

Abstract
Phage typing has been used for the epidemiological surveillance of Salmonella enterica serovar Enteritidis for over 2 decades. However, knowledge of the genetic and evolutionary relationships between phage types is very limited, making differences difficult to interpret. Here, single nucleotide polymorphisms (SNPs) identified from whole-genome comparisons were used to determine the relationships between some S. Enteritidis phage types (PTs) commonly associated with food-borne outbreaks in the United States. Emphasis was placed on the predominant phage types PT8, PT13a, and PT13 in North America. With >89,400 bp surveyed across 98 S. Enteritidis isolates representing 14 distinct phage types, 55 informative SNPs were discovered within 23 chromosomally anchored loci. To maximize the discriminatory and evolutionary partitioning of these highly homogeneous strains, sequences comprising informative SNPs were concatenated into a single combined data matrix and subjected to phylogenetic analysis. The resultant phylogeny allocated most S. Enteritidis isolates into two distinct clades (clades I and II) and four subclades. Synapomorphic (shared and derived) sets of SNPs capable of distinguishing individual clades/subclades were identified. However, individual phage types appeared to be evolutionarily disjunct when mapped to this phylogeny, suggesting that phage typing may not be valid for making phylogenetic inferences. Furthermore, the set of SNPs identified here represents useful genetic markers for strain differentiation of more clonal S. Enteritidis strains and provides core genotypic markers for future development of a SNP typing scheme with S. Enteritidis.
INTRODUCTION
Salmonella infection is the most commonly reported food-borne illness of humans and has the largest number of hospitalizations and deaths in the United States each year (1). Among the salmonellae, Salmonella enterica serovar Enteritidis is one of the serovars most commonly implicated in food-borne disease outbreaks (2). S. Enteritidis infections associated with the consumption of raw or undercooked eggs, for instance, have caused outbreaks affecting large and widely distributed populations (http://www.cdc.gov/salmonella/enteritidis/).
Phage typing is a phenotypic subtyping method which historically has played a central role in epidemiological studies of S. Enteritidis (3). Phage typing discriminates between strains belonging to the same serovar, requires basic laboratory equipment, is simple to implement, and provides rapid strain discrimination (4). Phage typing has been found to be useful for the tracking of spatial and temporal distributions of S. Enteritidis. For example, phage type 8 (PT8), PT13a, and PT13 are taken to be most common in North America (5, 6), while PT4 has been the dominant phage type in most countries of Western Europe (7–9). Additionally, PT14b represents a phage type that recently emerged in southern European countries in 2001 (10). The epidemiology of S. Enteritidis cases associated with international travel revealed distinct relationships between phage types and travel destinations (6, 11, 12). However, epidemiological comparisons based on phage types alone may not be enough when investigating outbreaks of endemic strains. This difficulty in examining endemic strains is routinely handled by the use of additional genotyping methods in conjunction with phage typing (13). Moreover, phage typing is also limited by the presence of ambiguous lysis reactions (4) and the occurrence of changes in phage type as a result of plasmid or phage acquisition, changes in lipopolysaccharide expression, or spontaneous mutations affecting phage receptor sites (14–18).
Little is known about the evolutionary relationships among phage types, and knowledge of the genetic basis for the variation in phage sensitivity is limited as well (19). The extraordinarily conserved nature of the S. Enteritidis genome has further confounded an accurate evolutionary framing of this serovar, making it difficult to find phylogenetic characters averse to homoplasious change. Conversely, however, genetic changes employed for molecular epidemiological purposes are not beholden to such a rigid evolutionary model and can come from other more diverged regions across the genome. For instance, cross-hybridization of phage genes by using microarrays revealed that S. Enteritidis isolates were grouped into two major lineages (20). By profiling of IS200, a Salmonella-specific DNA insertion element, 27 phage type strains of S. Enteritidis all fell into one of three band patterns, corresponding to three clonal lineages of recent evolutionary origin (21). Based on restriction fragment length polymorphism (RFLP) analysis using PvuII and PstI with the insertion element IS200, two major groups and six minor groups were observed among 33 S. Enteritidis strains with different PTs (22). Multilocus sequence typing (MLST) can be used to accurately identify bacterial lineages and reconstruct phylogenetic relationships within the population being studied (23). A recent study showed that all 30 S. Enteritidis isolates with 9 PTs in Japan collected between 1973 and 2004 belonged to a single sequence type (ST11) without any nucleotide differences across seven housekeeping genes used in the MLST typing scheme (24).
