

Abstract
Mutations in components of the Wnt/β-catenin signaling pathway are commonly found in colorectal cancers, and these mutations cause aberrant expression of genes controlled by Wnt-responsive DNA elements (WREs). While the c-Myc proto-oncogene (Myc) is required for intestinal phenotypes associated with pathogenic Wnt/β-catenin signaling in vivo, the WREs that control Myc expression in this setting have yet to be fully described. Previously, we demonstrated that the Myc 3′ WRE was required for intestinal homeostasis and intestinal repair in response to damage. Here, we tested the role of the Myc 3′ WRE in intestinal tumorigenesis using two independent mouse models. In comparison to ApcMin/+ mice, ApcMin/+ Myc 3′ WRE−/− mice contained 25% fewer tumors in the small intestine. Deletion of the Myc 3′ WRE−/− in the ApcMin/+ background resulted in 4-fold more colonic tumors. In a model of colitis-associated colorectal cancer, the Myc 3′ WRE suppressed colonic tumorigenesis, most notably within the cecum. Using chromatin immunoprecipitation and transcript analysis of purified colonic crypts, we found that the Myc 3′ WRE is required for the transcriptional regulation of Myc expression in vivo. These results emphasize the critical role of the Myc 3′ WRE in maintaining homeostatic Myc expression.
INTRODUCTION
The Wnt/β-catenin pathway governs cellular proliferation of the intestinal epithelium by controlling the expression of growth-promoting genes through Wnt-responsive DNA elements (WREs) (1, 2). The β-catenin transcriptional coactivator is the key regulatory protein in the Wnt pathway, and as such, its subcellular localization and protein levels are tightly regulated (3, 4). In the absence of an extracellular Wnt ligand, cytosolic β-catenin is targeted for proteasomal degradation by the multiprotein “destruction complex.” Under these conditions, members of the T-cell factor/lymphoid enhancer factor (TCF/Lef) (here called TCF) family of sequence-specific transcription factors recruit transducin-like enhancer (TLE) corepressors to repress Wnt target expression (5). In the presence of Wnt, the destruction complex is inactivated, and β-catenin is translocated into the nucleus, where it displaces corepressor complexes and recruits coactivator complexes to activate the expression of underlying gene targets. One critical and direct target is the c-MYC proto-oncogene (MYC) (6, 7). MYC is a transcription factor that promotes cell growth and cell cycle progression by predominantly activating the expression of genes whose products drive DNA replication, ribosome biogenesis, and metabolism (8, 9).
Mutations in components of the Wnt/β-catenin signaling pathway, most commonly in the adenomatous polyposis coli (APC) gene, are found in over 80% of spontaneously arising colorectal cancers (10). These mutations lead to the inappropriate expression of Wnt/β-catenin target genes. Several studies in mouse models of colorectal cancer indicated that Myc is required for the intestinal pathogenesis associated with deregulated Wnt/β-catenin signaling (11–15). However, those studies involved either heterozygous Myc mice or the removal of Myc coding sequences in the intestines and therefore did not evaluate the precise role of Wnt/β-catenin-dependent regulation of Myc expression and intestinal cancers. An understanding of this relationship requires defining the WREs that control Myc expression. The connection between Wnt/β-catenin signaling and MYC was made by using a screen designed to identify genes whose expression levels were influenced when full-length APC was restored in a human colorectal cancer (CRC) cell line (6). That same study mapped the first MYC WRE to the 5′-proximal promoter region of MYC approximately 600 bp upstream from the transcription start site (6). To search for additional WREs that control MYC expression, we conducted two genome-wide screens to identify β-catenin-bound regions in the genome of a human CRC cell line (16, 17). These screens found a robust β-catenin binding region that mapped 1.4 kb downstream from the MYC transcriptional stop site. We demonstrated that this β-catenin-bound region demarcated an enhancer element that we termed the MYC 3′ WRE (7). β-Catenin/TCF4 complexes that bound to the MYC 3′ WRE coordinated a chromatin loop with the proximal MYC 5′ promoter region to activate MYC expression in response to mitogen and Wnt/β-catenin signaling pathways (18).
Concurrent with our work on the MYC 3′ WRE, the discovery of a distal and upstream WRE was reported (19–21). This kb −335 WRE incorporates the single-nucleotide polymorphism (SNP) rs6983267. This particular SNP is associated with an increased risk of developing colorectal, breast, and prostate cancers, and it maps adjacent to a TCF binding motif (22–24). While this element has been shown to juxtapose to the MYC 5′ promoter region through a long-range chromatin loop, whether this conformation regulates MYC gene expression is controversial (19, 21). Since the discovery of this initial set of WREs, numerous WREs that regulate MYC expression in human CRC cells have been reported, and many form dynamic and cell-type-specific chromatin loops with the MYC promoter region (25–27). Of the many MYC WREs described to date, only two have been characterized in mice (28, 29). While the Myc −335 WRE was dispensable for intestinal development and homeostasis, ApcMin/+ Myc −335 WRE−/− mice contained fewer intestinal adenomas than did ApcMin/+ mice (29). Our group generated a Myc 3′ WRE−/− mouse and found that the deletion of this element mildly compromised the architecture of the small and large intestines (28). Analysis of Myc expression in the intestines of juvenile mice indicated that the primary role of the Myc 3′ WRE is to repress Myc expression (28). While Myc 3′ WRE−/− small intestines contained only a slight elevation in Myc mRNA levels, this effect was variable in littermates (28). In the colons, deletion of the Myc 3′ WRE caused a highly reproducible 2-fold increase in Myc mRNA levels and a 2.5-fold increase in MYC protein levels (28). In both the small intestines and colons of Myc 3′ WRE−/− mice, there was an increase in the number of proliferative cells and a decrease in the number of differentiated cells in comparison to the colons of wild-type (WT) littermates. When subjected to a model of acute colitis, the Myc 3′ WRE−/− colons displayed an enhanced regenerative response to repair the damaged tissue (28, 30).
In the present study, we hypothesized that the Myc 3′ WRE regulates intestinal tumorigenesis. To test this hypothesis, we bred Myc 3′ WRE−/− mice to ApcMin/+ mice and compared the numbers of adenomas in these mice to the numbers of adenomas in ApcMin/+ mice. In a separate line of experiments, we tested whether the Myc 3′ WRE influenced tumorigenesis using a chemically induced model of colorectal cancer in mice. Our results indicate that the Myc 3′ WRE suppresses colorectal carcinogenesis and that it is required for proper control of Myc gene expression in mouse colonic crypts.
MATERIALS AND METHODS
Mice.
ApcMin/+ mice were obtained from Jackson Laboratories, and the generation of Myc 3′ WRE−/− mice was described previously (28). ApcMin/+ mice were bred to Myc 3′ WRE−/− mice to obtain ApcMin/+ Myc 3′ WRE−/− mice. The Myc 3′ WRE−/− mice were backcrossed to C57/B6 mice for six generations prior to breeding to ApcMin/+ mice. DNA was isolated from tail biopsy specimens at weaning, and PCR was used to genotype the mice by using methods previously described (28). The following primers were used to genotype ApcMin/+ mice according to guidelines provided by Jackson Laboratories: oIMR0033 (5′-GCC ATC CCT TCA CGT TAG-3′), oIMR0034 (5′-TTC CAC TTT GGC ATA AGG C-3′), and oIMR0758 (5′-TTC TGA GAA AGA CAG AAG TTA-3′). Mice were housed in accordance with Penn State University College of Medicine Institutional Animal Care and Use Committee (PSUCOM IACUC) regulations. In addition, the PSUCOM IACUC approved all mouse experiments conducted in this study.
Assessment of polyps.
ApcMin/+ and ApcMin/+ Myc 3′ WRE−/− mice were examined at 14 weeks of age for polyp burden. This time point is sufficient to assess the formation of adenomas under a dissecting microscope, and it is within the range used in previous studies assessing the role of Myc in the ApcMin/+ background (12, 15). The small and large intestines were dissected, opened longitudinally, rinsed with 1× phosphate-buffered saline (PBS), and fixed overnight in 3.7% paraformaldehyde. The fixed sections were rinsed with deionized water, stained with 0.2% methylene blue, and then rinsed in ethanol. Polyps were counted at a ×20 magnification on a dissecting microscope and measured by using standard calipers.
Crypt isolation, reverse transcription, and real-time PCR analysis.
