs, potentially as a result of defects in Okazaki fragment processing (17, 33–39), suggests that defects in RNase H2 also cause increased genome instability due to defects in Okazaki fragment processing. Furthermore, ribonucleotides are misincorporated at significant levels by DNA polymerases during DNA replication (25). Since ribonucleotides are more susceptible to spontaneous cleavage than deoxyribonucleotides (40), loss of RER or the presence of aberrant RER due to Top1 (30) could lead to increased strand breaks and, potentially, genome instability. In S. cerevisiae, RNase H2 defects cause weak mutator and hyperrecombination phenotypes (7, 41–45), and RNase H2 null mouse cells have increased chromosomal rearrangements and a p53-dependent DNA damage response (40, 46). Interestingly, defects in RNase H2, as well as the Trex exonucleases, cause Aicardi-Goutieres syndrome, a neuroautoimmune disease that may result from inflammatory responses to aberrant DNA or RNA structures that accumulate as the result of these defects (10, 47).
RNase H2 defects cause only weak genome instability phenotypes, which is in contrast to the potential for substantially increased levels of genome instability caused by high levels of misincorporated ribonucleotides, errors in processing Okazaki fragments, and transcription intermediates thought to persist or occur in cells with RNase H2 defects (2–4, 7, 48, 49). Thus, we hypothesized that other DNA metabolism pathways compensate for RNase H2 defects. To investigate this and to gain a better understanding of the role of RNase H2 in genome maintenance, we conducted a targeted genetic interaction analysis of RNase H2 in S. cerevisiae. The results reported here indicate that defects in RNase H2 interact with defects in multiple pathways, including HR, resolution of branched HR intermediates, postreplication repair (PRR), stabilization and processing of stalled replication forks, sumoylation in response to DNA damage, and chromatin assembly. Furthermore, in a number of cases, the genes identified by these genetic interactions act to prevent GCRs from occurring as a result of RNase H2 defects. These results suggest that RNase H2 defects likely result in a higher burden of aberrant DNA and RNA structures than previously appreciated.
MATERIALS AND METHODS
S. cerevisiae strains.
The general methods and media used, including yeast extract-peptone-dextrose (YPD) and synthetic dropout media for propagating S. cerevisiae strains, have been described previously (34, 50). All strains used in this study were derivatives of S. cerevisiae strain S288c (see Table S4 in the supplemental material) and were grown at 30°C. Single mutant strains were made by deleting the gene of interest in RDKY3615 (MATα ura3-52 leu2Δ1 trp1Δ63 his3Δ200 lys2ΔBgl hom3-10 ade2::hisG ade8 hxt13::URA3) and RDKY7239 (MATα ura3-52 leu2Δ1 trp1Δ63 his3Δ200 lys2ΔBgl hom3-10 ade2::hisG ade8 hxt13::URA3) by HR-mediated integration of PCR-generated fragments according to standard methods. The single, double, and triple mutant strains used in the experiments described below were obtained by mating appropriate mutants and sporulating the resulting diploids. Freshly derived spore clones were frozen and subsequently genotyped and tested in doubling time experiments to ensure that the isolates of interest did not obtain growth rate suppressor mutations.
Fluctuation analysis and rnh203Δ mutation spectra.
The rates of accumulation of Thr+ revertants, Lys+ revertants, Canr mutations, and GCRs resulting in Canr–5-fluoroorotic acid resistance (5FOAr) mutations were determined by fluctuation analysis using the method of the median as previously described (50, 51). At least 14 independent cultures were analyzed in each experiment.
Mutation spectrum data were collected by making patches from individual colonies of RDKY7209 on YPD plates. Patches were replica plated onto selective media containing canavanine lacking arginine to select for Canr mutants. Canr mutants were allowed to grow at 30°C for 2 days, after which one Canr mutant per patch was restreaked on selective media. Chromosomal DNA was isolated from each Canr mutant and was then used as the template DNA for PCR amplification of the CAN1 gene, followed by identification of the inactivating mutation in the CAN1 gene essentially as has been previously described (50, 52–54). The CAN1 gene was amplified from each Canr mutant by using by PCR with the primers CAN1FX (5′-GTTGGATCCAGTTTTTAATCTGTCGTC-3′) and CAN1RX (5′-TTCGGTGTATGACTTATGAGGGTG-3′). The primers used for sequencing the PCR product were CAN1G (5′-CAGTGGAACTTTGTACGTCC-3′), CANSEQ3 (5′-TTCTGTCACGCAGTCCTTGG-3′), and CANSEQ5 (5′-AACTAGTTGGTATCACTGCT-3′). All DNA sequencing was performed by using an Applied Biosystems 3730XL DNA sequencer and standard chemistry. Sequence analysis was performed using Sequencher 4.2.2 (Gene Codes, Ann Arbor, MI). The rate of each type of mutation was obtained by multiplying the total Canr mutation rate, determined by fluctuation analysis, of the rnh203Δ strain by the proportion of each type of mutation found. The wild-type mutation spectrum was previously published (43).
Growth analysis.
For qualitative determination of growth rates, each mutant strain was streaked onto a YPD plate from a frozen stock and grown at 30°C for 2 to 3 days. Single colonies were then inoculated and grown to log phase at 30°C in 10 ml of YPD. Serial dilutions of the cell suspensions were then spotted onto a YPD plate, and the plates were grown at 30°C for approximately 30 h, after which the sizes of the resulting colonies were visualized. For quantitative measurements, cultures were inoculated at an optical density at 600 nm (OD600) of 0.05 by diluting the appropriate volume of an overnight culture into fresh YPD. Cultures were grown with shaking at 30°C, and OD600 measurements were taken every 30 to 45 min until the cells reached saturation. Doubling times were then calculated as previously described (55). The doubling times reported are the average of at least three cultures each inoculated with an independent single colony isolated by random spore analysis.
Budding index and quantitation of bud morphology.
The budding index, defined as the ratio of the number of individuals bearing buds to the total cell number, was determined microscopically, with individual cells bearing buds being counted as one cell (56). Cells were grown in YPD medium to log phase, sonicated, and examined by light microscopy, and the numbers of single cells, small-budded cells (bud smaller than one-third of the mother cell), large-budded cells (bud equal to or larger than one-third of the mother cell), and other cells (cells with protruded or multiple buds) were recorded. Two to four hundred cells were counted per experiment, and the experiments were generally performed independently 3 times.
RESULTS
Strains deficient in RNase H2 display a weak mutator phenotype.
Previous work indicated that strains with a deletion of genes encoding subunits of the RNase H2 protein complex displayed a weak mutator phenotype (42, 43, 45), which is consistent with a defect in DNA repair. To further investigate this, we generated and studied a set of isogenic mutant strains containing individual deletions of the genes encoding the subunits of the RNase H2 complex (see Table S1 in the supplemental material). The rnh201Δ, rnh202Δ, and rnh203Δ mutant strains were viable and had no growth phenotypes. Fluctuation analysis demonstrated that the rates of accumulating reversions of the lys2ΔBgl frameshift allele, Canr mutations, and GCRs in the RNase H2 mutant strains were similar to the rates in the wild-type strain. However, there was a 3- to 4-fold increase in the rate of reversion of the hom3-10 frameshift allele, the most sensitive mutator assay used, in each of the rnh201Δ, rnh202Δ, and rnh203Δ mutant strains compared to the rate in the wild-type strain (see Table S1). Our results, in combination with the weak mutator phenotype caused by RNase H2 defects demonstrated in other studies (7, 30, 42, 45), support the view that RNase H2 defects cause a weak mutator phenotype resulting in an increase in mutation rates that is less than 1% of the increased mutation rates seen in the absence of mismatch repair (50).
To further characterize the rnh203Δ mutator phenotype, a mutation spectrum analysis of the Canr mutations that occurred in the rnh203Δ mutant was performed (see Table S2 in the supplemental material). In the rnh203Δ mutant strain, we found a 3.5-fold increase in the frequency of AT-to-CG base substitutions and a >4.5-fold increase in the frequency of double base deletions, but only the increase in double base deletions was significant (P = 0.04, Fisher's exact test). The increase in double base deletions was consistent with the increased rate seen in the hom3-10 frameshift reversion assay (see Table S1) and with the published TOP1-dependent increase in double base deletions in an rnh201Δ mutant (30). Taken together, these data support the idea that RNase H2 plays a small role in suppressing both frameshift and base substitution mutations. We also observed a 4.4-fold decrease in the frequency of large deletions in the rnh203Δ mutant (P = 0.03, Fisher's exact test), suggesting that the cleavage of R-loops or misincorporated stretches of RNA by RNase H2 (24, 26, 48, 49, 57) might result in double-strand breaks and contribute to the formation of rare deletions.
Genetic interactions between RNase H2 defects and defects in other DNA metabolism pathways.
