

Abstract
Hepatitis D virus (HDV) is a satellite of hepatitis B virus (HBV), and infection with this virus aggravates acute and chronic liver disease. While HBV seroprevalence is very high across sub-Saharan Africa, much less is known about HDV in the region. In this study, almost 2,300 blood serum samples from Burkina Faso (n = 1,131), Nigeria (n = 974), Chad (n = 50), and the Central African Republic (n = 118) were screened for HBV and HDV. Among 743 HBsAg-positive serum samples, 74 were positive for HDV antibodies and/or HDV RNA, with considerable differences in prevalence, ranging from <2% (pregnant women from Burkina Faso) to 50% (liver patients from Central African Republic). HDV seems to be much more common in chronic liver disease patients in the Central African Republic (CAR) than in similar cohorts in Nigeria. In a large nested mother-child cohort in Burkina Faso, the prevalence of HDV antibodies was 10 times higher in the children than in their mothers, despite similar HBsAg prevalences, excluding vertical transmission as an important route of infection. The genotyping of 16 full-length and 8 partial HDV strains revealed clade 1 (17/24) in three of the four countries, while clades 5 (5/24) and 6 (2/24) were, at least in this study, confined to Central Nigeria. On the amino acid level, almost all our clade 1 strains exhibited a serine at position 202 in the hepatitis D antigen, supporting the hypothesis of an ancient African HDV-1 subgroup. Further studies are required to understand the public health significance of the highly varied HDV prevalences in different cohorts and countries in sub-Saharan Africa.
INTRODUCTION
Hepatitis D virus (HDV), a negative-strand RNA virus of 1.7 kb in size, is always associated with hepatitis B virus (HBV) infection, which it requires for proliferation. About one-fourth of the estimated 65 million chronic HBV carriers in Africa are suspected to be coinfected with HDV. Coinfection with these two viruses results in fulminant hepatitis more often than with HBV infection alone, and superinfection of HBV with HDV is associated with chronic HDV in up to 80% of carriers (1). Both HBV and HDV occur worldwide and are genetically highly diverse (2–7). While HBV is classified into nine genotypes, A to I (8), as well as a recently proposed genotype J (9), HDV strains have been separated into eight distinct clades 1 to 8 (5, 6).
HDV clade 1 is highly prevalent worldwide (10), while clades 2 and 4 have been described in East and Northeast Asia (11–13). In addition, clade 3 has been detected only in South America (14). Clades 5 to 8 were proposed in a study investigating 25 HDV strains from African immigrants in Europe. Of these, 15 were attributed to these supposedly “African” clades, while 10 strains clustered with clade 1 (5, 6). Despite their characterization as African clades, only few genotyping studies have been performed in Africa (Cameroon, clades 1, 5, 6, 7; Gabon, clades 1, 7, and 8; Mauritania, clades 1 and 5), and clade 1 dominated in all cohorts (2, 5, 6, 15–19).
In sub-Saharan Africa, HBV is highly prevalent, and early childhood transmission is thought to be the most important route of infection (20). While this results in a high proportion of chronic HBV carriers, HDV superinfections add considerably to the high burden of chronic liver disease (20, 21), since up to 70% of hepatitis B surface antigen (HBsAg)-positive carriers are also infected with HDV (15). In addition, concurrent infection with HDV complicates viral treatment, as regimens against HBV do not affect HDV replication. Furthermore, HDV infection suppresses HBV replication (22–24) and reduces HBV DNA in the serum to often undetectable levels, thus complicating the diagnosis and cogenotyping of HBV and HDV strains.
Here, we analyzed samples from Burkina Faso (BFA), the Central African Republic (CAR), and Chad (TCD), from where essentially no HDV antibody or genotyping data are available. In addition, we analyzed sera from different cohorts in Nigeria (NGA) and characterized, for the first time, HDV strains from this country. We analyzed >2,000 serum samples from these countries to determine the seroprevalence of HBV and HDV among different cohorts and analyzed the genetic diversity and spread of HBV and HDV variants. Our results revealed highly variable prevalences of both HBV and HDV in the different participating cohorts. Here, we substantially enlarge the database of indigenous African HDV sequences, providing important information to further understand the complex evolution of HDV on this continent.
MATERIALS AND METHODS
Specimens.
Blood serum samples were obtained from apparently healthy individuals (from BFA, TCD, and CAR), patients with symptomatic liver disease (from NGA and CAR), and HIV-positive individuals (from NGA) between 1998 and 2010. These were stored at −80°C. The characteristics of the donors are shown in Table 1. All analyses were approved by the competent ethics committees in each of the participating countries.
TABLE 1.
Prevalences of hepatitis B surface antigen, hepatitis D antibodies, and hepatitis D RNA in cohorts from different western and central African countries
| Country | Sampling location (yr) | Cohort type | Cohort size (n) | HBV/HDV prevalence (no. of positive patients/total no. of patients [%]) |
HDV clade present (no. of that clade) | |||
|---|---|---|---|---|---|---|---|---|
| HBsAg | HDV Aba | HDV RNAb | HDV RNA/HBsAg | |||||
| Burkina Faso | Bobo-Dioulasso (2001) | Mothers | 370 | 60/370 (16.2) | 1/40 (2.5) | 0/1 (0) | 0/40 (0) | |
| Bobo-Dioulasso (2001) | Children | 424 | 52/424 (12.2) | 9/44 (20.5) | 0/9 (0) | 0/44 (0) | ||
| Bobo-Dioulasso, Houndé (2007) | Pregnant women | 337 | 49/337 (14.5) | 0/49 (0) | 0/49 (0) | |||
| Nigeria | Ibadan (1998) | HIV+ patients | 106 | 11/69 (15.9) | 3/11 (27.3) | 0/3 (0) | 0/11 (0) | |
| Lagos (2004) | HIV+ patients | 319 | 45/308 (14.6) | 3/45 (6.7) | 2/3 (66.7) | 2/45 (4.4) | 1 (1); 6 (1) | |
| Ibadan (2003) | Liver patients | 93 | 44/70 (62.9) | 3/44 (6.8) | 0/3 (0) | 0/44 (0) | ||
| Ibadan (2006) | Liver patients | 126 | 103/126 (81.7) | 1/78 (1.3) | 1/1 (100) | 1/78 (1.3) | 1 (1) | |
| Abuja, Nasarawa state (2006) | HBsAg+ | 330 | 330/330 (100) | 40/326 (12.3) | 15/40 (37.5) | 15/326 (4.6) | 1 (6); 5 (5); 6 (1) | |
| Chad | Military camp (2007) | Military personnel | 50 | 14/50 (28)c | 3/14 (21.4)c | 3/14 (21.4)c | 1 (3) | |
| Central African Republic | Bangui (2007) | Children | 81 | 35/79 (44.3) | 1/35 (2.9) | 0/1 (0) | 0/35 (0) | |
| Bangui (2009) | Liver patients | 37 | 14/29 (48.3) | 7/14 (50) | 5/7 (71.4) | 5/14 (35.7) | 1 (6)d | |
Prevalence among HBsAg-positive samples, excluding equivocal samples.