Single nucleotide polymorphisms (SNPs) seem to be the most valuable molecular markers for studying the evolutionary relationships of isolates within homogeneous pathogenic clones (19, 25–29). During 2010, a large multistate outbreak of human S. Enteritidis infections associated with shell eggs occurred (http://www.cdc.gov/salmonella/enteritidis/). In response, the U.S. Food and Drug Administration (FDA) sequenced >100 environmental farm- and shell egg-associated outbreak food and clinical isolates to identify likely environmental sources causing egg contamination by using a novel whole-genome sequencing (WGS) approach (30, 31). With all of the available drafted genome sequences, we were able to explore mutational changes between several major phage types, including PT8, PT13a, and PT13, by targeted PCR resequencing. Here, a set of SNPs was discovered and used to examine the genetic diversity and phylogenetic relationships of a collection of 98 S. Enteritidis poultry-related and human clinical isolates representing 14 different phage types. Moreover, 31 S. Enteritidis strains, including 27 historical human clinical isolates originally from the American Type Culture Collection (ATCC), were evaluated with a rapid and simplified SNP typing scheme for the major clades identified in this study.
MATERIALS AND METHODS
Bacterial isolates and DNA extraction.
A total of 130 isolates from Salmonella enterica serovar Enteritidis (129 isolates) and serovar Berta (1 isolate) were used in this study. Strains were isolated from poultry, poultry-related sources, humans, ground beef, cat food, and mice and were obtained from the culture collections at the Center for Veterinary Medicine (CVM) and the Center for Food Safety and Applied Nutrition (CFSAN) of the U.S. Food and Drug Administration. A set of 98 S. Enteritidis strains, including 64 poultry-related isolates and 34 human clinical isolates, was used as the identification (ID) set for SNP validation (see Table S1 in the supplemental material). Genomic DNA was extracted from tryptic soy broth cultures grown overnight by using a Qiagen (Valencia, CA) DNeasy Blood & Tissue kit, according to the manufacturer's instructions, and stored at −20°C before use.
Phage typing.
All S. Enteritidis isolates were phage typed, according to previously described methods (32), at the National Microbiology Laboratory, Canadian Science Centre for Human and Animal Health, Winnipeg, Manitoba, Canada. Isolates that reacted with phages but retained unrecognizable lytic patterns were atypical and were designated atypical or RDNC (“react but do not conform” to designated phage types).
Resequencing.
One hundred forty-nine loci from the Salmonella chromosome and 11 loci from associated plasmids were used for SNP screening. These loci comprised 88 virulence-associated loci and 72 loci initially identified by comparative genome sequence (CGS) analysis between prevalent phage types in available genomes by using Mauve v.2.3.1 (33) (Fig. 1). Draft genomes used for CGS analysis included two strains each of PT4, PT1, PT13a, PT13, and PT21 and three PT8 S. Enteritidis strains. Variable loci identified in silico were PCR amplified over a partial set of S. Enteritidis isolates from the original ID strain set. The resulting amplicons were purified and sequenced in both directions by using the Sanger dideoxy method at Molecular Cloning Laboratories (MCLAB) (South San Francisco, CA). Loci containing potentially parsimony-informative (PI) SNPs were further sequenced over the rest of the S. Enteritidis isolates in the ID strain set.
FIG 1.

(Top) Circular map of 149 loci analyzed in this study relative to the Salmonella Enteritidis P125109 chromosome. The outer circle shows the genome scale. The fourth circle shows the ST64B-like phage insertion site. Triangles on the inner circles show locations of 149 loci analyzed, including 126 loci without an SNP(s) and 23 loci with an SNP(s). ORFs, open reading frames. (Bottom) Circular map of 11 virulence-related loci analyzed on the plasmid. The outer circle shows the plasmid scale. The inner circle shows 11 loci related to virulence analyzed in this study. Both panels were created by GenVision from DNASTAR Lasergene sequence analysis software (DNASTAR, Madison, WI).