RNase H2 is involved in multiple DNA metabolism pathways in which defects are known to cause the accumulation of DNA damage. One hypothesis that could explain the weak mutator and hyperrecombination phenotypes caused by RNase H2 defects is that RNase H2 defects cause DNA damage that is then compensated for by other DNA repair pathways. We therefore performed a targeted screen for genetic interactions between an rnh203Δ mutation and mutations affecting DNA metabolism. We tested mutations causing defects in HR, PRR, cleavage of branched DNAs, and processing of HR intermediates that are formed during DNA replication, checkpoints, and chromatin assembly and modification proteins that function in conjunction with DNA replication and DNA damage responses. Haploid double mutant strains were generated by mating and sporulation and then were used in spot tests to qualitatively compare growth rates (Fig. 1; see also Fig. S1 and S2 in the supplemental material). This screen identified 19 nonessential genes that contribute to the survival or fitness of an rnh203Δ mutant, as well as 34 mutations that had no effect. Eight genetic interactions were previously unreported (ASF1, ESC4/RTT107, MGS1, MPH1, RAD5, RAD6, RAD53, and RFA1), and 11 interactions with mutations in at least one gene encoding RNase H2 (6 with rnh203Δ) were previously identified (ESC2, CTF4, MRE11, MUS81, RAD27, RAD50, RAD51, RAD52, SGS1, TOP3, and XRS2) (20–24, 44, 58–63). These genetic interactions, as well as the lack of genetic interactions between an rnh203Δ mutation and mutations in other genes, were confirmed by measuring the doubling times of different double mutants and their respective single mutants (Fig. 2). With the exception of the 8 new genetic interactions identified here, our results agreed well with previously reported mutations that do or do not interact with deletions of genes encoding RNase H2 (44, 61, 64–71). In addition, other studies have indicated that strains lacking RNH203 require DEP1, DNA2, MMS4 (encodes a subunit of the Mus81-Mms4 complex), RMI1 (encodes a subunit of the Sgs1-Rmi1-Top3 complex), RNH1, and SMC6 for normal growth (20, 41, 44, 58, 72), although we did not investigate these interactions in the present study.
FIG 1.

Growth spot analysis for detection of RNase H2 genetic interactions. (A) Cultures were diluted to either 1 × 106 cells/ml or 1 × 105 cells/ml, and then 2-μl amounts from 10-fold serial dilutions were spotted in each row. Strains were spotted onto each plate as follows: top row, wild type; second row, the rnh203Δ single mutant; third row, the query single mutants (mutations identified at the bottom are represented at the right by mutXΔ); fourth through sixth rows, independent isolates of the double mutants. (B) Summary of RNase H2 defect-dependent growth interactions observed when an RNase H2 defect was combined with each of the 53 listed mutations. NHEJ, nonhomologous end joining.
FIG 2.

Cells lacking RNase H2 depend on different DNA metabolism genes for normal growth. The growth rates measured for the indicated single and double mutants are presented, along with error bars indicating the standard deviations. (A) Cases in which an RNase H2 defect causes a decrease in the growth rate. (B) Cases in which an RNase H2 defect has no effect on the growth rate.
RNase H2 defects cause alterations in the timing of cell cycle transitions.
We monitored the cell cycle distribution of log-phase cells of the slow-growing rnh203Δ double mutants and the corresponding single mutants to better characterize the growth defects. The cell cycle distribution was followed using the budding index, which uses the bud morphology of normal log-phase cells to identify cells in G1 phase (no bud), S phase (small bud), and G2/M phase (large bud). We also counted cells with aberrant morphology, which typically consist of cells with grossly elongated or multiple buds and have been observed in strains with alterations in the timing of cell cycle transitions, DNA replication defects, and uncoupling of replication defects from checkpoint responses (56, 64, 73, 74). We observed decreased proportions of G1- and S-phase cells, along with an increased fraction of cells with aberrant morphologies in strains where the rnh203Δ mutation was combined with the asf1Δ, esc2Δ, mgs1Δ, mph1Δ, mre11Δ, rad50Δ, rad52Δ, or xrs2Δ mutation (Fig. 3; see also Fig. S3 in the supplemental material). These results suggest that these double mutants accumulate replication-dependent DNA damage, consistent with prior studies demonstrating that RNase H2 deficiency in mouse cells causes cell cycle arrest mediated by p53-induced p21 expression (40, 46). In addition, a number of the rnh203Δ double mutants with slow-growth phenotypes did not have substantially altered cell cycle distributions or increased numbers of cells with aberrant morphology relative to the corresponding single mutant, including rnh203Δ ctf4Δ, rnh203Δ esc4Δ/rtt107Δ, rnh203Δ rad5Δ, rnh203Δ rad6Δ, rnh203Δ rad27Δ, rnh203Δ rad51Δ, rnh203Δ rad53Δ, rnh203Δ rfa1-t33, and rnh203Δ sgs1Δ double mutants (Fig. 3; see also Fig. S4).
FIG 3.
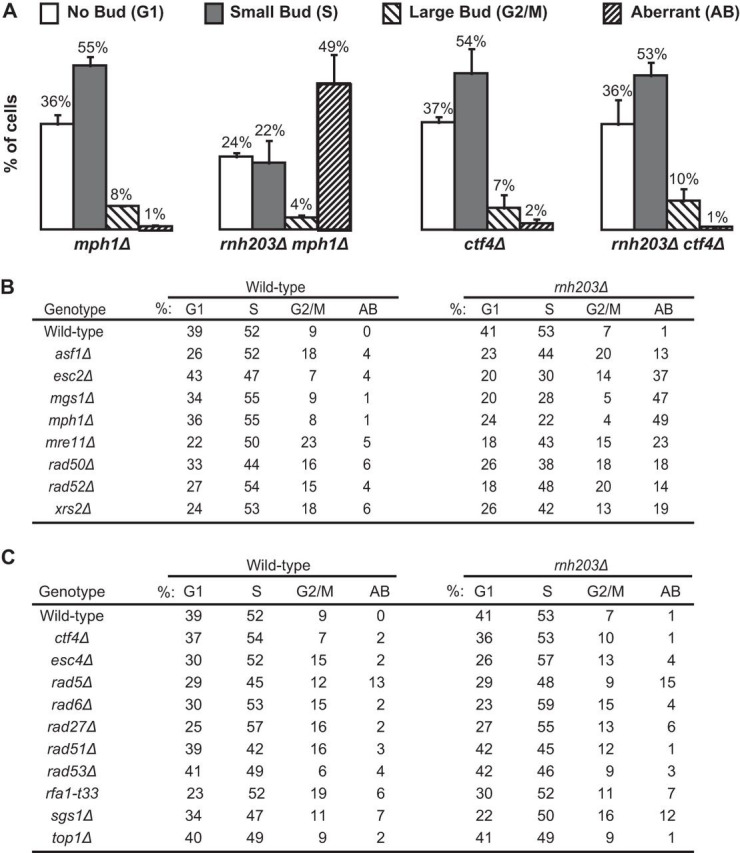
An RNase H2 defect can cause an accumulation of replication-dependent DNA damage that leads to an increase in an aberrant morphology phenotype. The budding index was measured to determine the percentage of cells in each phase of the cell cycle and the percentage of cells with an aberrant morphology phenotype. (A) Representative examples presented as histograms, with error bars indicating the standard deviations. (B, C) Summary of the budding index data for appropriate controls and mutant strains with an increase (B) and no increase (C) in aberrant morphology.
Defects in HR suppress the slow growth and aberrant morphology phenotypes of a subset of rnh203Δ double mutants.
Defects in HR can suppress the slow-growth phenotypes that are caused by HR-dependent formation of aberrant DNA structures during DNA replication in some DNA helicase-defective mutants (65, 69, 73, 75, 76). Previous work demonstrated that a rad51Δ mutation suppressed the slow-growth phenotype of the rnh201Δ sgs1Δ, rnh202Δ sgs1Δ, rnh203Δ sgs1Δ, and rnh202Δ mus81Δ double mutants (61, 68). This suggests that HR-dependent aberrant DNA structures may be formed in these double mutants. We therefore introduced a rad51Δ mutation into a series of rnh203Δ double mutant strains that had substantially increased doubling times, increased levels of cells with aberrant morphology, and in most cases, synergistic increases in GCR rates compared to those of the corresponding single mutants (Fig. 4).
FIG 4.
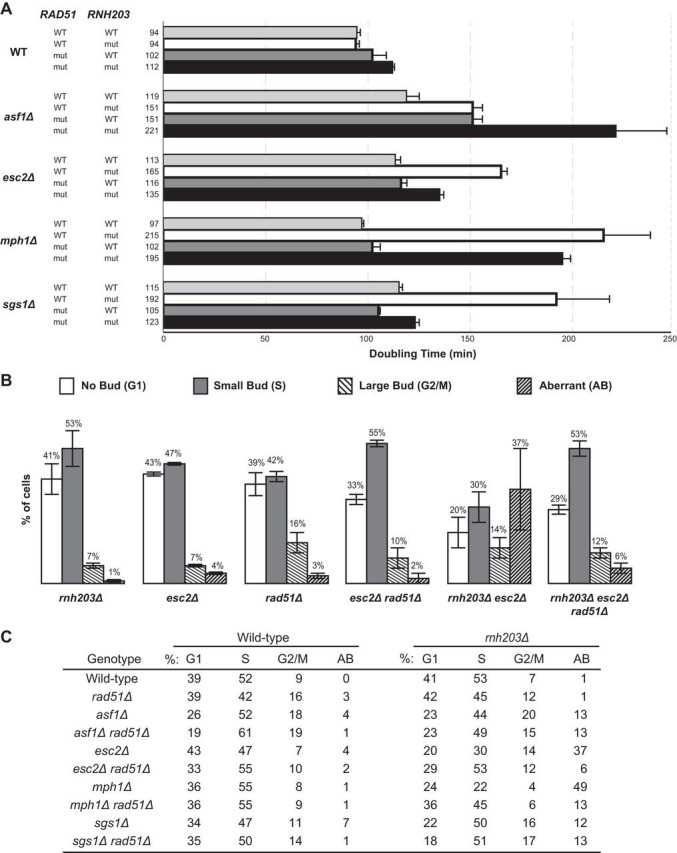
Defects in HR can suppress the slow growth phenotype and aberrant morphology phenotype of selected rnh203Δ double mutants. (A) Doubling times, with error bars indicating the standard deviations, are presented for the indicated single, double, and triple mutant strains. (B) The budding index was measured to determine the percentage of cells in each phase of the cell cycle and the percentage of cells with an aberrant morphology phenotype. Representative examples are presented as histograms, with error bars indicating the standard deviations. (C) Summary of the budding index data for mutant strains and controls.