Crypts were isolated from the colons of 10-week-old ApcMin/+ and ApcMin/+ Myc 3′ WRE−/− mice and 7-week-old WT and Myc 3′ WRE−/− mice by using a detailed protocol described previously by Sato et al. (31). Briefly, colons were dissected, opened longitudinally, rinsed with cold 1× PBS, and cut into 5-mm sections. The sections were rinsed two additional times in cold 1× PBS, followed by incubation for 30 min at 4°C in cold chelation buffer (5.6 mM Na2HPO4, 8 mM KH2PO4, 96.2 mM NaCl, 1.6 mM KCl, 43.4 mM sucrose, 54.9 mM d-sorbitol, 0.5 mM dithiothreitol [DTT]) containing 2 mM EDTA. The EDTA-containing chelation buffer was removed, and the crypts were resuspended in ice-cold chelation buffer. The crypts were separated mechanically by pipetting multiple times with a 10-ml pipette and then allowed to settle on a 10-cm dish under normal gravity. This resuspension, pipetting, and settling procedure was repeated 8 times, with the supernatant being collected and replaced with fresh chelation buffer after each repetition. The crypts were then passed through a 70-μm cell strainer to further purify them away from supportive tissue and collected by centrifugation at 200 × g for 3 min in a 50-ml conical tube. The crypts were incubated for 15 min at 37°C in 5 ml of TrypLE Express (Invitrogen). This treatment releases the upper differentiated portion of the crypts, while the lower proliferative portion is collected by centrifugation at 200 × g for 3 min. Total RNA was extracted from the proliferative portion by using TRIzol reagent, and cDNAs were synthesized with reverse transcriptase as previously described (28). For experiments involving mitogen treatments, the proliferative compartments of the crypts were resuspended in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS) and incubated on a rocking platform in the presence or absence of 100 ng/ml Wnt3A (catalog number 315-20; Peprotech), 500 ng/ml R-spondin1 (catalog number 3474-RS; R&D), and 20 ng/ml epidermal growth factor (EGF) (catalog number 315-09; Peprotech) for 1 or 3 h at 37°C. Gene expression levels were measured by quantitative real-time PCR using parameters previously described (28). Relative levels were determined by using the 2ΔCT method, with the β-actin gene serving as the reference gene. The results were confirmed by using 18S rRNA as the internal standard. Primer sequences are listed in Table S1 in the supplemental material.
Immunohistochemistry.
Tissue collection, processing, and immunohistochemistry were performed as previously described (28). The sections were incubated overnight at 4°C with primary antibodies diluted in 1% bovine serum albumin. MYC staining required a 4-day incubation period with primary antibody (28, 32). The following antibodies and dilutions were used: anti-cleaved caspase 3 (anti-CASP3) at 1:200 (catalog number 9661; Cell Signaling), anti-Ki67 at 1:200 (catalog number VP-RM04; Vector Laboratories), anti-MYC at 1:500 (catalog number SC-764; Santa Cruz), and anti-β-catenin at 1:200 (catalog number 9582; Cell Signaling).
Colitis-associated carcinogenesis.
Colitis-associated carcinogenesis (CAC) was induced in mice by using a protocol described previously by Greten et al. (33). Six-week-old WT and Myc 3′ WRE−/− mice were given a single intraperitoneal injection of 10 mg/kg of body weight of azoxymethane (AOM) (catalog number A5486; Sigma) dissolved in 200 μl 1× PBS. A week after the injection, the mice were administered 2.0% dextran sodium sulfate (DSS) (catalog number AB DB001-299; TdB Consulting) in their drinking water for 5 days, followed by 2 weeks of normal water. Chronic inflammation was induced by two subsequent cycles of 2.5% DSS for 5 days, with 2 weeks of normal water between cycles. Mice were euthanized 85 days after AOM injection and examined for colonic tumors as described above.
Chromatin immunoprecipitation.
Crypts were prepared and treated with or without recombinant Wnt3A, R-spondin1, and EGF for 3 h as described above for crypt preparation, reverse transcription, and real-time PCR. Crypts from one mouse provide sufficient material for two chromatin immunoprecipitation (ChIP) assays. The samples were then treated with 1% formaldehyde for 30 min, and cross-linking was stopped by the addition of 125 mM glycine. ChIP was performed as described previously by Mahmoudi et al. (34). The crypts were first washed twice in buffer B (0.25% Triton X-100, 1 mM EDTA, 0.5 mM EGTA, 20 mM HEPES [pH 7.6]) and twice in buffer C (150 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 20 mM HEPES [pH 7.6]). The crypts were then resuspended in resuspension buffer (0.3% SDS, 1% Triton X-100, 0.15 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 20 mM HEPES [pH 7.6]) containing freshly added protease inhibitors (10 μg/ml leupeptin, 10 μg/ml aprotinin, 1 mM phenylmethylsulfonyl fluoride [PMSF]). The cross-linked DNA was sheared by using a BioRupter sonicator (model UCD-200; Diagenode) with a total of 12 30-s bursts at the maximum setting. Insoluble material was removed by centrifugation at 21,000 × g for 10 min at 4°C. To precipitate chromatin, 5 μg of primary antibodies and a 20-μl bed of blocked protein A beads were added, and the samples were incubated overnight at 4°C on a rocking platform. Antibodies used for ChIP were antibodies to TCF4 (catalog number 05-511; Millipore), β-catenin (catalog number 610154; BD Transduction), RNA polymerase (RNAP) (catalog number 8WG16; Covance), H3K4me3 (catalog number 39159; Active Motif), TLE2 (catalog number SC-9123; Santa Cruz), and CDX2 (catalog number PA5-20891; Pierce). The precipitated complexes were washed twice with each of the following buffers: buffer 1 (0.1% SDS, 0.1% deoxycholate, 1% Triton X-100, 150 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 20 mM HEPES [pH 7.6]), buffer 2 (0.1% SDS, 0.1% deoxycholate, 1% Triton X-100, 0.5 M NaCl, 1 mM EDTA, 0.5 mM EGTA, 20 mM HEPES [pH 7.6]), buffer 3 (250 mM LiCl, 0.5% deoxycholate, 0.5% NP-40, 1 mM EDTA, 0.5 mM EGTA, 20 mM HEPES [pH 7.6]), and buffer 4 (1 mM EDTA, 0.5 mM EGTA, 20 mM HEPES [pH 7.6]). Following a single wash with water, the chromatin was released from the antibodies by two 15-min incubations in elution buffer (1% SDS, 0.1 M NaHCO3). The cross-links were reversed by incubating samples overnight at 65°C in elution buffer containing 200 mM NaCl. After removal of proteins via phenol-chloroform-isoamyl alcohol extraction and chloroform back extraction, the DNA was precipitated in ethanol. The DNA pellets were resuspended in 150 μl Tris-EDTA (TE), and the samples were subjected to quantitative real-time PCR as described previously (16). Oligonucleotides used to detect binding to the Myc 5′ and 3′ WREs as well as the control region are listed in Table S1 in the supplemental material.
Statistical analysis.
For the immunohistochemical analysis, stained cells were quantified from 25 crypts per animal for normal tissue or from 10 high-powered fields of view per animal for tumor tissue (n = 4 mice per genotype), and P values were determined by using the Mann-Whitney U test. For the experiments involving tumor load, tumor location, gene expression, and ChIP, significant differences in the data were assessed by using the Student t test. Ten mice per genotype were assessed in the ApcMin/+ experiments, and four mice per genotype were examined in the AOM/DSS experiments. For gene expression and ChIP experiments, three mice per genotype or condition were evaluated.
RESULTS
Deletion of the Myc 3′ WRE reduces the number of small intestinal tumors in ApcMin/+ mice.
The original adenomatous polyposis coli multiple intestinal neoplasia (ApcMin/+) mouse was generated by using an N-ethyl-N-nitrosourea (ENU) mutagenesis screen (35). A point mutation in Apc at codon 850 converts it from a leucine to a stop codon. The truncated APC protein produced from this allele is incapable of downregulating β-catenin, and as a result, numerous intestinal tumors develop in ApcMin/+ mice. For reasons not fully appreciated, adenomas form primarily in the small intestines of ApcMin/+ mice, with few tumors developing in the colon. To determine whether the Myc 3′ WRE regulated intestinal tumorigenesis, we crossed ApcMin/+ mice with Myc 3′ WRE−/− mice to generate ApcMin/+ Myc 3′ WRE−/− mice. We then harvested the small intestines of 14-week-old ApcMin/+ and ApcMin/+ Myc 3′ WRE−/− mice and quantified the number of adenomas using a dissecting microscope. The intestines of ApcMin/+ Myc 3′ WRE−/− mice contained a reduced number of tumors, most notably within the duodenum, compared to the intestines of ApcMin/+ mice (Fig. 1A and B). We next measured the size of the tumors using standard calipers and found that ApcMin/+ Myc 3′ WRE−/− mice contained fewer intermediate (0.5- to 2.0-mm) and large (>2.0-mm) tumors (Fig. 1C). While it appears that the deletion of the Myc 3′ WRE also reduced the number of small tumors (<0.5 mm) in an ApcMin/+ background, this effect did not reach statistical significance.
