

Abstract
The aryl hydrocarbon receptor (AhR) is a ligand-dependent transcription factor that can be activated by structurally diverse chemicals. To examine the mechanisms responsible for the promiscuity in AhR ligand binding, we determined the effects of mutations within the AhR ligand-binding domain (LBD) on the activity of diverse AhR ligands. Site-directed mutagenesis identified Ile319 of the mouse AhR and, to a lesser extent, Phe318 as residues involved in ligand-selective modulation of AhR transformation using a panel of 12 AhR ligands. These ligands could be categorized into four distinct structurally related groups based on their ability to activate AhR mutants at position 319 in vitro. The mutation I319K was selectively activated by FICZ and not by other examined ligands in vitro and in cell culture. F318L and F318A mutations resulted in the conversion of AhR agonists β-naphthoflavone and 3-methylcholanthrene, respectively, into partial agonists/antagonists. Hsp90 binding to the AhR was decreased with several mutations and was inversely correlated with AhR ligand-binding promiscuity. Together, these data define overlapping amino acid residues within the AhR LBD involved in the selectivity of ligand binding, the agonist or antagonist mode of ligand binding, and hsp90 binding and provide insights into the ligand diversity of AhR activators.
INTRODUCTION
The aryl hydrocarbon receptor (AhR) is a ligand-dependent nuclear receptor that mediates a broad spectrum of toxic and biological effects resulting from exposure to structurally diverse synthetic and natural compounds (1, 2). The prototypical and most studied AhR ligand is 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), an environmental contaminant and known human carcinogen (3). However, numerous other halogenated aromatic hydrocarbons (HAHs), polycyclic aromatic hydrocarbons (PAHs), and PAH-like compounds have also been described that possess high affinity for the AhR and can activate AhR-dependent signal transduction and produce diverse biological/toxic effects in various species (1, 4). The AhR-mediated toxic and biological effects are produced by metabolically stable dioxin-like HAH ligands, including TCDD, 2,3,7,8-tetrachlorodibenzofuran (TCDF), 3,3′,4,4′,5-pentachlorobiphenyl (PCB126), and others. In contrast, metabolically labile AhR ligands, including PAHs 3-methylcholanthrene (3MC), β-naphthoflavone (BNF), and 6-formylindolo[3,2-b]carbazole (FICZ), produce transient biological effects associated with AhR activation but no AhR-dependent toxicity, despite the fact that they also possess relatively high affinity for the AhR (5–7).
While the ligand-dependent mechanism of AhR signaling has many similarities to that of members of the steroid hormone nuclear receptor superfamily, the AhR is unrelated and it is a member of the basic helix-loop-helix Per-Arnt-Sim (bHLH-PAS) superfamily of regulatory proteins (8, 9). In its native state, the inactive unliganded AhR exists in the cytosol complexed with at least two molecules of hsp90 and the cochaperone proteins XAP2 and p23, and these proteins contribute to the cytosolic localization of unliganded AhR, protect it from degradation, and maintain it in a ligand and DNA binding-competent state (10–13). Following ligand binding to a site within the ligand-binding domain (LBD), the AhR protein complex translocates into the nucleus, hsp90 and cochaperones dissociate, and liganded AhR interacts with a homologous nuclear protein Ah receptor nuclear translocator (ARNT), leading to formation of the liganded AhR-ARNT dimer, which can bind to DNA with high affinity (14). The process by which ligand converts the AhR into its DNA binding form is commonly referred to as AhR transformation (15). Binding of the ligand-activated/transformed AhR-ARNT complex to its specific DNA recognition site, the dioxin response element (DRE), stimulates transcription from adjacent promoters/genes and ultimately results in AhR-dependent biological and toxic effects (16, 17).
While the mechanisms of AhR activation and signal transduction have been extensively characterized using TCDD as the ligand/agonist, several recent studies have suggested that the functional activity of the AhR can be differentially altered by the specific ligand to which it is bound (18–20). This idea is supported by gene microarray studies which have reported the ability of different AhR ligands to not only induce expression of a common set of gene products but to also induce a distinctly different, ligand-specific set of AhR-dependent gene products (21–23). Ligand-specific modulation of the AhR signaling pathway has been suggested to result from ligand-dependent changes in the AhR which could allow it to interact with different nuclear factors or dimerization partners, to bind to unconventional DREs or unique DNA sequences, and/or to be bound by different coactivators (24–27). For these changes to occur, significant differences in the binding interactions between ligands and amino acids within the AhR LBD must occur, and these differences must translate into alterations in AhR structure/function that ultimately lead to distinct ligand- and AhR-dependent biological responses (1, 2).
How might the AhR bind and be activated by structurally diverse ligands? Similar to the AhR, the pregnane X receptor (PXR) can bind and be activated by a structurally diverse collection of ligands. The results of 3-dimensional protein structure analysis of PXR ligand binding has revealed that the dramatic structural promiscuity of PXR ligands can be explained by the characteristics of its binding pocket, which allows structurally diverse ligands to bind in a variety of different orientations and positions within the PXR LBD (28–30). By analogy, the structural diversity of AhR ligands may also result from differential interactions of ligands or classes of ligands with different residues within the AhR LBD (1, 2). Consistent with this hypothesis is the identification of AhR antagonists that appear to be ligand selective in that they can preferentially inhibit the ability of some ligands (i.e., TCDD and related HAHs) to bind to and activate the AhR, while other AhR ligands (i.e., PAHs, flavonoids, indirubin) are not affected (31). Although progress in understanding the details of the molecular mechanisms of ligand binding and ligand-dependent AhR activation/transformation has been hampered by the lack of a 3-dimensional structure of the AhR LBD, site-directed mutagenesis studies and structure-function analysis using a homology model of the AhR LBD have produced some insights (32, 33).
Relatively few studies have directly examined the effects of AhR LBD mutagenesis on the binding of and/or activation by structurally diverse ligands. However, several specific residues whose mutation resulted in ligand-specific differences in AhR functionality and agonist activity have been identified (1, 34–36). Interestingly, when these specific residues (H285, F289, F318, and H320) are mapped onto the mouse AhR LBD homology model (32, 33), they are located in proximity to each other, suggesting that they may encompass a distinct region of the LBD involved in ligand-specific activation of the AhR. This region is also adjacent to amino acids previously implicated in ligand binding (Ala375) and hsp90 binding (Phe281), indicating possible involvement of ligand-dependent hsp90 displacement in ligand-selective effects (10, 37). As suggested by these studies, ligand-selective interactions with the AhR remain to be more clearly identified and characterized. Accordingly, here we describe the results of studies using a combination of site-directed mutagenesis and functional analysis to investigate the mechanism of AhR ligand selectivity.
MATERIALS AND METHODS
Plasmid constructs.
mβAhR/pcDNA3 and mβArnt/pcDNA3 have been previously described (38, 39). Point mutations of mβAhR/pcDNA3 were carried out using the QuikChange technique (Stratagene). All constructs were verified by sequencing.
In vitro expression.
Wild-type (wt) and mutant AhRs were synthesized in vitro in the presence of l-methionine or [35S]l-methionine (PerkinElmer) using the TNT Quick coupled transcription/translation rabbit reticulocyte lysate kit (Promega). To compare relative expression levels of each mutant AhR, aliquots of in vitro synthesized 35S-labeled wt and mutant AhR protein were analyzed by SDS-PAGE as previously described (39). Unlabeled AhR and ARNT were used for functional analysis studies.
Gel retardation assay.
Wild-type and mutant AhRs and ARNT were synthesized in vitro in the presence of unlabeled l-methionine using the TNT Quick coupled transcription/translation rabbit reticulocyte lysate kit (Promega). The resulting AhR and ARNT translation mixtures were mixed in a 1:1:8 (vol/vol/vol) ratio with 150 mM KCl MEDG buffer (25 mM MOPS [morpholinepropanesulfonic acid; pH 7.5], 10% [vol/vol] glycerol, 15 mM KCl, 1 mM EDTA, 1 mM dithiothreitol [DTT]) and incubated with the indicated concentration of TCDD or 1% (vol/vol) dimethyl sulfoxide (DMSO; the solvent control) for the indicated periods of time at room temperature. Annealed double-stranded oligonucleotides containing the AhR-ARNT DNA binding site (DRE3) from the murine CYP1A1 upstream regulatory sequence were 32P labeled, and gel retardation analysis was conducted with the transformed AhR reactions as detailed previously (39). Gels were visualized using Fujifilm imaging plate (IP) analysis (FLA9000 and BAS-SR imaging plates) and quantitated with Fujifilm MultiGauge software.
Hydroxyapatite (HAP) ligand binding assay.
[3H]TCDD (13 Ci/mmol) was obtained from Steven Safe (Texas A&M University). Determination of [3H]TCDD binding to the in vitro synthesized proteins diluted in 150 mM KCl MEDG buffer (8:92, vol/vol) was as previously described (33). For competitive displacement experiments, the indicated concentrations of 3MC or BNF were added to the binding reactions. Equivalent amounts of unprogrammed in vitro synthesized reactions were used as a nonspecific binding control (40). For affinity measurements, the in vitro transformation reactions and nonspecific binding controls were incubated in the presence of increasing concentrations (1 to 20 nM) of [3H]TCDD.
Coimmunoprecipitation and Western blotting assays.
COS-1 cells were transiently transfected with AhR expression vectors using Lipofectamine 2000 (Invitrogen) at an 8 μg/20 μl ratio in 10-mm tissue culture plates. Cell lysis and hsp90 coimmunoprecipitation analysis were performed as previously described (10). The anti-hsp90 antibody 3G3 was a kind gift of Gary Perdew (Pennsylvania State University). Western blotting was carried out using a 1:400 dilution of the anti-AhR M20 antibody (Santa Cruz).
Reporter gene induction assays.
