

Abstract
We have recently shown that genetic replacement of VE-cadherin by a VE-cadherin–α-catenin fusion construct strongly impairs opening of endothelial cell contacts during leukocyte extravasation and induction of vascular permeability in adult mice. Here we show that this mutation leads to lethality at midgestation on a clean C57BL/6 background. Investigating the reasons for embryonic lethality, we observed a lack of fetal liver hematopoiesis and severe lymphedema but no detectable defects in blood vessel formation and remodeling. As for the hematopoiesis defect, VE-cadherin–α-catenin affected neither the generation of hematopoietic stem and progenitor cells (HSPCs) from hemogenic endothelium nor their differentiation into multiple hematopoietic lineages. Instead, HSPCs accumulated in the fetal circulation, suggesting that their entry into the fetal liver was blocked. Edema formation was caused by disturbed lymphatic vessel development. Lymphatic progenitor cells of VE-cadherin–α-catenin-expressing embryos were able to leave the cardinal vein and migrate to the site of the first lymphatic vessel formation, yet subsequently, these cells failed to form large lumenized lymphatic vessels. Thus, stabilizing endothelial cell contacts by a covalent link between VE-cadherin and α-catenin affects recruitment of hematopoietic progenitors into the fetal liver and the development of lymph but not blood vessels.
INTRODUCTION
In the embryo proper, both the hematopoietic lineage and lymphatic endothelial cells (LECs) originate from blood endothelial cells (BECs). In the aorta-gonad-mesonephros (AGM) region, BECs within the dorsal aorta act as a source of hematopoietic progenitor cells (1, 2), which are generated from this hemogenic endothelium in a process called “endothelial to hematopoietic transition” (EHT). Hematopoietic progenitor cells start to express hematopoietic markers, detach from the endothelium, and subsequently colonize the fetal liver (3–7).
Lymphatic endothelial cells (LECs) are specified in the dorsal roof of the cardinal vein (CV) (8) and after an endothelial to mesenchymal transition (EndMT) migrate away as strings of loosely connected spindle-shaped cells (9, 10). LECs reorganize dorsolaterally of the CV into the first, large lumenized lymphatic structures, from which lymphatics emerge through sprouting lymphangiogenesis (9).
The endothelial cell-specific adhesion molecule VE-cadherin is crucial for the formation of endothelial cell junctions. VE-cadherin is indispensable for the generation of a hierarchical and lumenized vasculature and the regulation of endothelial barrier function in established vessels (11, 12). The adhesive strength of VE-cadherin is critically dependent on and regulated by intracellular binding partners, the catenins. The very C terminus of VE-cadherin binds to β- and γ-catenins, which by binding to α-catenin link the whole complex to the actin cytoskeleton. This linkage is essential for proper adhesive function of VE-cadherin.
Targeted disruption of VE-cadherin severely impairs remodeling and maturation of the first primitive vascular plexus in the mouse yolk sac and embryo proper, leading to lethality at embryonic day 9.5 (E9.5) (13, 14). However, formation of a primitive vascular plexus, even in its absence, demonstrates that VE-cadherin is not required for the specification and differentiation of BECs during vasculogenesis but for vessel maintenance (15). Indeed, Carmeliet and colleagues suggested that VE-cadherin modulates vascular endothelial growth factor (VEGF)-mediated endothelial survival via complex formation with β-catenin, VEGF receptor 2 (VEGFR-2), and phosphoinositide 3-kinase (PI3-kinase) (13). Furthermore, VE-cadherin has been shown to be important during lumen formation in the developing mouse aorta (16). In contrast to this essential function during vascular development, VE-cadherin is dispensable for the generation of hematopoietic progenitor cells in the yolk sac of VE-cadherin null embryos (17).
Recently, we have generated mice that express a covalent fusion of VE-cadherin to α-catenin (VEC-α-C), which strongly enhances junctional stability (18). In these mice, vascular permeability is no longer induced by inflammatory stimuli, such as VEGF and histamine. Also, leukocyte extravasation was strongly reduced in various tissues, establishing the junctional route as a major pathway over alternative transcellular routes in both processes (18). However, it also raised the question as to what extent junctional stabilization of endothelial cells would affect embryonic vascular development.
Since in the Cdh5VEC-α-C allele, the VEC-α-C fusion genetically replaces endogenous VE-cadherin, it faithfully reflects the VE-cadherin expression pattern and level. On a mixed B6/129 genetic background, homozygous Cdh5VEC-α-C/VEC-α-C mice are viable and, although underrepresented, show no overt morphological abnormalities when raised under specific-pathogen-free conditions. Based on fluorescence recovery after photobleaching (FRAP) analysis, we showed that VEC-α-C more efficiently associated with the actin cytoskeleton in endothelial cells than endogenous VE-cadherin, providing an explanation of why VEC-α-C stabilizes endothelial junctions. Although VEC-α-C contains a VE-cadherin truncation that lacks the β-catenin binding site, VEGFR-2 signaling and Wnt-induced translocation of β-catenin to the nucleus were not altered in endothelial cells expressing VEC-α-C. Furthermore, fusion of VE-cadherin to α-catenin affected neither association with the phosphatase VE-PTP or p120 nor endocytosis of VE-cadherin (18).
Here, we analyzed whether stabilization of endothelial junctions via replacement of VE-cadherin by VEC-α-C would affect development of the vascular system. We found that Cdh5VEC-α-C/VEC-α-C mice die as fetuses on a pure C57BL/6 background. At E10.5, embryos were viable and inconspicuous, demonstrating that VEC-α-C rescued the lethality caused by deletion of the β-catenin binding site in VE-cadherin. While angiogenesis progressed normally, the VEC-α-C mutation affected hematopoietic colonization of the fetal liver and caused severe edema. Whereas the generation and differentiation of primitive hematopoietic progenitor cells were normal, VEC-α-C inhibited their homing into the fetal liver. Edema formation in Cdh5VEC-α-C/VEC-α-C fetuses was caused by disrupted lymph vessel development due to a failure of normally specified and migrating lymphatic progenitor cells to develop into ordered large lumenized vessels.
MATERIALS AND METHODS
Antibodies and reagents.
The following antibodies were used: anti-CD3e 145-2C11 (BD Biosciences), anti-CD45R/B220 RA3-6B2 (BD Biosciences), anti-Ter119 (BD Biosciences), anti-CD11b (Mac-1) M1/70 (BD Biosciences), anti-Gr-1 RB6-8C5 (BD Biosciences), anti-PECAM-1 1G5.1 and 5D2.6 (19), anti-VEGFR-3 (AF743; R&D Systems), anti-Prox1 (102-PA32; ReliaTech), anti-LYVE1 (no. 223322; R&D Systems), antipodoplanin (8.1.1; our antibody), antipodoplanin-phycoerythrin (PE) (8.1.1; eBiosciences), anti-CD117 c-Kit (2B8; BD Biosciences), anti-CD117 c-Kit (D13A2; Cell Signaling), and antiendomucin (7C7.1; our antibody), anti-VE-cadherin (VE42; produced by us). All Alexa Fluor-coupled antibodies were purchased from Invitrogen, and horseradish peroxidase (HRP)-coupled antibodies were obtained from Dianova.
Constructs.
The VEC-α-C construct has been described previously (18). The VECΔ75 construct was generated by deletion of the 75 C-terminal amino acids (aa) of VE-cadherin. To this end, a C-to-T mutation was inserted at bp 2130 (with bp 1 being the first of the start codon ATG) of the mouse VE-cadherin cDNA (20) giving rise to a new ClaI site. Ligated oligonucleotides 5′ CGGCTAGGAATTCCTGCAGCCCGGGACTAGTT 3′ and 5′ CTAGAACTAGTCCCG GGCTGCAGGAATTCCTAGC 3′ were cloned into the ClaI and XbaI sites of this plasmid, thus creating a truncated molecule with base pairs ATG (Met, aa 708 of VE-cadherin), ATC (Ile, aa 709 of VE-cadherin), and GGC (Gly, additional amino acid) in analogy to the VEC-α-C fusion molecule.
Mice.
VEC-α-C mice have been described previously (18) and were backcrossed with C57BL/6 mice at least 12 times. VECΔ75 mice were generated analogous to the VEC-α-C mice by using a VECΔ75 cDNA, which was inserted into the endogenous VE-cadherin locus via recombinase-mediated cassette exchange (RMCE). A Southern blot for positive ES cells is shown in Fig. 1. VE-cadherin conditional knockout mice have been generated in our laboratory and will be described elsewhere. Vav-iCre mice have been described previously (21). For time-phased matings, mice were mated in the evening and separated the morning thereafter. All animal experiments were performed in accordance with the local animal protection legislation (Bezirksregierung Muenster).
FIG 1.