FIG 1.
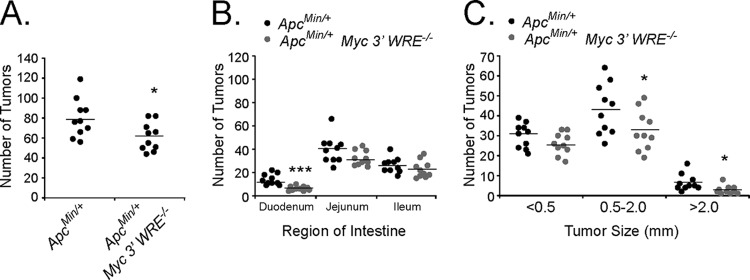
Deletion of the Myc 3′ WRE reduces the number of small intestinal tumors in ApcMin/+ mice. (A) Number of tumors in the small intestines of 14-week-old ApcMin/+ and ApcMin/+ Myc 3′ WRE−/− mice. (B) Number of tumors in the indicated region of the small intestines. (C) Size of tumors quantified within the small intestines of mice with the indicated genotypes. In panels A to C, tumors were quantified in 10 ApcMin/+ and 10 ApcMin/+ Myc 3′ WRE−/− mice. Errors are standard errors of the means (*, P < 0.05; ***, P < 0.001).
Deletion of the Myc 3′ WRE increases the number of colonic tumors in ApcMin/+ mice.
A previous study indicated that approximately half of a panel of Wnt/β-catenin target genes required Myc for expression within mouse intestines (13). In addition, MYC has been implicated as a global amplifier of gene expression in mouse embryonic stem cells, mouse lymphocytes, and human cancer cell lines (36, 37). As the deletion of the Myc 3′ WRE causes a 2.5-fold increase in colonic MYC protein levels, we wished to determine whether the Myc 3′ WRE regulated tumorigenesis in the colon (28). To initiate these studies, we first examined the expression of a panel of Wnt/β-catenin target genes in the colons of ApcMin/+ and ApcMin/+ Myc−/− mice at a time prior to adenoma formation (10 weeks of age). RNAs were isolated from purified colonic crypt preparations, and cDNAs were synthesized by using reverse transcriptase. We assessed gene expression levels using gene-specific oligonucleotides and quantitative real-time PCR analysis. As expected, Myc expression levels were higher in the colonic crypts of ApcMin/+ Myc 3′ WRE−/− mice than in those of ApcMin/+ mice (Fig. 2). Moreover, expression levels of 14 of the 25 targets analyzed (56%) were also elevated, indicating that the deletion of the Myc 3′ WRE, and the subsequent increase in MYC protein levels, activates the expression of a subset of Wnt/β-catenin target genes. We next analyzed colonic adenomas in ApcMin/+ Myc 3′ WRE−/− and ApcMin/+ mice. In a cohort of 10 mice per genotype (14 weeks of age), we found that the colons of ApcMin/+ Myc 3′ WRE−/− mice harbored an average of 4.5 tumors and that the colons of ApcMin/+ mice contained an average of 1.5 tumors (Fig. 3A). In fact, the colons of 70% of ApcMin/+ Myc 3′ WRE−/− mice contained three or more tumors, whereas this tumor burden was observed in only 10% of ApcMin/+ mouse colons (Fig. 3B). Upon closer examination, deletion of the Myc 3′ WRE in an ApcMin/+ background caused increased numbers of small (<0.5-mm) and intermediate (0.5- to 2.0-mm) tumors but decreased the formation of larger tumors (>2.0 mm) in mice at this time point (Fig. 3C). Together, these results indicate that the Myc 3′ WRE suppresses colorectal tumors that are caused by a mutation in Apc.
FIG 2.
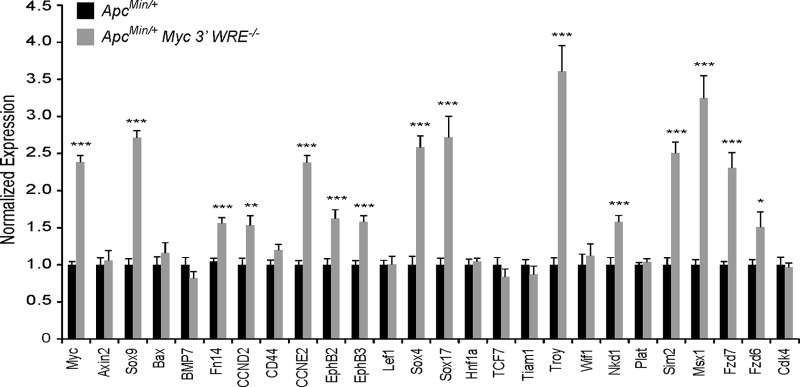
Expression of Wnt/β-catenin target genes in the colons of preneoplastic ApcMin/+ and ApcMin/+ Myc 3′ WRE−/− mice. RNAs were isolated from purified colonic crypts, and cDNAs were synthesized with reverse transcriptase. Expression levels of the indicated genes were assessed by using gene-specific oligonucleotides in quantitative and real-time PCRs (n = 3 mice per genotype and 12 PCR replicates per gene). The data are normalized to β-actin gene levels. The data are presented as relative expression levels in ApcMin/+ Myc 3′ WRE−/− crypts, with levels in ApcMin/+ crypts set to 1. Errors are standard errors of the means (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
FIG 3.
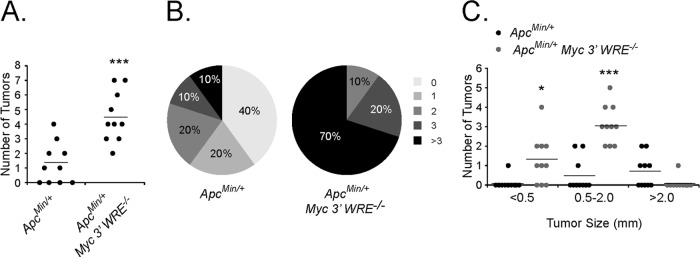
Deletion of the Myc 3′ WRE increases the number of colonic tumors in ApcMin/+ mice. (A) Number of tumors in the colons of 14-week-old ApcMin/+ and ApcMin/+ Myc 3′ WRE−/− mice (n = 10 mice analyzed per genotype). (B) Percentage of mice in each group that contained the indicated number of tumors. (C) Size of tumors quantified within the colons of mice with the indicated genotypes (n = 10 mice analyzed per genotype). In panels A and C, errors are standard errors of the means (*, P < 0.05; ***, P < 0.001).
The dosage of MYC protein levels is critical to intestinal tumorigenesis caused by Apc deficiencies in mouse models (11–15). The removal of either a single copy or both copies of Myc reduces the small intestinal tumor burden in ApcMin/+ mice (12, 15). MYC is a potent regulator of cellular proliferation and growth; however, at elevated levels, MYC can also induce apoptosis (38, 39). To determine whether the Myc 3′ WRE was regulating proliferation or apoptosis of the ApcMin/+ colonic epithelium, we conducted immunohistochemical analyses on colonic sections prepared from preneoplastic (10-week-old) ApcMin/+ Myc 3′ WRE−/− and ApcMin/+ mice. Sections were stained with antibodies directed against Ki67, to identify proliferative cells, and antibodies against cleaved caspase 3 (CASP3), to identify cells undergoing apoptosis. On average, and in comparison to ApcMin/+ mice, the colonic epithelium of ApcMin/+ Myc 3′ WRE−/− mice contained 4.5-fold-higher levels of Ki67+ cells per crypt (Fig. 4A and B). In addition, the colonic crypts of ApcMin/+ Myc 3′ WRE−/− mice displayed a 3-fold increase in the number of Ki67+ cells that occupied the middle and upper regions of the crypt. We noted no difference in the numbers of apoptotic cells. Next, we analyzed sections of colonic tumors prepared from age-matched (14-week-old) mice. While the tumor sections of both ApcMin/+ Myc 3′ WRE−/− and ApcMin/+ mice contained higher numbers of Ki67+ cells than those in preneoplastic colonic epithelia, the deletion of the Myc 3′ WRE only slightly increased the number of proliferative cells in an ApcMin/+ background (Fig. 4A and C). Colonic tumors in mice of both genotypes contained 6-fold-higher numbers of CASP3+ cells than those in uninvolved colonic mucosa; however, the numbers of apoptotic cells did not differ in the tumors of ApcMin/+ Myc 3′ WRE−/− and ApcMin/+ mice (Fig. 4A and C).