We found that a rad51Δ mutation suppressed or partially suppressed the slow growth and aberrant cell morphology phenotypes of the rnh203Δ esc2Δ and rnh203Δ mph1Δ double mutants, respectively. In addition, the slow-growth but not the aberrant morphology phenotype of the rnh203Δ sgs1Δ double mutant was suppressed by a rad51Δ mutation. These results suggest that the slow-growth phenotype and, in some cases, the aberrant cell morphology phenotype of the rnh203Δ esc2Δ, rnh203Δ mph1Δ, and rnh203Δ sgs1Δ double mutants is caused by the accumulation of HR-dependent DNA structures. This could be because RNase H2 normally prevents DNA damage that underlies the formation of HR-dependent DNA structures or causes defects in the processing of such structures, so they persist for longer times.
In contrast, the growth defect of the rnh203Δ asf1Δ double mutant was enhanced by the rad51Δ mutation, whereas the aberrant cell morphology phenotype was not affected. These results suggest that potential DNA damage accumulating in the rnh203Δ asf1Δ double mutant is formed independently of Rad51 and is likely repaired by Rad51-dependent HR.
RNase H2 double mutants have increased rates of accumulating GCRs.
Since the accumulation of DNA damage can lead to genome instability, we analyzed a series of rnh203Δ double mutants and their respective single mutants by utilizing an established assay that measures the rate of accumulating GCRs, including translocations, deletion of chromosome arms coupled to healing by de novo telomere addition and end-to-end chromosome fusions, and other types of dicentric translocations (32, 51, 77). In many cases, combining a deletion of RNH203 with mutations in genes of interest known to suppress GCRs had no effect on the GCR rate (see Table S3 in the supplemental material); this was true even though many of the double mutants analyzed had growth defects and/or morphology defects. However, synergistic increases in GCR rates were observed when the rnh203Δ mutation was combined with defects in chromatin remodeling (78) (asf1Δ, P = 0.1 [borderline significance]), HR and processing of HR intermediates (79–82) (mre11Δ, P = 0.001; mus81Δ, P = 0.001; esc4Δ/rtt107Δ, P = 0.003; sgs1Δ, P < 0.001; esc2Δ, P < 0.001), and PRR (83) (pol30-119 [eliminates the major ubiquitination/sumoylation site on PCNA {84}], P < 0.0001; rad6Δ, P = 0.036) (all P values determined by Mann-Whitney test) (Table 1). Taken together, these results suggest that RNase H2 defects cause DNA damage whose repair requires processing by chromatin remodeling, HR, and PRR to prevent the formation of GCRs.
TABLE 1.
Effects of an RNase H2 mutation on the rates of accumulating GCRsa
| Genotype | RDKY no., GCR rate (with 95% CI) (fold increase relative to GCR rate of wild type) |
|
|---|---|---|
| Wild type | rnh203Δ mutant | |
| Wild type | 7239, 3.6 (1.9–14.0) × 10−10 (1) | 7209, 2.0 (0.0–8.9) × 10−10 (0.5) |
| asf1Δ | 7211, 1.4 (0.1–2.4) × 10−8 (39) | 7213, 4.1 (0.2–16.5) × 10−8 (114) |
| cac1Δ | 7214, 3.9 (2.3–7.6) × 10−8 (108) | 7216, 1.0 (0.6–7.6) × 10−9 (3) |
| esc2Δ | 7248, 1.7 (1.0–2.7) × 10−8 (48) | 7250, 2.1 (1.1–6.4) × 10−7 (592) |
| esc4Δ | 7252, 1.3 (1.9–7.3) × 10−8 (36) | 7254, 5.9 (2.4–14.1) × 10−8 (163) |
| exo1Δ | 7256, 1.1 (0.3–2.9) × 10−8 (29) | 7258, 3.4 (0.8–11.7) × 10−9 (9) |
| mre11Δ | 7273, 4.3 (1.7–5.4) × 10−7 (1,194) | 7275, 1.4 (0.5–2.0) × 10−6 (3,972) |
| mus81Δ | 7294, 8.4 (4.5–16.0) × 10−9 (23) | 7296, 2.5 (1.2–4.1) × 10−8 (69) |
| pol30-119 | 7316, 2.9 (1.1–6.7) × 10−9 (8) | 7318, 7.0 (5.3–15.0) × 10−8 (147) |
| rad6Δ | 7385, 2.3 (1.9–7.7) × 10−9 (6) | 7386, 6.5 (2.4–53.0) × 10−9 (18) |
| sgs1Δ | 7414, 6.9 (2.7–11.4) × 10−9 (19) | 7416, 7.6 (6.2–11.7) × 10−8 (211) |
| tel1Δ | 7434, 5.9 (2.0–11.1) × 10−9 (16) | 7436, 8.0 (4.8–54.0) × 10−10 (2) |
All strains are isogenic to wild-type RDKY7239 (MATα ura3-52 leu2Δ1 trp1Δ63 his3Δ200 lys2ΔBgl hom3-10 ade2::hisG ade8 hxt13::URA3). 95% CI, 95% confidence interval.
Surprisingly, combining an rnh203Δ mutation with a cac1Δ mutation reduced the increased GCR rate caused by the cac1Δ mutation (P = 0.0003, Mann-Whitney test) (56) to a level that was not different from that of either the rnh203Δ single mutant or a wild-type strain (Table 1). Analysis of the rnh203Δ exo1Δ and rnh203Δ tel1Δ double mutants showed a similar but weaker effect (P = 0.047 and P = 0.028, respectively, Mann-Whitney test) (Table 1). These results suggest that in some cases, the normal action of RNase H2 might result in DNA damage that can lead to GCRs in the presence of some genetic defects.
To further characterize the mutants with HR-dependent growth and morphology defects, we analyzed the effect of a rad51Δ mutation on the rate of accumulating GCRs in the rnh203Δ asf1Δ, rnh203Δ esc2Δ, rnh203Δ mph1Δ, and rnh203Δ sgs1Δ double mutant strains (Table 2), which also showed growth and morphology defects. The synergistic increase in the GCR rate seen in the rnh203Δ esc2Δ double mutant was reduced 22-fold (P = 0.0004, Mann-Whitney test) by the rad51Δ mutation, even though a rad51Δ mutation significantly increased the GCR rate of the rnh203Δ single mutant and wild-type strains (Table 2). In contrast, a rad51Δ mutation did not significantly reduce the GCR rate of the rnh203Δ asf1Δ double mutants (P = 0.98, Mann-Whitney test) and may have weakly reduced the GCR rate of the rnh203Δ sgs1Δ double mutant (P = 0.1 [borderline significance], Mann-Whitney test). The rad51Δ mutation did not affect the GCR rate of the rnh203Δ mph1Δ double mutant, although it should be noted that this double mutant did not show an increased GCR rate compared to that of the single mutants. These results suggest that in the absence of RNase H2 and either Esc2 or possibly Sgs1, HR-dependent formation of aberrant DNA structures likely underlies the formation of GCRs, as well as the slow growth and/or morphology defects observed. This observation further supports a role of RNase H2 in DNA repair such that RNase H2 defects either result in DNA damage that can underlie the formation of HR-dependent DNA structures or cause defects in the processing of such structures so that they persist for longer times.
TABLE 2.
Effects of a rad51Δ mutation on the GCR rates of rnh203Δ mutantsa
| Genotype | RDKY no., GCR rate (with 95% CI) (fold increase relative to GCR rate of wild type) |
|
|---|---|---|
| Wild type | rnh203Δ | |
| Wild type | 7239, 3.6 (1.9–14.0) × 10−10 (1) | 7209, 2.0 (0.0–8.9) × 10−10 (0.5) |
| rad51Δ | 7344, 3.5 (1.5–7.9) × 10−9 (10) | 7346, 3.7 (1.8–21.6) × 10−9 (10) |
| asf1Δ | 7211, 1.4 (0.1–2.4) × 10−8 (39) | 7213, 4.1 (0.2–16.5) × 10−8 (114) |
| asf1Δ rad51Δ | 7501, <1.9 (0.0–30.0) × 10−9 (5) | 7503, 2.6 (1.1–7.9) × 10−8 (72) |
| esc2Δ | 7248, 1.7 (1.0–2.7) × 10−8 (48) | 7250, 2.1 (1.1–6.4) × 10−7 (592) |
| esc2Δ rad51Δ | 7507, 1.9 (0.0–3.1) × 10−9 (5) | 7509, 1.0 (0.0–3.3) × 10−8 (27) |
| mph1Δ | 7286, 1.1 (0.6–5.6) × 10−9 (3) | 7288, <1.2 (0.0–2.8) × 10−9 (3) |
| mph1Δ rad51Δ | 7511, 9.9 (0.0–44.3) × 10−10 (3) | 7513, 2.3 (1.2–3.5) × 10−9 (6) |
| sgs1Δ | 7414, 6.9 (2.7–11.4) × 10−9 (19) | 7416, 7.6 (6.2–11.7) × 10−8 (211) |
| sgs1Δ rad51Δ | 7515, 1.4 (0.3–2.0) × 10−8 (39) | 7517, 2.3 (0.5–19.0) × 10−8 (64) |
All strains are isogenic to wild-type RDKY7239 (MATα ura3-52 leu2Δ1 trp1Δ63 his3Δ200 lys2ΔBgl hom3-10 ade2::hisG ade8 hxt13::URA3). 95% CI, 95% confidence interval.