Generation of VECΔ75 knock-in mice. (A) Schematic illustration of the targeting strategy used for the generation of VECΔ75 mice. Mouse ES cells heterozygous for the previously described VE-cadherin RMCE targeting locus (containing two incompatible LoxP/Lox2272 sites flanking exon [Ex2] and a neomycin resistance cassette [Neo], shown in panel A) were cotransfected with recombination vector DNA harboring the same two incompatible LoxP/Lox2272 sites flanking a replacement cassette and a Cre recombinase expression vector. The replacement cassette comprised the cDNA coding for VECΔ75 followed by a transcriptional stop (TS) cassette and an FLP recombination target (FRT)-flanked hygromycin resistance cassette. In the resulting knock-in mice, the starting codon of VECΔ75 was exactly in the position of the original starting codon in exon 2 of the genomic sequence. Positive recombination events were identified by Southern blotting. Expected fragment sizes, the position of a 5′ external probe, and binding sites for primers used for genotyping are indicated. (B) Southern blot analysis of genomic DNA of a wild-type mouse (+/+) and a heterozygous knock-in mouse (+/VECΔ75) hybridized with the 5′ probe generating a 7.6-kb fragment for the RMCE locus and a 9.9-kb fragment for the wild-type locus. (C) Western blot of Triton X lysates of endothelioma cells generated from VEC WT and VECΔ75 knock-in mice.
Preparation of fetal liver.
Fetal livers of embryos from staged matings were extracted with tissue forceps from embryo bulks. Fetal livers were weighed and then singularized in phosphate-buffered saline (PBS) using a P200 pipette (Eppendorf), and total cell numbers were determined using a Neubauer counting chamber.
FACS analysis of fetal blood.
Fluorescence-activated cell sorter (FACS) analysis of fetal blood was performed as described previously (22). Erythrocytes were depleted from total fetal blood using a density gradient centrifugation consisting of 4 ml of cell suspension in PBS–5% fetal calf serum (FCS) on top of 4 ml of Histopaque 1077 for 40 min at 400 × g and 20°C (22, 23). The expression of c-Kit and lineage markers (CD45R/B220, CD3, Ter119, Mac-1, and Gr-1) to identify hematopoietic stem and progenitor cells (HSPCs) was analyzed by flow cytometry. Propidium iodide or 7-aminoactinomycin D (7AAD) was used to exclude dead cells.
CFU assay determining the number of HSPCs.
Cells from fetal blood or fetal livers were plated in 35-mm-diameter bacterial dishes at concentrations ranging from 3 × 104 to 3 × 105 cells per ml of Iscove's modified Dulbecco's medium (IMDM) (42200-014; GIBCO) containing 1% (wt/vol) methylcellulose (Shin-Etsu-Chemical), 10% heat-inactivated fetal bovine serum (FBS) (A15-043; PAA), 1% deionized and delipidated bovine serum albumin (BSA) (StemCell), 3% X63-IL3 cell-conditioned medium, 200 μg/ml iron-saturated transferrin (Sigma), 10 μg/ml cholesterol (L4646; Sigma), 25 μg/ml linoleic acid-oleic acid (L9655; Sigma), 5 U/ml erythropoietin (Epo) (Janssen Cilag), 10 μg/ml recombinant human insulin (I9278; Sigma), 12.5 ng/ml recombinant mouse stem cell factor (rmSCF) (CS-C2044; Cell Systems), 10 ng/ml recombinant human interleukin-6 (rhIL-6) (CS-C1066; Cell Systems), 100 nM β-mercaptoethanol (Sigma), and 15% 5637 cell-conditioned medium. The number and composition of total CFU and CFU for erythroid cells as well as CFU for granulocytes, granulocytes plus macrophages, macrophages, and erythroid cells plus granulocytes plus macrophages (i.e. multilineage) were analyzed after 8 or 14 days, respectively. Cytospin preparations of picked and May-Grünwald-Giemsa-stained colonies were performed to confirm correct classification. Samples were analyzed with a Zeiss Axioscope2.
Preparation of mouse embryos for whole-mount stainings.
Whole-mount stainings of embryos were done as previously described (24) using the Vectastain kit and SIGMAFAST 3,3′-diaminobenzidine tetrahydrochloride (DAB) with a metal enhancer tablet set according to the manufacturer's instructions.
Preparation of mouse embryos for analysis using ultramicroscopy.
Embryos analyzed using ultramicroscopy were stained and cleared as described previously (9) using anti-PECAM1, anti-VEGFR-3, anti-Prox1, and anti-c-Kit (D13A2) as primary antibodies. Cleared samples were subsequently imaged on a LaVision ultramicroscope (LaVision BioTec, Bielefeld, Germany). Three-dimensional (3D) reconstruction and analysis of image stacks were performed using IMARIS software (version 7.6.1; Bitplane).
Whole-mount staining of skin preparations from embryos.
Embryos from staged matings were removed from the uterus, and whole-mount staining of skin preparations was performed as previously described (25) using anti-PECAM1, anti-VE-cadherin, antiendomucin, anti-VEGFR-3, and anti-Prox1 as primary antibodies. Skins were imaged with a Zeiss LSM 780 confocal laser-scanning microscope (Carl Zeiss, Inc.). Surface rendering of endomucin-stained blood vessels was performed using IMARIS software (version 7.6.1; Bitplane).
Whole-mount staining of skin preparations from adult mice.
Murine ear stamps were dissected from adult mice, fixed in 4% paraformaldehyde (PFA), and digested with collagenase I (Roche). After permeabilization with 0.5% Triton X-100 and blocking with 1% BSA in 0.5% PBS–Tween, the samples were stained for 72 h with anti-VE-cadherin and anti-PECAM-1 antibodies. Primary antibodies were detected by 48 h of incubation with Alexa Fluor fluorophore-coupled antibodies. Ear stamps were mounted with fluorescent mounting medium (Dako) and visualized with a LSM780 confocal laser-scanning microscope (Carl Zeiss, Inc.).
Preparation of paraffin sections.
For histology of aortic clusters, fetal liver, and heart in paraffin-embedded sections, mouse embryos were fixed overnight in 4% PFA in PBS at 4°C. After dehydration, samples were embedded in paraffin and sectioned on a microtome in 5- to 6-μm-thick consecutive sections. Mounting was performed on poly-l-lysine-coated slides (Menzel-Glaeser). Sections were dewaxed, rehydrated, and subsequently stained with Mayer's hematoxylin (E10.5) or additionally counterstained with eosin (E13.5) and mounted with Entellan. Aortic clusters were identified and imaged using a Zeiss Axioscope2, and fetal liver sections were visualized with a Zeiss Axio Imager (20× numeric aperture, 0.5; 40×, 1.3) and analyzed with Volocity software (Improvision).
Isolation and culture of mouse lymphatic endothelial cells.
Primary endothelial cells were isolated from lungs of 7- to 9-week-old Cdh5wt/wt or Cdh5VECαC/VECαC mice as previously described (26). CD31-positive endothelial cells were additionally sorted by flow cytometry for lymphatic identity using a PE coupled podoplanin antibody (BD Biosciences). For immunofluorescence, murine LECs (MLECs) were grown on gelatin-coated Transwell filters (Corning, Inc.) and stained with anti-VE-cadherin, anti-LYVE1, and antipodoplanin antibodies, followed by detection with Alexa Fluor fluorophore-coupled secondary antibodies. Immunostained MLECs were analyzed with a LSM780 confocal laser-scanning microscope (Carl Zeiss, Inc.). For analysis of colony growth, MLECs were seeded on gelatin-coated 48-well plates at 1 × 104 cells/well and visualized with an Axio Observer (Carl Zeiss, Inc.) in a chamber incubated at 37°C with 5% CO2. All movies taken were analyzed with Volocity software (Improvision).
Statistical analysis.
Data sets were checked for normality (Shapiro-Wilk) and equal variance. P values were determined by Student's t test if allowed or otherwise by Mann-Whitney rank sum test. Analysis was performed using SigmaPlot 11.0. Values are presented as means ± standard errors of means (SEM).
RESULTS
Direct fusion of VE-cadherin to α-catenin rescues the lethality caused by deletion of the β-catenin binding site in VE-cadherin and allows unperturbed angiogenesis.
Having generated mice with strongly stabilized endothelial junctions due to replacement of VE-cadherin by a VEC-α-C fusion protein raised the question of whether this would affect embryonic development. Indeed, we found that Cdh5VEC-α-C/VEC-α-C mice died late during fetal development on a pure C57BL/6 background.
Since VE-cadherin in VEC-α-C lacks its C-terminal 75 amino acids containing the β-catenin binding site, we speculated whether this could cause the fetal lethality, as a C-terminal truncation of VE-cadherin by 82 amino acids results in embryonic lethality due to defective vascular remodeling (13). Mice with an endothelial cell-specific deletion of β-catenin show impaired vascular development as early as E9.5 (27–29) and severe heart defects at E10.5 due to a failure of the EndMT of the endocardium required for cushion formation in the developing heart (27).