Prevalence among HBsAg-positive HDV Ab-positive samples.
Prevalence calculations based on HBV and HDV TaqMan PCR. Samples positive for at least one assay were considered HBV positive.
Includes one sample that was not tested for HBsAg and therefore not included in the previous columns.
Serology.
Hepatitis B surface antigen (HBsAg) was tested using either the Murex HBsAg version 3 enzyme-linked immunosorbent assay (ELISA) (Abbott, Ottignies-Louvain-La-Neuve, Belgium) (samples from BFA, NGA, and CAR) or the Smart Check HBsAg test (GlobalMed, Alexandria, VA, USA) (samples from NGA). Hepatitis D virus antibody (HDV Ab) status was determined using the Murex Anti-Delta (Abbott) or the ETI-AB-DELTAK-2 ELISA (DiaSorin, Brussels, Belgium). All analyses were performed according to the manufacturers' instructions. Equivocal samples were retested, if possible, and the result of the second test was accepted.
RNA and DNA isolation, RT-PCR, PCR, and sequencing.
RNA was extracted using 140 μl of serum in the QIAamp viral RNA minikit (Qiagen, Venlo, the Netherlands). Reverse transcription-PCR (RT-PCR) was carried out under the following conditions: 5 μl extracted RNA was denatured with 45 ng random primers and 10 nmol nucleotides for 5 min at 72°C. The reaction was then performed for 80 min at 50°C using 200 U SuperScript III reverse transcriptase and 40 U RNaseOUT recombinant RNase inhibitor (all reagents from Invitrogen, Leek, the Netherlands). The complete genome was amplified in three overlapping fragments, covering nucleotides (nt) 307 to 870, 715 to 1302, and 868 to 483 (Table 2). PCR products were purified using the JetQuick purification kit (Genomed, Antwerp, Belgium) and sequenced using the ABI Prism BigDye Terminator cycle sequencing reaction kit (Applied Biosystems, Nieuwerkerk aan den IJssel, the Netherlands) in an ABI 3130xl genetic analyzer (Applied Biosystems).
TABLE 2.
| Virus | Primer name | Primer orientation | PCR fragment/position (nt) | Binding site (length) (nt) | Sequence (5′ to 3′)c | Annealing step (°C, s) | Elongation step (°C, s) | Reference |
|---|---|---|---|---|---|---|---|---|
| HDV | 320ds | Forward | 307–870 | 307–328 (22) | CCAGAGRAMCCCTTCARCGAAC | 66, 30 | 72, 35 | Adapted from 12 |
| n320ds | Forward | 307–870 | 307–328 (24) | CCAGAGRAMCCCTCTCCARCGAAC | 66, 30 | 72, 35 | ||
| rv900 | Reverse | 307–870 | 870–853 (18) | GTCCGACCTGGGCATCCG | 66, 30 | 72, 35 | ||
| 710s | Forward | 715–1302 | 715–732 (18) | CGCCGGCTGGGCAACATT | 66, 30 | 72, 35 | ||
| 1302das | Reverse | 715–1302 | 1321–1302 (20) | GGNTTCACCGACRAGGAGAG | 66, 30 | 72, 35 | ||
| fw900_2a | Forward | 868–483 | 868–888 (21) | GACCGCGRGGAGGTGGAGATG | 62, 30 | 72, 50 | ||
| 480as | Reverse | 868–483 | 483–466 (18) | CCGGGATAAGCCTCACTC | 62, 30 | 72, 50 | ||
| Delta-F | Forward | TaqMan | 691–707 (17) | GCATGGTCCCAGCCTCC | 60, 60 | 46 | ||
| Delta-R | Reverse | TaqMan | 905–889 (17) | TCTTCGGGTCGGCATGG | 60, 60 | 46 | ||
| Delta-Probe | Forward | TaqMan | 856–870 (15) | FAM-ATGCCCAGGTCGGAC-MGB | 60, 60 | 46 | ||
| HBV | HBV-QTqm-F | Forward | TaqMan | 333–352 (20) | ACTCACCAACCTCTTGTCCT | 60, 60 | ||
| HBV-QTqm-R | Reverse | TaqMan | 476–457 (20) | GACAAACGGGCAACATACCT | 60, 60 | |||
| HBV-QTqm-Probe | Forward | TaqMan | 368–391 (24) | FAM-TATCGCTGGATGTGTCTGCGGCGT-TAMRA | 60, 60 |
HDV genotyping PCR conditions: 98°C for 30 s, 40 cycles at 98°C for 10 s (specific annealing and elongation conditions), and final elongation at 72°C for 7 min.
TaqMan PCR conditions: 50°C for 2 min; 95°C for 10 min; 45 cycles (HDV) or 50 cycles (HBV) at 95°C for 15 s, and elongation at 60°C.
FAM, 6-carboxyfluorescein; MGB, minor groove binder; TAMRA, 6-carboxytetramethylrhodamine.
HDV quantification was performed using 5 μl of cDNA template material under the conditions given in Table 2. The TaqMan PCR product of sample 36286 was cloned using the TOPO TA cloning kit (Invitrogen). Concentrations were measured using the ND-1000 spectrophotometer (Isogen Life Science, De Meern, the Netherlands), and the copy numbers were calculated. A 10-fold dilution series of the plasmid, ranging from 107 to 10 copies, was used as a standard in the quantification reaction. In all reactions, the correlation coefficient of the standard series was >0.998.
HBV DNA was extracted from HDV TaqMan PCR-positive sera of sufficient volume using the QIAamp DNA blood minikit (Qiagen), according to the manufacturer's instructions. HBV genotyping was performed on at least one of the HBV preS-, S-, X-, and C-gene regions, as previously described (25). Quantitative real-time PCR was performed on 5 μl of the extracted DNA (Table 2) and compared to a 10-fold dilution series of WHO International Standard 97/750 (National Institute for Biological Standards and Control [NIBSC], Hertfordshire, United Kingdom). The detection limit was 100 copies per ml of serum.
Phylogenetic analysis.
Raw sequences were edited using SeqScape version 2.5 (Applied Biosystems), aligned with the L-INS-i option of MAFFT version 6 for Windows (26, 27), and manually corrected. Neighbor-joining (NJ) trees and genetic distances were calculated using the MEGA5 software (28) with Kimura 2-parameter, the pairwise deletion option, and 1,000 bootstrap replicates. Statistical tests were performed using SigmaStat v3.11 (Systat Software, Erkrath, Germany). All full-length HDV strains available on NCBI GenBank were included in the analysis.
Nucleotide sequence accession numbers.
Sequences were submitted to EMBL/GenBank/DDGJ under accession no. JX888098 through JX888135.