Deletion of TOP1 suppresses the increased GCR rate of the rnh203Δ esc2Δ double mutant.
Recent studies have shown that some of the mutations, particularly two base deletions, that arise in rnh201Δ mutants are due to DNA strand breaks generated by topoisomerase 1 at the site of misincorporated ribonucleotides (30). To determine whether topoisomerase 1-dependent DNA strand breaks promote the increased GCRs seen in rnh203Δ double mutants, we tested the effect of a top1Δ mutation on the GCR rate of two of the double mutants, rnh203Δ esc2Δ and rnh203Δ sgs1Δ, that had the largest synergistic increases in GCR rates compared to the GCR rates of the respective single mutants. We found that top1Δ mutation strongly suppressed the increased GCR rate of the rnh203Δ esc2Δ double mutant (P = 0.001, Mann-Whitney test) but did not suppress the GCR rate of the rnh203Δ sgs1Δ double mutant (P = 0.24, Mann-Whitney test) (Table 3). The top1Δ mutation had no effect on the growth rates of the rnh203Δ esc2Δ and rnh203Δ sgs1Δ double mutants (Fig. 5A). Finally, the top1Δ mutation completely suppressed the aberrant morphology phenotype of the rnh203Δ esc2Δ double mutant but had no effect on the aberrant morphology phenotype of the rnh203Δ sgs1Δ double mutant (Fig. 5B and C). These results show that in some but not all rnh203Δ double mutants, the action of topoisomerase 1 on misincorporated ribonucleotides likely underlies the increased GCRs seen.
TABLE 3.
Effects of a top1Δ mutation on the GCR rates of rnh203Δ mutantsa
| Genotype | RDKY no., GCR rate (with 95% CI) (fold increase relative to GCR rate of wild type) |
|
|---|---|---|
| Wild type | rnh203Δ | |
| Wild type | 7239, 3.6 (1.9–14.0) × 10−10 (1) | 7209, 2.0 (0.0–8.9) × 10−10 (0.5) |
| top1Δ | 8143, 7.6 (1.6–56.6) × 10−10 (2) | 8141, 5.6 (2.5–30.1) × 10−10 (3) |
| esc2Δ | 7248, 1.7 (1.0–2.7) × 10−8 (48) | 7250, 2.1 (1.1–6.4) × 10−7 (592) |
| esc2Δ top1Δ | 8145, 1.1 (0.5–2.6) × 10−8 (30) | 8147, 4.6 (0.0–21) × 10−9 (13) |
| sgs1Δ | 7414, 6.9 (2.7–11.4) × 10−9 (19) | 7416, 7.6 (6.2–11.7) × 10−8 (211) |
| sgs1Δ top1Δ | 8149, 2.2 (0.7–29.6) × 10−8 (61) | 8151, 8.3 (1.3–50.0) × 10−8 (230) |
All strains are isogenic to wild-type RDKY7239 (MATα ura3-52 leu2Δ1 trp1Δ63 his3Δ200 lys2ΔBgl hom3-10 ade2::hisG ade8 hxt13::URA3). 95% CI, 95% confidence interval.
FIG 5.

Topoisomerase 1 defects can suppress the slow-growth phenotype and aberrant morphology phenotype of selected rnh203Δ double mutants. (A) Doubling times, with error bars indicating the standard deviations, are presented for the indicated single, double, and triple mutant strains. (B) The budding index was measured to determine the percentage of cells in each phase of the cell cycle and the percentage of cells with an aberrant morphology phenotype. Representative examples are presented as histograms, with error bars indicating the standard deviations. (C) Summary of the budding index data for mutant strains and controls.
DISCUSSION
We surveyed a carefully selected group of DNA metabolism genes for genetic interactions with the genes encoding RNase H2. We found that RNase H2 defects alone caused a weak mutator phenotype, including a small increase in the rate of reversion of the hom3-10 frameshift allele, an increase in AT-to-CG base substitutions, and an increase in double base deletion mutations, consistent with previous studies (7, 30, 42, 43, 45). We also found that, although RNase H2 defects did not increase the rate of accumulating GCRs, they did cause synergistic increases in GCR rates when combined with 8 mutations affecting DNA metabolism and a synthetic growth defect when combined with 19 mutations affecting DNA metabolism (Table 4). Furthermore, RNase H2 defects caused an increased percentage of cells with aberrant morphology when combined with 8 of the 19 interacting mutations (Table 4). In some of the cases tested, deletion of RAD51 or TOP1 suppressed some combination of the double mutant phenotypes (Table 4). In aggregate, the results from our analysis suggest that RNase H2 defects cause an accumulation of mutagenic damage in DNA that is then repaired or compensated for by different DNA metabolism pathways.
TABLE 4.
Summary of observed genetic interactions with the rnh203Δ mutation
| Mutation | Effect of interacting mutation ona: |
|||
|---|---|---|---|---|
| Growth | Aberrant morphology | GCR rate | Growth in Biogridb | |
| esc2Δ | Strong (rad51Δ suppressed, top1Δ unaffected) | Increased (rad51Δ suppressed, top1Δ suppressed) | Increased (rad51Δ suppressed, top1Δ suppressed) | 1, 2, 3 |
| mgs1Δ | Strong | Increased | Same | — |
| mph1Δ | Strong (rad51Δ suppressed) | Increased (rad51Δ suppressed) | Same (rad51Δ unaffected) | — |
| mre11Δ | Strong | Increased | Increased | 1, 2, 3 |
| rad5Δ | Strong | Same | Same | — |
| rad6Δ | Strong | Same | Same | — |
| rad27Δ | Strong | Same | Same | 1, 2, 3 |
| rad50Δ | Strong | Increased | NDc | 1, 2, 3 |
| rad52Δ | Strong | Increased | Same | 1, 2 |
| rad53Δ | Strong | Same | Same | — |
| rfa1-t33 | Strong | Same | Same | — |
| sgs1Δ | Strong (rad51Δ suppressed, top1Δ unaffected) | Same (rad51Δ unaffected, top1Δ unaffected) | Increased (rad51Δ unaffected, top1Δ unaffected) | 1, 2, 3 |
| top3Δ | Strong | NDc | Same | 2 |
| xrs2Δ | Strong | Increased | NDc | 1, 3 |
| asf1Δ | Moderate (rad51Δ enhanced) | Increased (rad51Δ enhanced) | Increased (rad51Δ unaffected) | — |
| esc4Δ | Moderate | Same | Increased | — |
| ctf4Δ | Weak | Same | ND | 1, 2 |
| mus81Δ | Weak | ND | Increased | 2 |
| rad51Δ | Weak | Same | Same | 1, 2 |
| cac1Δ | None | ND | Reduced | — |
| exo1Δ | None | ND | Reduced | — |
| pol30-119 | None | ND | Increased | — |
| siz1Δ | None | ND | Increased | — |
| tel1Δ | None | ND | Reduced | — |
Boldface shows those cases where a genetic interaction was observed. ND, not determined.
1, interaction with rnh201Δ; 2, interaction with rnh202Δ; 3, interaction with rnh203Δ; —, no data reported in Biogrid.
Interaction was not determined for the specified mutation as a mutation in another gene encoding a subunit in the same protein complex was tested.
The rad51Δ-mediated suppression of the slow-growth phenotype of specific rnh203Δ double mutants indicates that the loss of RNase H2 activity in these mutants causes increased DNA damage during DNA replication, leading to the formation of aberrant DNA structures (44, 55, 65, 75). Defects in HR, sister chromatid HR and end resection (rad52Δ, mre11Δ, rad50Δ, xrs2Δ, esc2Δ, smc6, and potentially, esc4Δ/rtt107Δ and ctf4Δ) (85–88), and resolution of branched HR intermediates (mus81Δ, top3Δ, sgs1Δ, mms4Δ, and mph1Δ) (61, 69) all caused slow-growth interactions with RNase H2 defects. The weak or no interaction observed between an rnh203Δ mutation and rad51Δ and rad59Δ mutations, respectively, compared to the strong interaction with a rad52Δ mutation suggest that the Rad51 and Rad59 HR subpathways are redundant in the repair of the DNA damage caused by RNase H2 defects (89). The interactions between RNase H2 defects and defects in the Rad5-dependent error-free branch of PRR (rad5Δ, rad6Δ; lack of an interaction with bre1Δ [83, 90–92]), which in vitro can regress stalled replication forks (93), is consistent with the idea that aberrant DNA structures causing slow-growth phenotypes are generated by DNA replication of templates containing single-strand breaks and other types of damage. Consistent with this, a prior study demonstrated that rnh1Δ rnh201Δ double mutant cells exhibit high levels of constitutive mono- and polyubiquitylated PCNA, indicative of chronic PRR activation in the absence of RNase H activity (72). Interactions between an rnh203Δ mutation and mutations in RAD27, DNA2, RNH1, and potentially, MGS1 are consistent with either the production of rnh203Δ-dependent strand breaks due to errors in Okazaki fragment maturation or partial redundancy in pathways that process misincorporated ribonucleotides and R-loops (2–4, 7, 48, 49, 57, 72). The fact that an rnh203Δ mutation caused a growth defect when combined with a RAD53 deletion but not with deletions of the MEC1 or TEL1 genes encoding upstream checkpoint components is consistent with known redundancies between the checkpoint kinases (for a discussion, see reference 94). Similarly, the lack of interactions with mutations affecting upstream signaling components suggests that multiple checkpoint pathways may detect the DNA damage that occurs as a result of RNase H2 defects.