To test the developmental consequences of the shorter 75-amino-acid deletion of the VE-cadherin cytoplasmic tail, we generated knock-in mice with wild-type (WT) VE-cadherin replaced by a 75-amino-acid truncation (Cdh5VECΔ75) (Fig. 1 and 2A). The Δ75 truncation, as was described for the Δ82 truncation by Carmeliet and colleagues, led to embryonic lethality of Cdh5VECΔ75/VECΔ75 embryos at E9.5 (Table 1 and Fig. 2B). In contrast, Cdh5VEC-α-C/VEC-α-C embryos (VEC-α-C embryos) were indistinguishable from wild-type littermates at E9.5 (Table 1 and Fig. 2B), and at E10.5, we detected no gross abnormalities in angiogenesis (Fig. 2C). Additionally, coverage of fetal skin with endomucin-positive vessels at E13.5 and E14.5 did not differ between VEC-α-C and control embryos (Fig. 2D and E) (data not shown).
FIG 2.

A VE-cadherin-α-catenin fusion protein compensates for the lack of junctional β-catenin and does not affect blood vessel formation. (A) Schematic illustration of VE-cadherin (VEC), β-catenin (β-cat), α-catenin (α-cat), VE-cadherin–α-catenin fusion protein (VEC-α-C), and VE-cadherin C-terminal 75-amino-acid truncation mutant (VECΔ75). Illustrated are the five extracellular domains, the juxtamembrane domain (JMD), and the catenin binding domain (CBD) of VE-cadherin (blue). (B) Embryos homozygous for the Cdh5VECΔ75 allele died at E9.5 (right panel), while homozygous Cdh5VEC-α-C embryos (center panel) were indistinguishable from wild-type and heterozygous littermates at this stage (left panel) (Table 1). (C) PECAM-1 whole-mount immunostaining of E10.5 embryos revealed no major differences in the vasculature of wild-type (left) and homozygously VEC-α-C-expressing (right) embryos. Head and trunk regions are enlarged in the small panels. (D) Whole-mount immunostaining for endomucin in the skin of E13.5 VEC-α-C and control fetuses. Scale bars, 50 μm. (E) Analysis of the area covered by endomucin-positive vessels in the skin of E13.5 fetuses revealed no difference in angiogenesis. For quantification, skin preparations from five VEC WT fetuses and four VEC-α-C fetuses were analyzed by evaluation of maximum intensity projections of at least seven stacks per fetus (Zeiss LSM 780; IMARIS). n.s., not significant; ctrl, control.
TABLE 1.
Analysis of progeny from Cdh5+/VECΔ75 and Cdh5+/VEC-α-C intercrosses
| Intercross and stage | No. of litters | No. of progenya |
|||
|---|---|---|---|---|---|
| Total | WT | het | hom viable/total (%) | ||
| Cdh5+/VECΔ75 | |||||
| E9.5 | 9 | 78 | 23 | 37 | 0/19 (0) |
| Cdh5+/VEC-α-C | |||||
| E9.5 | 4 | 27 | 7 | 14 | 6/6 (100) |
| E10.5 | 32 | 235 | 54 | 123 | 58/58 (100) |
| E11.5 | 4 | 32 | 7 | 14 | 11/11 (100) |
| E12.5 | 7 | 58 | 12 | 34 | 12/12 (100) |
| E13.5 | 31 | 247 | 59 | 138 | 44/50 (88) |
| E14.5 | 15 | 117 | 25 | 58 | 21/34 (62) |
| E16.5 | 9 | 62 | 13 | 30 | 13/18 (72) |
| P21 | 17 | 87 | 34 | 53 | 0/0 |
WT, wild type (genotype Cdh5+/+); het, heterozygous (genotype Cdh5+/VECΔ75 or Cdh5+/VEC-α-C); hom, homozygous (genotype Cdh5VECΔ75/VECΔ75 or Cdh5VEC-α-C/VEC-α-C).
Interestingly, the heart defects in cushion formation that were detected in embryos lacking endothelial β-catenin (22), were not detected in E11.0 VEC-α-C embryos (Fig. 3). These defects had been explained by impaired β-catenin/T cell factor (TCF)/Lef transcriptional activity during EndMT of the endocardium (22).
FIG 3.

EndMT of endocardial cells is not affected in VEC-α-C embryos. (A) Paraffin sections of VEC WT and homozygous VEC-α-C E11.0 embryos stained with hematoxylin and eosin. The formation of cushions (asterisks) was normal in the VEC-α-c embryo. Scale bars, 300 μm. (B) The number of mesenchymal cells within cushions was determined from four to six serial 6-μm sections of two VEC-α-C and two VEC WT embryos, and the results are represented as mean values ± standard deviation (P = 0.45 by t test). A, atrium, left atrial chamber; V, ventricle, common ventricular chamber; n.s., not significant.
Thus, the fusion of α-catenin to a truncated form of VE-cadherin lacking its β-catenin binding site fully rescued defects caused by the lack of junctional β-catenin and furthermore caused no transcription-related signaling deficiencies as described for the complete lack of endothelial β-catenin (27). Our results therefore demonstrate that the cytosolic β-catenin pool is sufficient and junction-associated β-catenin is not required for these transcription-regulating processes.
Defective fetal liver colonization in VEC-α-C embryos.
Despite apparently normal development to midgestation, we did not identify homozygous VEC-α-C offspring when mice were bred on a C57BL/6 background. Timed matings revealed lethality of homozygous VEC-α-C embryos beginning around E13.5 (Table 1), often associated with a strongly reduced fetal liver size (Fig. 4A). During embryonic development, the fetal liver is one of the main sites of hematopoietic differentiation. Starting from late E9 onwards, hematopoietic stem and progenitor cells (HSPCs) originating from hemogenic endothelium of the yolk sac, AGM, or placenta colonize the fetal liver (6). Between E13 and E14, the hematopoietic compartment accounts for approximately 75% of the hepatic parenchyma (30).
FIG 4.

Defective fetal liver colonization in VE-cadherin–α-catenin embryos. (A) Embryos (E14.5) homozygously expressing VEC-α-C show variably reduced fetal liver size (circled areas) in comparison to WT littermates. (B) Organ weight and cellularity of whole fetal liver isolates from VEC-α-C or control embryos at E13.5 (n = 24, VEC WT; n = 15, VEC-α-C), and E14.5 (n = 22, VEC WT; n = 27, VEC-α-C). Data were normalized for each litter to the average fetal liver weight or cellularity of the respective control embryos. ***, P < 0.001. (C) H&E-stained paraffin sections demonstrating the reduced fetal liver cellularity in E13.5 VEC-α-C compared to control fetuses. Hepatoblasts (white arrows), hematopoietic cells (black arrows), and endothelial cells (arrowheads) are marked. Scale bars, 25 μm. Additional details are provided in Table 1. (D) The numbers of hepatoblasts and hematopoietic cells were assessed in 9 to 12 distinct areas (80 by 80 μm) for each VEC-α-C (n = 5) or VEC WT (n = 3) embryo and are presented as means ± SD.
Quantification of fetal liver weight and cellularity in VEC-α-C fetuses revealed a significant reduction compared to wild-type littermates (Fig. 4B). At E14.5, the liver size in all VEC-α-C fetuses was reduced—in half of them even down to 20% to 50% of the weight/cellularity of the average control littermate. Concomitantly, paraffin sections revealed a reduced ratio of hematopoietic to hepatoblastic cells in VEC-α-C fetal livers corresponding to the reduced organ weight and cellularity (Fig. 4C and D). Thus, stabilization of endothelial junctions by covalently linking VE-cadherin to α-catenin strongly impaired hematopoietic development.
Hematopoietic stem and progenitor cells emerge normally in aortic clusters in VEC-α-C embryos.
Definitive HSPCs arise in clusters from the endothelium of the dorsal aorta, as well as the vitelline and umbilical arteries (31). To test whether VEC-α-C inhibits this EHT, we analyzed serial paraffin sections of E10.5 embryos and detected aortic clusters in comparable numbers in VEC-α-C embryos and wild-type littermates (Fig. 5A). For a quantitative analysis, we analyzed whole-mount-stained (c-Kit and PECAM-1) intact E10.5 embryos by planar illumination microscopy (ultramicroscopy) (Fig. 5B to D) (32). Volume reconstruction along the length of the thoracic dorsal aortae allowed quantitation of all emerging clusters (Fig. 5B), demonstrating that c-Kit-expressing cells delaminated from and were in direct contact with the PECAM1-positive endothelium (Fig. 5C). In transversally oriented reconstructions, cluster appearance was virtually indistinguishable from classical hematoxylin and eosin (H&E)-stained sections (Fig. 5A and D). The number of c-Kit+ aortic clusters in VEC-α-C embryos was not different from that in control littermates (Fig. 5E).
FIG 5.