RESULTS
Hepatitis B surface antigen serology.
Of 2,273 leftover blood serum samples from apparently healthy adults (from BFA and TCD), children (from BFA and CAR), patients with symptomatic liver disease (from NGA and CAR), and HIV-positive individuals (from NGA), 2,145 samples were tested for HBsAg, while for the remaining 128, there was not sufficient serum available for this test. Of the 2,145 samples, 743 were HBsAg positive, while 3 were equivocal and excluded from prevalence calculations.
The observed HBsAg seroprevalence rates ranged from 12.2% in children from Burkina Faso up to 81.7% in a liver patient cohort from Nigeria (Table 1). While the HBsAg prevalence was similar in healthy adults from Burkina Faso (14.5% for a cohort size of 337, and 16.2% for a cohort size of 370) and HIV-positive donors from Nigeria (14.6% for a cohort size of 308, and 15.9% for a cohort size of 69), it was significantly different in children from Burkina Faso (12.2%, n = 424) and from the CAR (44.3%, n = 79). Interestingly, the latter was similar to the prevalence observed in liver patients from the same country (48.3%, n = 29), while even higher prevalences were found in liver patients from Nigeria (62.9% for a cohort size of 70 and 81.7% for a cohort size of 126) (Table 1).
One hundred twenty-eight serum samples were not tested with HBsAg ELISA, but the burden of infection was assessed using the results from the HDV Ab ELISA and the quantitative HBV and HDV PCRs. In particular, none of the 50 serum samples obtained from Chadian military personnel were tested for HBsAg.
Among these 50 serum samples from Chad, 13 were found to be HBV DNA positive. In addition, one sample was HDV RNA positive, while HBV DNA was below the detection limit, resulting in an overall HBV prevalence of 28% (14/50) in Chad (Table 1).
HDV Ab serology.
An HDV Ab ELISA was performed on 695 of the 743 HBsAg-positive serum samples and 98 of the 128 samples that had not been tested for HBsAg. The remaining 48 and 30 serum samples could not be screened with HDV Ab ELISA due to low sample volume. Sixty-eight of the 695 HBsAg-positive samples and 3 of the 98 serum samples of unknown HBsAg status were HDV Ab positive, while 9 and 1 samples, respectively, were equivocal and excluded from prevalence calculations.
The prevalences of HDV Ab among HBsAg-positive sera from the different cohorts varied substantially (Table 1). They ranged from 0% in pregnant women from Burkina Faso (n = 49) to 50% in liver patients from the CAR (n = 14) (Table 1). In Burkina Faso, the HDV Ab prevalence in healthy adults was generally low (0% for a cohort size of 49, and 2.5% for a cohort size of 40), while a surprising 20.5% of children (n = 44) were found to be positive for HDV Ab. In the CAR, in contrast, the HDV Ab prevalence was low in children (2.9%, n = 35), while 50% of HBsAg-positive liver patients (n = 14, plus one of unknown HBsAg status) had antibodies against HDV (Table 1).
Also in Nigeria, HDV Ab prevalences varied between the different cohorts (Table 1). In HIV-positive/HBsAg-positive donors from Lagos (n = 45) and Ibadan (n = 11), 6.7% and 27.3% were HDV Ab positive, respectively, while in liver patients from Ibadan (cohort sizes of 78 and 44), HDV Ab were found in only 1.3% (plus one HBsAg-negative patient) and 6.8% of patients. Furthermore, an HDV Ab prevalence of 12.3% was found in HBsAg-positive sera from patients in the states of Abuja and Nasarawa State.
Of 50 serum samples from apparently healthy military personnel from Chad, none of which were analyzed for HBsAg, 31 were screened for HDV Ab, while the remaining 19 were directly subjected to RNA extraction. Two of the 31 unselected samples (6.5%) were found to be HDV Ab positive. All HDV Ab-positive samples and 30 untested samples underwent RNA extraction.
Thus, 50 of 504 HBsAg-positive patients in Nigeria, 10 of 133 of HBsAg positives in Burkina Faso, 2 of 31 unselected samples in Chad, and 8 of 49 HBsAg positives in the CAR were HDV Ab positive (Table 1).
HDV quantification and genotyping.
HDV quantification of 88 samples with complete HBsAg and HDV serology and 40 samples with incomplete or missing serology revealed 24 and 4 samples, respectively, to be positive for HDV RNA. The viral load (detection limit, 1 × 103 copies/ml serum) of the 28 HDV TaqMan-positive samples ranged from 8.3 × 103 to 1.6 × 109 copies/ml, without notable geographic differences in RNA titers.
Twenty-two of the HDV TaqMan-positive samples, as well as 2 that were repeatedly TaqMan negative, were PCR positive for at least one HDV fragment. Fragment 307 to 870 was obtained for 19 samples (and partially for 3 samples), fragment 715 to 1302 for 21 samples, and fragment 868 to 483 for 18 samples (plus 3 partial fragments). Taken together, the region 907 to 1265, which is commonly used for genotyping, was obtained in 22 samples, and for 19 of these, the complete large hepatitis D antigen (L-HDAg) open reading frame (nucleotides 954 to 1598) was available (and partially for 1 sample lacking 21 nucleotides [nt]). For 16/24 samples, the full HDV genome was obtained. However, all strains in this study were genotyped and revealed a predominance of clade 1 (17/24 [71%]) and the presence of clades 5 (5/24 [21%]) and 6 (2/24 [8%]), the latter two mainly from the Nasarawa State and the capital Abuja (Table 1).
In this study, clade 1 strains were found in sera from patients in Nigeria (8/15 [53%]), the CAR (6/6 [100%]), and Chad (3/3 [100%]), while clades 5 (5/15 [33%]) and 6 (2/15 [13%]) were, at least in this study, found only in Nigerian samples.
Phylogenetic analysis of HDV.