FIG 4.
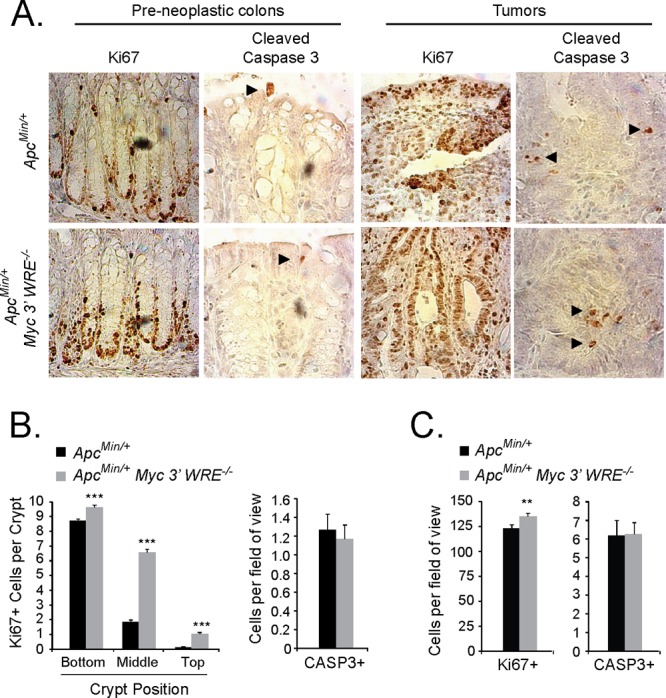
The Myc 3′ WRE controls proliferation but not apoptosis in the colons of ApcMin/+ mice. (A) Ki67- and cleaved caspase 3 (CASP3)-stained sections of preneoplastic colons and colonic tumors from ApcMin/+ (top) and ApcMin/+ Myc 3′ WRE−/− (bottom) mice. Representative images are shown (n = 4 mice analyzed per genotype). Arrowheads indicate CASP3+ cells. (B) Quantification of Ki67+ (left) and CASP3+ (right) cells in preneoplastic colons of ApcMin/+ and ApcMin/+ Myc 3′ WRE−/− mice (n = 4 mice analyzed, with cells counted in a total of 100 crypts per genotype). (C) Same as panel B except that Ki67+ and CASP3+ cells were analyzed in tumor sections prepared from mice with the indicated genotypes (n = 4 mice, with 40 fields of view examined per genotype). In panels B and C, errors are standard errors of the means (**, P < 0.01; ***, P < 0.001).
Deletion of the Myc 3′ WRE increases the number of colitis-associated colonic tumors.
We next determined whether the Myc 3′ WRE influenced tumorigenesis using the azoxymethane/dextran sodium sulfate (AOM/DSS) model of colitis-associated carcinogenesis (CAC) (33). Wild-type (WT) and Myc 3′ WRE−/− mice were first given a single intraperitoneal injection of the AOM mutagen (Fig. 5A). After a recovery period, the mice were given DSS in their drinking water for 5 days and were then returned to normal drinking water. Following two additional DSS/recovery cycles and an extended recovery period, mice were sacrificed, and the colons were harvested. Deletion of the Myc 3′ WRE caused a higher number of colonic tumors in this model, which were primarily of intermediate size (size, 0.5 to 2.0 mm) (Fig. 5B to D).
FIG 5.
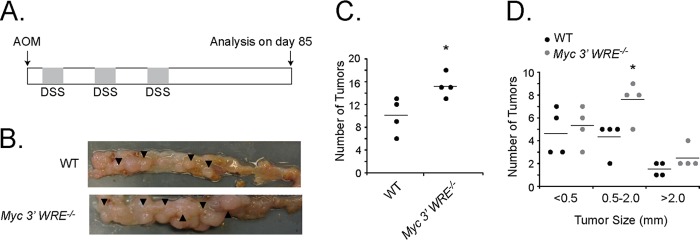
The Myc 3′ WRE suppresses colitis-associated carcinogenesis (CAC). (A) Schematic of the CAC protocol. Myc 3′ WRE−/− mice and WT littermates were given a single intraperitoneal injection of azoxymethane (AOM) and then subjected to 3 cycles of DSS-induced colitis. After 85 days, mice were sacrificed, and tumors within the distal colon were tallied. (B) Representative images of colons isolated from WT and Myc 3′ WRE−/− mice subjected to the AOM/DSS protocol. (C) Numbers of tumors in the colons of WT and Myc 3′ WRE−/− mice subjected to the AOM/DSS protocol. (D) Size of tumors quantified within the colons of mice with the indicated genotypes. In panels C and D, 4 mice were examined per genotype, and errors are standard errors of the means (*, P < 0.05).
The cecum is located immediately posterior from the ileum of the small intestines, and it marks the beginning of the large intestines. We noticed that the ceca of Myc 3′ WRE−/− mice subjected to the CAC protocol contained an average of 20 tumors, whereas none were found in this region of WT mice (Fig. 6A and B). We then prepared cecal sections, stained them with hematoxylin and eosin, and found that while the epithelial architecture of WT ceca was largely intact, Myc 3′ WRE−/− ceca displayed a highly irregular and disorganized phenotype (Fig. 6C). We then conducted a series of immunohistochemical analyses to further characterize the cecal sections (Fig. 6B). Interestingly, β-catenin localized to the nucleus in cells dispersed throughout the cecal crypt in WT mice. This is in contrast to the expected location of β-catenin in the small intestines and descending colon, where its nuclear localization is found predominantly in cells that receive a Wnt signal along the lower one-third of the crypt (40). Most cells within the tumor sections prepared from Myc 3′ WRE−/− mice stained positive for nuclear β-catenin (Fig. 6B). MYC-expressing cells, on the other hand, were largely confined to base of cecal crypts in WT mice. Similar to the β-catenin staining pattern, most cells dispersed throughout the Myc 3′ WRE−/− tumors contained nuclear MYC. Together, these results suggest that nuclear β-catenin and nuclear MYC may contribute to tumorigenesis within the cecum in mice subjected to CAC.
FIG 6.

Myc 3′ WRE−/− mice subjected to the AOM/DSS carcinogenesis protocol develop numerous tumors in the cecum. (A) Representative images of ceca isolated from WT and Myc 3′ WRE−/− mice following AOM and DSS treatment. (B) Quantification of cecal tumors in mice with the indicated genotypes (n = 4 mice examined per group). Errors are standard errors of the means (**, P < 0.01). (C) Hematoxylin and eosin (H&E)-stained sections and immunohistochemical analysis of β-catenin- and MYC-expressing cells in the ceca of mice with the indicated genotypes. The arrowheads in the enlarged panels identify a subset of positive cells. Shown are representative images from 4 mice examined per genotype.
The Myc 3′ WRE is required for proper control of Myc gene expression in colonic crypts.
To gain a better understanding of how the Myc 3′ WRE functions to repress colorectal carcinogenesis, we conducted a series of experiments to analyze this element in the colonic epithelium. In our previous study, we reported that the colons of juvenile Myc 3′ WRE−/− mice contained elevated levels of Myc mRNA and MYC protein in comparison to the colons of WT littermates (28). Those experiments involved RNAs and proteins prepared from gross colonic sections, which contain a heterogeneous population of cells. To determine whether the Myc 3′ WRE functioned in the colonic crypts, we first purified these structures from the colons of 7-week-old Myc 3′ WRE−/− and WT mice. RNAs were isolated from the proliferative compartments of the crypts, cDNAs were synthesized, and Myc transcript levels were measured by using quantitative real-time PCR. Deletion of the Myc 3′ WRE caused a 2.5-fold increase in the levels of Myc mRNAs relative to the control (Fig. 7A).
FIG 7.