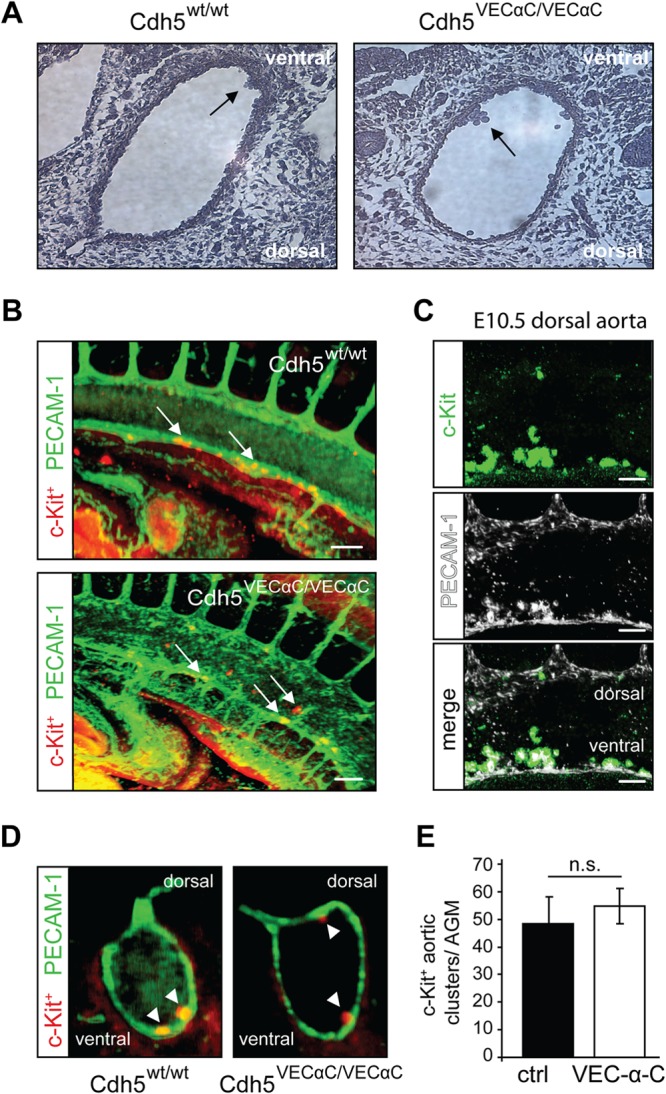
Endothelial to hematopoietic transition in the dorsal aorta is not impaired in VE-cadherin–α-catenin embryos. (A) At E10.5, aortic hematopoietic clusters (arrows) were visualized in serial hematoxylin-stained paraffin sections through the dorsal aorta (DA) of wild-type (n = 3) and VEC-α-C (n = 2) littermates. Single sections are depicted. (B) Identification of c-Kit+ aortic clusters in the DA of whole-mount immunostained (c-Kit+ and PECAM-1) E10.5 control and VEC-α-C littermates. Depicted are 3D renderings of image stacks acquired from optically cleared embryos using light sheet microscopy. The orientation corresponds to a sagittal section of the DA. Scale bars, 100 μm. (C) Magnification of c-Kit+ intraaortic clusters (green; top panel) from a VE-cadherin wild-type embryo at E10.5. Clusters are preferentially located at the ventral side of the DA but are also found at the dorsal side of the DA, although at lower frequency in both genotypes. Of note, c-Kit+ clusters stained positive for PECAM-1 (white; middle panel) in both genotypes. Scale bars, 100 μm. (D) As in panel B, but the sectional plane through the DA corresponds to a transversal orientation. (E) Enumeration of c-Kit+ aortic clusters in the DA of control and VEC-α-C embryos (IMARIS, Bitplane). No difference was observed between embryos of both genotypes (n = 6, control; n = 6 VEC-α-C). AGM, aorta-gonad-mesonephros.
Hematopoietic progenitor cells accumulate in the circulation of VEC-α-C embryos.
Next, we asked whether progenitor cells were able to detach from the endothelium and thus were detectable in the fetal circulation. Flow cytometric analysis of E13.5 fetal blood (14 VEC-α-C and 15 wild-type fetuses) surprisingly revealed an increase of c-Kit-positive, lineage marker-negative (c-Kit+ lin−) progenitors in VEC-α-C compared to wild-type fetuses (Fig. 6A and B). Thus, detachment of c-Kit+ lin− progenitors from the hemogenic endothelium in VEC-α-C embryos is not decreased, and early progenitor cells apparently accumulate in the fetal circulation.
FIG 6.

Primitive hematopoietic progenitor cells accumulate in the fetal circulation of VE-cadherin–α-catenin embryos. (A) Flow cytometric analysis of blood after exsanguination of a representative E13.5 wild-type fetus and VEC-α-C fetus. Gates in the dot plot define c-Kit+ and lineage marker-positive (lin+) cells (CD45R/B220, CD3, Ter119, Mac-1, and Gr-1 cells). (B) Quantification of c-Kit+ lin− absolute cell numbers (primitive hematopoietic progenitors) in the blood of wild-type (n = 15) and VEC-α-C (n = 14) fetuses. (C) To confirm the correct classification, colonies of myeloid cells were picked from semisolid medium, washed in PBS, and spread by cytospin. After heat fixation, cells were subjected to panchromatic staining, mounted, and scored using an upright microscope. Shown are representative examples of granulocyte, monocyte, and granulocyte-monocyte colonies. (D, E, and F) Fetal blood or liver cells were plated into methylcellulose supplemented for myeloid growth and CFU, giving rise to erythroid (E), granulocyte (G), macrophage (M), granulocyte-macrophage bilineage (GM), and large multilineage (ML) colonies, which were counted after 8 and 14 days (control, n = 5; VEC-α-C, n = 5). The frequency of clonogenic progenitor cells in homozygous VEC-α-C fetuses compared to wild-type fetuses was significantly elevated in blood (D) and reduced in fetal liver (E). Compared to control fetuses, the relative abundance of different progenitor types was not changed in the liver of VEC-α-C embryos (F). *, P < 0.05; **, P < 0.01; ***, P < 0.001. n.s., not significant.
We investigated the myeloid differentiation potential of hematopoietic progenitors from the fetal blood and liver of VEC-α-C fetuses in semisolid methocel cultures and confirmed colony identity in panchromatically stained cytospin preparations (Fig. 6C). As expected, the frequency of clonogenic progenitors (CFU) in fetal blood from wild-type or heterozygote control littermates was very low (total of approximately 0.0025%) (Fig. 6D). Surprisingly, the CFU frequency in the circulation of VEC-α-C fetuses, in particular of progenitors with multilineage potential was 10-fold increased. In sharp contrast, the number of CFU within the liver was strongly reduced in VEC-α-C compared to control fetuses (Fig. 6E). Interestingly, the differentiation potential and lineage distribution of the few progenitor cells that succeeded in colonizing the fetal liver of VEC-α-C fetuses were unaltered (Fig. 6F).
Because c-Kit+ lin− early progenitors that have the potential to differentiate into all myeloid lineages accumulate in the circulation of VEC-α-C fetuses but are dramatically reduced in their liver tissue, we conclude that recruitment of these progenitors from the circulation into the liver is impaired.
Hematopoietic stem and progenitor cells do not require VE-cadherin for colonization of the fetal liver.
As HSPCs arise from hemogenic endothelium, they still weakly express VE-cadherin at their earliest differentiation stages (33, 34). Therefore, we wanted to investigate a potential role of VE-cadherin during differentiation of HSPCs, which could be affected by VEC-α-C. To this end, we conditionally deleted VE-cadherin by crossing VE-cadherin conditional knockout mice (Cdh5fl/fl) to Vav-iCre mice (Vav-iCre+/T) that express Cre under an early hematopoietic promoter (21, 35). We analyzed the effect of VE-cadherin deletion on hematopoietic cells in Vav-iCre transgene-expressing mice that were either homozygous (Cdh5fl/fl) or heterozygous (Cdh5fl/del) for a floxed (shown as “fl”) VE-cadherin allele. In the latter case, the second VE-Cad allele was null. Such mice were viable and fertile and showed no fetal liver colonization defects at E13.5 (Fig. 7A). We controlled the efficiency of Cre-mediated recombination in a R26R-YFP mouse reporter line, in which Vav-iCre induced permanent yellow fluorescent protein (YFP) expression from the Rosa26 locus (36). To this end, we analyzed the Vav-iCre activity in c-Kit+ fetal liver cells of double heterozygous fetuses (R26R+/YFP;Vav-iCre+/T) at E12.5 and E13.5. Representative FACS analyses of c-Kit+ cells from Vav-iCre negative (control) or positive R26R+/YFP fetuses at E13.5 are shown in Fig. 7B. While YFP-positive cells were never detected in Vav-iCre negative controls, 98.3% ± 0.5% of c-Kit+ cells in the fetal liver of Vav-iCre-expressing fetuses were YFP positive (n = 4) (Fig. 7B, inset bar diagram). Thus, we demonstrated functional Vav promoter-driven Cre activity in the overwhelming majority of early fetal liver progenitor cells and conclude that VE-cadherin is not required on hematopoietic cells for colonization of and differentiation within the fetal liver.