COS-1 cells were transiently transfected in 96-well plates using the following amounts per well: 0.5 μl Lipofectamine 2000 (Invitrogen), 20 ng wt mβAhR/pcDNA3 or 60 ng of mutant AhR expression vectors, and 100 ng pGudLuc6.1 (41) and pcDNA3.1+ (Invitrogen), the latter added to adjust the mixture to a total DNA content of 200 ng. Twenty-four hours after transfection, cells were incubated with DMSO (0.1%, vol/vol) or the indicated concentration of ligand for 18 to 22 h, washed cells were lysed using passive lysis buffer (Promega), and aliquots were analyzed for firefly luciferase activity using the luciferase reporter assay system (Promega) and an Orion microplate luminometer (Berthold Detection Systems). Aliquots (5 to 10 μl) of cell lysates were analyzed for protein concentrations using the Bio-Rad Bradford protein assay as described by the manufacturer.
Statistical analysis.
Analysis of statistical significance of differences of experimental values was conducted using the Student t test in Excel or SigmaPlot. Determination of ligand binding affinity was conducted by regression analysis of the saturating binding curves in SigmaPlot.
AhR PASB LBD homology model.
AhR Per-Arnt-Sim B (PASB) LBD homology model pictures were generated in PyMol (version 1) using the Protein Data Bank (PDB) file kindly provided by Laura Bonati (University of Milano-Bicocca, Italy).
RESULTS
Amino acid residues 318 to 320 are involved in ligand-specific activation.
Site-directed mutational analysis studies previously identified a small number of amino acid residues within the LBD that appear to be involved in ligand-specific AhR activation (1, 34–36). When mapped onto the PASB LBD homology model, these residues (H285, F289, F318, and H320) delineate a smaller region of the LBD, which also contains a previously uncharacterized Ile319 (Fig. 1, yellow residues). Given the central position of Ile319 in the LBD region of interest (Fig. 1), this residue may be involved in ligand selectivity of AhR activation. To examine the role of each of these targeted amino acids (H285, F289, F318, I319, and H320) in ligand-selective AhR activation, we determined the effect of their mutation to alanine on ligand-dependent transformation/DNA binding of in vitro synthesized AhR after confirming that the wt and mutant AhR proteins were expressed at comparable levels in vitro (Fig. 2A).
FIG 1.
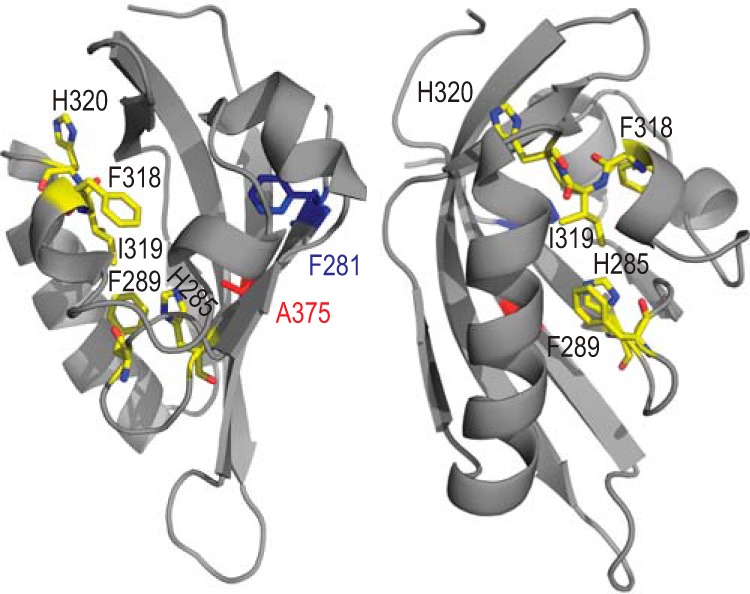
Structural model of the mouse AhR (mAhR) PASB domain. Residues presumably involved in ligand-specific AhR activation are indicated in yellow. The residue involved in hsp90 binding (Phe281) is indicated in blue, and ligand (TCDD) binding (Ala375) is indicated in red.
FIG 2.

Specific region of the mAhR PASB is involved in ligand-specific activation. (A) Protein expression of AhR mutants. Indicated AhR mutants were synthesized in vitro in the presence of [35S]methionine, separated with SDS-PAGE, and analyzed by autoradiography. A representative gel is shown. (B) In vitro synthesized wild-type or mutant AhRs and ARNT were incubated in the presence of solvent control DMSO (1%, vol/vol) or AhR agonists TCDD (20 nM), 3MC (1 μM), BNF (1 μM), indirubin (1 μM), or ANF (10 μM) for 2 h and analyzed by the gel retardation assay. Representative gels are shown. SC, specific complex (liganded AhR-ARNT-DRE). (C) Quantitation of gels shown in panel B. Values represent means ± standard deviations from three independent reactions. Asterisks indicate those values that are significantly higher than DMSO controls (not shown) at P values of <0.05 as determined by the Student t test and demonstrate an at least 2-fold increase over the background (DMSO) reaction values. (D) [3H]TCDD binding to select mAhR mutations. In vitro synthesized wild-type or mutant AhRs were incubated in the presence of 10 nM [3H]TCDD and in the presence or absence of 1 μM 3MC for 30 min, and [3H]TCDD bound to the protein fraction was determined by the hydroxyapatite assay. Specific binding was calculated as total binding (in the absence of 3MC) normalized to binding in the unprogrammed TNT reaction. Values represent means ± standard deviations from three independent reactions. Asterisks indicate the values that are significantly lower than corresponding [3H]TCDD reactions at P values of <0.05 as determined by the Student t test. (A to D) Results are representative of three independent experiments.
Several AhR ligands were chosen for the preliminary analysis, including TCDD, 3MC, BNF, indirubin, and partial agonist/antagonist α-naphthoflavone (ANF). These compounds were used at equipotent concentrations (20 nM, 1 μM, 1 μM, 1 μM, and 10 μM, respectively) at which maximal DNA binding response is produced (50% effective concentrations [EC50s] for TCDD, 3MC, BNF, indirubin, and ANF were 3.3 nM, 2.8 nM, 8.4 nM, 3.1 nM, and 0.15 μM, respectively).
The wild type (wt) and mutant proteins were synthesized in vitro, incubated in the presence of ligands, and analyzed for DNA binding using the gel retardation assay. In this experiment, the wtAhR was activated by all ligands to approximately similar levels, while AhRs containing H285A and F289A were essentially inactive (Fig. 2B). In contrast, the AhRs with Ala substitutions at positions 318 to 320 could transform and bind to DNA in a ligand-specific manner (Fig. 2B).
The specific AhR-ARNT-DRE bands were quantitated, adjusted to the background (DMSO) levels, and normalized to the wtAhR/TCDD level (Fig. 2C). In this and consequent experiments in this study, the background (DMSO) levels of DNA binding of the wtAhR were consistently at 5 to 10% of total complex formation in the presence of TCDD. These DMSO values were used to calculate the specific complex formation and were not presented separately in most experiments.
While H320A was activated by all compounds in the examined panel except ANF, AhR containing I319A exhibited the most selective response, with 3MC being the only ligand that could activate DNA binding by this mutant AhR to a level significantly greater than background (Fig. 2C). [3H]TCDD binding experiments confirmed that I319A did not bind TCDD (Fig. 2D). In agreement with a previous study (35), F318A was activated by TCDD (and to a smaller extent by 3MC) but not by BNF (Fig. 2C). The [3H]TCDD binding by this mutant relative to that of the wtAhR (40%; Fig. 2D) was similar to its DNA binding level relative to that of the wtAhR (Fig. 2C).
Mutations affect EC50s and antagonist properties of AhR ligands.
To further investigate the TCDD-specific activation of F318A and the 3MC-specific activation of I319A, their EC50s were determined from concentration curves (Fig. 3A and B). While the maximal level of F318A/TCDD activation decreased relative to that of the wtAhR (Fig. 3A), the measured EC50 was little changed (6.9 nM for F318A compared to 2.8 nM for wtAhR). In contrast, the I319A mutation resulted in a much larger increase in the EC50 (86.1 nM for I319A compared to 3.3 nM for wtAhR) (Fig. 3B).
FIG 3.

Mutations affect maximal level, EC50s, and partial antagonist potential of AhR agonists. (A and B) In vitro synthesized wild-type or mutant AhRs and ARNT were incubated in the presence of increasing concentrations of TCDD (A) or 3MC (B) and analyzed by the gel retardation assay. Values represent means ± standard deviations from three independent reactions. (C) In vitro synthesized wild-type or mutant AhRs and ARNT were incubated in the presence of solvent control DMSO (1%, vol/vol) and/or AhR agonists TCDD (10 nM), 3MC (1 μM), ANF (10 μM), and BNF (1 μM) as indicated for 2 h and analyzed by the gel retardation assay. Representative gels are shown. (D) Quantitation of gels shown in panel C. Values represent means ± standard deviations from three independent reactions. Asterisks indicate those antagonist (compound and TCDD) values that are significantly lower than TCDD-alone reactions at P values of <0.05 as determined by the Student t test.
Since 3MC could fully displace [3H]TCDD from F318A (Fig. 2D) but only activated its DNA binding to half the level of TCDD-dependent activation (Fig. 2C), this compound may possess some antagonist properties with the TCDD-activated F318A. DNA binding of F318A was analyzed following transformation in the presence of TCDD and/or 3MC, partial agonist/antagonist ANF, or BNF, which is a chemical isomer of ANF and, reportedly, an AhR agonist. All three compounds (3MC, ANF, and BNF) activated DNA binding of F318A to significantly lower levels than TCDD and displayed antagonist-like properties in decreasing TCDD-dependent DNA binding of this mutant (Fig. 3D). In contrast, only ANF demonstrated antagonist properties with the TCDD-activated wtAhR (Fig. 3D). ANF is termed partial agonist/antagonist of the AhR since it is capable of partially activating AhR on its own (Fig. 3D) in addition to antagonism in the presence of a stronger agonist (TCDD). These results demonstrated that both 3MC and BNF behave as partial agonist/antagonists of F318A (Fig. 3D). Unlike F318A, the I319A mutation did not display any antagonist properties with inactive compounds (TCDD, BNF, and ANF) (data not shown).