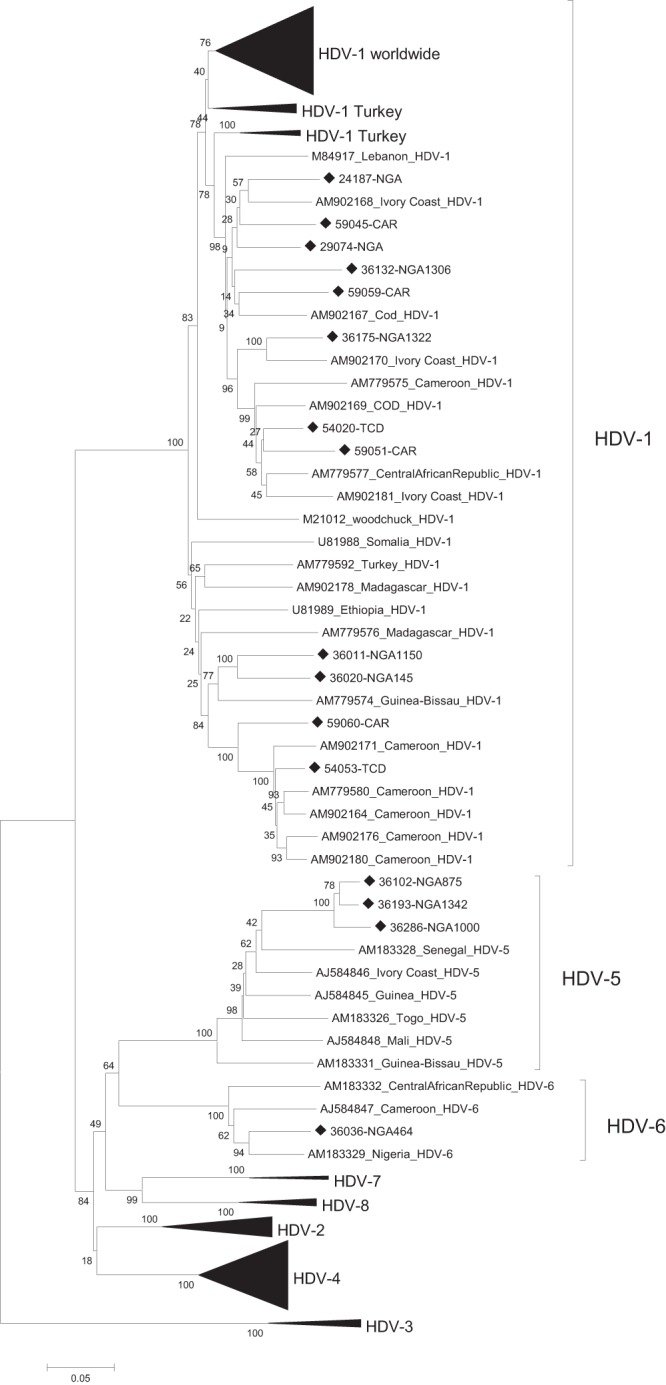
Phylogenetic analyses of the 87 available full-length HDV-1 genome sequences from public databases (including the 12 strains from this study) revealed that HDV-1 formed multiple clusters in the phylogenetic tree (Fig. 1). Interestingly, one cluster consisted of strains from all over the world, while 2 smaller clusters contained predominantly African strains, including the strains from this study (Fig. 1). This apparent segregation of clade 1 was reproducible only when the full-length genome was considered and could not be reproduced when analyzing only the region nt 907 to 1265, which is typically used for HDV genotyping (Fig. 2). Furthermore, the genetic variability over the full genome within the African clusters (mean genetic diversity within the individual clusters, 12.7% and 14%; maximum genetic distance within the individual clusters, 18.1% and 18.8%) was similar to the one observed in all other HDV-1 strains (mean, 12.8%; maximum, 21.3%). The overall HDV-1 mean genetic diversity was 15.2% (maximum genetic distance, 23.4%).
FIG 1.
Phylogenetic clustering of all available full-length HDV strains. The strains from this study are indicated by diamonds. The scale bar indicates nucleotide substitutions per site. Bootstrap numbers next to the branches represent nucleotide substitutions per site. COD, Democratic Republic of the Congo; NGA, Nigeria.
FIG 2.
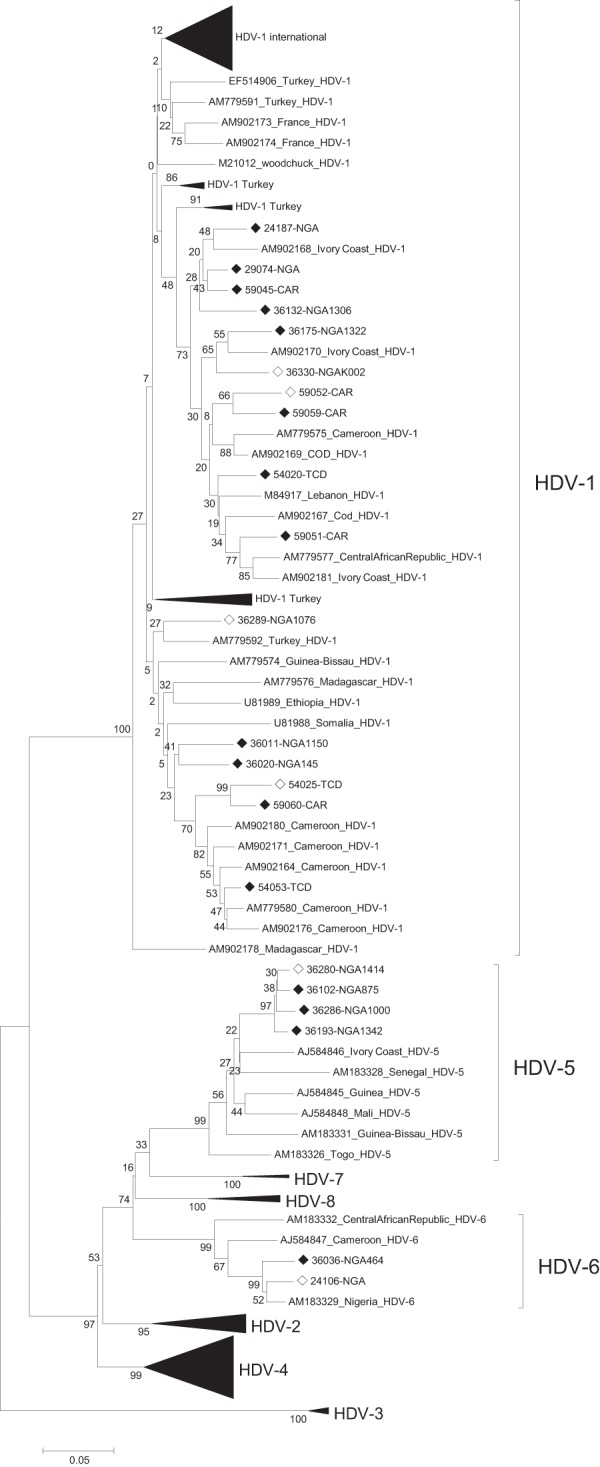
Phylogenetic analysis of the genotyping region 907 to 1265 with sequences for which the full-length genomes were available. Open diamonds indicate partial-length strains obtained in this study. Strains for which the full-length sequences are available (see Fig. 1) are indicated by solid diamonds. The scale bar indicates nucleotide substitutions per site. Bootstrap numbers next to the branches represent nucleotide substitutions per site. COD, Democratic Republic of the Congo; NGA, Nigeria.
Clade 5 was found in 33% of genotyped strains and, at least in our study, only in Nigeria. These formed a separate cluster apart from the clade 5 strains from other West African countries (Fig. 1 and 2). The overall mean intraclade diversity of the full-length strains was 13% (maximum genetic distance, 16.9%). The intragroup diversity of the subgroups varied and was 3.9% (maximum genetic distance, 4.6%) in the Nigerian cluster and 12.8% (maximum genetic distance, 16.9%) in the remaining strains.