FIG 7.

VE-cadherin is not required on hematopoietic progenitors for the colonization of the fetal liver. (A) At E13.5, fetal liver weight and cellularity were determined from isolates of Cdh5fl/+; Vav-iCreT/+ or Cdh5fl/fl fetuses (n = 44) that served as controls and Cdh5fl/−; Vav-iCreT/+ or Cdh5fl/fl; Vav-iCreT/+ fetuses (n = 16) that were deficient for VE-cadherin on hematopoietic cells. Fetal liver weight and cellularity were comparable in both groups (P = 0.465), indicating that VE-cadherin is not required on hematopoietic progenitor cells for colonization of the fetal liver. (B) To demonstrate the efficacy of iCre-mediated recombination in fetal hematopoietic cells, c-Kit+ fetal livers from R26R-YFP; Vav-iCreT/+ fetuses were analyzed for YFP expression by flow cytometry. A representative histogram of one fetus at E13.5 is shown. (The number indicates the percentage of cells in the depicted expression gate.) The bar diagram in the inset depicts the average from 4 analyzed individuals.
VEC-α-C embryos suffer from severe edema and display disruption of the developing dermal lymphatic vessels.
Additionally to the fetal liver defect on the C57BL/6 background, we noticed edema formation of various severities starting at E13.5 in homozygous VEC-α-C mice (Fig. 8A). Quantification revealed about 64% of VEC-α-C fetuses at E13.5 and about 80% at E14.5 presented with edema (Fig. 8B). Because the severity of edema formation and fetal liver defect often did not coincide, both phenotypes appeared to arise independently.
FIG 8.

Fusion of VE-cadherin to α-catenin impairs lymphatic vessel integrity, leading to edema. (A) From E13.5 onwards, VEC-α-C fetuses developed edema with various severities. Depicted are two VEC-α-C fetuses with either mild or severe edema. (B) Penetrance of edema formation in VEC-α-C fetuses compared to that in wild-type littermates. (The numbers indicate the total numbers of fetuses analyzed.) At E14.5, 80% of VEC-α-C embryos displayed visible edema. (C) At the indicated developmental stages, skin whole mounts were immunostained for Prox-1 and VEGFR-3 to visualize lymphatic endothelium and for the panendothelial marker PECAM-1, demonstrating progressive lymphatic vessel deterioration in VEC-α-C fetuses. Scale bars, 50 μm.
We further investigated development of the lymphatic vasculature using whole-mount immunostaining of back skin from VEC-α-C or littermate control fetuses. Whereas at E13.5, the superficial lymphatics in control fetuses had developed into a contiguous plexus, VEC-α-C fetuses showed disrupted lymphatic vessels (Fig. 8C). Lymphatic endothelial cells were present but did not form a branched network as in the controls. One day later at E14.5, this phenotype became more pronounced, with only isolated patches of lymphatic cells detectable in the skin of VEC-α-C fetuses, while the lymphatic vascular plexus of control fetuses had further matured. At both time points, the blood vasculatures of VEC-α-C and control littermates were indistinguishable (Fig. 8C, upper panel), and also VE-cadherin staining of lymphatic junctions was indistinguishable between VEC-α-C and control fetuses (Fig. 9A). Thus, a lack of plasticity of the VE-cadherin–catenin complex impairs the formation of a network of connected superficial lymphatics in the skin.
FIG 9.

VE-cadherin staining in embryonic skin, adult skin, and primary MLECs. (A) Whole-mount skin preparations from E13.5 VEC WT and VEC-α-C embryos were stained for VE-cadherin and VEGFR-3. Note that islands of disconnected lymphatic endothelial cells were only detected in the skin of VEC-α-C embryos. Scale bars, 50 μm. (B) Whole-mount preparations of ear skin from adult VEC-WT and VEC-α-C mice revealed no differences for the characteristic VE-cadherin and PECAM-1 staining of button-like junctions in initial lymphatics. Scale bars, 20 μm. (C) Primary MLECs were stained for VE-cadherin and the lymphatic markers podoplanin and LYVE-1. Scale bars, 20 μm. (D) The colony growth and cell movement of primary MLECs from Cdh5wt/wt and Cdh5VECαC/VECαC mice were visualized over 24 h (see Movies S1 and S2 in the supplemental material) on gelatin-coated 48-well plates. Depicted here are VEC WT (upper panel) and VEC-α-C (lower panel) MLEC colonies shortly after seeding.
In adult mice, a special form of junctions has recently been described in initial lymphatics with overlapping flaps at borders of oak leaf-shaped lymphatic endothelial cells, which lacked VE-cadherin-containing junctions at the tips of the flaps and were anchored on the sides by VE-cadherin-containing button-like junctions (37). Analysis of whole mounts of ear stamps of adult VEC-α-C mice of mixed 129/BL6 genetic background for the distribution of VE-cadherin and PECAM-1 revealed the same button-like junctions that were found in control mice (Fig. 9B).
To test whether VEC-α-C modulates the rigidity of junctions or the growth of LECs in clusters, we isolated LECs from lungs of 7- to 9-week-old Cdh5wt/wt or Cdh5VECαC/VECαC mice and cultured them on gelatin-coated surfaces. Staining of these cells for VE-cadherin showed no difference in the recruitment of VEC-α-C and VE-cadherin to junctions (Fig. 9C). However, LECs from VEC-α-C mice were more widespread than cells from control mice (Fig. 9D). Video imaging of these cells over a period of 24 h further documented the more intense spreading of VEC-α-C LECs. The motilities of both types of LECs, however, were indistinguishable as determined by comparing the motilities of 40 cells of each genotype (data not shown).
Covalent linkage of VE-cadherin to α-catenin does not interfere with the specification and release of initial LECs but impairs the formation of the first lumenized lymphatic structures.
To determine whether earlier steps in lymphatic development also were affected in VEC-α-C mice, we visualized formation of the first lymphatic vascular structures between E11.0 and E12.0 by ultramicroscopy (32). We have recently shown that the first or initial Prox1+ lymphatic endothelial cells (iLECs), which are specified in the CV, leave their venous origin as streaks of cells and form two large, lumenized, lymphatic vessels (9). These are the dorsolaterally situated peripheral longitudinal lymphatic vessel (PLLV) and the more ventrally localized primordial thoracic duct (pTD).
At E11.0, the frequency and distribution of Prox1+ iLECs were indistinguishable in VEC-α-C embryos and control littermates. Comparable numbers of iLECs emigrated from their venous origin (Fig. 10A and B) and condensed to initiate PLLV and pTD formation (Fig. 10A and B; schematically depicted in panel C). Thus, plasticity of the VE-cadherin–catenin complex is necessary for neither the specification of iLECs nor their emigration from their venous sources prior to and during initiation of PLLV and pTD formation. At E12.0, however, in control littermates, the pTD and PLLV had developed into contiguous large lumenized structures, while in VEC-α-C embryos, they were composed of multiple smaller, sometimes discontinuous lumina (Figure 10D to F; schematically depicted in panels G and H), which is particularly prominent from the reconstruction depicting VEGFR-3 only (Fig. 10E) and best appreciated in individual transversally oriented, optical planes (Fig. 10F).
FIG 10.

The initial development of lymphatic vessels proceeds normally, but the formation of larger lumenized lymphatic structures is impaired in VE-cadherin–α-catenin fetuses. (A) Embryos at E11.0 were whole-mount immunostained for Prox-1, VEGFR-3, and PECAM-1 and analyzed by light sheet microscopy (ultramicroscopy). Volume reconstruction of the resulting image stacks revealed no abnormalities of the emerging lymphatic endothelial cells in VEC-α-C embryos compared to control littermates. Shown is a sagittal projection of the newly forming lymphatic system on one side at the level of the thorax. Scale bars, 200 μm. (B) Same field of view as in panel A; however, for reduced complexity of the projection, only Prox-1 and VEGFR-3, representing venous and lymphatic vascular structures, are shown. Scale bars, 200 μm. (C) Scheme of a sagittal section through one common cardinal vein (CV) and the discharging intersegmental veins (blue) at E11.0. Prox1+ cells are depicted with yellow nuclei, and LECs are shown in green. Newly emerging lymphatics start to form the peripheral lymphatic vessel (PLLV) and primordial thoracic duct (pTD). (D) VEC WT and VEC-α-C littermates at E12.0, whole-mount immunostained as described in panel A, reveal severely impaired lumenization of the largest lymphatic structures, pTD and PLLV. VEC-α-C fetuses presented with a discontinuous lumen of the pTD, characterized by multiple constrictions. Shown is a sagittal projection of a volume reconstruction of ultramicroscopic image stacks. Scale bars, 300 μm. (E) As in panel D; however, for clarity, only the channel representing VEGFR-3 is shown. (F) Fragmentation of the pTD is particularly obvious in a single image plane from a homozygous VEC-α-C fetus. Scale bars, 200 μm. (G and H) Models of the newly forming lymphatic vessels at E12.0 in a VEC WT fetus (G) and VEC-α-C fetus (H) (elements as in panel C). Depicted are the altered structures of lymphatic vessels sprouting from the PLLV and the fragmentation of the pTD.