The Myc 3′ WRE is required for proper regulation of Myc gene expression in purified colonic crypts. (A) Relative levels of Myc mRNA, as assessed by quantitative reverse transcription-PCR, in purified colonic crypts from WT and Myc 3′ WRE−/− mice. (B) Schematic of the Myc gene locus, with its position on chromosome 15 indicated above and the Myc 5′ and 3′ WREs represented as white rectangles. Gray rectangles indicate the positions of the amplicons produced in the PCRs. (C to G) Colonic crypts were isolated from 7-week-old Myc 3′ WRE−/− mice and WT littermates and fixed in formaldehyde. ChIP assays were conducted by using the antibodies indicated, and the precipitated DNA was measured by using specific oligonucleotides to produce the indicated amplicons in quantitative and real-time PCR assays. Control primer sets 1 and 9 were used to monitor the level of background in the ChIP assays. In panels A and C to G, 3 mice per genotype were examined, with 12 total PCR replicates per sample. Errors are standard errors of the means (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
Next, we conducted chromatin immunoprecipitation (ChIP) experiments to analyze the Myc proximal promoter region. The precipitated DNA was measured in quantitative real-time PCR assays using oligonucleotides designed against the Myc 5′ WRE and regions 1.8 kb upstream from the Myc 5′ WRE and 8.5 kb downstream from the Myc 3′ WRE as controls (Fig. 7B). Increased levels of RNA polymerase 2 (RNAP) were found at the Myc promoter in the crypts prepared from Myc 3′ WRE−/− mice relative to levels seen in WT crypts (Fig. 7C). Minimal signals were detected at the control regions, which attests to the specificity of the ChIP assays. Histone H3 that is trimethylated on lysine 4 (H3K4me3) is commonly found within the promoter regions of actively transcribed genes, including MYC (7, 41, 42). Deletion of the Myc 3′ WRE caused increased levels of H3K4me3 at the Myc promoter region (Fig. 7D).
To gain a better understanding of the underlying mechanisms that account for the increased Myc transcriptional activity in Myc 3′ WRE−/− colons, we interrogated transcriptional regulatory factors that control the Wnt response. TCFs are sequence-specific transcription factors that bind WREs and function as platforms to mediate the exchange of corepressor complexes with coactivator complexes (1, 5). TCF4 (also referred to as TCF7L2) is the major TCF member expressed in intestinal epithelial cells (43), and we noted no difference in TCF4 occupancy at the Myc 5′ WRE in knockout versus WT colons (Fig. 7E). In contrast, we found that the deletion of the Myc 3′ WRE caused a 2.5-fold increase in the levels of β-catenin at the Myc 5′-proximal promoter region (Fig. 7F). Using anti-TLE2 antibodies in the ChIP assay, we found reduced levels of this transcriptional corepressor at the Myc 5′ WRE within Myc 3′ WRE−/− colons (Fig. 7G). Together, these results indicate that the elevated levels of Myc transcript in Myc 3′ WRE colons correlate with increased recruitment of β-catenin and RNAP and a decrease in TLE2 occupancy at the Myc proximal promoter.
We next tested whether the Myc 3′ WRE was required for the induction of Myc gene expression in response to mitogens. Purified crypts from WT and Myc 3′ WRE−/− mice were treated with Wnt3A, epidermal growth factor (EGF), and R-spondin1 for 1 and 3 h. R-spondin1 is a recently described ligand for the LGR5 receptor that marks intestinal stem cells, and it is commonly used as a cofactor in intestinal organoid cultures in vitro (44). In response to these mitogens, Myc mRNA was steadily induced in crypts prepared from WT mice (Fig. 8A). In contrast, Myc mRNA was not induced in Myc 3′ WRE−/− crypts. A similar trend was also seen in WT and Myc 3′ WRE−/− crypts treated with Wnt3A and R-spondin1 or with EGF alone (Fig. 8B and C). Thus, deletion of the Myc 3′ WRE results in a more transcriptionally active Myc locus in colonic crypts under basal conditions, and the Myc 3′ WRE is required for the induction of Myc gene expression in response to Wnt3A, EGF, and R-spondin1.
FIG 8.
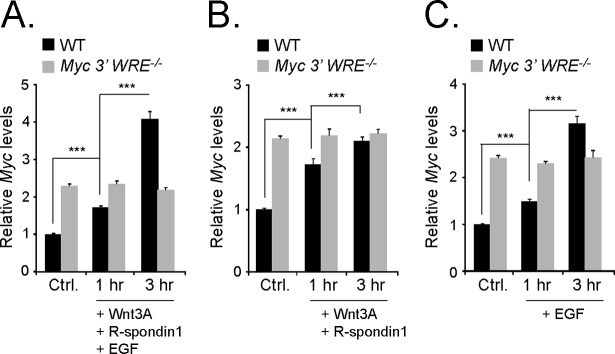
The Myc 3′ WRE is required for mitogen-induced Myc gene expression. The proliferative regions of colonic crypts were isolated from 7-week-old Myc 3′ WRE−/− mice and WT littermates and were not treated (Ctrl.) or treated for 1 or 3 h with recombinant Wnt3A, R-spondin1, and EGF (A), Wnt3A and R-spondin1 (B), or EGF (C). RNAs were isolated, cDNAs were synthesized, and Myc expression levels were evaluated by using quantitative real-time PCR. Myc mRNA levels were normalized to β-actin gene levels (n = 3 mice analyzed per genotype, with 12 total PCR replicates per sample). Errors are standard errors of the means (***, P < 0.001).
Mitogens induce an exchange of transcriptional regulatory complexes at Myc 5′ and 3′ Wnt-responsive elements.
We performed a series of ChIP experiments in untreated (control) and mitogen-treated (Wnt3A, EGF, and R-spondin1) WT colonic crypts to ascertain the underlying mechanisms that regulate Myc gene expression. Oligonucleotides that anneal to the Myc 5′ and 3′ WREs were used to interrogate factors that bound to these elements and histones that occupied these regions (Fig. 9A). First, we precipitated RNAP and found that mitogens induced RNAP recruitment to the Myc 5′ WRE (Fig. 9B). RNAP binding was not observed at the Myc 3′ WRE. Mitogens also induced H3K4me3 levels within the Myc 5′ promoter region (Fig. 9C). Using TCF4-specific antibodies in ChIP assays, we observed static TCF4 binding to both the Myc 5′ and 3′ WREs (Fig. 9D). Next, we analyzed β-catenin and found that mitogen treatment caused 6-fold and 9-fold increases in the level of β-catenin that was bound to the Myc 5′ and 3′ WREs, respectively (Fig. 9E). We found that TLE2 occupied the Myc 5′ and 3′ WREs in mouse colonic crypts and that mitogen treatment reduced TLE2 binding to both elements (Fig. 9F). Thus, β-catenin displaces TLE2 complexes bound to TCF4 to activate Myc expression in response to mitogens.
FIG 9.

Mitogens induce an exchange of transcriptional regulatory complexes at Myc 5′ and 3′ Wnt-responsive elements. (A) Diagram of the Myc locus, with the Myc 5′ and 3′ WREs indicated by white rectangles. Gray rectangles indicate the positions of the amplicons produced in quantitative real-time PCRs in panels B to G. (B) The proliferative region of colonic crypts were isolated from 7-week-old WT mice and were untreated (Ctrl.) or treated with recombinant Wnt3A, EGF, and R-spondin1 (mitogens) for 3 h. ChIP assays were performed by using antibodies directed against RNA polymerase 2 (RNAP), and the precipitated DNA was measured by using quantitative real-time PCR with oligonucleotides that hybridized to the regions indicated. Colonic crypts were isolated from 3 mice, and 12 PCR replicates were prepared for each sample. Errors are standard errors of the means (**, P < 0.01; ***, P < 0.001). (C to G) Same as panel B except that ChIP assays were conducted with antibodies directed against H3K4me3 (C), TCF4 (D), β-catenin (E), TLE2 (F), or CDX2 (G).
The fact that the deletion of the Myc 3′ WRE causes increased Myc gene expression in the colons of mice, and an imbalance of proliferative and differentiated cells within the crypts, indicates that the repressive role of this element is critical to homeostasis (28). Moreover, we were intrigued that this effect was not as strong in the small intestines. As TCFs and TLEs are expressed in both the small and large intestines, we hypothesized that a more tissue-specific transcription factor could be responsible for repression via the Myc 3′ WRE. The homeodomain transcription factor CDX2 is required for development of the intestines, and it is highly expressed in the colon (45, 46). TCF and CDX2 have also been found to cooccupy a large number of cis-regulatory elements within the intestinal genome (47). We therefore tested whether CDX2 occupied the Myc locus and found that its binding was enriched at the Myc 3′ WRE in untreated crypts, with lower levels being detected at the Myc 5′ WRE (Fig. 9G). Whereas mitogens reduced CDX2 binding to the 5′ WRE only slightly, they caused a 5-fold reduction of CDX2 binding to the 3′ WRE. Taken together, results from these experiments demonstrate that a dynamic exchange of β-catenin coactivators with TLE corepressors at TCF4 factors bound to proximal WREs occurs to activate Myc expression in colonic crypt explants in response to Wnt3A, EGF, and R-spondin1 signaling.