Mutations of Ile319 demonstrate ligand-selective properties.
Since I319A demonstrated the most ligand-selective characteristics in the preliminary experiments, a strategy was designed to generate a series of mutations of Ile319 in which this residue was converted into all possible amino acids (except Pro or Gly, which would likely disrupt the overall PASB domain fold) and then to examine the ability of these mutant AhRs to be activated by an expanded set of structurally diverse AhR ligands/agonists. Initial in vitro expression analysis revealed that the mutations did not negatively affect the AhR protein expression levels (data not shown).
In addition to TCDD, 3MC, BNF, and indirubin, the AhR agonist panel was expanded to include dibenz[a,h]anthracene (DBA) and benzo[a]pyrene (BaP) as prototypical PAHs and TCDF and PCB126 as additional HAHs, as well as the following compounds. FICZ (Fig. 4A) is a potent AhR agonist and a proposed endogenous ligand (42, 43). THS-017 (Fig. 4D) is an atypical AhR ligand that also binds to HIF2α and was used to further refine the homology model of the AhR PASB LBD (32, 44). YH439 (Fig. 4D) is an atypical ligand that was used to identify ligand-specific differences with a His285 mutation (36). Leflunomide (Fig. 4A) is an atypical AhR agonist recently shown to be an activator of zebrafish AhR1a (an AhR which does not bind TCDD) in vivo (45, 46). Since direct binding of leflunomide to the AhR has not been previously reported, we confirmed its ability to compete with [3H]TCDD in ligand binding experiments using guinea pig hepatic cytosolic AhR and a hydroxyapatite binding assay (data not shown).
FIG 4.
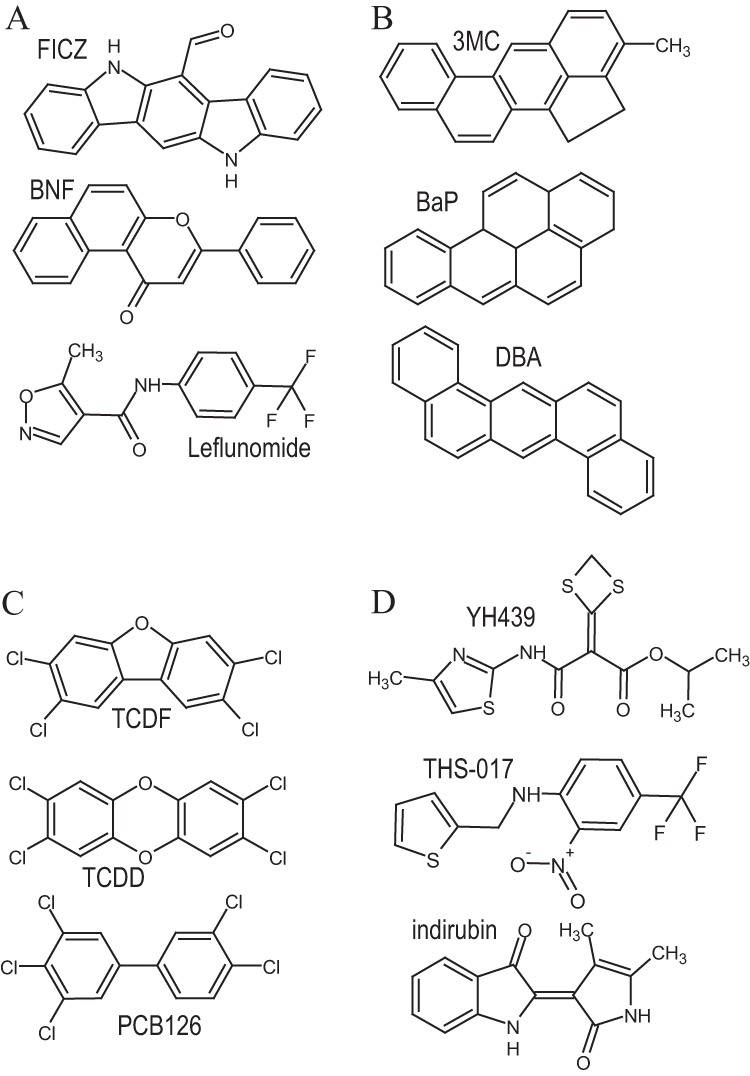
Structurally diverse AhR agonists examined in these studies. (A to D) AhR ligand classification based on similarity of responses in transformation/DNA binding with Ile319 mutations. Details of ligand classification are described in the text.
To facilitate analysis, the agonist panel was divided into four groups based in part on the structural characteristics of the compounds (Fig. 4A to D). This classification of AhR agonists was also based on the results of functional analysis, as detailed below. Groups 2 and 3 included PAHs and HAHs, respectively, while groups 1 and 4 included heteroatom-containing PAHs and atypical AhR ligands, respectively.
We examined the ability of each of the 12 structurally diverse AhR ligands/agonists (Fig. 4A to D) to stimulate the transformation and DNA binding of the wt and the Ile319 mutant AhRs using maximal activating concentrations of each compound (determined in preliminary studies). The results of these DNA binding studies are presented in Fig. 5A to D and Table 1. AhRs containing mutations of Ile319 to charged residues (R, D, E, or H) or to aromatic residues (Y or W) failed to transform and bind to DNA in response to any ligand. Mutations of Ile319 to hydrophobic residues (C, V, M, and L), which were expected to result in minimal structural changes, were activated by all ligands (Table 1). In contrast, mutation of Ile319 to the remaining seven amino acids (K, N, Q, S, T, A, and F) revealed a dramatic range in ligand-specific response to different ligands (Table 1 and Fig. 5A to D). Analysis of the results of these experiments have allowed categorization of the 12 tested AhR ligands/agonists (Fig. 4A to D) into four groups based on similarities or differences in their ability to stimulate transformation and DNA binding of AhRs containing particular Ile319 mutations, as described below.
FIG 5.

Ligand-specific activation of Ile319 mutations. In vitro synthesized wild-type or mutant mAhRs and ARNT were incubated in the presence of solvent control DMSO (1%, vol/vol) or FICZ (0.1 μM), BNF (1 μM), or leflunomide (100 μM) (A), 3MC (1 μM), DBA (0.1 μM), or BaP (1 μM) (B), TCDF (0.1 μM), TCDD (20 nM), or PCB126 (1 μM) (C), or YH439 (10 μM), THS-017 (50 μM), or indirubin (1 μM) (D) for 2 to 2.5 h and analyzed by the gel retardation assay. Gels were visualized and specific bands were quantitated. Values represent means ± standard deviations from three independent reactions.
TABLE 1.
Ligand-selective DNA binding of Ile319-mutated AhRs
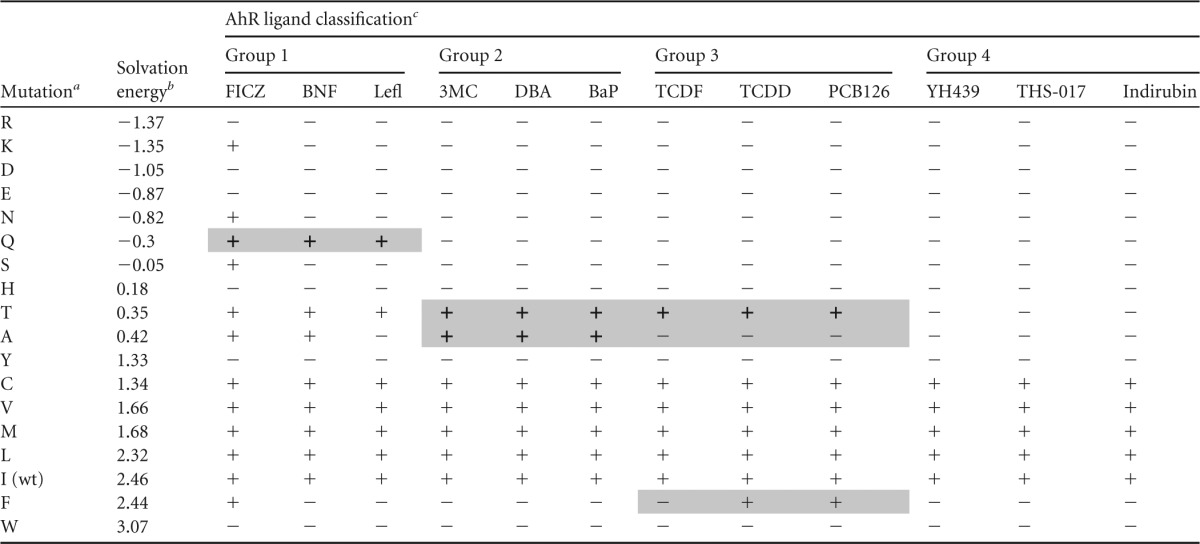
Ile319 was mutated to the indicated residue.
Observed free energy of solvation, kcal mol−1 (61).
+ indicates mutations active with a given ligand in the DNA binding assay; − indicates inactive mutation/ligand combinations. Mutation/ligand activities specific for a given group of ligands are shaded.
Group 1 ligands.
Only compounds in this group (FICZ, BNF, leflunomide) (Fig. 4A) could stimulate transformation/DNA binding of I319Q (Fig. 5A). While there are no obvious major structural similarities between the compounds in this group, each does have a single double-bonded oxygen (Fig. 4A) not found in most other tested AhR ligand agonists, suggesting unique ligand-protein interactions within the LBD that would not be observed with other ligands. Interestingly, FICZ was the only ligand agonist that could stimulate transformation and DNA binding of I319K, and it was consistently more potent than BNF or leflunomide in activating DNA binding of AhRs containing I319S, I319T, I319A, or I319C substitutions (Fig. 5A, Table 1, and data not shown). Based on the likely similarity with the wtAhR/FICZ activation, one can assume that FICZ directly binds to and activates I319K; however, this cannot be experimentally tested due to the lack of radiolabeled FICZ for binding studies.