Collectively, these results revealed that stabilization of endothelial junctions through replacement of VE-cadherin by a VEC-α-C fusion interferes with the morphogenesis of the first large lymphatic vessels, pTD and PLLV.
DISCUSSION
In this study, we analyzed the consequences of stabilizing VE-cadherin-mediated adhesion on the development of mice in which VE-cadherin was genetically replaced by a covalent, irreversible fusion between VE-cadherin and α-catenin. Although this caused fetal lethality, we detected no defects in blood vessel formation, suggesting that stability regulation of the VE-cadherin–catenin complex is dispensable during the formation of an endothelial vascular plexus and subsequent vascular remodeling. In contrast, lymphatic vessel development was strongly impaired, revealing unexpected differential requirements for junctional plasticity and/or the regulation of junctional rearrangements during blood and lymphatic vessel formation. Remarkably, stabilization of endothelial junctions by VEC-α-C blocked the entry of hematopoietic progenitors into the fetal liver and thereby fetal liver hematopoiesis. Thus, diapedesis of embryonic HSPCs is strongly affected by enhancing VE-cadherin-mediated adhesion.
We showed previously that VEC-α-C mice are viable and fertile when bred on a mixed genetic background (B6/129), although they were not born at Mendelian frequency (18). It is at present not clear why almost all embryos die on a pure B6 genetic background. The phenomenon that the genetic background affects survival of targeted mutations is well known in mice, and in most cases, like the one reported here, the modifying genes that affect survival are unknown.
The VEC-α-C fusion rendered adult healthy mice (mixed B6/129 genetic background), fully resistant to the induction of vascular leaks and strongly inhibited leukocyte migration into several, although not all, inflamed tissues (18, 38), establishing that VEC-α-C indeed stabilizes endothelial junctions and impairs opening of endothelial cell contacts during inflammation in the adult animal.
Our new results on impaired HSPC entry into the fetal liver extend this finding to the homing of hematopoietic progenitors in the embryo. Since vascular remodeling during angiogenesis likely requires junctional plasticity, and endothelial cells indeed rearrange their position within sprouts and migrate along each other (39), we had expected that fetal lethality in our mice would at least partly be caused by vascular remodeling defects. However, there is a precedent in Drosophila development demonstrating that various DE-cadherin–α-catenin fusions were also unable to affect the migration of border cells during oogenesis, although these cells interact with the neighboring cells via DE-cadherin (40). Thus, an irreversible link between a cadherin and α-catenin seems not to affect the plasticity of cadherin-mediated contacts in embryonic cell migration. Such processes can be considered rather slow compared to the rapid opening of endothelial contacts during leukocyte diapedesis, which occurs within 1 or 2 min (41). These kinetic differences are probably reflected by different mechanisms that regulate cadherin adhesion. We believe that the adhesive function of VEC-α-C is probably still regulatable: for example, via internalization or through modulation of the association with the actin cytoskeleton downstream of α-catenin. Indeed, we showed previously that binding of VEC-α-C to p120 and its endocytotic internalization do not differ from those of wild-type VE-cadherin (18).
Deletion of the β-catenin binding site from VE-cadherin abolishes its proper adhesive activity and its association with VEGFR-2 (42), which causes enhanced apoptosis and disturbs maintenance of the primary vascular plexus, leading to embryonic lethality at E9.5 (13). Both insufficient adhesion and impaired VEGFR-2 survival and signaling were fully rescued in our VEC-α-C embryos, which displayed a normal-appearing blood vasculature. Furthermore, we have shown before that VEC-α-C retains VEGFR-2 binding and normal VEGF-induced signaling (18). Thus, our results reveal that the described signaling defects caused by the lack of junctional β-catenin are actually due to defective VE-cadherin–catenin anchorage at junctions, rather than the lack of β-catenin per se. In agreement with this, defects attributed to the loss of endothelial β-catenin expression, like reduced contact stability and defective heart cushion formation due to impaired EndMT of the endocardium (27, 28), were not found in VEC-α-C mice. EndMT depends on proper β-catenin/TCF/Lef transcriptional activity, and the absence of EndMT defects in VEC-α-C embryos allows the conclusion that junctional β-catenin is dispensable and instead cytosolic β-catenin pools fully suffice for EndMT-inducing transcriptional events. Concordantly, we showed before that VEC-α-C does not affect Wnt-induced β-catenin translocation into the nucleus (18).
We detected a severe impairment to initiate fetal liver hematopoiesis in our VEC-α-C mice. After HSPCs have detached from the hemogenic endothelium in the yolk sac, AGM, and placenta (6), they migrate via the circulation into the fetal liver parenchyme, which they seed starting at late E9. We excluded formation of c-Kit+ hematopoietic cell clusters in the dorsal aorta as the mechanism that affects fetal liver development in VEC-α-C mice. Hematopoietic cells were released, easily detectable in the fetal circulation, and differentiated into all major myeloid lineages in vitro. Remarkably, however, the frequency of c-Kit+ lin− hematopoietic cells was dramatically increased in the circulation and decreased in the liver of VEC-α-C embryos. The few HSPCs that succeeded in entering the fetal liver had a normal capacity to proliferate and differentiate. Since Vav-iCre-driven deletion of VE-cadherin in HSPCs did not result in hematopoietic abnormalities, we excluded that low-level VE-cadherin described on the earliest hematopoietic populations (33, 34) was relevant for fetal liver colonization. Thus, stabilization of endothelial junctions via the VEC-α-C fusion does not affect the generation or differentiation of hematopoietic cells but appears to inhibit transendothelial migration of HSPCs into the fetal liver.
The molecular mechanisms mediating homing of hematopoietic cells to the fetal liver are poorly defined. Hematopoietic stem cell (SC) homing to the adult bone marrow has been more intensely analyzed, and various adhesive mechanisms, such as endothelial selectins, integrins, and the chemokine SDF-1 and its receptor CXCR4, were reported to be involved (43). The only adhesion molecules known to be relevant for fetal liver homing of HSPCs are β1-integrins, which are required on the HSPC surface for fetal liver entry (22). In agreement with our results, impaired entry into the fetal liver led to an increase of β1-integrin-deficient progenitors in the circulation. On the endothelial side, the molecular requirements for the transmigration of hematopoietic progenitors across fetal liver endothelium have not yet been analyzed. Based on in vitro assays, it was shown that inhibition of VE-cadherin by antibodies increased the transendothelial migration of CD34+ hematopoietic progenitor cells through cultured human bone marrow endothelial cells (44). In agreement with these in vitro results, our results show that VE-cadherin is a gatekeeper for the junctional transmigration pathway during homing of HSPCs into the fetal liver in vivo.
Edema formation was observed in VEC-α-C fetuses from stage E13.5 onwards. Rudimentary lymphatic vessel structures were present in the skin of these embryos, but they were disrupted and deteriorated further, leading to disconnected islands of lymphatic cells at E14.5. Specification and release of nascent LECs from their venous origin were unperturbed in VEC-α-C embryos; however, the formation of the large lumenized structures PLLV and pTD was impaired. Both first lymphatic vessels presented with multiple, smaller, often fragmented lumina, while blood vascular lumen formation was unperturbed in VEC-α-C embryos.
Until now, the molecular process of lumen formation in lymphatic vessels has been poorly investigated. In blood vessels, multiple mechanisms of lumen formation have been proposed, including intercellular fusion of intracellular vacuoles (45), hollowing of a nonlumenized endothelial cell cord (16), or cell rearrangements and extensive junctional remodeling (46). During multicellular lumen formation, VE-cadherin is required to establish and maintain apical/basal polarity (16, 47).
The first lymphatic vessels PLLV and pTD are more irregularly shaped and larger than the dorsal aorta (DA) and CV (9) but are not stabilized by blood flow. Interestingly, starting at midgestation, lymphatic vessels were shown to become functional (48). Therefore, the lymphatic plexus of the skin but also the collecting vessels PLLV and pTD are likely to experience rapid and large volume changes resulting from the onset of lymph flow. The remarkable difference we noted in blood and lymph vessels of VEC-α-C mice toward the changed plasticity of the VE-cadherin–catenin complex may therefore be caused by the higher lumen size fluctuation of lymphatic vessels compared to blood vessels during physiological activity. Besides more extensive spatial remodeling, rapid volume changes are also likely to require fast changes in junctional arrangement, which may be impaired by the VEC-α-C fusion.