Only a subset of the slow-growing rnh203Δ double mutants accumulated cells with an aberrant morphology. The aberrant cell morphology seen is consistent with protracted checkpoint activation or uncoupling of the morphology checkpoint from cell cycle delay caused by DNA damage during DNA replication or defects in DNA replication (56, 64, 73, 74). Mutations affecting HR, including sister chromatid HR and processing of HR intermediates (rad52Δ, mre11Δ, rad50Δ, xrs2Δ, esc2Δ, and mph1Δ) caused aberrant morphology when combined with the rnh203Δ mutation. This observation suggests that HR and HR-related processes likely convert the rnh203Δ-dependent DNA damage into products that constitutively activate DNA damage signaling. In the case of the asf1Δ rnh203Δ double mutant, it is possible that the failure to appropriately recover from checkpoint activation (95, 96) results in uncoupling of the morphology checkpoint and increased levels of cells with aberrant morphology. The double mutants that have slow-growth phenotypes but normal or largely normal morphology might have increased but not persistent DNA damage, which in some cases can result in increased GCR rates (sgs1Δ, esc4Δ).
Additionally, only some mutations causing a growth defect in combination with the rnh203Δ mutation also resulted in a synergistic increase in GCR rates. The observation that mre11Δ, rad50Δ, and xrs2Δ mutations but not rad51Δ and rad52Δ mutations caused increased GCR rates when combined with an rnh203Δ mutation could reflect the role of Mre11-Rad50-Xrs2 but not Rad51 or Rad52 in stabilizing and processing stalled replication forks (97, 98). Similarly, Sgs1, Esc2, Esc4/Rtt107, the Mus81-Mms4 complex, and the sumoylation of PCNA, which is blocked by the pol30-119 mutation, play roles in the processing of stalled replication forks (69, 76, 80, 87). These observations support the idea that, while RNase H2 defects cause increased levels of DNA damage that is repaired by a variety of pathways, defects in processing of stalled replication forks are likely the primary source of the increased GCRs seen in RNase H2 double mutants.
The rad51Δ mutation suppressed the growth defects of the rnh203Δ esc2Δ, rnh203Δ sgs1Δ, and rnh203Δ mph1Δ double mutants. This suppression is reminiscent of the ability of a rad51Δ mutation to suppress growth defects caused by toxic HR intermediates that accumulate when mutations in the SGS1, RRM3, and SRS2 genes are combined (65, 75). Consistent with the effect of the rad51Δ mutation, Esc2 and Sgs1 act in parallel pathways that prevent the accumulation of toxic Rad51-dependent HR intermediates (87, 99). Furthermore, Mph1 has been shown to act in a redundant pathway that produces toxic HR intermediates during DNA replication of esc2Δ mutants in the presence of methyl methane sulfonate (100). The effect of the rad51Δ mutation suggests that defects in RNase H2 could increase the level of the DNA damage that is processed by Esc2, Sgs1, and Mph1. However, the rad51Δ mutation had differing effects on the increases in aberrant cell morphology and GCR rates in the rnh203Δ esc2Δ, rnh203Δ sgs1Δ, and rnh203Δ mph1Δ double mutants. In general, all of the phenotypes of the rnh203Δ esc2Δ and rnh203Δ mph1Δ double mutants were suppressed by the rad51Δ mutation, with the caveat that the rnh203Δ mph1Δ double mutants did not have an increased GCR rate in the GCR assay used here. In contrast, the aberrant cell morphology and increased GCR rate of the rnh203Δ sgs1Δ double mutant were not suppressed by the rad51Δ mutation, which could reflect roles of Sgs1 in preventing the formation of HR intermediates (101), as opposed to roles of Sgs1, Mph1, and Esc2 in later processing steps (87, 99, 100).
The rnh203Δ asf1Δ double mutant differed substantially from the rnh203Δ esc2Δ, rnh203Δ sgs1Δ, and rnh203Δ mph1Δ double mutants. The rad51Δ mutation exacerbated the growth defect of the rnh203Δ asf1Δ double mutant but did not increase the GCR rate or the fraction of cells with aberrant morphology. These results, in combination with the effect of the rad51Δ mutation on the growth rates of the rnh203Δ and asf1Δ single mutants, are consistent with the possibility that defects in both RNH203 and Asf1-dependent chromatin assembly (56) independently cause DNA damage where the repair of asf1Δ-induced DNA damage but not the repair of the rnh203Δ-dependent DNA damage is normally mediated by Rad51-dependent HR. This conclusion is consistent with the effect of HR-defective mutations on the growth defect of an rnh203Δ mus81Δ double mutant (44).
A critical question is what type of DNA damage caused by RNase H2 defects leads to the slow growth, aberrant cell morphology, and/or increased GCR phenotypes of the rnh203Δ double mutants. One possibility is that defects in RNase H1 and H2 can lead to the accumulation of transcriptional intermediates that lead to double-strand breaks and genome instability, which can be suppressed by overexpression of RNase H1 (49, 57, 102). However, overexpression of RNase H1 did not suppress the increased genome instability caused by HR and repair defects (57), suggesting that the accumulation of transcriptional intermediates cannot account for the results obtained here. RNase H2 defects can also lead to a failure to remove misincorporated ribonucleotides from DNA, resulting in dinucleotide instability induced by the cleavage of misincorporated ribonucleotides by topoisomerase 1 (30). In the case of the rnh203Δ esc2Δ double mutant, our results are consistent with the idea that misincorporated ribonucleotides due to RNase H2 defects that are cleaved by topoisomerase 1 can lead to increased rates of accumulating GCRs and aberrant cell morphology. In contrast, deletion of TOP1 had no effect on the rnh203Δ sgs1Δ double mutant, suggesting that inefficient Okazaki fragment processing underlies this genetic interaction. This conclusion seems likely given the observed genetic interaction between defects in RNase H2 and Rad27/FEN1 (7, 20–23), a key endonuclease that functions in Okazaki fragment processing and suppression of GCRs. However, it should be noted that there are parallels between removing ribonucleotides at the end of an Okazaki fragment and from DNA once a strand break is made at the site of a misincorporated ribonucleotide (26). Genetic and enzymatic studies have shown that RNase H2 is not absolutely required for Okazaki fragment processing, but rather, it improves the efficiency of cleavage of the 5′ ends of Okazaki fragments (103). This suggests that even a subtle effect, such as a change in the kinetics of this reaction, is sufficient to trigger a requirement of other pathways to prevent aberrant cell growth and genome instability.
Previous studies, including qualitative genome-wide screens, identified 11 genes in which mutations caused a growth defect when combined with an RNase H2 defect (6 with rnh203Δ) (20–24, 44, 58–63). Our studies, using a targeted hypothesis-driven approach that permitted more detailed growth analysis, identified an additional 8 genes in which mutations cause a growth interaction with an RNase H2 defect, in this case an rnh203Δ mutation. A unique feature of our screen was the analysis of GCR rates in the double mutants, allowing us to identify at least 8 genes in which mutations caused synergistic increases in GCR rates in combination with RNase H2 defects. These results indicate that the levels of DNA damage in RNase H2-defective strains are much higher than might be predicted from the weak phenotypes caused by individual RNase H2 defects and that this damage is compensated for by other pathways. This might explain how an apparently weak defect may underlie a disease like Aicardi-Goutieres syndrome; the level of DNA damage caused by the absence of RNase H2 might be high enough to cause an autoimmune or autoinflammatory response (10, 47) and the more deleterious effects of this DNA damage might be suppressed by other pathways, as demonstrated here. This finding also suggests that performing a broad genetic screen for defects that, in combination with RNase H2 defects, cause a synergistic increase in GCR rates in different assays and subsequent analysis of suppressors of the increased GCR rates (e.g., rad51Δ, top1Δ, and other mutations) would facilitate the identification of the types of DNA damage that occur in RNase H2-defective strains and the pathways that compensate for this damage.
ACKNOWLEDGMENTS
We thank members of the Kolodner laboratory for helpful discussions, Nikki Bowen for help with some of the growth rate experiments, and Karen Arden and Anjana Srivatsan for helpful comments on the manuscript.
This work was supported by NIH grant GM26017 to R.D.K.
SUPPLEMENTAL MATERIAL
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00960-13.