DISCUSSION
Studies have shown that Myc is required for intestinal tumors that form in ApcMin/+ mice and for phenotypes associated with acute deletion of Apc within the intestines (11–14). Results from those studies lend support to the hypothesis that the increased level of Myc expression driven by pathogenic Wnt/β-catenin signaling contributes to intestinal tumorigenesis. The Myc 3′ WRE is a critical regulator of intestinal homeostasis. Our previous work indicated that the deletion of the Myc 3′ WRE did not impact Myc expression significantly in the small intestine, but the deletion of this element caused a 2.5-fold increase MYC protein levels in the colons of juvenile (7-week-old) mice (28). Because this level of induction is within the range observed in human CRC tissues compared to uninvolved mucosa, Myc 3′ WRE−/− mice afforded us an opportunity to test whether a clinically relevant level of MYC impacts colorectal tumorigenesis (48–50). Using both genetic (ApcMin/+) and chemically induced (AOM/DSS) models of intestinal cancer, we find that the Myc 3′ WRE suppresses colorectal carcinogenesis. As tumors that develop using the AOM/DSS model of colitis-associated carcinogenesis frequently harbor mutations in CTNNB1 that stabilize the expression of the β-catenin protein produced from this allele (51), and ApcMin/+ mice express a truncated APC protein (35), our findings suggest that a 2.5-fold increase in MYC cooperates with aberrant Wnt/β-catenin signaling to drive colorectal cancers.
Our mRNA analysis of a panel of Wnt/β-catenin target genes found that the expression levels of over half of these genes were elevated in the preneoplastic ApcMin/+ Myc 3′ WRE−/− versus ApcMin/+ colons. These findings support those of Sansom et al., who reported that expression levels of approximately half of the Wnt/β-catenin target genes analyzed in the small intestines of Apc−/− mice were downregulated upon the concomitant removal of the Myc gene (13). Together, these results suggest that MYC and TCF transcription factors share a set of direct target genes. However, recent work has supported a model where MYC elevates the expression levels of actively transcribed genes universally, rather than inducing the expression of a new set of genes (36, 37). Our data do not support this model, as the expressions of several Wnt/β-catenin target genes were not influenced by the increase in Myc expression when the Myc 3′ WRE was deleted in an ApcMin/+ background (Fig. 2). There are numerous differences between our study and those reported by Nie et al. and Lin et al. that could account for these discrepancies. These differences include, but are not limited to, cell types, methods used to induce MYC protein levels, levels of MYC expression, and tissues analyzed.
Our results indicate that the colons of Myc 3′ WRE−/− mice are predisposed to developing colitis-associated colorectal cancers, with a large number of tumors localizing to the cecum. We previously reported, using a model of DSS-induced acute colitis, that Myc 3′ WRE−/− colons displayed a greater regenerative response during the recovery phase of the protocol (28). Interestingly, we found that an enhanced neutrophil response accompanied the recovery of these mice, which likely facilitated the restoration of the colonic epithelium by removing bacteria that penetrated into the lamina propria and submucosa (30). On the other hand, neutrophils have been shown to promote tumorigenesis in chronic models of inflammation (52). We suspect that the enhanced neutrophil infiltration in response to colonic damage seen in Myc 3′ WRE−/− colons may be beneficial in the short term but detrimental over longer periods of time. Regardless, deregulated MYC expression and chronic inflammation contribute to tumor formation within the cecum, which is where 13% of human colorectal carcinomas localize (53).
Our findings suggest that the CDX2 homeodomain transcription factor plays an important role in repressing Myc gene expression through the Myc 3′ WRE in the colon. The phenotypic similarities of the intestines of Cdx2 genetic mouse models and those of Myc 3′ WRE−/− mice support this assertion. Cdx2 heterozygous mice develop tumors predominantly within the distal colon and cecum, with few tumors forming in the small intestine (54). Apc+/Δ716 Cdx2+/− contained a reduced number of small intestinal tumors and an increased number of colonic tumors compared to Apc+/Δ716 mice (55). Finally, Hinoi et al. reported that using a CDX2 promoter-driven NLS-Cre transgene to remove Apc in the intestines of Apc+/loxP mice led to the development of colorectal adenomas (45). These phenotypes are in contrast to those predominantly seen in Apc models where adenomas form predominantly in the small intestine (56). Thus, CDX2-dependent repression of Myc gene expression may be an underlying reason why tumors are relatively rare within the colons of ApcMin/+ mice.
Concurrent with our work, Sur and colleagues reported a role for the Myc −335 WRE in intestinal tumorigenesis in mice (29). They found that although the deletion of this element did not affect Myc expression within the duodenum, when Myc −335−/− mice were crossed with ApcMin/+ mice, the deletion of the Myc −335 element caused a dramatic reduction of tumors within the small intestines (29). ApcMin/+ Myc −335−/− mice also contained a reduced number of colonic tumors in comparison to ApcMin/+ mice, although the effect was not as severe as that seen in the small intestines. Thus, the Myc −335 WRE promotes intestinal tumorigenesis. Similarly, deletion of the Myc 3′ WRE did not significantly affect Myc expression in the small intestines of juvenile mice (28). Likewise, whereas the deletion of the Myc 3′ WRE suppressed tumorigenesis within the small intestines of ApcMin/+ mice, this effect was not as drastic as those reported by Sur et al. (29). However, in contrast to ApcMin/+ Myc −335 WRE colons, ApcMin/+ Myc 3′ WRE−/− colons contained 4-fold more tumors than seen within the colons of ApcMin/+ mice. Therefore, in terms of Wnt/β-catenin-driven intestinal tumors, these two WREs have opposing roles. Mice lacking the Myc −335 WRE are resistant to tumorigenesis, primarily in the small intestine, and mice lacking the Myc 3′ WRE are more susceptible to tumorigenesis in the colon.
MYC is a ubiquitous transcription factor that is required for cellular proliferation. The fact that its expression level is elevated in over 30% of cancers, including CRC, argues that it is an attractive candidate for the development of therapeutics to treat these diseases (57). This strategy has gained traction as impairing MYC function, or its expression, has proven efficacious in mitigating disease phenotypes in culture systems and mouse models (58, 59). Drugs such as the JQ1 inhibitor (60), which reduces oncogenic MYC expression, and 10058-F4 (61, 62), which impairs MYC's function as a transcription factor, have not yet been extensively evaluated in mouse models of CRC. Our work indicates that Myc 3′ WRE−/− mice are valuable tools to test current drugs, and to evaluate novel drug candidates, for their efficacy as therapeutic agents for the treatment of colorectal cancers that develop in the anatomically correct microenvironment.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Yochum laboratory for helpful discussions and Kang Li in the Morphological and Molecular Pathology Core Research Laboratory (Penn State University College of Medicine) for his help with embedding tissue sections and preparing slides for immunohistochemistry.
This research was supported by National Institutes of Health grant R01DK080805 (G.S.Y.).
Footnotes
Published ahead of print 24 February 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00969-13.