Group 2 ligands.
This group contains the prototypical PAHs: 3MC, DBA, and BaP (Fig. 4B). Unlike AhR ligands from other groups, group 2 compounds demonstrate robust activation of AhRs containing I319A or I319T substitutions (Fig. 5B). Based on these criteria alone, FICZ could also be included in this group (Fig. 5A); however, its ability to activate a distinct subset of other mutant AhRs (I319K, I319N, I319Q) suggests that it interacts in the binding pocket in a unique manner and thus it was not included in this group. The compounds in this group share obvious structural similarities in that they represent a series of fused aromatic rings.
Group 3 ligands.
Compounds included in this group consist of the prototypical HAH AhR ligands (TCDD, TCDF, and PCB126) (Fig. 4C) and are unable to activate I319A, in addition to being the only ligand group (TCDD and PCB126) that could activate I319F (Fig. 5C). Interestingly, TCDD was a significantly more efficacious activator of I319T than the other HAH ligands tested (TCDF and PCB126) (Fig. 5C).
Group 4 ligands.
Compounds within this group (YH439, THS-017, indirubin) (Fig. 4D) were unique in that they failed to activate AhRs containing either I319T or I319A and, overall, activated the fewest number of mutant AhRs (Fig. 5D). Only AhR containing Ile319 mutations to C, V, M, and L could be transformed and bind to DNA in response to these compounds. The compounds in this group comprise mostly novel atypical AhR ligand/agonist structures (Fig. 4D), which may explain their high sensitivity to mutational changes.
Overall, these results reveal significant differences in the ability and/or potency/efficacy of different AhR ligand agonists to stimulate AhR transformation and DNA binding of AhRs containing different mutations (Fig. 5A to D). These results are consistent with significant differences in the specific binding of different ligands or classes of ligands to the AhR. Moreover, they suggest differences in ligand-dependent mechanisms of AhR transformation and argue against a common set of amino acid interactions within the AhR LBD for all ligand agonists.
Expanded mutagenesis of Phe318.
Given the success with the Ile319 mutations, a similar series of Phe318 mutations to all amino acids (except Gly and Pro) was generated, and the level of in vitro protein expression of all of these Phe318 mutations was similar to wtAhR (data not shown). Overall, the Phe318 mutagenesis resulted in several inactive mutants (S, D, Q, K, and R), while the remaining mutations demonstrated two common phenotypes, as follows.
The first type of Phe318 mutants included mutations to E, N, H, T, C, V, and I and demonstrated a functional profile reminiscent of the F318A mutant. Specifically, similar to F318A (Fig. 2C), these mutants demonstrated higher activation with TCDD than other ligands of the preliminary agonist panel (F318N, F318H, F318C, F318I) (Fig. 6A). However, with the expanded agonist panel, these mutants also demonstrated generally higher activation with FICZ and DBA, as demonstrated for F318E in Fig. 6B.
FIG 6.
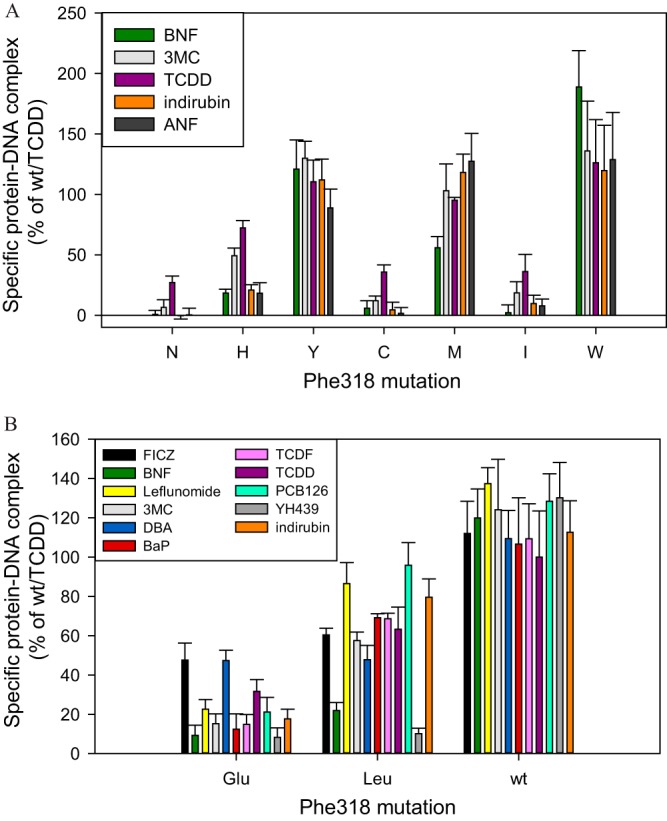
Ligand-specific activation of Phe318 mutations. In vitro synthesized wild-type or mutant mAhRs and ARNT were incubated in the presence of solvent control DMSO (1%, vol/vol) or TCDD (20 nM), 3MC (1 μM), BNF (1 μM), indirubin (1 μM), ANF (10 μM), DBA (0.1 μM), BaP (1 μM), FICZ (0.1 μM), PCB126 (1 μM), TCDF (0.1 μM), YH439 (10 μM), or leflunomide (100 μM) for 2 to 2.5 h and analyzed by gel retardation assay. Gels were visualized and specific bands were quantitated. Values represent means ± standard deviations from three independent reactions.
The second type of Phe318 mutants included mutations to Y, M, W, and L and demonstrated comparable DNA-binding levels with all ligands studied (Fig. 6A and B), with few exceptions. Specifically, F318L appeared to be much less active with BNF and YH439 compared to all other ligands examined (Fig. 6B).
Overall, Phe318 mutations did not present much interest for developing ligand-selective AhR variants. Unlike the variable patterns for the Ile319 mutants (Table 1), the ligand-selective Phe318 mutants appear to predictably favor activation by certain ligands (FICZ, DBA, and TCDD).
BNF is a partial agonist/antagonist of F318L.
The disproportionate reduction in the ability of BNF and YH439 to stimulate transformation and DNA binding of F318L was further examined using BNF. One possible explanation for the reduced activity of these ligands with F318L is that this substitution selectively reduced binding of these ligands. Accordingly, we examined the concentration-dependent ability of BNF to compete with [3H]TCDD for binding to both wt and F318L-containing AhRs. These competitive binding experiments not only demonstrate that the presence of the F318L mutation does not eliminate the ability of BNF to effectively compete with [3H]TCDD for binding to the AhR, but they indicate that BNF is only a slightly weaker competitor for the mutant AhR than it is for the wtAhR (Fig. 7A).
FIG 7.

BNF is a partial antagonist of an AhR containing an F318L substitution. (A) In vitro synthesized mAhR(F318L) was incubated in the presence of 10 nM [3H]TCDD and indicated concentrations of BNF for 30 min, and [3H]TCDD bound to the protein fraction was measured by the hydroxyapatite assay. The unprogrammed TNT lysate was used as a nonspecific binding control, and specific binding was calculated as a difference between the total and nonspecific reactions. Values are presented as means ± standard deviations from three independent reactions. Asterisks indicate the values that are significantly lower than the no-competitor reaction, as indicated by the Student t test with P values of <0.05. (B) In vitro synthesized wt or F318L mAhR (with added ARNT) was incubated with increasing concentrations of BNF for 2 to 2.5 h and analyzed by the gel retardation assay. Gels were visualized, and specific bands were quantitated. Values represent means ± standard deviations from three independent reactions. The curves were fitted using the nonlinear regression analysis (SigmaPlot). (C) In vitro synthesized F318L mAhR was transformed in the presence of increasing concentrations of BNF alone or in the presence of 10 nM TCDD and analyzed as described for panel B. (D) In vitro synthesized F318L mAhR was transformed in the presence of increasing concentrations of ANF alone or in the presence of 10 nM TCDD and analyzed as described for panel B. (C and D) Asterisks indicate the values that are significantly lower than the TCDD reaction at P values of <0.05 as determined by the Student t test. (A to D) Results are representative of three independent experiments.
Next, the concentration-dependent ability of BNF to stimulate AhR transformation and DNA binding was compared for the wtAhR and F318L. The results of these experiments (Fig. 7B) reveal that the F318L mutant AhR requires more than 400 times more BNF to produce a DNA binding response than that observed with the wtAhR (EC50s for wtAhR and F318L were 9.03 ± 0.36 nM and 3.8 ± 2.0 μM, respectively). These results demonstrate that the reduced potency of BNF as an agonist of F318L is due to a change in its efficacy as an activator of AhR transformation and DNA binding and not due to its inability to bind to the AhR, suggesting possible AhR antagonist potential. Moreover, the F318L mutation appears to alter or restrict the ability of BNF to interact with particular residues within or adjacent to the LBD important for AhR transformation.
The BNF antagonist potential with F318L was examined using increasing BNF concentrations in the presence of TCDD in the AhR transformation reaction (Fig. 7C). While BNF was capable of activating DNA binding of F318L in a concentration-dependent manner (agonist activity), in the presence of TCDD, it significantly decreased the TCDD-dependent activity (Fig. 7C), which constitutes antagonism. The resulting U-shaped dose-response antagonist curve (Fig. 7C) is characteristic for partial AhR agonist/antagonists, such as ANF and MNF, which previously demonstrated antagonist activity at lower concentrations and full agonist activity at higher concentrations (47–50).