REFERENCES
- 1.Waga S, Stillman B. 1994. Anatomy of a DNA replication fork revealed by reconstitution of SV40 DNA replication in vitro. Nature 369:207–212. [DOI] [PubMed] [Google Scholar]
- 2.Zheng L, Shen B. 2011. Okazaki fragment maturation: nucleases take centre stage. J. Mol. Cell Biol. 3:23–30. 10.1093/jmcb/mjq048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bae SH, Bae KH, Kim JA, Seo YS. 2001. RPA governs endonuclease switching during processing of Okazaki fragments in eukaryotes. Nature 412:456–461. 10.1038/35086609. [DOI] [PubMed] [Google Scholar]
- 4.MacNeill SA. 2001. DNA replication: partners in the Okazaki two-step. Curr. Biol. 11:R842–R844. 10.1016/S0960-9822(01)00500-0. [DOI] [PubMed] [Google Scholar]
- 5.Huang L, Kim Y, Turchi JJ, Bambara RA. 1994. Structure-specific cleavage of the RNA primer from Okazaki fragments by calf thymus RNase HI. J. Biol. Chem. 269:25922–25927. [PubMed] [Google Scholar]
- 6.Murante RS, Henricksen LA, Bambara RA. 1998. Junction ribonuclease: an activity in Okazaki fragment processing. Proc. Natl. Acad. Sci. U. S. A. 95:2244–2249. 10.1073/pnas.95.5.2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qiu J, Qian Y, Frank P, Wintersberger U, Shen B. 1999. Saccharomyces cerevisiae RNase H(35) functions in RNA primer removal during lagging-strand DNA synthesis, most efficiently in cooperation with Rad27 nuclease. Mol. Cell. Biol. 19:8361–8371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turchi JJ, Huang L, Murante RS, Kim Y, Bambara RA. 1994. Enzymatic completion of mammalian lagging-strand DNA replication. Proc. Natl. Acad. Sci. U. S. A. 91:9803–9807. 10.1073/pnas.91.21.9803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jeong HS, Backlund PS, Chen HC, Karavanov AA, Crouch RJ. 2004. RNase H2 of Saccharomyces cerevisiae is a complex of three proteins. Nucleic Acids Res. 32:407–414. 10.1093/nar/gkh209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E, Ali M, Semple C, Aicardi J, Babul-Hirji R, Baumann C, Baxter P, Bertini E, Chandler KE, Chitayat D, Cau D, Dery C, Fazzi E, Goizet C, King MD, Klepper J, Lacombe D, Lanzi G, Lyall H, Martinez-Frias ML, Mathieu M, McKeown C, Monier A, Oade Y, Quarrell OW, Rittey CD, Rogers RC, Sanchis A, Stephenson JB, Tacke U, Till M, Tolmie JL, Tomlin P, Voit T, Weschke B, Woods CG, Lebon P, Bonthron DT, Ponting CP, Jackson AP. 2006. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutieres syndrome and mimic congenital viral brain infection. Nat. Genet. 38:910–916. 10.1038/ng1842. [DOI] [PubMed] [Google Scholar]
- 11.Flores-Rozas H, Kolodner RD. 2000. Links between replication, recombination and genome instability in eukaryotes. Trends Biochem. Sci. 25:196–200. 10.1016/S0968-0004(00)01568-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goulian M, Richards SH, Heard CJ, Bigsby BM. 1990. Discontinuous DNA synthesis by purified mammalian proteins. J. Biol. Chem. 265:18461–18471. [PubMed] [Google Scholar]
- 13.Kao HI, Bambara RA. 2003. The protein components and mechanism of eukaryotic Okazaki fragment maturation. Crit. Rev. Biochem. Mol. Biol. 38:433–452. 10.1080/10409230390259382. [DOI] [PubMed] [Google Scholar]
- 14.Burgers PM. 2009. Polymerase dynamics at the eukaryotic DNA replication fork. J. Biol. Chem. 284:4041–4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henry RA, Balakrishnan L, Ying-Lin ST, Campbell JL, Bambara RA. 2010. Components of the secondary pathway stimulate the primary pathway of eukaryotic Okazaki fragment processing. J. Biol. Chem. 285:28496–28505. 10.1074/jbc.M110.131870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rossi ML, Bambara RA. 2006. Reconstituted Okazaki fragment processing indicates two pathways of primer removal. J. Biol. Chem. 281:26051–26061. 10.1074/jbc.M604805200. [DOI] [PubMed] [Google Scholar]
- 17.Tishkoff DX, Filosi N, Gaida GM, Kolodner RD. 1997. A novel mutation avoidance mechanism dependent on S. cerevisiae RAD27 is distinct from DNA mismatch repair. Cell 88:253–263. 10.1016/S0092-8674(00)81846-2. [DOI] [PubMed] [Google Scholar]
- 18.Tran PT, Erdeniz N, Dudley S, Liskay RM. 2002. Characterization of nuclease-dependent functions of Exo1p in Saccharomyces cerevisiae. DNA Repair 1:895–912. 10.1016/S1568-7864(02)00114-3. [DOI] [PubMed] [Google Scholar]
- 19.Frank P, Braunshofer-Reiter C, Wintersberger U. 1998. Yeast RNase H(35) is the counterpart of the mammalian RNase HI, and is evolutionarily related to prokaryotic RNase HII. FEBS Lett. 421:23–26. 10.1016/S0014-5793(97)01528-7. [DOI] [PubMed] [Google Scholar]
- 20.Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JL, Toufighi K, Mostafavi S, Prinz J, St Onge RP, VanderSluis B, Makhnevych T, Vizeacoumar FJ, Alizadeh S, Bahr S, Brost RL, Chen Y, Cokol M, Deshpande R, Li Z, Lin ZY, Liang W, Marback M, Paw J, San Luis BJ, Shuteriqi E, Tong AH, van Dyk N, Wallace IM, Whitney JA, Weirauch MT, Zhong G, Zhu H, Houry WA, Brudno M, Ragibizadeh S, Papp B, Pal C, Roth FP, Giaever G, Nislow C, Troyanskaya OG, Bussey H, Bader GD, Gingras AC, Morris QD, Kim PM, Kaiser CA, Myers CL, Andrews BJ, Boone C. 2010. The genetic landscape of a cell. Science 327:425–431. 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dixon SJ, Fedyshyn Y, Koh JL, Prasad TS, Chahwan C, Chua G, Toufighi K, Baryshnikova A, Hayles J, Hoe KL, Kim DU, Park HO, Myers CL, Pandey A, Durocher D, Andrews BJ, Boone C. 2008. Significant conservation of synthetic lethal genetic interaction networks between distantly related eukaryotes. Proc. Natl. Acad. Sci. U. S. A. 105:16653–16658. 10.1073/pnas.0806261105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loeillet S, Palancade B, Cartron M, Thierry A, Richard GF, Dujon B, Doye V, Nicolas A. 2005. Genetic network interactions among replication, repair and nuclear pore deficiencies in yeast. DNA Rep. 4:459–468. 10.1016/j.dnarep.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 23.Pan X, Ye P, Yuan DS, Wang X, Bader JS, Boeke JD. 2006. A DNA integrity network in the yeast Saccharomyces cerevisiae. Cell 124:1069–1081. 10.1016/j.cell.2005.12.036. [DOI] [PubMed] [Google Scholar]
- 24.Chon H, Sparks JL, Rychlik M, Nowotny M, Burgers PM, Crouch RJ, Cerritelli SM. 2013. RNase H2 roles in genome integrity revealed by unlinking its activities. Nucleic Acids Res. 41:3130–3143. 10.1093/nar/gkt027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nick McElhinny SA, Watts BE, Kumar D, Watt DL, Lundstrom EB, Burgers PM, Johansson E, Chabes A, Kunkel TA. 2010. Abundant ribonucleotide incorporation into DNA by yeast replicative polymerases. Proc. Natl. Acad. Sci. U. S. A. 107:4949–4954. 10.1073/pnas.0914857107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sparks JL, Chon H, Cerritelli SM, Kunkel TA, Johansson E, Crouch RJ, Burgers PM. 2012. RNase H2-initiated ribonucleotide excision repair. Mol. Cell 47:980–986. 10.1016/j.molcel.2012.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arudchandran A, Cerritelli S, Narimatsu S, Itaya M, Shin DY, Shimada Y, Crouch RJ. 2000. The absence of ribonuclease H1 or H2 alters the sensitivity of Saccharomyces cerevisiae to hydroxyurea, caffeine and ethyl methanesulphonate: implications for roles of RNases H in DNA replication and repair. Genes Cells 5:789–802. 10.1046/j.1365-2443.2000.00373.x. [DOI] [PubMed] [Google Scholar]
- 28.Cerritelli SM, Crouch RJ. 2009. Ribonuclease H: the enzymes in eukaryotes. FEBS J. 276:1494–1505. 10.1111/j.1742-4658.2009.06908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stith CM, Sterling J, Resnick MA, Gordenin DA, Burgers PM. 2008. Flexibility of eukaryotic Okazaki fragment maturation through regulated strand displacement synthesis. J. Biol. Chem. 283:34129–34140. 10.1074/jbc.M806668200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim N, Huang SN, Williams JS, Li YC, Clark AB, Cho JE, Kunkel TA, Pommier Y, Jinks-Robertson S. 2011. Mutagenic processing of ribonucleotides in DNA by yeast topoisomerase I. Science 332:1561–1564. 10.1126/science.1205016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sekiguchi J, Shuman S. 1997. Site-specific ribonuclease activity of eukaryotic DNA topoisomerase I. Mol. Cell 1:89–97. 10.1016/S1097-2765(00)80010-6. [DOI] [PubMed] [Google Scholar]
- 32.Kolodner RD, Putnam CD, Myung K. 2002. Maintenance of genome stability in Saccharomyces cerevisiae. Science 297:552–557. 10.1126/science.1075277. [DOI] [PubMed] [Google Scholar]
- 33.Budd ME, Reis CC, Smith S, Myung K, Campbell JL. 2006. Evidence suggesting that Pif1 helicase functions in DNA replication with the Dna2 helicase/nuclease and DNA polymerase delta. Mol. Cell. Biol. 26:2490–2500. 10.1128/MCB.26.7.2490-2500.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen C, Kolodner RD. 1999. Gross chromosomal rearrangements in Saccharomyces cerevisiae replication and recombination defective mutants. Nat. Genet. 23:81–85. 10.1038/12687. [DOI] [PubMed] [Google Scholar]
- 35.Greene AL, Snipe JR, Gordenin DA, Resnick MA. 1999. Functional analysis of human FEN1 in Saccharomyces cerevisiae and its role in genome stability. Hum. Mol. Genet. 8:2263–2273. 10.1093/hmg/8.12.2263. [DOI] [PubMed] [Google Scholar]