REFERENCES
- 1.Archbold HC, Yang YX, Chen L, Cadigan KM. 2012. How do they do Wnt they do? Regulation of transcription by the Wnt/beta-catenin pathway. Acta Physiol. (Oxf.) 204:74–109. 10.1111/j.1748-1716.2011.02293.x [DOI] [PubMed] [Google Scholar]
- 2.Yeung TM, Chia LA, Kosinski CM, Kuo CJ. 2011. Regulation of self-renewal and differentiation by the intestinal stem cell niche. Cell. Mol. Life Sci. 68:2513–2523. 10.1007/s00018-011-0687-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clevers H, Nusse R. 2012. Wnt/beta-catenin signaling and disease. Cell 149:1192–1205. 10.1016/j.cell.2012.05.012 [DOI] [PubMed] [Google Scholar]
- 4.MacDonald BT, Tamai K, He X. 2009. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev. Cell 17:9–26. 10.1016/j.devcel.2009.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mosimann C, Hausmann G, Basler K. 2009. Beta-catenin hits chromatin: regulation of Wnt target gene activation. Nat. Rev. Mol. Cell Biol. 10:276–286. 10.1038/nrm2654 [DOI] [PubMed] [Google Scholar]
- 6.He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. 1998. Identification of c-MYC as a target of the APC pathway. Science 281:1509–1512. 10.1126/science.281.5382.1509 [DOI] [PubMed] [Google Scholar]
- 7.Yochum GS, Cleland R, Goodman RH. 2008. A genome-wide screen for beta-catenin binding sites identifies a downstream enhancer element that controls c-Myc gene expression. Mol. Cell. Biol. 28:7368–7379. 10.1128/MCB.00744-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dang CV, O'Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F. 2006. The c-Myc target gene network. Semin. Cancer Biol. 16:253–264. 10.1016/j.semcancer.2006.07.014 [DOI] [PubMed] [Google Scholar]
- 9.Eilers M, Eisenman RN. 2008. Myc's broad reach. Genes Dev. 22:2755–2766. 10.1101/gad.1712408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fearon ER. 2011. Molecular genetics of colorectal cancer. Annu. Rev. Pathol. 6:479–507. 10.1146/annurev-pathol-011110-130235 [DOI] [PubMed] [Google Scholar]
- 11.Athineos D, Sansom OJ. 2010. Myc heterozygosity attenuates the phenotypes of APC deficiency in the small intestine. Oncogene 29:2585–2590. 10.1038/onc.2010.5 [DOI] [PubMed] [Google Scholar]
- 12.Ignatenko NA, Holubec H, Besselsen DG, Blohm-Mangone KA, Padilla-Torres JL, Nagle RB, de Alboranc IM, Guillen RJ, Gerner EW. 2006. Role of c-Myc in intestinal tumorigenesis of the ApcMin/+ mouse. Cancer Biol. Ther. 5:1658–1664. 10.4161/cbt.5.12.3376 [DOI] [PubMed] [Google Scholar]
- 13.Sansom OJ, Meniel VS, Muncan V, Phesse TJ, Wilkins JA, Reed KR, Vass JK, Athineos D, Clevers H, Clarke AR. 2007. Myc deletion rescues Apc deficiency in the small intestine. Nature 446:676–679. 10.1038/nature05674 [DOI] [PubMed] [Google Scholar]
- 14.Wilkins JA, Sansom OJ. 2008. C-Myc is a critical mediator of the phenotypes of Apc loss in the intestine. Cancer Res. 68:4963–4966. 10.1158/0008-5472.CAN-07-5558 [DOI] [PubMed] [Google Scholar]
- 15.Yekkala K, Baudino TA. 2007. Inhibition of intestinal polyposis with reduced angiogenesis in ApcMin/+ mice due to decreases in c-Myc expression. Mol. Cancer Res. 5:1296–1303. 10.1158/1541-7786.MCR-07-0232 [DOI] [PubMed] [Google Scholar]
- 16.Bottomly D, Kyler SL, McWeeney SK, Yochum GS. 2010. Identification of beta-catenin binding regions in colon cancer cells using ChIP-Seq. Nucleic Acids Res. 38:5735–5745. 10.1093/nar/gkq363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yochum GS, McWeeney S, Rajaraman V, Cleland R, Peters S, Goodman RH. 2007. Serial analysis of chromatin occupancy identifies beta-catenin target genes in colorectal carcinoma cells. Proc. Natl. Acad. Sci. U. S. A. 104:3324–3329. 10.1073/pnas.0611576104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yochum GS, Sherrick CM, MacPartlin M, Goodman RH. 2010. A beta-catenin/TCF-coordinated chromatin loop at MYC integrates 5′ and 3′ Wnt responsive enhancers. Proc. Natl. Acad. Sci. U. S. A. 107:145–150. 10.1073/pnas.0912294107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pomerantz MM, Ahmadiyeh N, Jia L, Herman P, Verzi MP, Doddapaneni H, Beckwith CA, Chan JA, Hills A, Davis M, Yao KL, Kehoe SM, Lenz HJ, Haiman CA, Yan CL, Henderson BE, Frenkel B, Barretina J, Bass A, Tabernero J, Baselga J, Regan MM, Manak JR, Shivdasani R, Coetzee GA, Freedman ML. 2009. The 8q24 cancer risk variant rs6983267 shows long-range interaction with MYC in colorectal cancer. Nat. Genet. 41:882–884. 10.1038/ng.403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tuupanen S, Turunen M, Lehtonen R, Hallikas O, Vanharanta S, Kivioja T, Bjorklund M, Wei GH, Yan J, Niittymaki I, Mecklin JP, Jarvinen H, Ristimaki A, Di-Bernardo M, East P, Carvajal-Carmona L, Houlston RS, Tomlinson I, Palin K, Ukkonen E, Karhu A, Taipale J, Aaltonen LA. 2009. The common colorectal cancer predisposition SNP rs6983267 at chromosome 8q24 confers potential to enhanced Wnt signaling. Nat. Genet. 41:885–890. 10.1038/ng.406 [DOI] [PubMed] [Google Scholar]
- 21.Wright JB, Brown SJ, Cole MD. 2010. Upregulation of c-MYC in cis through a large chromatin loop linked to a cancer risk-associated single-nucleotide polymorphism in colorectal cancer cells. Mol. Cell. Biol. 30:1411–1420. 10.1128/MCB.01384-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haiman CA, Le Marchand L, Yamamato J, Stram DO, Sheng X, Kolonel LN, Wu AH, Reich D, Henderson BE. 2007. A common genetic risk factor for colorectal and prostate cancer. Nat. Genet. 39:954–956. 10.1038/ng2098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tomlinson I, Webb E, Carvajal-Carmona L, Broderick P, Kemp Z, Spain S, Penegar S, Chandler I, Gorman M, Wood W, Barclay E, Lubbe S, Martin L, Sellick G, Jaeger E, Hubner R, Wild R, Rowan A, Fielding S, Howarth K, Silver A, Atkin W, Muir K, Logan R, Kerr D, Johnstone E, Sieber O, Gray R, Thomas H, Peto J, Cazier B, Houlston R, Consortium C. 2007. A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nat. Genet. 39:984–988. 10.1038/ng2085 [DOI] [PubMed] [Google Scholar]
- 24.Zanke BW, Greenwood CMT, Rangrej J, Kustra R, Tenesa A, Farrington SM, Prendergast J, Olschwang S, Chiang T, Crowdy E, Ferretti V, Laflamme P, Sundararajan S, Roumy S, Olivier JF, Robidoux F, Sladek R, Montpetit A, Campbell P, Bezieau S, O'Shea AM, Zogopoulos G, Cotterchio M, Newcomb P, McLaughlin J, Younghusband B, Green R, Green J, Porteous MEM, Campbell H, Blanche H, Sahbatou M, Tubacher E, Bonaiti-Pellie C, Buecher B, Riboli E, Kury S, Chanock SJ, Potter J, Thomas G, Gallinger S, Hudson TJ, Dunlop MG. 2007. Genome-wide association scan identifies a colorectal cancer susceptibility locus on chromosome 8q24. Nat. Genet. 39:989–994. 10.1038/ng2089 [DOI] [PubMed] [Google Scholar]
- 25.Ahmadiyeh N, Pomerantz MM, Grisanzio C, Herman P, Jia L, Almendro V, He HH, Brown M, Liu XS, Davis M, Caswell JL, Beckwith CA, Hills A, MacConaill L, Coetzee GA, Regan MM, Freedman ML. 2010. 8q24 prostate, breast, and colon cancer risk loci show tissue-specific long-range interaction with MYC. Proc. Natl. Acad. Sci. U. S. A. 107:9742–9746. 10.1073/pnas.0910668107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sotelo J, Esposito D, Duhagon MA, Banfield K, Mehalko J, Liao HL, Stephens RM, Harris TJR, Munroe DJ, Wu XL. 2010. Long-range enhancers on 8q24 regulate c-Myc. Proc. Natl. Acad. Sci. U. S. A. 107:3001–3005. 10.1073/pnas.0906067107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yochum GS. 2011. Multiple Wnt/beta-catenin responsive enhancers align with the MYC promoter through long-range chromatin loops. PLoS One 6:e18966. 10.1371/journal.pone.0018966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Konsavage WM, Jin G, Yochum GS. 2012. The Myc 3′ Wnt-responsive element regulates homeostasis and regeneration in the mouse intestinal tract. Mol. Cell. Biol. 32:3891–3902. 10.1128/MCB.00548-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sur IK, Hallikas O, Vaharautio A, Yan J, Turunen M, Enge M, Taipale M, Karhu A, Aaltonen LA, Taipale J. 2012. Mice lacking a Myc enhancer that includes human SNP rs6983267 are resistant to intestinal tumors. Science 338:1360–1363. 10.1126/science.1228606 [DOI] [PubMed] [Google Scholar]