In fact, the BNF biphasic antagonist response curve for TCDD-activated F318L (Fig. 7C) is essentially identical to that obtained for this mutant with TCDD in the presence of increasing concentrations of ANF, a well-established partial AhR agonist/antagonist (Fig. 7D). Since BNF did not demonstrate antagonism with wtAhR/TCDD at 1 μM (Fig. 3D) but was an antagonist at this concentration with F318L (Fig. 7C), this specific mutation was responsible for converting BNF into a partial agonist/antagonist. It appears that the F318L mutation also improved the ANF antagonist properties, since ANF (at 1 μM) decreased TCDD-dependent wtAhR DNA binding by 35% (Fig. 3D) compared to a decrease of 55% with F318L (Fig. 7D).
Although partial agonist/antagonist is an accepted term for AhR ligands with this type of activity, it is important to realize that these compounds act as agonists and antagonists likely through two distinct mechanisms, possibly binding in two distinct but overlapping sites within the AhR LBD with a resulting agonist or antagonist mode of interaction. In this mechanism, higher-affinity binding in the antagonist site would result in antagonist properties at lower concentrations and the characteristic U-shaped curve.
Effects of mutations on TCDD ligand binding affinity and hsp90 binding.
The effect of select TCDD-responsive Phe318 and Ile319 mutations, including F318E, F318L, F318A, and I319V on [3H]TCDD binding affinity was examined using nonlinear saturating binding curve analysis with an in vitro synthesized mutant and wtAhR proteins. The calculated [3H]TCDD affinity values (as averages ± standard deviations of triplicate measurements) were 3.2 ± 0.5 nM, 3.2 ± 0.4 nM, 3.1 ± 0.1 nM, and 2.5 ± 0.6 nM for F318E, F318L, F318A, and I319V, respectively. There were no significant differences among these values and that of the wtAhR (2.6 ± 0.8 nM), indicating that the examined mutations at positions Phe318 and Ile319 have little effect on the affinity of TCDD for the AhR. However, certain mutations at these positions, such as I319A, completely eliminated TCDD binding altogether (Fig. 2D), and this “all or nothing” effect with regard to ligand binding suggests that insertion of specific amino acids in these positions may result in significant alterations within the ligand binding pocket that contribute to steric constraints and loss of ligand binding.
In addition to ligand binding, the PASB domain mediates AhR binding to hsp90, and residues within the binding pocket have been shown to be involved (9, 39, 51). Since hsp90 maintains the AhR in a ligand-binding competent state (52), mutation-mediated changes in AhR-hsp90 association may result in variable effects on binding and activation by different ligands, potentially underlying ligand-specific mechanisms. Therefore, AhR mutants with various degrees of ligand promiscuity were examined for hsp90 binding. These included I319K (activated only by FICZ in vitro), F318E (activated by most compounds but with preferential activation by FICZ, DBA, and TCDD), and F318L (activated by most compounds equally well to TCDD).
Hsp90 binding to these mutants and wtAhR was examined in transient transfections in COS-1 cells (Fig. 8A). Although similar amounts of AhR protein were produced from each of the expression constructs used in these studies (see inputs in Fig. 8A), hsp90 binding levels varied significantly between the mutant AhRs. Specifically, hsp90 binding to the I319K AhR was the lowest, binding to F318L was similar to that of wtAhR, and binding to F318E was intermediate to that of I319K and F318L (Fig. 8B). Interestingly, the hsp90 binding values were inversely correlated with ligand promiscuity of these mutations.
FIG 8.
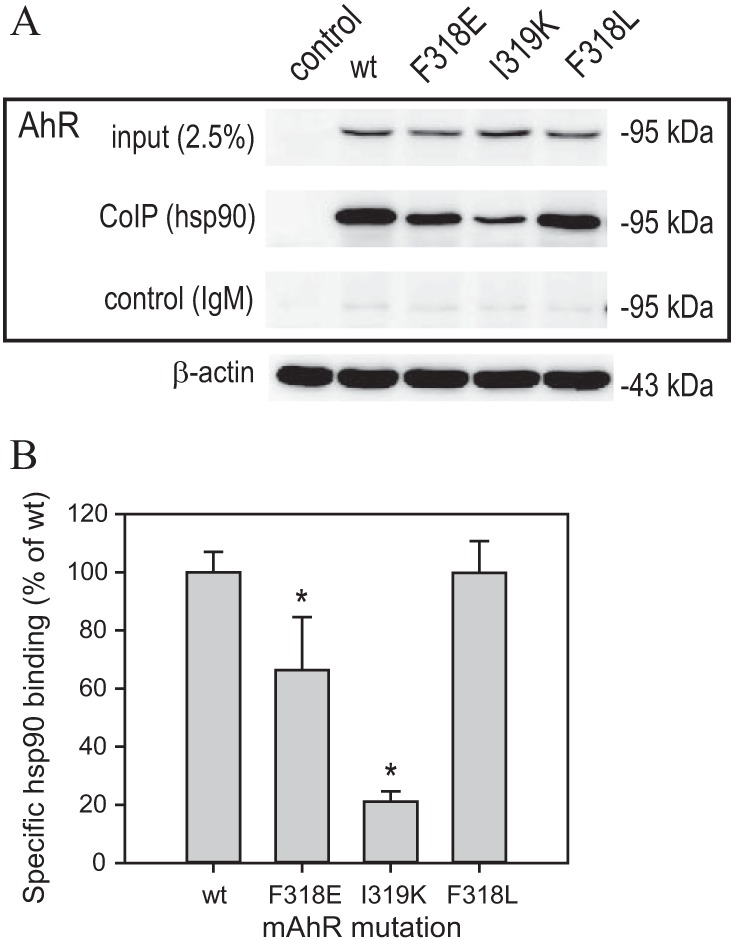
Hsp90 binding analysis with select mAhR mutations. (A) COS-1 cells were transfected with indicated mAhR constructs and 20 h later lysed using RIPA lysis buffer. Lysates were immunoprecipitated using anti-hsp90 antibody (3G3) or IgM control, and the precipitated proteins were analyzed by Western assay using anti-AhR or anti-beta-actin antibodies. Representative gels are shown. (B) Quantitation of gels shown in panel A. Values represent means ± standard deviations from three independent reactions. Asterisks indicate the values that are significantly different from the wild type (wt) at P values of <0.05 as determined by the Student t test.
Activation of ligand-selective mutations in cell culture.
Select AhR mutations were further characterized in reporter assays in cell culture. AhR constructs and an AhR-responsive luciferase reporter pGudLuc6.1 (41) were transiently transfected into COS-1 cells. Cells were incubated with a panel of ligands for 20 h, and the luciferase activity of cell lysates was measured. Transient transfections were optimized for equal levels of AhR proteins (Fig. 9A). Overall, the patterns of ligand-specific activation in the reporter assay (Fig. 9B) matched those observed with the in vitro transformation/DNA binding assays. Specifically, AhRs containing I319Q, I319F, or I319K demonstrated highly ligand-specific activation with both the in vitro and cell culture-based assays (Fig. 5A to D, 9B).
FIG 9.

FICZ-specific mutation is not activated by other AhR ligands in cell culture. COS-1 cells transiently transfected with wild-type or mutant mAhR (or control plasmid pcDNA3.1+ [ctrl]) and DRE-containing reporter pGudLuc6.1 were treated with solvent control DMSO (0.1%, vol/vol), TCDD (10 nM), FICZ (0.1 μM), PCB126 (1 μM), DBA (0.1 μM), BaP (1 μM), indirubin (1 μM), or 3MC (0.1 μM) for 18 to 20 h. (A) Cells were lysed, and lysates were analyzed for firefly luciferase activity. Results are representative of three experiments. (B) Aliquots of lysate in panel A were analyzed by Western assay with anti-AhR antibody (M20). Values represent means ± standard deviations from three independent reactions.
However, several differences between the in vitro and cell-based experimental systems were also observed. I319Q was not activated by indirubin in vitro (Fig. 5D) but was readily activated in cell culture (Fig. 9B). It is possible that indirubin may be metabolically converted into a different AhR ligand with a different ligand-specific profile, namely, with specificity similar to group 1 ligands. Chemical transformation in cell culture into AhR ligands/agonists has been previously observed for other indole-containing compounds, including the formation of FICZ (43).
While I319T mutant AhR was readily activated into its DNA binding form by TCDD in vitro (Fig. 5C), this was not observed in the reporter assay (Fig. 9B). In contrast, PCB126 was more active with this particular mutation in the reporter assay (compared to TCDD) than with the in vitro DNA binding assay (Fig. 5C and 9). The mechanisms underlying these differences remain to be determined.
The results from the representative Phe318 mutation F318E are demonstrated in Fig. 9B. Similar to the DNA-binding assay (Fig. 6B), this mutation was most active with FICZ and DBA but was also active with other compounds. Other Phe318 mutations, such as F318A, revealed similar ligand specificity in reporter assays (data not shown). Due to differences in antagonist mechanisms between the TNT-based and COS-1 cell experimental systems, as well as due to BNF toxicity at required concentrations, antagonist properties of Phe318 mutations (including F318L) have not been studied in reporter assays.
DISCUSSION
The classical AhR-dependent mechanism of activation of gene expression has been generally well characterized, and the linearity of this pathway implies that various AhR agonist ligands would trigger similar AhR-mediated biological responses. However, this point of view has been challenged by multiple studies. Ligand-specific biological responses have been observed in some instances of AhR-dependent transcription, including that of T cell differentiation and the effects of selective AhR modulators (18, 24–27, 53–57). To account for these ligand-specific differences, modifications of the classical pathway have been suggested, with most involving ligand-selective binding of the AhR complex to unconventional DRE sequences (24, 25, 58) or binding by different subsets of coactivators (27). These mechanisms imply ligand-dependent differences in the conformation of the AhR complex, and therefore distinct ligand-specific mechanisms of binding of ligands to the AhR would likely exist. In fact, different AhR ligands may bind within the ligand-binding domain (LBD) in distinct sites (59).
Mutations of Ile319 reveal a diversity of ligand-selective mechanisms.