We know that VEC-α-C interacts more efficiently with the actin cytoskeleton, likely causing the higher stability of endothelial junctions in VEC-α-C mice (18). However, exactly why the VEC-α-C interaction with the actin cytoskeleton is enhanced remains an open question. This could be due to the irreversibility of the VEC-α-C fusion, since it was reported that junction-destabilizing factors trigger the dissociation of the VE-cadherin catenin complex (49, 50). Alternatively, truncation of the N-terminal third of α-catenin in our VEC-α-C fusion could lead to allosteric changes that enhance direct actin binding or expose additional binding sites for actin linker molecules, such as vinculin (51, 52).
In conclusion, this study reveals that the stabilization of endothelial cell junctions by covalently linking VE-cadherin to α-catenin, thereby circumventing junctional β-catenin, causes fetal lethality of mice on a clean C57BL/6 background. The VEC-α-C mutation inhibited homing of hematopoietic progenitors into the fetal liver, leading to impaired hematopoiesis. Despite the inhibitory effect of VEC-α-C on the opening of endothelial junctions, the plasticity of junctions was still sufficient to allow normal angiogenesis and remodeling of blood vessels. Transcription-related signaling defects known to arise from β-catenin deletion in endothelial cells were compensated for by the fusion of VE-cadherin to α-catenin, demonstrating that cytosolic β-catenin is sufficient and junctional β-catenin not required for these signaling processes. Finally, the deficits in the formation of large luminal lymphatic structures and the failure to form a properly connected lymphatic network in the skin reveal that lymphatic vessel formation has different requirements for junctional plasticity than blood vessel formation.
Supplementary Material
ACKNOWLEDGMENTS
We thank Barbara Waschk for help with the colony assays and Stefan Volkery for help and advice with microscopy.
This work was supported by funds of the DFG SFB629 and Excellence Cluster 1003 to F.K. and D.V. and SFB656 to F.K. and by support from the Max Planck Society to F.K. and D.V.
We declare we have no conflicts of interest.
Footnotes
Published ahead of print 24 February 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01526-13.
REFERENCES
- 1.Chen MJ, Yokomizo T, Zeigler BM, Dzierzak E, Speck NA. 2009. Runx1 is required for the endothelial to haematopoietic cell transition but not thereafter. Nature 457:887–891. 10.1038/nature07619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zovein AC, Hofmann JJ, Lynch M, French WJ, Turlo KA, Yang Y, Becker MS, Zanetta L, Dejana E, Gasson JC, Tallquist MD, Iruela-Arispe ML. 2008. Fate tracing reveals the endothelial origin of hematopoietic stem cells. Cell Stem Cell 3:625–636. 10.1016/j.stem.2008.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Medvinsky A, Dzierzak E. 1996. Definitive hematopoiesis is autonomously initiated by the AGM region. Cell 86:897–906. 10.1016/S0092-8674(00)80165-8 [DOI] [PubMed] [Google Scholar]
- 4.Samokhvalov IM, Samokhvalova NI, Nishikawa S. 2007. Cell tracing shows the contribution of the yolk sac to adult haematopoiesis. Nature 446:1056–1061. 10.1038/nature05725 [DOI] [PubMed] [Google Scholar]
- 5.Gekas C, Dieterlen-Lievre F, Orkin SH, Mikkola HK. 2005. The placenta is a niche for hematopoietic stem cells. Dev. Cell 8:365–375. 10.1016/j.devcel.2004.12.016 [DOI] [PubMed] [Google Scholar]
- 6.Dzierzak E, Speck NA. 2008. Of lineage and legacy: the development of mammalian hematopoietic stem cells. Nat. Immunol. 9:129–136. 10.1038/ni1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boisset JC, van Cappellen W, Andrieu-Soler C, Galjart N, Dzierzak E, Robin C. 2010. In vivo imaging of haematopoietic cells emerging from the mouse aortic endothelium. Nature 464:116–120. 10.1038/nature08764 [DOI] [PubMed] [Google Scholar]
- 8.Tammela T, Alitalo K. 2010. Lymphangiogenesis: molecular mechanisms and future promise. Cell 140:460–476. 10.1016/j.cell.2010.01.045 [DOI] [PubMed] [Google Scholar]
- 9.Hagerling R, Pollmann C, Andreas M, Schmidt C, Nurmi H, Adams RH, Alitalo K, Andresen V, Schulte-Merker S, Kiefer F. 2013. A novel multistep mechanism for initial lymphangiogenesis in mouse embryos based on ultramicroscopy. EMBO J. 32:629–644. 10.1038/emboj.2012.340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang Y, Garcia-Verdugo JM, Soriano-Navarro M, Srinivasan RS, Scallan JP, Singh MK, Epstein JA, Oliver G. 2012. Lymphatic endothelial progenitors bud from the cardinal vein and intersomitic vessels in mammalian embryos. Blood 120:2340–2348. 10.1182/blood-2012-05-428607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vestweber D, Broermann A, Schulte D. 2010. Control of endothelial barrier function by regulating vascular endothelial-cadherin. Curr. Opin. Hematol. 17:230–236. 10.1097/MOH.0b013e328338664b [DOI] [PubMed] [Google Scholar]
- 12.Dejana E, Vestweber D. 2013. The role of VE-cadherin in vascular morphogenesis and permeability control. Prog. Mol. Biol. Transl. Sci. 116:119–144. 10.1016/B978-0-12-394311-8.00006-6 [DOI] [PubMed] [Google Scholar]
- 13.Carmeliet P, Lampugnani M-G, Moons L, Breviario F, Compernolle V, Bono F, Balconi G, Spagnuolo R, Oosthuyse B, Dewerchin M, Zanetti A, Angellilo A, Mattot V, Nuyens D, Lutgens E, Clotman F, de Ruiter MC, Gittenberger-de Groot A, Poelmann R, Lupu F, Herbert J-M, Collen D, Dejana E. 1999. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell 98:147–157. 10.1016/S0092-8674(00)81010-7 [DOI] [PubMed] [Google Scholar]
- 14.Gory-Faure S, Prandini MH, Pointu H, Roullot V, Pignot-Paintrand I, Vernet M, Huber P. 1999. Role of vascular endothelial-cadherin in vascular morphogenesis. Development 126:2093–2102 [DOI] [PubMed] [Google Scholar]
- 15.Crosby CV, Fleming PA, Argraves WS, Corada M, Zanetta L, Dejana E, Drake CJ. 2005. VE-cadherin is not required for the formation of nascent blood vessels but acts to prevent their disassembly. Blood 105:2771–2776. 10.1182/blood-2004-06-2244 [DOI] [PubMed] [Google Scholar]
- 16.Strilic B, Kucera T, Eglinger J, Hughes MR, McNagny KM, Tsukita S, Dejana E, Ferrara N, Lammert E. 2009. The molecular basis of vascular lumen formation in the developing mouse aorta. Dev. Cell 17:505–515. 10.1016/j.devcel.2009.08.011 [DOI] [PubMed] [Google Scholar]
- 17.Rampon C, Huber P. 2003. Multilineage hematopoietic progenitor activity generated autonomously in the mouse yolk sac: analysis using angiogenesis-defective embryos. Int. J. Dev. Biol. 47:273–280 [PubMed] [Google Scholar]
- 18.Schulte D, Kuppers V, Dartsch N, Broermann A, Li H, Zarbock A, Kamenyeva O, Kiefer F, Khandoga A, Massberg S, Vestweber D. 2011. Stabilizing the VE-cadherin-catenin complex blocks leukocyte extravasation and vascular permeability. EMBO J. 30:4157–4170. 10.1038/emboj.2011.304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wegmann F, Petri B, Khandoga AG, Moser C, Khandoga A, Volkery S, Li H, Nasdala I, Brandau O, Fassler R, Butz S, Krombach F, Vestweber D. 2006. ESAM supports neutrophil extravasation, activation of Rho, and VEGF-induced vascular permeability. J. Exp. Med. 203:1671–1677. 10.1084/jem.20060565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Breier G, Breviario F, Caveda L, Berthier R, Schnurch H, Gotsch U, Vestweber D, Risau W, Dejana E. 1996. Molecular cloning and expression of murine vascular endothelial-cadherin in early stage development of cardiovascular system. Blood 87:630–641 [PubMed] [Google Scholar]
- 21.de Boer J, Williams A, Skavdis G, Harker N, Coles M, Tolaini M, Norton T, Williams K, Roderick K, Potocnik AJ, Kioussis D. 2003. Transgenic mice with hematopoietic and lymphoid specific expression of Cre. Eur. J. Immunol. 33:314–325. 10.1002/immu.200310005 [DOI] [PubMed] [Google Scholar]
- 22.Potocnik AJ, Brakebusch C, Fassler R. 2000. Fetal and adult hematopoietic stem cells require beta1 integrin function for colonizing fetal liver, spleen, and bone marrow. Immunity 12:653–663. 10.1016/S1074-7613(00)80216-2 [DOI] [PubMed] [Google Scholar]