- 30.Konsavage WM, Jr, Roper JN, Ishmael FT, Yochum GS. 2013. The Myc 3′ Wnt responsive element regulates neutrophil recruitment after acute colonic injury in mice. Dig. Dis. Sci. 58:2858–2867. 10.1007/s10620-013-2686-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sato T, Stange DE, Ferrante M, Vries RG, Van Es JH, Van den Brink S, Van Houdt WJ, Pronk A, Van Gorp J, Siersema PD, Clevers H. 2011. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett's epithelium. Gastroenterology 141:1762–1772. 10.1053/j.gastro.2011.07.050 [DOI] [PubMed] [Google Scholar]
- 32.Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, Danenberg E, Clarke AR, Sansom OJ, Clevers H. 2009. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 457:608–611. 10.1038/nature07602 [DOI] [PubMed] [Google Scholar]
- 33.Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, Karin M. 2004. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 118:285–296. 10.1016/j.cell.2004.07.013 [DOI] [PubMed] [Google Scholar]
- 34.Mahmoudi T, Li VS, Ng SS, Taouatas N, Vries RG, Mohammed S, Heck AJ, Clevers H. 2009. The kinase TNIK is an essential activator of Wnt target genes. EMBO J. 28:3329–3340. 10.1038/emboj.2009.285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Su LK, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C, Gould KA, Dove WF. 1992. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 256:668–670. 10.1126/science.1350108 [DOI] [PubMed] [Google Scholar]
- 36.Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, Young RA. 2012. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 151:56–67. 10.1016/j.cell.2012.08.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, Wang R, Green DR, Tessarollo L, Casellas R, Zhao K, Levens D. 2012. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 151:68–79. 10.1016/j.cell.2012.08.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Finch AJ, Soucek L, Junttila MR, Swigart LB, Evan GI. 2009. Acute overexpression of Myc in intestinal epithelium recapitulates some but not all the changes elicited by Wnt/beta-catenin pathway activation. Mol. Cell. Biol. 29:5306–5315. 10.1128/MCB.01745-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murphy DJ, Junttila MR, Pouyet L, Karnezis A, Shchors K, Bui DA, Brown-Swigart L, Johnson L, Evan GI. 2008. Distinct thresholds govern Myc's biological output in vivo. Cancer Cell 14:447–457. 10.1016/j.ccr.2008.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sancho E, Batlle E, Clevers H. 2004. Signaling pathways in intestinal development and cancer. Annu. Rev. Cell Dev. Biol. 20:695–723. 10.1146/annurev.cellbio.20.010403.092805 [DOI] [PubMed] [Google Scholar]
- 41.Heintzman ND, Stuart RK, Hon G, Fu YT, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu CX, Ching KA, Wang W, Weng ZP, Green RD, Crawford GE, Ren B. 2007. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 39:311–318. 10.1038/ng1966 [DOI] [PubMed] [Google Scholar]
- 42.Sierra J, Yoshida T, Joazeiro CA, Jones KA. 2006. The APC tumor suppressor counteracts beta-catenin activation and H3K4 methylation at Wnt target genes. Genes Dev. 20:586–600. 10.1101/gad.1385806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H. 1997. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC−/− colon carcinoma. Science 275:1784–1787. 10.1126/science.275.5307.1784 [DOI] [PubMed] [Google Scholar]
- 44.Clevers H. 2013. The intestinal crypt, a prototype stem cell compartment. Cell 154:274–284. 10.1016/j.cell.2013.07.004 [DOI] [PubMed] [Google Scholar]
- 45.Hinoi T, Akyol A, Theisen BK, Ferguson DO, Greenson JK, Williams BO, Cho KR, Fearon ER. 2007. Mouse model of colonic adenoma-carcinoma progression based on somatic Apc inactivation. Cancer Res. 67:9721–9730. 10.1158/0008-5472.CAN-07-2735 [DOI] [PubMed] [Google Scholar]
- 46.Hryniuk A, Grainger S, Savory JG, Lohnes D. 2012. Cdx function is required for maintenance of intestinal identity in the adult. Dev. Biol. 363:426–437. 10.1016/j.ydbio.2012.01.010 [DOI] [PubMed] [Google Scholar]
- 47.Verzi MP, Hatzis P, Sulahian R, Philips J, Schuijers J, Shin H, Freed E, Lynch JP, Dang DT, Brown M, Clevers H, Liu XS, Shivdasani RA. 2010. TCF4 and CDX2, major transcription factors for intestinal function, converge on the same cis-regulatory regions. Proc. Natl. Acad. Sci. U. S. A. 107:15157–15162. 10.1073/pnas.1003822107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stewart J, Evan G, Watson J, Sikora K. 1986. Detection of the c-myc oncogene product in colonic polyps and carcinomas. Br. J. Cancer 53:1–6. 10.1038/bjc.1986.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sugio K, Kurata S, Sasaki M, Soejima J, Sasazuki T. 1988. Differential expression of c-myc gene and c-fos gene in premalignant and malignant tissues from patients with familial polyposis coli. Cancer Res. 48:4855–4861 [PubMed] [Google Scholar]
- 50.Tsuboi K, Hirayoshi K, Takeuchi K, Sabe H, Shimada Y, Ohshio G, Tobe T, Hatanaka M. 1987. Expression of the c-myc gene in human gastrointestinal malignancies. Biochem. Biophys. Res. Commun. 146:699–704. 10.1016/0006-291X(87)90585-7 [DOI] [PubMed] [Google Scholar]
- 51.Takahashi M, Nakatsugi S, Sugimura T, Wakabayashi K. 2000. Frequent mutations of the beta-catenin gene in mouse colon tumors induced by azoxymethane. Carcinogenesis 21:1117–1120. 10.1093/carcin/21.6.1117 [DOI] [PubMed] [Google Scholar]
- 52.Jamieson T, Clarke M, Steele CW, Samuel MS, Neumann J, Jung A, Huels D, Olson MF, Das S, Nibbs RJ, Sansom OJ. 2012. Inhibition of CXCR2 profoundly suppresses inflammation-driven and spontaneous tumorigenesis. J. Clin. Invest. 122:3127–3144. 10.1172/JCI61067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rosenberg JM, Welch JP. 1985. Carcinoid tumors of the colon. A study of 72 patients. Am. J. Surg. 149:775–779 [DOI] [PubMed] [Google Scholar]
- 54.Chawengsaksophak K, James R, Hammond VE, Kontgen F, Beck F. 1997. Homeosis and intestinal tumours in Cdx2 mutant mice. Nature 386:84–87. 10.1038/386084a0 [DOI] [PubMed] [Google Scholar]
- 55.Aoki K, Tamai Y, Horiike S, Oshima M, Taketo MM. 2003. Colonic polyposis caused by mTOR-mediated chromosomal instability in Apc+/Delta716 Cdx2+/− compound mutant mice. Nat. Genet. 35:323–330. 10.1038/ng1265 [DOI] [PubMed] [Google Scholar]
- 56.Taketo MM. 2006. Mouse models of gastrointestinal tumors. Cancer Sci. 97:355–361. 10.1111/j.1349-7006.2006.00190.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Prochownik EV. 2004. c-Myc as a therapeutic target in cancer. Expert Rev. Anticancer Ther. 4:289–302. 10.1586/14737140.4.2.289 [DOI] [PubMed] [Google Scholar]
- 58.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, Chesi M, Schinzel AC, McKeown MR, Heffernan TP, Vakoc CR, Bergsagel PL, Ghobrial IM, Richardson PG, Young RA, Hahn WC, Anderson KC, Kung AL, Bradner JE, Mitsiades CS. 2011. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146:904–917. 10.1016/j.cell.2011.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zirath H, Frenzel A, Oliynyk G, Segerstrom L, Westermark UK, Larsson K, Munksgaard Persson M, Hultenby K, Lehtio J, Einvik C, Pahlman S, Kogner P, Jakobsson PJ, Arsenian Henriksson M. 2013. MYC inhibition induces metabolic changes leading to accumulation of lipid droplets in tumor cells. Proc. Natl. Acad. Sci. U. S. A. 110:10258–10263. 10.1073/pnas.1222404110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S, Bradner JE. 2010. Selective inhibition of BET bromodomains. Nature 468:1067–1073. 10.1038/nature09504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hammoudeh DI, Follis AV, Prochownik EV, Metallo SJ. 2009. Multiple independent binding sites for small-molecule inhibitors on the oncoprotein c-Myc. J. Am. Chem. Soc. 131:7390–7401. 10.1021/ja900616b [DOI] [PubMed] [Google Scholar]
- 62.Wang H, Hammoudeh DI, Follis AV, Reese BE, Lazo JS, Metallo SJ, Prochownik EV. 2007. Improved low molecular weight Myc-Max inhibitors. Mol. Cancer Ther. 6:2399–2408. 10.1158/1535-7163.MCT-07-0005 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.