Ile319 has been found to be a key residue in ligand specificity since mutations of this residue resulted in a wide variety of ligand-selective effects (Table 1). In contrast, mutations of Phe318 presented a more predictable pattern, in general favoring certain ligands (TCDD, DBA, and FICZ) over others (Fig. 6A and B). Both these amino acid residues are predicted to line the LBD cavity and are therefore termed fingerprint residues (33, 60). In tern AhR, Val present in the position homologous to mouse Ile319 may contribute to the lower affinity of the tern AhR for TCDD (40). However, since [3H]TCDD binding affinity was not changed with several Phe318 and Ile319 mutations in this study (F318A, F318L, F318E, I319V), the contribution of these positions to the affinity of bound ligand in mouse AhR appears to be minor.
In contrast, the nature of the amino acid at position 319 appears to have a critical effect on the AhR ligand promiscuity (Table 1). The observed activation patterns correlate to some extent with solvation energy (61) of the substituted amino acids. Several hydrophilic residues (with low solvation energy) (Table 1) in this position favor AhR activation by group 1 compounds, while only substitutions to hydrophobic residues (with high solvation energy) (Table 1) retain activity with the group 4 compounds. Substitutions to Ala or Phe appear to differentiate the group 2 and group 3 compounds (Table 1), indicating that the size of the hydrophobic side chain may determine subtle differences in ligand binding.
Mutations of Phe318 result in changed agonist and antagonist modes of activation.
This study has identified Phe318 as an amino acid residue involved in determination of agonist/antagonist behavior of AhR ligands and exerting a lesser effect on ligand selectivity than Ile319.
The F318L mutation dramatically decreased BNF-dependent agonism (Fig. 7B), while it only slightly affected BNF binding (Fig. 7A). Thus, BNF antagonist activity may be due to BNF binding to the AhR, while the F318L mutations would interfere with the BNF binding in the agonist conformation/site. Thus, Phe318 mutations (F318L and F318A, as discussed above) may distinguish between the agonist and antagonist binding sites for compounds like BNF and 3MC. To our knowledge, this is the first report of an individual residue within the PASB involved in AhR transformation. Unlike Phe318 mutations, no antagonist activity was found for Ile319 mutations with inactive compounds, indicating distinct structural roles for these residues.
Currently, there are several hypotheses regarding the mechanism of antagonist action on AhR functionality. In one possible mechanism, antagonists would bind to the AhR but, unlike agonists, would be unable to displace hsp90 from the PASB domain binding site (10). In this mechanism, Phe318 would facilitate hsp90 displacement by BNF through destabilization of AhR-hsp90 interactions, and the F318L substitution would interfere with this effect. However, hsp90 binding to F318L was similar to that of the wtAhR (Fig. 8B), arguing against this mechanism. Moreover, considering that some hsp90 binding determinants are located within the LBD and close to Ala375 (10) (Fig. 1), binding of any ligand, agonist or antagonist, would be expected to displace hsp90 from the AhR LBD. Accordingly, the mechanism of action of AhR antagonists will require further study.
Bound hsp90 may maintain AhR ligand promiscuity.
Hsp90 binding studies revealed an inverse correlation between levels of hsp90 binding and ligand promiscuity of AhR mutations. Specifically, levels of hsp90 binding were lowest with I319K, intermediate with F318E, and greatest with F318L and wtAhR, while ligand promiscuity was lowest with I319K (activated only by one ligand [FICZ]) and greatest with F318L and wtAhR (activated by most or all ligands tested) (Fig. 8B). This correlation was further observed for hsp90 binding studies with F318A and I319V (data not shown). Since hsp90 has been proposed to maintain the AhR LBD in an open ligand-binding competent state (13), it is possible that decreased hsp90 binding would result in a diminished ability of most AhR ligands to bind within the AhR LBD, and only the most optimal ligands (such as FICZ, TCDD, and DBA) would be able to bind to and activate these AhR mutants. In this mechanism, reduced hsp90 binding would invariably result in fewer active ligands (i.e., reduced ligand promiscuity with a given AhR mutation).
Table 2 summarizes and compares the functional properties of select AhR LBD residues implicated in ligand binding and characterized in this and previous (10, 37) studies. One common thread from the comparison of these data appears to be the interconnection of hsp90 binding properties with the agonist/antagonist modes and ligand promiscuity of AhR activation. This interdependence confirms the overlap of hsp90-binding and ligand-binding regions (10) with the antagonist/agonist region, which was suggested by the structural proximity of the involved amino acid residues (Fig. 1).
TABLE 2.
Ligand-binding and hsp90-binding properties of select AhR LBD residues
| Residue | Function | Mutation | Change in function with mutation | Additional properties of mutations | Reference |
|---|---|---|---|---|---|
| Region of aa 280 to 287 | Mediates hsp90 binding | To various residues | Decreased hsp90 binding | Changed ANF agonist/antagonist potential | 10 |
| Phe318 | Mediates agonist/antagonist switch | To Ala, Leu | Converted 3MC and/or BNF into an antagonist | Decrease in hsp90 binding | This study |
| Ile319 | Mediates ligand-specific agonist binding | To K, Q, A, T, F | Conferred various degrees of ligand-selective activation | Decrease in hsp90 binding | This study |
| Ala375 | Mediates agonist binding | To Val | Decreased AhR affinity for TCDD | 37 |
Consistent with this common overlap of functional properties (Table 2), Ala375, previously shown to stabilize bound TCDD and whose mutation can dramatically decrease TCDD binding affinity (33, 37), may itself demonstrate ligand-selective preferences in ligand binding. In fact, indirubin is a more potent activator of the human AhR, which contains a Val in position 375, than the mouse AhR, which contains an Ala in this position (62).
The validity of the classical mechanism of AhR activation by structurally diverse chemicals.
Taken together, the mutagenesis and functional activity results presented here and elsewhere clearly demonstrate that the AhR can directly bind and be activated by structurally diverse chemicals (1, 2, 36, 63–66). These conclusions are in striking contrast to a recent publication by Wincent and coworkers (42) that proposed that the structural diversity of AhR ligands was an artifact due to inhibition of cytochrome P450-mediated degradation of an extremely potent but metabolically labile AhR agonist (FICZ) that was reported to be present in culture media. In this mechanism, it was proposed that these structurally diverse AhR ligands inhibit the P450 enzymes that degrade FICZ, resulting in its stabilization, and as such, FICZ was proposed to be the true ligand responsible for activation of AhR-dependent transcription by these compounds (42). At least two major lines of evidence argue against this FICZ degradation-based mechanism and support the ability of the AhR to bind and be directly activated by structurally diverse chemicals.
First, in the mutagenesis studies described here, we identified an AhR mutant (I319K) that was activated only by FICZ in cells in culture and not by any other tested AhR ligand (Fig. 9B). If the hypothesis by Wincent et al. (42) was correct, then at least some of the structurally diverse ligands that were tested here would have been expected to indirectly activate the I319K-dependent reporter gene expression by inhibiting FICZ degradation, but this was not observed. Therefore, the hypothesis that FICZ-dependent AhR transcription would mediate transcriptional activation by diverse AhR ligands is incorrect. Wincent et al. (42) based their conclusions on the indirect evidence comprising correlations of AhR transcriptional activity with inhibition of CYP1 cytochromes by AhR ligands. In this, they did not present direct experimental evidence against the mechanism of AhR activation by diverse ligands. In contrast, our results present direct experimental evidence against the FICZ-mediated mechanism that supports the activation of the AhR by these diverse chemicals. Moreover, the well-documented ability of structurally diverse chemicals to competitively bind to and/or stimulate AhR-dependent transformation and DNA binding in vitro using cytosolic extracts (reviewed in reference 1) also argues that these diverse chemicals are indeed AhR ligands/agonists. Even if FICZ was present in the cytosol in these in vitro studies, it would have no impact on the results beyond contributing to an elevation in background activity. Taken together, these results strongly support the ability of the AhR to be directly bound and be activated by structurally diverse chemicals.
Numerous investigators have proposed a wide variety of structurally diverse endogenous ligands for the AhR, including FICZ, kynurenine, ITE, bilirubin, biliverdin, prostaglandins, leukotriene metabolites, 7-ketocholesterol, and others (63–66). Identification of ligand-selective mutations that can be activated by these compounds would provide the means to verify their identity as endogenous ligands in biological models, such as insertion of these ligand-selective AhRs into transgenic AhR-knockout mice. Furthermore, ligand-selective mutations can be used as biological probes for functional classifications of AhR agonists, in order to elucidate the mechanisms of binding and toxicity of dioxin-like compounds, as well as to study endogenous AhR-dependent pathways. Finally, development of ligand-specific AhR bioassays will be useful in environmental and pharmacological screening and will further our understanding of the mechanisms of AhR-dependent toxic and biological effects.
ACKNOWLEDGMENTS
This research was supported by the National Institute of Environmental Health Sciences (R01ES07685), an NIEHS Superfund Research Grant (P42ES004699), and the California Agricultural Experiment Station.
We thank Steven Safe (Texas A&M University) for TCDD and [3H]TCDD, Gary Perdew (Pennsylvania State University) for 3G3 antibody, and Laura Bonati (University of Milano-Bicocca) for the PASB model files and helpful discussions of results.