- 36.Kang MJ, Lee CH, Kang YH, Cho IT, Nguyen TA, Seo YS. 2010. Genetic and functional interactions between Mus81-Mms4 and Rad27. Nucleic Acids Res. 38:7611–7625. 10.1093/nar/gkq651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kang YH, Lee CH, Seo YS. 2010. Dna2 on the road to Okazaki fragment processing and genome stability in eukaryotes. Crit. Rev. Biochem. Mol. Biol. 45:71–96. 10.3109/10409230903578593. [DOI] [PubMed] [Google Scholar]
- 38.Liu Y, Zhang H, Veeraraghavan J, Bambara RA, Freudenreich CH. 2004. Saccharomyces cerevisiae flap endonuclease 1 uses flap equilibration to maintain triplet repeat stability. Mol. Cell. Biol. 24:4049–4064. 10.1128/MCB.24.9.4049-4064.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Myung K, Chen C, Kolodner RD. 2001. Multiple pathways cooperate in the suppression of genome instability in Saccharomyces cerevisiae. Nature 411:1073–1076. 10.1038/35082608. [DOI] [PubMed] [Google Scholar]
- 40.Reijns MA, Rabe B, Rigby RE, Mill P, Astell KR, Lettice LA, Boyle S, Leitch A, Keighren M, Kilanowski F, Devenney PS, Sexton D, Grimes G, Holt IJ, Hill RE, Taylor MS, Lawson KA, Dorin JR, Jackson AP. 2012. Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell 149:1008–1022. 10.1016/j.cell.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Budd ME, Tong AH, Polaczek P, Peng X, Boone C, Campbell JL. 2005. A network of multi-tasking proteins at the DNA replication fork preserves genome stability. PLoS Genet. 1:e61. 10.1371/journal.pgen.0010061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ghodgaonkar MM, Lazzaro F, Olivera-Pimentel M, Artola-Boran M, Cejka P, Reijns MA, Jackson AP, Plevani P, Muzi-Falconi M, Jiricny J. 2013. Ribonucleotides misincorporated into DNA act as strand-discrimination signals in eukaryotic mismatch repair. Mol. Cell 50:323–332. 10.1016/j.molcel.2013.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang ME, Rio AG, Nicolas A, Kolodner RD. 2003. A genomewide screen in Saccharomyces cerevisiae for genes that suppress the accumulation of mutations. Proc. Natl. Acad. Sci. U. S. A. 100:11529–11534. 10.1073/pnas.2035018100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ii M, Ii T, Mironova LI, Brill SJ. 2011. Epistasis analysis between homologous recombination genes in Saccharomyces cerevisiae identifies multiple repair pathways for Sgs1, Mus81-Mms4 and RNase H2. Mut. Res. 714:33–43. 10.1016/j.mrfmmm.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lujan SA, Williams JS, Clausen AR, Clark AB, Kunkel TA. 2013. Ribonucleotides are signals for mismatch repair of leading-strand replication errors. Mol. Cell 50:437–443. 10.1016/j.molcel.2013.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hiller B, Achleitner M, Glage S, Naumann R, Behrendt R, Roers A. 2012. Mammalian RNase H2 removes ribonucleotides from DNA to maintain genome integrity. J. Exp. Med. 209:1419–1426. 10.1084/jem.20120876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, Black DN, van Bokhoven H, Brunner HG, Hamel BC, Corry PC, Cowan FM, Frints SG, Klepper J, Livingston JH, Lynch SA, Massey RF, Meritet JF, Michaud JL, Ponsot G, Voit T, Lebon P, Bonthron DT, Jackson AP, Barnes DE, Lindahl T. 2006. Mutations in the gene encoding the 3′–5′ DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat. Genet. 38:917–920. 10.1038/ng1845. [DOI] [PubMed] [Google Scholar]
- 48.Aguilera A, Garcia-Muse T. 2012. R loops: from transcription byproducts to threats to genome stability. Mol. Cell 46:115–124. 10.1016/j.molcel.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 49.Huertas P, Aguilera A. 2003. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol. Cell 12:711–721. 10.1016/j.molcel.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 50.Marsischky GT, Filosi N, Kane MF, Kolodner R. 1996. Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair. Genes Dev. 10:407–420. 10.1101/gad.10.4.407. [DOI] [PubMed] [Google Scholar]
- 51.Chen C, Umezu K, Kolodner RD. 1998. Chromosomal rearrangements occur in S. cerevisiae rfa1 mutator mutants due to mutagenic lesions processed by double-strand-break repair. Mol. Cell 2:9–22. 10.1016/S1097-2765(00)80109-4. [DOI] [PubMed] [Google Scholar]
- 52.Das Gupta R, Kolodner RD. 2000. Novel dominant mutations in Saccharomyces cerevisiae MSH6. Nat. Genet. 24:53–56. 10.1038/71684. [DOI] [PubMed] [Google Scholar]
- 53.Flores-Rozas H, Kolodner RD. 1998. The Saccharomyces cerevisiae MLH3 gene functions in MSH3-dependent suppression of frameshift mutations. Proc. Natl. Acad. Sci. U. S. A. 95:12404–12409. 10.1073/pnas.95.21.12404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Harrington JM, Kolodner RD. 2007. Saccharomyces cerevisiae Msh2-Msh3 acts in repair of base-base mispairs. Mol. Cell. Biol. 27:6546–6554. 10.1128/MCB.00855-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schmidt KH, Kolodner RD. 2004. Requirement of Rrm3 helicase for repair of spontaneous DNA lesions in cells lacking Srs2 or Sgs1 helicase. Mol. Cell. Biol. 24:3213–3226. 10.1128/MCB.24.8.3213-3226.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kats ES, Albuquerque CP, Zhou H, Kolodner RD. 2006. Checkpoint functions are required for normal S-phase progression in Saccharomyces cerevisiae RCAF- and CAF-I-defective mutants. Proc. Natl. Acad. Sci. U. S. A. 103:3710–3715. 10.1073/pnas.0511102103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wahba L, Amon JD, Koshland D, Vuica-Ross M. 2011. RNase H and multiple RNA biogenesis factors cooperate to prevent RNA:DNA hybrids from generating genome instability. Mol. Cell 44:978–988. 10.1016/j.molcel.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Collins SR, Miller KM, Maas NL, Roguev A, Fillingham J, Chu CS, Schuldiner M, Gebbia M, Recht J, Shales M, Ding H, Xu H, Han J, Ingvarsdottir K, Cheng B, Andrews B, Boone C, Berger SL, Hieter P, Zhang Z, Brown GW, Ingles CJ, Emili A, Allis CD, Toczyski DP, Weissman JS, Greenblatt JF, Krogan NJ. 2007. Functional dissection of protein complexes involved in yeast chromosome biology using a genetic interaction map. Nature 446:806–810. 10.1038/nature05649. [DOI] [PubMed] [Google Scholar]
- 59.Decourty L, Saveanu C, Zemam K, Hantraye F, Frachon E, Rousselle JC, Fromont-Racine M, Jacquier A. 2008. Linking functionally related genes by sensitive and quantitative characterization of genetic interaction profiles. Proc. Natl. Acad. Sci. U. S. A. 105:5821–5826. 10.1073/pnas.0710533105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hegnauer AM, Hustedt N, Shimada K, Pike BL, Vogel M, Amsler P, Rubin SM, van Leeuwen F, Guenole A, van Attikum H, Thoma NH, Gasser SM. 2012. An N-terminal acidic region of Sgs1 interacts with Rpa70 and recruits Rad53 kinase to stalled forks. EMBO J. 31:3768–3783. 10.1038/emboj.2012.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ii M, Brill SJ. 2005. Roles of SGS1, MUS81, and RAD51 in the repair of lagging-strand replication defects in Saccharomyces cerevisiae. Curr. Genet. 48:213–225. 10.1007/s00294-005-0014-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tong AH, Lesage G, Bader GD, Ding H, Xu H, Xin X, Young J, Berriz GF, Brost RL, Chang M, Chen Y, Cheng X, Chua G, Friesen H, Goldberg DS, Haynes J, Humphries C, He G, Hussein S, Ke L, Krogan N, Li Z, Levinson JN, Lu H, Menard P, Munyana C, Parsons AB, Ryan O, Tonikian R, Roberts T, Sdicu AM, Shapiro J, Sheikh B, Suter B, Wong SL, Zhang LV, Zhu H, Burd CG, Munro S, Sander C, Rine J, Greenblatt J, Peter M, Bretscher A, Bell G, Roth FP, Brown GW, Andrews B, Bussey H, Boone C. 2004. Global mapping of the yeast genetic interaction network. Science 303:808–813. 10.1126/science.1091317. [DOI] [PubMed] [Google Scholar]
- 63.Wilmes GM, Bergkessel M, Bandyopadhyay S, Shales M, Braberg H, Cagney G, Collins SR, Whitworth GB, Kress TL, Weissman JS, Ideker T, Guthrie C, Krogan NJ. 2008. A genetic interaction map of RNA-processing factors reveals links between Sem1/Dss1-containing complexes and mRNA export and splicing. Mol. Cell 32:735–746. 10.1016/j.molcel.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Enserink JM, Smolka MB, Zhou H, Kolodner RD. 2006. Checkpoint proteins control morphogenetic events during DNA replication stress in Saccharomyces cerevisiae. J. Cell Biol. 175:729–741. 10.1083/jcb.200605080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fabre F, Chan A, Heyer WD, Gangloff S. 2002. Alternate pathways involving Sgs1/Top3, Mus81/ Mms4, and Srs2 prevent formation of toxic recombination intermediates from single-stranded gaps created by DNA replication. Proc. Natl. Acad. Sci. U. S. A. 99:16887–16892. 10.1073/pnas.252652399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hishida T, Ohya T, Kubota Y, Kamada Y, Shinagawa H. 2006. Functional and physical interaction of yeast Mgs1 with PCNA: impact on RAD6-dependent DNA damage tolerance. Mol. Cell. Biol. 26:5509–5517. 10.1128/MCB.00307-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kaufman PD, Cohen JL, Osley MA. 1998. Hir proteins are required for position-dependent gene silencing in Saccharomyces cerevisiae in the absence of chromatin assembly factor I. Mol. Cell. Biol. 18:4793–4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ooi SL, Shoemaker DD, Boeke JD. 2003. DNA helicase gene interaction network defined using synthetic lethality analyzed by microarray. Nat. Genet. 35:277–286. 10.1038/ng1258. [DOI] [PubMed] [Google Scholar]