Also, our two clade 6 strains from Nigeria clustered with a Nigerian strain (Fig. 1 and 2). In analyzing all HDV-6 sequences, we found a mean genetic diversity of 11.9% (maximum genetic distance, 14.6%).
Hepatitis B virus quantification and genotyping.
HBV quantification was performed in 23 samples with a known HDV genotype. HBV DNA was undetectable in 11 of these (47.8%), possibly reflecting an inhibition of HBV replication by HDV or a clearance of HBV DNA. In the remaining 12 samples, 2.51 × 103 to 5.32 × 109 HBV copies/ml were detected. In 4 of these, the HBV load was even higher than that of HDV.
In 58.8% (10/17) of the HDV-1 positive serum samples, HBV was detectable, while in only 20% (1/5) of HDV-5-positive serum samples, HBV DNA was found. HDV-1-positive sera seemed to have a higher HBV load (mean, 4.79 × 108 copies/ml; median, 2.92 × 103 copies/ml) than HDV-5 sera (mean, 2.48 × 103 copies/ml; median, 0 copies/ml), although this was not statistically significant (P = 0.311, Fisher's exact test). Furthermore, one HDV-6 sample was found to have 4.12 × 103 HBV copies/ml.
Although HBV genotyping proved difficult due to the low viral load, seven HDV sequences correlated with HBV genotypes. HBV-E and HDV-1 were found in 4 of the 7 samples, while the combinations HBV-A/HDV-1, HBV-D/HDV-1, and HBV-E/HDV-6 were observed in one sample each. However, due to the small number of samples that could be genotyped for both HBV and HDV, these correlations may not necessarily be fully representative of the association of HBV genotypes and HDV clades.
Hepatitis delta antigen.
For 19 samples for which the full HDAg open reading frame was obtained, the predicted amino acid (aa) sequence was analyzed. For 17 of these, the large HDAg was 214 aa long, while one sample each harbored an insertion (no. 59045) or a deletion (sample no. 36036) of one amino acid. None of the strains were observed to have a change in the open reading frame.
Clade 1 was previously described to have a more efficient virus assembly than other clades (29, 30). Indeed, all our clade-1 strains harbored proline at position 205, while in our strains of the other clades, arginine was found in this position. Similarly, the clathrin box, thought to be important for virion packaging (31, 32), varied between clade 1 (LFPSD) and our strains of the other clades (LPLLE). Furthermore, the isoprenylation signal, which is important for virion maturation (31), was CRPQ in clade 1 and CTPQ in our non-HDV-1 strains.
In addition, aa 202 had been proposed as a marker to characterize the geographic origin of HDV-1, with African strains having a serine and Eurasian strains an arginine in this position (33). Indeed, our clade 1 strains had a serine at this position, except one strain from Chad that had a proline at position 202.
DISCUSSION
In the present study, we characterized 743 HBsAg-positive serum samples from Burkina Faso, the CAR, and Chad, from where no HDV data were available, as well as from Nigeria, and we characterized, for the first time, HDV strains from these countries. Among the 743 HBsAg-positive serum samples, 74 were positive for either HDV antibodies or HDV RNA, with considerable differences in prevalences, ranging from 0% (in pregnant women from Burkina Faso) to 50% (in liver patients from the Central African Republic). Furthermore, as so far only 34 full-length HDV strains from Africa are available, our 16 full-genome sequences considerably enlarge the genotype information available from this continent.
In all our analyzed cohorts, HBsAg was highly endemic, but its prevalence varied widely, ranging from 12.2% in apparently healthy patients to 81.7% in symptomatic liver patients. Although it is difficult to compare the different cohorts, the HBsAg prevalence was similar in healthy adults and HIV carriers from Burkina Faso (BFA) and Nigeria (NGA) (14.5% to 16.2%). In contrast, differences in prevalences were substantial between children from Burkina Faso (12.2%) and the CAR (44.3%). Although the cohort from the CAR included children with a higher age (2 to 15 years) than in BFA (2 to 6 years), HBsAg prevalences in children from the CAR were relatively constant and >40%, irrespective of the age group analyzed (2 to 5, 6 to 10, or >10 years of age), indicating a high burden and an excessive risk of early infection with HBV in children from the CAR. This high prevalence is also surprising in light of HBsAg prevalences of only 14% and 15.5% in young adults and high school students, respectively, that were previously reported from the CAR (34, 35). Furthermore, the observed HBV prevalence in the children was almost as high as in liver patients from the same country (48.3%). The children were part of an unselected fever rash cohort attending different hospitals in and around Bangui in 2007, of which 58% and 31% were IgM positive for either measles or rubella. Thus, these children seem to be from the general population, and it is difficult to explain the HBsAg carrier status by an obvious sampling bias. Although no HBV sequences are available for this cohort, this excessive burden of disease indicates an unusual route of transmission for this group of children.
Also, in samples from military personnel from the Republic of Chad, a high HBV prevalence of 28% was observed. As the HBV burden in the Chadian cohort was estimated based on HBV DNA and HDV RNA positivity, the HBV prevalence may even be underestimated in this group. Although military personnel may have higher HBV infection rates in Africa than unselected populations (18), this first HBV prevalence study in Chad seems to suggest that HBV is highly endemic in this country.
The rare HDV seroprevalence studies that have been performed during the past 15 years and the recent reports of the African clades 5 to 8 (5, 6) in strains from Cameroon (17.6% anti-HDV antibody prevalence among HBsAg-positive individuals), Gabon (15.6% to 70.6%), Mauritania (14.7% to 33.1%), Mozambique (0%), Nigeria (0% to 12.5%), and Senegal (3.2%), revealed various HDV antibody rates (2, 15–19, 36–40). Interestingly, also in our study, the HDV Ab prevalences varied widely and ranged from 0% to 27.3% in asymptomatic carriers and from 1.3% to 50% in liver patients. Surprisingly, 20.5% of HBsAg-positive children from Burkina Faso (12.2% HBsAg prevalence) were also HDV Ab positive, a prevalence almost 10 times higher than the one observed in their mothers. Furthermore, in mother-child pairs, seropositivity for HBsAg and anti-HDV, respectively, did not correlate between the two cohorts, confirming horizontal rather than vertical routes of transmission for both HBV and HDV. Furthermore, vast HDV Ab differences were observed in liver patients from Nigeria and the CAR. In Nigeria, a large proportion of liver patients were HBsAg positive, and only 1.3% and 6.8% of these were HDV Ab positive, with prevalences similar to or lower than those observed in the other Nigerian cohorts (6.7% to 27.3% HDV Ab prevalence). On the other hand, 50% of liver patients from the CAR were HDV Ab positive, indicating that in the CAR, in contrast to Nigeria, HDV superinfection might be the cause of chronic hepatitis with frequent severe liver conditions.