Footnotes
Published ahead of print 3 March 2014
REFERENCES
- 1.DeGroot DE, He G, Fraccalvieri D, Bonati L, Pandini A, Denison MS. 2011. AhR ligands: promiscuity in binding and diversity in response, p 63–79 In Pohjanvirta R. (ed), The AH receptor in biology and toxicology. Wiley, Hoboken, NJ [Google Scholar]
- 2.Denison MS, Soshilov AA, He G, DeGroot DE, Zhao B. 2011. Exactly the same but different: promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor. Toxicol. Sci. 124:1–22. 10.1093/toxsci/kfr218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.NTP. 2011. 12th report on carcinogens. Department of Health, Human Services, National Toxicology Program, Research Triangle Park, NC [Google Scholar]
- 4.White SS, Birnbaum LS. 2009. An overview of the effects of dioxins and dioxin-like compounds on vertebrates, as documented in human and ecological epidemiology. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 27:197–211. 10.1080/10590500903310047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berghard A, Gradin K, Toftgård R. 1992. The stability of dioxin-receptor ligands influences cytochrome P450IA1 expression in human keratinocytes. Carcinogenesis 13:651–655. 10.1093/carcin/13.4.651 [DOI] [PubMed] [Google Scholar]
- 6.Kim MJ, Pelloux V, Guyot E, Tordjman J, Bui LC, Chevallier A, Forest C, Benelli C, Clement K, Barouki R. 2012. Inflammatory pathway genes belong to major targets of persistent organic pollutants in adipose cells. Environ. Health Perspect. 120:508–514. 10.1289/ehp.1104282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Riddick DS, Huang Y, Harper PA, Okey AB. 1994. 2,3,7,8-Tetrachlorodibenzo-p-dioxin versus 3-methylcholanthrene: comparative studies of Ah receptor binding, transformation, and induction of CYP1A1. J. Biol. Chem. 269:12118–12128 [PubMed] [Google Scholar]
- 8.Coumailleau P, Poellinger L, Gustafsson JA, Whitelaw ML. 1995. Definition of a minimal domain of the dioxin receptor that is associated with Hsp90 and maintains wild type ligand binding affinity and specificity. J. Biol. Chem. 270:25291–25300. 10.1074/jbc.270.42.25291 [DOI] [PubMed] [Google Scholar]
- 9.Fukunaga BN, Probst MR, Reisz-Porszasz S, Hankinson O. 1995. Identification of functional domains of the aryl hydrocarbon receptor. J. Biol. Chem. 270:29270–29278. 10.1074/jbc.270.49.29270 [DOI] [PubMed] [Google Scholar]
- 10.Soshilov A, Denison MS. 2011. Ligand displaces heat shock protein 90 from overlapping binding sites within the aryl hydrocarbon receptor ligand-binding domain. J. Biol. Chem. 286:35275–35282. 10.1074/jbc.M111.246439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen HS, Singh SS, Perdew GH. 1997. The Ah receptor is a sensitive target of geldanamycin-induced protein turnover. Arch. Biochem. Biophys. 348:190–198. 10.1006/abbi.1997.0398 [DOI] [PubMed] [Google Scholar]
- 12.Henry EC, Gasiewicz TA. 1993. Transformation of the aryl hydrocarbon receptor to a DNA-binding form is accompanied by release of the 90 kDa heat-shock protein and increased affinity for 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biochem. J. 294(Part 1):95–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pongratz I, Mason GG, Poellinger L. 1992. Dual roles of the 90-kDa heat shock protein hsp90 in modulating functional activities of the dioxin receptor. Evidence that the dioxin receptor functionally belongs to a subclass of nuclear receptors which require hsp90 both for ligand binding activity and repression of intrinsic DNA binding activity. J. Biol. Chem. 267:13728–13734 [PubMed] [Google Scholar]
- 14.Probst MR, Reisz-Porszasz S, Agbunag RV, Ong MS, Hankinson O. 1993. Role of the aryl hydrocarbon receptor nuclear translocator protein in aryl hydrocarbon (dioxin) receptor action. Mol. Pharmacol. 44:511–518 [PubMed] [Google Scholar]
- 15.Denison MS, Phelps CL, Dehoog J, Kim HJ, Bank PA, Yao EF. 1991. Species variation in Ah receptor transformation and DNA binding, p 337–349 In Gallo MA, Scheuplein RJ, Van Der, Heijden KA. (ed), Banbury report no. 35: biological basis of risk assessment of dioxins and related compounds. Cold Spring Harbor Press, Cold Spring Harbor, NY [Google Scholar]
- 16.Denison MS, Fisher JM, Whitlock JP., Jr 1988. The DNA recognition site for the dioxin-Ah receptor complex. Nucleotide sequence and functional analysis. J. Biol. Chem. 263:17221–17224 [PubMed] [Google Scholar]
- 17.Bunger MK, Glover E, Moran SM, Walisser JA, Lahvis GP, Hsu EL, Bradfield CA. 2008. Abnormal liver development and resistance to 2,3,7,8-tetrachlorodibenzo-p-dioxin toxicity in mice carrying a mutation in the DNA-binding domain of the aryl hydrocarbon receptor. Toxicol. Sci. 106:83–92. 10.1093/toxsci/kfn149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, Caccamo M, Oukka M, Weiner HL. 2008. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 453:65–71. 10.1038/nature06880 [DOI] [PubMed] [Google Scholar]
- 19.Boitano AE, Wang J, Romeo R, Bouchez LC, Parker AE, Sutton SE, Walker JR, Flaveny CA, Perdew GH, Denison MS, Schultz PG, Cooke MP. 2010. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science 329:1345–1348. 10.1126/science.1191536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gramatzki D, Pantazis G, Schittenhelm J, Tabatabai G, Kohle C, Wick W, Schwarz M, Weller M, Tritschler I. 2009. Aryl hydrocarbon receptor inhibition downregulates the TGF-beta/Smad pathway in human glioblastoma cells. Oncogene 28:2593–2605. 10.1038/onc.2009.104 [DOI] [PubMed] [Google Scholar]
- 21.Goodale BC, Tilton SC, Corvi MM, Wilson GR, Janszen DB, Anderson KA, Waters KM, Tanguay RL. 2013. Structurally distinct polycyclic aromatic hydrocarbons induce differential transcriptional responses in developing zebrafish. Toxicol. Appl. Pharmacol. 272:656–670. 10.1016/j.taap.2013.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hrubá E, Vondráček J, Líbalová H, Topinka J, Bryja V, Souček K, Machala M. 2011. Gene expression changes in human prostate carcinoma cells exposed to genotoxic and nongenotoxic aryl hydrocarbon receptor ligands. Toxicol. Lett. 206:178–188. 10.1016/j.toxlet.2011.07.011 [DOI] [PubMed] [Google Scholar]
- 23.Ovando BJ, Ellison CA, Vezina CM, Olson JR. 2010. Toxicogenomic analysis of exposure to TCDD, PCB126, and PCB153: identification of genomic biomarkers of exposure to AhR ligands. BMC Genomics 11:583. 10.1186/1471-2164-11-583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gouedard C, Barouki R, Morel Y. 2004. Dietary polyphenols increase paraoxonase 1 gene expression by an aryl hydrocarbon receptor-dependent mechanism. Mol. Cell. Biol. 24:5209–5222. 10.1128/MCB.24.12.5209-5222.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matikainen T, Perez GI, Jurisicova A, Pru JK, Schlezinger JJ, Ryu HY, Laine J, Sakai T, Korsmeyer SJ, Casper RF, Sherr DH, Tilly JL. 2001. Aromatic hydrocarbon receptor-driven Bax gene expression is required for premature ovarian failure caused by biohazardous environmental chemicals. Nat. Genet. 28:355–360. 10.1038/ng575 [DOI] [PubMed] [Google Scholar]
- 26.Murray IA, Morales JL, Flaveny CA, DiNatale BC, Chiaro C, Gowdahalli K, Amin S, Perdew GH. 2010. Evidence for ligand-mediated selective modulation of aryl hydrocarbon receptor activity. Mol. Pharmacol. 77:247–254. 10.1124/mol.109.061788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang S, Rowlands C, Safe S. 2008. Ligand-dependent interactions of the Ah receptor with coactivators in a mammalian two-hybrid assay. Toxicol. Appl. Pharmacol. 227:196–206. 10.1016/j.taap.2007.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ngan C-H, Beglov D, Rudnitskaya AN, Kozakov D, Waxman DJ, Vajda S. 2009. The structural basis of pregnane X receptor binding promiscuity. Biochemistry 48:11572–11581. 10.1021/bi901578n [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ekins S, Chang C, Mani S, Krasowski MD, Reschly EJ, Iyer M, Kholodovych V, Ai N, Welsh WJ, Sinz M, Swaan PW, Patel R, Bachmann K. 2007. Human pregnane X receptor antagonists and agonists define molecular requirements for different binding sites. Mol. Pharmacol. 72:592–603. 10.1124/mol.107.038398 [DOI] [PubMed] [Google Scholar]
- 30.Mani S, Dou W, Redinbo MR. 2013. PXR antagonists and implication in drug metabolism. Drug Metab. Rev. 45:60–72. 10.3109/03602532.2012.746363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao B, Degroot DE, Hayashi A, He G, Denison MS. 2010. CH223191 is a ligand-selective antagonist of the Ah (Dioxin) receptor. Toxicol. Sci. 117:393–403. 10.1093/toxsci/kfq217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Motto I, Bordogna A, Soshilov AA, Denison MS, Bonati L. 2011. New aryl hydrocarbon receptor homology model targeted to improve docking reliability. J. Chem. Inf. Model. 51:2868–2881. 10.1021/ci2001617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pandini A, Soshilov AA, Song Y, Zhao J, Bonati L, Denison MS. 2009. Detection of the TCDD binding-fingerprint within the Ah receptor ligand binding domain by structurally driven mutagenesis and functional analysis. Biochemistry 48:5972–5983. 10.1021/bi900259z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Backlund M, Ingelman-Sundberg M. 2004. Different structural requirements of the ligand binding domain of the aryl hydrocarbon receptor for high- and low-affinity ligand binding and receptor activation. Mol. Pharmacol. 65:416–425. 10.1124/mol.65.2.416 [DOI] [PubMed] [Google Scholar]