- 69.Schmidt KH, Kolodner RD. 2006. Suppression of spontaneous genome rearrangements in yeast DNA helicase mutants. Proc. Natl. Acad. Sci. U. S. A. 103:18196–18201. 10.1073/pnas.0608566103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shor E, Gangloff S, Wagner M, Weinstein J, Price G, Rothstein R. 2002. Mutations in homologous recombination genes rescue top3 slow growth in Saccharomyces cerevisiae. Genetics 162:647–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vance JR, Wilson TE. 2002. Yeast Tdp1 and Rad1-Rad10 function as redundant pathways for repairing Top1 replicative damage. Proc. Natl. Acad. Sci. U. S. A. 99:13669–13674. 10.1073/pnas.202242599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lazzaro F, Novarina D, Amara F, Watt DL, Stone JE, Costanzo V, Burgers PM, Kunkel TA, Plevani P, Muzi-Falconi M. 2012. RNase H and postreplication repair protect cells from ribonucleotides incorporated in DNA. Mol. Cell 45:99–110. 10.1016/j.molcel.2011.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Heyer WD, Rao MR, Erdile LF, Kelly TJ, Kolodner RD. 1990. An essential Saccharomyces cerevisiae single-stranded DNA binding protein is homologous to the large subunit of human RP-A. EMBO J. 9:2321–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zettel MF, Garza LR, Cass AM, Myhre RA, Haizlip LA, Osadebe SN, Sudimack DW, Pathak R, Stone TL, Polymenis M. 2003. The budding index of Saccharomyces cerevisiae deletion strains identifies genes important for cell cycle progression. FEMS Microbiol. Lett. 223:253–258. 10.1016/S0378-1097(03)00384-7. [DOI] [PubMed] [Google Scholar]
- 75.Gangloff S, Soustelle C, Fabre F. 2000. Homologous recombination is responsible for cell death in the absence of the Sgs1 and Srs2 helicases. Nat. Genet. 25:192–194. 10.1038/76055. [DOI] [PubMed] [Google Scholar]
- 76.Torres JZ, Schnakenberg SL, Zakian VA. 2004. Saccharomyces cerevisiae Rrm3p DNA helicase promotes genome integrity by preventing replication fork stalling: viability of rrm3 cells requires the intra-S-phase checkpoint and fork restart activities. Mol. Cell. Biol. 24:3198–3212. 10.1128/MCB.24.8.3198-3212.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Myung K, Datta A, Kolodner RD. 2001. Suppression of spontaneous chromosomal rearrangements by S phase checkpoint functions in Saccharomyces cerevisiae. Cell 104:397–408. 10.1016/S0092-8674(01)00227-6. [DOI] [PubMed] [Google Scholar]
- 78.Tyler JK, Adams CR, Chen SR, Kobayashi R, Kamakaka RT, Kadonaga JT. 1999. The RCAF complex mediates chromatin assembly during DNA replication and repair. Nature 402:555–560. 10.1038/990147. [DOI] [PubMed] [Google Scholar]
- 79.Bai Y, Davis AP, Symington LS. 1999. A novel allele of RAD52 that causes severe DNA repair and recombination deficiencies only in the absence of RAD51 or RAD59. Genetics 153:1117–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chin JK, Bashkirov VI, Heyer WD, Romesberg FE. 2006. Esc4/Rtt107 and the control of recombination during replication. DNA Repair 5:618–628. 10.1016/j.dnarep.2006.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mazon G, Lam AF, Ho CK, Kupiec M, Symington LS. 2012. The Rad1-Rad10 nuclease promotes chromosome translocations between dispersed repeats. Nat. Struct. Mol. Biol. 19:964–971. 10.1038/nsmb.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mimitou EP, Symington LS. 2008. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 455:770–774. 10.1038/nature07312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Putnam CD, Hayes TK, Kolodner RD. 2010. Post-replication repair suppresses duplication-mediated genome instability. PLoS Genet. 6:e1000933. 10.1371/journal.pgen.1000933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S. 2002. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 419:135–141. 10.1038/nature00991. [DOI] [PubMed] [Google Scholar]
- 85.Hwang JY, Smith S, Ceschia A, Torres-Rosell J, Aragon L, Myung K. 2008. Smc5-Smc6 complex suppresses gross chromosomal rearrangements mediated by break-induced replications. DNA Repair 7:1426–1436. 10.1016/j.dnarep.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Paull TT. 2010. Making the best of the loose ends: Mre11/Rad50 complexes and Sae2 promote DNA double-strand break resection. DNA Repair 9:1283–1291. 10.1016/j.dnarep.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sollier J, Driscoll R, Castellucci F, Foiani M, Jackson SP, Branzei D. 2009. The Saccharomyces cerevisiae Esc2 and Smc5-6 proteins promote sister chromatid junction-mediated intra-S repair. Mol. Biol. Cell 20:1671–1682. 10.1091/mbc.E08-08-0875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ullal P, Vilella-Mitjana F, Jarmuz A, Aragon L. 2011. Rtt107 phosphorylation promotes localisation to DNA double-stranded breaks (DSBs) and recombinational repair between sister chromatids. PLoS One 6:e20152. 10.1371/journal.pone.0020152. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 89.Krogh BO, Symington LS. 2004. Recombination proteins in yeast. Annu. Rev. Genet. 38:233–271. 10.1146/annurev.genet.38.072902.091500. [DOI] [PubMed] [Google Scholar]
- 90.Hwang JY, Smith S, Myung K. 2005. The Rad1-Rad10 complex promotes the production of gross chromosomal rearrangements from spontaneous DNA damage in Saccharomyces cerevisiae. Genetics 169:1927–1937. 10.1534/genetics.104.039768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kats ES, Enserink JM, Martinez S, Kolodner RD. 2009. The Saccharomyces cerevisiae Rad6 postreplication repair and Siz1/Srs2 homologous recombination-inhibiting pathways process DNA damage that arises in asf1 mutants. Mol. Cell. Biol. 29:5226–5237. 10.1128/MCB.00894-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Myung K, Smith S. 2008. The RAD5-dependent postreplication repair pathway is important to suppress gross chromosomal rearrangements. J. Nat. Cancer Inst. Monogr. 2008(39):12–15. 10.1093/jncimonographs/lgn019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Blastyak A, Pinter L, Unk I, Prakash L, Prakash S, Haracska L. 2007. Yeast Rad5 protein required for postreplication repair has a DNA helicase activity specific for replication fork regression. Mol. Cell 28:167–175. 10.1016/j.molcel.2007.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jaehnig EJ, Kuo D, Hombauer H, Ideker TG, Kolodner RD. 2013. Checkpoint kinases regulate a global network of transcription factors in response to DNA damage. Cell Rep. 4:174–188. 10.1016/j.celrep.2013.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chen CC, Carson JJ, Feser J, Tamburini B, Zabaronick S, Linger J, Tyler JK. 2008. Acetylated lysine 56 on histone H3 drives chromatin assembly after repair and signals for the completion of repair. Cell 134:231–243. 10.1016/j.cell.2008.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kim JA, Haber JE. 2009. Chromatin assembly factors Asf1 and CAF-1 have overlapping roles in deactivating the DNA damage checkpoint when DNA repair is complete. Proc. Natl. Acad. Sci. U. S. A. 106:1151–1156. 10.1073/pnas.0812578106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bentsen IB, Nielsen I, Lisby M, Nielsen HB, Gupta SS, Mundbjerg K, Andersen AH, Bjergbaek L. 2013. MRX protects fork integrity at protein-DNA barriers, and its absence causes checkpoint activation dependent on chromatin context. Nucleic Acids Res. 41:3173–3189. 10.1093/nar/gkt051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mott C, Symington LS. 2011. RAD51-independent inverted-repeat recombination by a strand-annealing mechanism. DNA Repair 10:408–415. 10.1016/j.dnarep.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mankouri HW, Ngo HP, Hickson ID. 2009. Esc2 and Sgs1 act in functionally distinct branches of the homologous recombination repair pathway in Saccharomyces cerevisiae. Mol. Biol. Cell 20:1683–1694. 10.1091/mbc.E08-08-0877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Choi K, Szakal B, Chen YH, Branzei D, Zhao X. 2010. The Smc5/6 complex and Esc2 influence multiple replication-associated recombination processes in Saccharomyces cerevisiae. Mol. Biol. Cell 21:2306–2314. 10.1091/mbc.E10-01-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sugawara N, Goldfarb T, Studamire B, Alani E, Haber JE. 2004. Heteroduplex rejection during single-strand annealing requires Sgs1 helicase and mismatch repair proteins Msh2 and Msh6 but not Pms1. Proc. Natl. Acad. Sci. U. S. A. 101:9315–9320. 10.1073/pnas.0305749101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li X, Manley JL. 2005. Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell 122:365–378. 10.1016/j.cell.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 103.Bubeck D, Reijns MA, Graham SC, Astell KR, Jones EY, Jackson AP. 2011. PCNA directs type 2 RNase H activity on DNA replication and repair substrates. Nucleic Acids Res. 39:3652–3666. 10.1093/nar/gkq980. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.