The HDV viral load varied widely in the analyzed samples, ranging from 103 to 109 copies/ml. Since the infection with HDV may inhibit HBV replication, and (noninfectious HDV) RNA particles may, in contrast to HBV, assemble even in the absence of large HBsAg (24, 41), it was not unexpected that HBV DNA was undetectable in almost 50% (11/23) of the HDV RNA-positive donors. Although the primer binding sites are highly conserved in genotypes E and A, the dominant genotypes in sub-Saharan Africa, we cannot totally exclude that individual strains might have been missed due to rare mutations. This would, however, not explain the absence of HBV DNA in the large number of HDV carriers. Furthermore, this does not seem to be the result of more efficient or abundant HDV replication, as the HBV DNA-negative samples had HDV loads (mean, 3.68 × 108 copies/ml; median, 3.18 × 107 copies/ml) that were similar to the HBV DNA-positive ones (12/23; mean, 1.54 × 108 copies/ml; median, 1.06 × 107 copies/ml). While on average, a higher HDV load was observed in clade 1 strains (mean, 2.5 × 108 copies/ml; median, 1.3 × 107 copies/ml) than in clade 5 strains (mean, 4.19 × 107 copies/ml; median, 8.98 × 106 copies/ml) or one clade 6 strain (5.9 × 106 copies/ml), the low numbers of clade 5 and 6 strains may not be representative enough even when taken together. In fact, 71% (17/24) of the genotyped strains were assigned to clade 1, with strains originating from Nigeria (n = 8), CAR (n = 6), and Chad (n = 3). Furthermore, 21% and 8% of our strains belonged to clades 5 (5/24) and 6 (2/24), respectively, and all of these strains originated from (mostly Central) Nigeria. While clade 5 was also found across West Africa, clade 6 seems to be more geographically confined (Fig. 1 and 2).
Phylogenetic analysis of clade 1 strains revealed several clusters, although not necessarily with high bootstrap support, with strains from this study largely clustering together with other African strains (Fig. 1). As the clustering could not be reproduced when using the genotyping region only (Fig. 2), the analysis of geographic clusters would generally benefit from extending the sequencing window in phylogenetic analyses. In addition, previous studies indicated an ancestral origin of clade 1 strains in Africa (42). However, the observed genetic diversity of HDV-1 was similar in width in the African clusters (mean genetic diversity within individual clusters, 12.7% and 14%; maximum genetic distance within individual clusters, 18.1% and 18.8%) and the non-African HDV-1 strains (mean, 12.8%; maximum, 21.3%).
On the amino acid level, almost all our clade 1 strains revealed a serine at position 202 in the HDAg, which is thought to be characteristic of African clade 1 strains (in contrast to Eurasian strains, which have alanine at position 202) (33). This seems to support the hypothesis that most African HDV-1 strains may represent an ancient subgroup from this continent. Together with the recently characterized HDV clades 5 to 8 (5, 6), HDV sequences are overall more divergent in Africa than anywhere else in the world (31), indicating that, similar to hepatitis B virus, HDV might have originated in Africa (5, 43). Nevertheless, the true origin and emergence of HDV remain unclear. While it has been discussed that HDV may have co-originated with the most simple duck hepatitis B viruses (DHBV) (31), HDV has so far been found only in humans. Thus, it may be speculated that HDV originated in ancestral humans, possibly in Africa.
All studies on HDV in sub-Saharan Africa revealed a predominance of HDV-1 in a region where HBV genotype E (and to a lower extent, the African HBV-A subgenotypes 3 to 7) predominates. However, a close coevolution of HDV-1 and HBV-E seems unlikely, since HDV-1 can be found worldwide, whereas HBV-E is largely confined to Africa and is thought to have a short evolutionary history (8, 44, 45). Moreover, several lines of evidence suggest that HBV-E was only recently introduced into the African population (8, 44, 45). A recent study in Turkey found that all HDV-1 variants were predominantly associated with HBV genotype D (33). More than 50% of these belonged to the putative African group (serine 202), while the remaining strains harbored alanine at position 202 and clustered in the Eurasian group (33). The high prevalence of HDV-1, however, may be explained by the more efficient spread of this clade. Indeed, HDV-1 displays some unique nucleotide and amino acid features in the large HDAg compared with the other HDV clades, potentially leading to more efficient viral reproduction (29). However, this is largely speculative, and more detailed studies are required.
In conclusion, our study revealed high prevalences of both HBV and HDV in multiple countries and cohorts from sub-Saharan Africa. For instance, conspicuous differences in HDV prevalence were found in cohorts with severe chronic hepatitis from Nigeria and CAR and in mother-child pairs from Burkina Faso. While the ubiquitous clade 1 may have originated in Africa, coevolution with the predominant HBV genotype E in this region seems unlikely. Clades 5 and 6 seem to be less common and/or more geographically confined. The high genetic variability and prevalences of HDV in different cohorts and countries, its complex evolution and transmission, and its impact on hepatitis progression found in this and other studies are in contrast to the findings of the few studies on HDV in sub-Saharan Africa.
ACKNOWLEDGMENTS
We thank Emilie Charpentier and Aurélie Sausy for their technical expertise and help in performing the experiments. We also thank Barbara Gärtner from the University of the Saarland, Germany, for providing positive HDV controls.
This work was supported by the Centre de Recherche Public de la Santé and the Ministry of Cooperation of the Grand-Duchy of Luxembourg. I.E.A. was supported by a fellowship from the National Research Fund, Luxembourg (TR-PHD-BFR07-129).
We declare no conflicts of interest.