- 35.Goryo K, Suzuki A, Carpio CAD, Siizaki K, Kuriyama E, Mikami Y, Kinoshita K, Yasumoto K-I, Rannug A, Miyamoto A, Fujii-Kuriyama Y, Sogawa K. 2007. Identification of amino acid residues in the Ah receptor involved in ligand binding. Biochem. Biophys. Res. Commun. 354:396–402. 10.1016/j.bbrc.2006.12.227 [DOI] [PubMed] [Google Scholar]
- 36.Whelan F, Hao N, Furness SGB, Whitelaw ML, Chapman-Smith A. 2010. Amino acid substitutions in the aryl hydrocarbon receptor ligand binding domain reveal YH439 as an atypical AhR activator. Mol. Pharmacol. 77:1037–1046. 10.1124/mol.109.062927 [DOI] [PubMed] [Google Scholar]
- 37.Poland A, Palen D, Glover E. 1994. Analysis of the four alleles of the murine aryl hydrocarbon receptor. Mol. Pharmacol. 46:915–921 [PubMed] [Google Scholar]
- 38.Fukunaga BN, Hankinson O. 1996. Identification of a novel domain in the aryl hydrocarbon receptor required for DNA binding. J. Biol. Chem. 271:3743–3749. 10.1074/jbc.271.7.3743 [DOI] [PubMed] [Google Scholar]
- 39.Soshilov A, Denison MS. 2008. Role of the Per/Arnt/Sim domains in ligand-dependent transformation of the aryl hydrocarbon receptor. J. Biol. Chem. 283:32995–33005. 10.1074/jbc.M802414200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karchner SI, Franks DG, Kennedy SW, Hahn ME. 2006. The molecular basis for differential dioxin sensitivity in birds: role of the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. U. S. A. 103:6252–6257. 10.1073/pnas.0509950103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rushing SR, Denison MS. 2002. The silencing mediator of retinoic acid and thyroid hormone receptors can interact with the aryl hydrocarbon (Ah) receptor but fails to repress Ah receptor-dependent gene expression. Arch. Biochem. Biophys. 403:189–201. 10.1016/S0003-9861(02)00233-3 [DOI] [PubMed] [Google Scholar]
- 42.Wincent E, Bengtsson J, Mohammadi Bardbori A, Alsberg T, Luecke S, Rannug U, Rannug A. 2012. Inhibition of cytochrome P4501-dependent clearance of the endogenous agonist FICZ as a mechanism for activation of the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. U. S. A. 109:4479–4484. 10.1073/pnas.1118467109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rannug U, Rannug A, Sjoberg U, Li H, Westerholm R, Bergman J. 1995. Structure elucidation of two tryptophan-derived, high affinity Ah receptor ligands. Chem. Biol. 2:841–845 [DOI] [PubMed] [Google Scholar]
- 44.Scheuermann TH, Tomchick DR, Machius M, Guo Y, Bruick RK, Gardner KH. 2009. Artificial ligand binding within the HIF2alpha PAS-B domain of the HIF2 transcription factor. Proc. Natl. Acad. Sci. U. S. A. 106:450–455. 10.1073/pnas.0808092106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goodale BC, La Du JK, Bisson WH, Janszen DB, Waters KM, Tanguay RL. 2012. Ahr2 mutant reveals functional diversity of aryl hydrocarbon receptors in zebrafish. PLoS One 7:e29346. 10.1371/journal.pone.0029346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O'Donnell EF, Saili KS, Koch DC, Kopparapu PR, Farrer D, Bisson WH, Mathew LK, Sengupta S, Kerkvliet NI, Tanguay RL, Kolluri SK. 2010. The anti-inflammatory drug leflunomide is an agonist of the aryl hydrocarbon receptor. PLoS One 5:e13128. 10.1371/journal.pone.0013128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blank JA, Tucker AN, Sweatlock J, Gasiewicz TA, Luster MI. 1987. Alpha-naphthoflavone antagonism of 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced murine lymphocyte ethoxyresorufin-O-deethylase activity and immunosuppression. Mol. Pharmacol. 32:169–172 [PubMed] [Google Scholar]
- 48.Santostefano M, Merchant M, Arellano L, Morrison V, Denison MS, Safe S. 1993. Alpha-naphthoflavone-induced CYP1A1 gene expression and cytosolic aryl hydrocarbon receptor transformation. Mol. Pharmacol. 43:200–206 [PubMed] [Google Scholar]
- 49.Lu YF, Santostefano M, Cunningham BD, Threadgill MD, Safe S. 1995. Identification of 3′-methoxy-4′-nitroflavone as a pure aryl hydrocarbon (Ah) receptor antagonist and evidence for more than one form of the nuclear Ah receptor in MCF-7 human breast cancer cells. Arch. Biochem. Biophys. 316:470–477. 10.1006/abbi.1995.1062 [DOI] [PubMed] [Google Scholar]
- 50.Zhou J, Gasiewicz TA. 2003. 3′-Methoxy-4′-nitroflavone, a reported aryl hydrocarbon receptor antagonist, enhances Cyp1a1 transcription by a dioxin responsive element-dependent mechanism. Arch. Biochem. Biophys. 416:68–80. 10.1016/S0003-9861(03)00274-1 [DOI] [PubMed] [Google Scholar]
- 51.Perdew GH. 1988. Association of the Ah receptor with the 90-kDa heat shock protein. J. Biol. Chem. 263:13802–13805 [PubMed] [Google Scholar]
- 52.Antonsson C, Whitelaw ML, McGuire J, Gustafsson JA, Poellinger L. 1995. Distinct roles of the molecular chaperone hsp90 in modulating dioxin receptor function via the basic helix-loop-helix and PAS domains. Mol. Cell. Biol. 15:756–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gouedard C, Barouki R, Morel Y. 2004. Induction of the paraoxonase-1 gene expression by resveratrol. Arterioscler. Thromb Vasc. Biol. 24:2378–2383. 10.1161/01.ATV.0000146530.24736.ce [DOI] [PubMed] [Google Scholar]
- 54.Murray IA, Flaveny CA, Chiaro CR, Sharma AK, Tanos RS, Schroeder JC, Amin SG, Bisson WH, Kolluri SK, Perdew GH. 2011. Suppression of cytokine-mediated complement factor gene expression through selective activation of the Ah receptor with 3′,4′-dimethoxy-alpha-naphthoflavone. Mol. Pharmacol. 79:508–519. 10.1124/mol.110.069369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Murray IA, Krishnegowda G, DiNatale BC, Flaveny C, Chiaro C, Lin JM, Sharma AK, Amin S, Perdew GH. 2010. Development of a selective modulator of aryl hydrocarbon (Ah) receptor activity that exhibits anti-inflammatory properties. Chem. Res. Toxicol. 23:955–966. 10.1021/tx100045h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen I, McDougal A, Wang F, Safe S. 1998. Aryl hydrocarbon receptor-mediated antiestrogenic and antitumorigenic activity of diindolylmethane. Carcinogenesis 19:1631–1639. 10.1093/carcin/19.9.1631 [DOI] [PubMed] [Google Scholar]
- 57.McDougal A, Wilson C, Safe S. 1997. Inhibition of 7,12-dimethylbenz[a]anthracene-induced rat mammary tumor growth by aryl hydrocarbon receptor agonists. Cancer Lett. 120:53–63. 10.1016/S0304-3835(97)00299-1 [DOI] [PubMed] [Google Scholar]
- 58.Huang G, Elferink CJ. 2012. A novel nonconsensus xenobiotic response element capable of mediating aryl hydrocarbon receptor-dependent gene expression. Mol. Pharmacol. 81:338–347. 10.1124/mol.111.075952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Petkov PI, Rowlands JC, Budinsky R, Zhao B, Denison MS, Mekenyan O. 2010. Mechanism-based common reactivity pattern (COREPA) modelling of aryl hydrocarbon receptor binding affinity. SAR QSAR Environ. Res. 21:187–214. 10.1080/10629360903570933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fraccalvieri D, Soshilov AA, Karchner SI, Franks DG, Pandini A, Bonati L, Hahn ME, Denison MS. 2013. Comparative analysis of homology models of the Ah receptor ligand binding domain: verification of structure-function predictions by site-directed mutagenesis of a nonfunctional receptor. Biochemistry 52:714–725. 10.1021/bi301457f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Eisenberg D, McLachlan AD. 1986. Solvation energy in protein folding and binding. Nature 319:199–203. 10.1038/319199a0 [DOI] [PubMed] [Google Scholar]
- 62.Flaveny CA, Perdew GH. 2009. Transgenic humanized AhR mouse reveals differences between human and mouse AhR ligand selectivity. Mol. Cell. Pharmacol. 1:119–123. 10.4255/mcpharmacol.09.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, Schumacher T, Jestaedt L, Schrenk D, Weller M, Jugold M, Guillemin GJ, Miller CL, Lutz C, Radlwimmer B, Lehmann I, von Deimling A, Wick W, Platten M. 2011. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 478:197–203. 10.1038/nature10491 [DOI] [PubMed] [Google Scholar]
- 64.Henry EC, Bemis JC, Henry O, Kende AS, Gasiewicz TA. 2006. A potential endogenous ligand for the aryl hydrocarbon receptor has potent agonist activity in vitro and in vivo. Arch. Biochem. Biophys. 450:67–77. 10.1016/j.abb.2006.02.008 [DOI] [PubMed] [Google Scholar]
- 65.Savouret J-F, Antenos M, Quesne M, Xu J, Milgrom E, Casper RF. 2001. 7-Ketocholesterol is an endogenous modulator for the arylhydrocarbon receptor. J. Biol. Chem. 276:3054–3059. 10.1074/jbc.M005988200 [DOI] [PubMed] [Google Scholar]
- 66.Phelan D, Winter GM, Rogers WJ, Lam JC, Denison MS. 1998. Activation of the Ah receptor signal transduction pathway by bilirubin and biliverdin. Arch. Biochem. Biophys. 357:155–163. 10.1006/abbi.1998.0814 [DOI] [PubMed] [Google Scholar]