- 23.Rodewald HR, Kretzschmar K, Takeda S, Hohl C, Dessing M. 1994. Identification of pro-thymocytes in murine fetal blood: T lineage commitment can precede thymus colonization. EMBO J. 13:4229–4240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baumer S, Keller L, Holtmann A, Funke R, August B, Gamp A, Wolburg H, Wolburg-Buchholz K, Deutsch U, Vestweber D. 2006. Vascular endothelial cell-specific phosphotyrosine phosphatase (VE-PTP) activity is required for blood vessel development. Blood 107:4754–4762. 10.1182/blood-2006-01-0141 [DOI] [PubMed] [Google Scholar]
- 25.Bohmer R, Neuhaus B, Buhren S, Zhang D, Stehling M, Bock B, Kiefer F. 2010. Regulation of developmental lymphangiogenesis by Syk(+) leukocytes. Dev. Cell 18:437–449. 10.1016/j.devcel.2010.01.009 [DOI] [PubMed] [Google Scholar]
- 26.Schniedermann J, Rennecke M, Buttler K, Richter G, Städtler A-M, Norgall S, Badar M, Barleon B, May T, Weich HA. 2010. Mouse lung contains endothelial progenitors with high capacity to form blood and lymphatic vessels. BMC Cell Biol. 11. 10.1038/emboj.2012.340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liebner S, Cattelino A, Gallini R, Rudini N, Iurlaro M, Piccolo S, Dejana E. 2004. Beta-catenin is required for endothelial-mesenchymal transformation during heart cushion development in the mouse. J. Cell Biol. 166:359–367. 10.1083/jcb.200403050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cattelino A, Liebner S, Gallini R, Zanetti A, Balconi G, Corsi A, Bianco P, Wolburg H, Moore R, Oreda B, Kemler R, Dejana E. 2003. The conditional inactivation of the beta-catenin gene in endothelial cells causes a defective vascular pattern and increased vascular fragility. J. Cell Biol. 162:1111–1122. 10.1083/jcb.200212157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Corada M, Nyqvist D, Orsenigo F, Caprini A, Giampietro C, Taketo MM, Iruela-Arispe ML, Adams RH, Dejana E. 2010. The Wnt/beta-catenin pathway modulates vascular remodeling and specification by upregulating Dll4/Notch signaling. Dev. Cell 18:938–949. 10.1016/j.devcel.2010.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sasaki K, Sonoda Y. 2000. Histometrical and three-dimensional analyses of liver hematopoiesis in the mouse embryo. Arch. Histol. Cytol. 63:137–146. 10.1679/aohc.63.137 [DOI] [PubMed] [Google Scholar]
- 31.Yokomizo T, Dzierzak E. 2010. Three-dimensional cartography of hematopoietic clusters in the vasculature of whole mouse embryos. Development 137:3651–3661. 10.1242/dev.051094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dodt HU, Leischner U, Schierloh A, Jahrling N, Mauch CP, Deininger K, Deussing JM, Eder M, Zieglgansberger W, Becker K. 2007. Ultramicroscopy: three-dimensional visualization of neuronal networks in the whole mouse brain. Nat. Methods 4:331–336. 10.1038/nmeth1036 [DOI] [PubMed] [Google Scholar]
- 33.Kim I, Yilmaz OH, Morrison SJ. 2005. CD144 (VE-cadherin) is transiently expressed by fetal liver hematopoietic stem cells. Blood 106:903–905. 10.1182/blood-2004-12-4960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oberlin E, Fleury M, Clay D, Petit-Cocault L, Candelier JJ, Mennesson B, Jaffredo T, Souyri M. 2010. VE-cadherin expression allows identification of a new class of hematopoietic stem cells within human embryonic liver. Blood 116:4444–4455. 10.1182/blood-2010-03-272625 [DOI] [PubMed] [Google Scholar]
- 35.Ogilvy S, Metcalf D, Gibson L, Bath ML, Harris AW, Adams JM. 1999. Promoter elements of vav drive transgene expression in vivo throughout the hematopoietic compartment. Blood 94:1855–1863 [PubMed] [Google Scholar]
- 36.Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F. 2001. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 1:4. 10.1186/1471-213X-1-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baluk P, Fuxe J, Hashizume H, Romano T, Lashnits E, Butz S, Vestweber D, Corada M, Molendini C, Dejana E, McDonald DM. 2007. Functionally specialized junctions between endothelial cells of lymphatic vessels. J. Exp. Med. 204:2349–2362. 10.1084/jem.20062596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Küppers V, Vestweber D, Schulte D. 2013. Locking endothelial junctions blocks leukocyte extravasation, but not in all tissues. Tissue Barriers 1:e23805. 10.4161/tisb.23805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jakobsson L, Franco CA, Bentley K, Collins RT, Ponsioen B, Aspalter IM, Rosewell I, Busse M, Thurston G, Medvinsky A, Schulte-Merker S, Gerhardt H. 2010. Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat. Cell Biol. 12:943–953. 10.1038/ncb2103 [DOI] [PubMed] [Google Scholar]
- 40.Pacquelet A, Rorth P. 2005. Regulatory mechanisms required for DE-cadherin function in cell migration and other types of adhesion. J. Cell Biol. 170:803–812. 10.1083/jcb.200506131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Woodfin A, Voisin MB, Beyrau M, Colom B, Caille D, Diapouli FM, Nash GB, Chavakis T, Albelda SM, Rainger GE, Meda P, Imhof BA, Nourshargh S. 2011. The junctional adhesion molecule JAM-C regulates polarized transendothelial migration of neutrophils in vivo. Nat. Immunol. 12:761–769. 10.1038/ni.2062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grazia Lampugnani M, Zanetti A, Corada M, Takahashi T, Balconi G, Breviario F, Orsenigo F, Cattelino A, Kemler R, Daniel TO, Dejana E. 2003. Contact inhibition of VEGF-induced proliferation requires vascular endothelial cadherin, beta-catenin, and the phosphatase DEP-1/CD148. J. Cell Biol. 161:793–804. 10.1083/jcb.200209019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sahin AO, Buitenhuis M. 2012. Molecular mechanisms underlying adhesion and migration of hematopoietic stem cells. Cell Adhes. Migration 6:39–48. 10.4161/cam.18975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Buul JD, Voermans C, van den Berg V, Anthony EC, Mul FP, van Wetering S, van der Schoot CE, Hordijk PL. 2002. Migration of human hematopoietic progenitor cells across bone marrow endothelium is regulated by vascular endothelial cadherin. J. Immunol. 168:588–596 [DOI] [PubMed] [Google Scholar]
- 45.Kamei M, Saunders WB, Bayless KJ, Dye L, Davis GE, Weinstein BM. 2006. Endothelial tubes assemble from intracellular vacuoles in vivo. Nature 442:453–456. 10.1038/nature04923 [DOI] [PubMed] [Google Scholar]
- 46.Herwig L, Blum Y, Krudewig A, Ellertsdottir E, Lenard A, Belting HG, Affolter M. 2011. Distinct cellular mechanisms of blood vessel fusion in the zebrafish embryo. Curr. Biol. 21:1942–1948. 10.1016/j.cub.2011.10.016 [DOI] [PubMed] [Google Scholar]
- 47.Wang Y, Kaiser MS, Larson JD, Nasevicius A, Clark KJ, Wadman SA, Roberg-Perez SE, Ekker SC, Hackett PB, McGrail M, Essner JJ. 2010. Moesin1 and Ve-cadherin are required in endothelial cells during in vivo tubulogenesis. Development 137:3119–3128. 10.1242/dev.048785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Planas-Paz L, Strilic B, Goedecke A, Breier G, Fassler R, Lammert E. 2012. Mechanoinduction of lymph vessel expansion. EMBO J. 31:788–804. 10.1038/emboj.2011.456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen XL, Nam JO, Jean C, Lawson C, Walsh CT, Goka E, Lim ST, Tomar A, Tancioni I, Uryu S, Guan JL, Acevedo LM, Weis SM, Cheresh DA, Schlaepfer DD. 2012. VEGF-induced vascular permeability is mediated by FAK. Dev. Cell 22:146–157. 10.1016/j.devcel.2011.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Monaghan-Benson E, Burridge K. 2009. The regulation of vascular endothelial growth factor-induced microvascular permeability requires Rac and reactive oxygen species. J. Biol. Chem. 284:25602–25611. 10.1074/jbc.M109.009894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huveneers S, Oldenburg J, Spanjaard E, van der Krogt G, Grigoriev I, Akhmanova A, Rehmann H, de Rooij J. 2012. Vinculin associates with endothelial VE-cadherin junctions to control force-dependent remodeling. J. Cell Biol. 196:641–652. 10.1083/jcb.201108120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yonemura S, Wada Y, Watanabe T, Nagafuchi A, Shibata M. 2010. Alpha-catenin as a tension transducer that induces adherens junction development. Nat. Cell Biol. 12:533–542. 10.1038/ncb2055 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.