Footnotes
Published ahead of print 5 March 2014
REFERENCES
- 1.World Health Organization. 2001. Hepatitis Delta fact sheet (WHO/CDS/CSR/NCS/2001.1). World Health Organization, Geneva, Switzerland: http://www.who.int/csr/disease/hepatitis/HepatitisD_whocdscsrncs2001_1.pdf?ua=1 [Google Scholar]
- 2.Foupouapouognigni Y, Noah DN, Sartre MT, Njouom R. 2011. High prevalence and predominance of hepatitis delta virus genotype 1 infection in Cameroon. J. Clin. Microbiol. 49:1162–1164. 10.1128/JCM.01822-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hadziyannis SJ. 2011. Natural history of chronic hepatitis B in Euro-Mediterranean and African countries. J. Hepatol. 55:183–191. 10.1016/j.jhep.2010.12.030 [DOI] [PubMed] [Google Scholar]
- 4.Rossi C, Shrier I, Marshall L, Cnossen S, Schwartzman K, Klein MB, Schwarzer G, Greenaway C. 2012. Seroprevalence of chronic hepatitis B virus infection and prior immunity in immigrants and refugees: a systematic review and meta-analysis. PLoS One 7:e44611. 10.1371/journal.pone.0044611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Radjef N, Gordien E, Ivaniushina V, Gault E, Anais P, Drugan T, Trinchet JC, Roulot D, Tamby M, Milinkovitch MC, Dény P. 2004. Molecular phylogenetic analyses indicate a wide and ancient radiation of African hepatitis delta virus, suggesting a deltavirus genus of at least seven major clades. J. Virol. 78:2537–2544. 10.1128/JVI.78.5.2537-2544.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Le Gal F, Gault E, Ripault MP, Serpaggi J, Trinchet JC, Gordien E, Dény P. 2006. Eighth major clade for hepatitis delta virus. Emerg. Infect. Dis. 12:1447–1450. 10.3201/eid1209.060112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alvarado-Mora MV, Romano CM, Gomes-Gouvêa MS, Gutierrez MF, Carrilho FJ, Pinho JR. 2011. Dynamics of hepatitis D (delta) virus genotype 3 in the Amazon region of South America. Infect. Genet. Evol. 11:1462–1468. 10.1016/j.meegid.2011.05.020 [DOI] [PubMed] [Google Scholar]
- 8.Mulders MN, Venard V, Njayou M, Edorh AP, Bola Oyefolu AO, Kehinde MO, Muyembe Tamfum JJ, Nebie YK, Maiga I, Ammerlaan W, Fack F, Omilabu SA, Le Faou A, Muller CP. 2004. Low genetic diversity despite hyperendemicity of hepatitis B virus genotype E throughout West Africa. J. Infect. Dis. 190:400–408. 10.1086/421502 [DOI] [PubMed] [Google Scholar]
- 9.Tatematsu K, Tanaka Y, Kurbanov F, Sugauchi F, Mano S, Maeshiro T, Nakayoshi T, Wakuta M, Miyakawa Y, Mizokami M. 2009. A genetic variant of hepatitis B virus divergent from known human and ape genotypes isolated from a Japanese patient and provisionally assigned to new genotype J. J. Virol. 83:10538–10547. 10.1128/JVI.00462-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hughes SA, Wedemeyer H, Harrison PM. 2011. Hepatitis delta virus. Lancet 378:73–85. 10.1016/S0140-6736(10)61931-9 [DOI] [PubMed] [Google Scholar]
- 11.Imazeki F, Omata M, Ohto M. 1991. Complete nucleotide sequence of hepatitis delta virus RNA in Japan. Nucleic Acids Res. 19:5439. 10.1093/nar/19.19.5439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ivaniushina V, Radjef N, Alexeeva M, Gault E, Semenov S, Salhi M, Kiselev O, Dény P. 2001. Hepatitis delta virus genotypes I and II cocirculate in an endemic area of Yakutia, Russia. J. Gen. Virol. 82:2709–2718 [DOI] [PubMed] [Google Scholar]
- 13.Wu JC, Chiang TY, Sheen IJ. 1998. Characterization and phylogenetic analysis of a novel hepatitis D virus strain discovered by restriction fragment length polymorphism analysis. J. Gen. Virol. 79(Pt 5):1105–1113 [DOI] [PubMed] [Google Scholar]
- 14.Casey JL, Brown TL, Colan EJ, Wignall FS, Gerin JL. 1993. A genotype of hepatitis D virus that occurs in northern South America. Proc. Natl. Acad. Sci. U. S. A. 90:9016–9020. 10.1073/pnas.90.19.9016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Makuwa M, Mintsa-Ndong A, Souquiere S, Nkoghé D, Leroy EM, Kazanji M. 2009. Prevalence and molecular diversity of hepatitis B virus and hepatitis delta virus in urban and rural populations in northern Gabon in central Africa. J. Clin. Microbiol. 47:2265–2268. 10.1128/JCM.02012-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Makuwa M, Caron M, Souquiere S, Malonga-Mouelet G, Mahe A, Kazanji M. 2008. Prevalence and genetic diversity of hepatitis B and delta viruses in pregnant women in Gabon: molecular evidence that hepatitis delta virus clade 8 originates from and is endemic in central Africa. J. Clin. Microbiol. 46:754–756. 10.1128/JCM.02142-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mansour W, Malick FZ, Sidiya A, Ishagh E, Chekaraou MA, Veillon P, Ducancelle A, Brichler S, Le Gal F, Lo B, Gordien E, Lunel-Fabiani F. 2012. Prevalence, risk factors, and molecular epidemiology of hepatitis B and hepatitis delta virus in pregnant women and in patients in Mauritania. J. Med. Virol. 84:1186–1198. 10.1002/jmv.23336 [DOI] [PubMed] [Google Scholar]
- 18.Mansour W, Bollahi MA, Hamed CT, Brichler S, Le Gal F, Ducancelle A, Lô B, Gordien E, Rosenheim M, Lunel F. 2012. Virological and epidemiological features of hepatitis delta infection among blood donors in Nouakchott, Mauritania. J. Clin. Virol. 55:12–16. 10.1016/j.jcv.2012.05.011 [DOI] [PubMed] [Google Scholar]
- 19.Lunel-Fabiani F, Mansour W, Amar AO, Aye M, Le Gal F, Malick FZ, Baïdy L, Brichler S, Veillon P, Ducancelle A, Gordien E, Rosenheim M. 2013. Impact of hepatitis B and delta virus co-infection on liver disease in Mauritania: a cross sectional study. J. Infect. 67:448–457. 10.1016/j.jinf.2013.06.008 [DOI] [PubMed] [Google Scholar]
- 20.World Health Organization. 2002. Hepatitis B fact sheet (WHO/CDS/CSR/LYO/2002.2). World Health Organization, Geneva, Switzerland: http://www.who.int/csr/disease/hepatitis/HepatitisB_whocdscsrlyo2002_2.pdf?ua=1 [Google Scholar]
- 21.Smedile A, Rosina F, Saracco G, Chiaberge E, Lattore V, Fabiano A, Brunetto MR, Verme G, Rizzetto M, Bonino F. 1991. Hepatitis B virus replication modulates pathogenesis of hepatitis D virus in chronic hepatitis D. Hepatology 13:413–416. 10.1002/hep.1840130305 [DOI] [PubMed] [Google Scholar]
- 22.Cross TJ, Rizzi P, Horner M, Jolly A, Hussain MJ, Smith HM, Vergani D, Harrison PM. 2008. The increasing prevalence of hepatitis delta virus (HDV) infection in South London. J. Med. Virol. 80:277–282. 10.1002/jmv.21078 [DOI] [PubMed] [Google Scholar]
- 23.Jardi R, Rodriguez F, Buti M, Costa X, Cotrina M, Galimany R, Esteban R, Guardia J. 2001. Role of hepatitis B, C, and D viruses in dual and triple infection: influence of viral genotypes and hepatitis B precore and basal core promoter mutations on viral replicative interference. Hepatology. 34:404–410. 10.1053/jhep.2001.26511 [DOI] [PubMed] [Google Scholar]
- 24.Mathurin P, Thibault V, Kadidja K, Ganne-Carrié N, Moussalli J, El Younsi M, Di Martino V, Lunel F, Charlotte F, Vidaud M, Opolon P, Poynard T. 2000. Replication status and histological features of patients with triple (B, C, D) and dual (B, C) hepatic infections. J. Viral Hepat. 7:15–22. 10.1046/j.1365-2893.2000.00195.x [DOI] [PubMed] [Google Scholar]
- 25.Olinger CM, Venard V, Njayou M, Oyefolu AO, Maïga I, Kemp AJ, Omilabu SA, le Faou A, Muller CP. 2006. Phylogenetic analysis of the precore/core gene of hepatitis B virus genotypes E and A in West Africa: new subtypes, mixed infections and recombinations. J. Gen. Virol. 87:1163–1173. 10.1099/vir.0.81614-0 [DOI] [PubMed] [Google Scholar]
- 26.Katoh K, Toh H. 2008. Recent developments in the MAFFT multiple sequence alignment program. Brief. Bioinform. 9:286–298. 10.1093/bib/bbn013 [DOI] [PubMed] [Google Scholar]
- 27.Katoh K, Asimenos G, Toh H. 2009. Multiple alignment of DNA sequences with MAFFT. Methods Mol. Biol. 537:39–64. 10.1007/978-1-59745-251-9_3 [DOI] [PubMed] [Google Scholar]
- 28.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739. 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shih HH, Shih C, Wang HW, Su CW, Sheen IJ, Wu JC. 2010. Pro-205 of large hepatitis delta antigen and Pro-62 of major hepatitis B surface antigen influence the assembly of different genotypes of hepatitis D virus. J. Gen. Virol. 91:1004–1012. 10.1099/vir.0.017541-0 [DOI] [PubMed] [Google Scholar]
- 30.Lee CH, Chang SC, Wu CH, Chang MF. 2001. A novel chromosome region maintenance 1-independent nuclear export signal of the large form of hepatitis delta antigen that is required for the viral assembly. J. Biol. Chem. 276:8142–8148. 10.1074/jbc.M004477200 [DOI] [PubMed] [Google Scholar]
- 31.Huang CR, Lo SJ. 2010. Evolution and diversity of the human hepatitis d virus genome. Adv. Bioinformatics 2010:323654. 10.1155/2010/323654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang C, Chang SC, Yu IC, Tsay YG, Chang MF. 2007. Large hepatitis delta antigen is a novel clathrin adaptor-like protein. J. Virol. 81:5985–5994. 10.1128/JVI.02809-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Le Gal F, Badur S, Hawajri NA, Akyüz F, Kaymakoglu S, Brichler S, Zoulim F, Gordien E, Gault E, Dény P. 2012. Current hepatitis delta virus type 1 (HDV1) infections in central and eastern Turkey indicate a wide genetic diversity that is probably linked to different HDV1 origins. Arch. Virol. 157:647–659. 10.1007/s00705-011-1212-8 [DOI] [PubMed] [Google Scholar]
- 34.Pawlotsky JM, Bélec L, Grésenguet G, Deforges L, Bouvier M, Duval J, Dhumeaux D. 1995. High prevalence of hepatitis B, C, and E markers in young sexually active adults from the Central African Republic. J. Med. Virol. 46:269–272. 10.1002/jmv.1890460318 [DOI] [PubMed] [Google Scholar]
- 35.Komas NP, Baï-Sepou S, Manirakiza A, Léal J, Béré A, Le Faou A. 2010. The prevalence of hepatitis B virus markers in a cohort of students in Bangui, Central African Republic. BMC Infect. Dis. 10:226–2334-10-226. 10.1186/1471-2334-10-226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cunha L, Plouzeau C, Ingrand P, Gudo JP, Ingrand I, Mondlane J, Beauchant M, Agius G. 2007. Use of replacement blood donors to study the epidemiology of major blood-borne viruses in the general population of Maputo, Mozambique. J. Med. Virol. 79:1832–1840. 10.1002/jmv.21010 [DOI] [PubMed] [Google Scholar]
- 37.Nwokediuko SC, Ijeoma U. 2009. Seroprevalence of antibody to HDV in Nigerians with hepatitis B virus-related liver diseases. Niger. J. Clin. Pract. 12:439–442 [PubMed] [Google Scholar]
- 38.Olal SO, Akere A, Otegbayo JA, Odaibo GN, Olaleye DO, Afolabi NB, Bamgboye EA. 2012. Are patients with primary hepatocellular carcinoma infectious of hepatitis B, C and D viruses? Afr. J. Med. Med. Sci. 41(Suppl):187–191 [PubMed] [Google Scholar]
- 39.Onyekwere CA, Audu RA, Duro-Emmanuel F, Ige FA. 2012. Hepatitis D infection in Nigeria. Indian J. Gastroenterol. 31:34–35. 10.1007/s12664-011-0158-9 [DOI] [PubMed] [Google Scholar]
- 40.Diop-Ndiaye H, Touré-Kane C, Etard JF, Lô G, Diaw P, Ngom-Gueye NF, Gueye PM, Ba-Fall K, Ndiaye I, Sow PS, Delaporte E, Mboup S. 2008. Hepatitis B, C seroprevalence and delta viruses in HIV-1 Senegalese patients at HAART initiation (retrospective study). J. Med. Virol. 80:1332–1336. 10.1002/jmv.21236 [DOI] [PubMed] [Google Scholar]
- 41.Wang CJ, Chen PJ, Wu JC, Patel D, Chen DS. 1991. Small-form hepatitis B surface antigen is sufficient to help in the assembly of hepatitis delta virus-like particles. J. Virol. 65:6630–6636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang YY, Tsega E, Hansson BG. 1996. Phylogenetic analysis of hepatitis D viruses indicating a new genotype I subgroup among African isolates. J. Clin. Microbiol. 34:3023–3030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taylor J, Pelchat M. 2010. Origin of hepatitis delta virus. Future Microbiol. 5:393–402. 10.2217/fmb.10.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andernach IE, Hubschen JM, Muller CP. 2009. Hepatitis B virus: the genotype E puzzle. Rev. Med. Virol. 19:231–240. 10.1002/rmv.618 [DOI] [PubMed] [Google Scholar]
- 45.Andernach IE, Nolte C, Pape JW, Muller CP. 2009. Slave trade and hepatitis B virus genotypes and subgenotypes in Haiti and Africa. Emerg. Infect. Dis. 15:1222–1228. 10.3201/eid1508.081642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Le Gal F, Gordien E, Affolabi D, Hanslik T, Alloui C, Dény P, Gault E. 2005. Quantification of hepatitis delta virus RNA in serum by consensus real-time PCR indicates different patterns of virological response to interferon therapy in chronically infected patients. J. Clin. Microbiol. 43:2363–2369. 10.1128/JCM.43.5.2363-2369.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]