Genetic analysis.
Consensus sequences for each isolate from the bidirectional sequence reads were assembled with the Sequencher v.4.9 program (Gene Codes Corporation, Ann Arbor, MI). Contigs for each locus exported from Sequencher were aligned by using the default settings of Clustal W in Molecular Evolutionary Genetic Analysis (MEGA 5.10) software (34). Alignments were exported as FASTA files and then converted to interleaved NEXUS files by using the SequenceMatrix program (35). A matrix including a concatenation of sequences from 23 loci was created for 100 S. Enteritidis isolates and one closely related S. Berta strain, which was used as the outgroup in all analyses. Basic population genetic parameters were estimated by using the DNA Sequence Polymorphism (DnaSP 5.10) program (36), and levels of nucleotide diversity were estimated by using π (37). To test the neutral equilibrium model, Tajima's D test was employed by using the DnaSP program, in which the test statistics were compared with the distributions generated from 10,000 coalescent simulations. In addition, model-based cluster analysis was done on the combined data set by using STRUCTURE (38, 39), in which isolates are assigned to clusters probabilistically, assuming Hardy-Weinberg equilibrium and linkage equilibrium within populations.
Phylogenetic analyses.
Phylogenetic analyses were initially executed in PAUP* (Phylogenetic Analysis Using Parsimony) v.4b10 (40), using the maximum parsimony (MP) criterion under heuristic search methods, with random addition of taxa (n = 100 iterations) and tree bisection-reconnection (TBR) search methods in effect. The shortest trees were then used as the starting topology for the evaluation of 16 nested models of sequence evolution (41–43). The best-fit model of substitution was determined by the likelihood score of each tree under a χ2 approximation of the null distribution (44). The GARLI (Genetic Algorithm for Rapid Likelihood Inference) v0.951 program (45) was then used to perform heuristic phylogenetic searches under the K80 (K2P) + γ + I (Kimura two-parameter test with gamma-distributed rate heterogeneity and a proportion of invariant sites) nucleotide substitution model, where topologies were evaluated based on their likelihood. The best tree was found by running the GARLI program on the original data matrix using default settings. Multiple runs were performed (n = 100) to ensure that results were consistent, as the algorithm is inherently stochastic (45). To estimate support for each node, phylogenies were created for 10,00 bootstrap replicates of the data set from GARLI (46). Grid computing through the Lattice Project (47) was used to decrease the computational time required to complete the best tree search and the bootstrap replicates for the data set. The GARLI executable was converted to a grid service such that batches of bootstrap replicates were distributed among hundreds of computers, where they were conducted asynchronously and in parallel (48, 49).
Congruence (i.e., phylogenetic concordance) between loci was assessed by using the incongruence length difference (ILD) test (50). ILD tests were done by using the partition homogeneity test available in PAUP* v.4b10, with 1,000 data partitions, under simple heuristic searches with 100 random taxon additions and TBR branch swapping in effect.
Pyrosequencing.
Winclada v1.00.08 (51) was used to identify signature nucleotide substitutions unique to a specific clade of S. Enteritidis strains. Pyrosequencing was used to evaluate the SNPs in the 31 S. Enteritidis nonpoultry isolates. Briefly, oligonucleotide primers for amplification and sequencing of SNP-containing regions were designed by using PSQ Assay Design software (Qiagen, Valencia, CA) (see Table S2 in the supplemental material). Biotinylated and nonbiotinylated primers were obtained from Integrated DNA Technologies (Coralville, IA). After amplification, pyrosequencing assays were performed by using a PyroMark Gold Q96 Reagents kit (Qiagen, Valencia, CA) and a PyroMark Q96 ID system (Qiagen, Valencia, CA) according to the manufacturer's instructions. Briefly, 20 μl of biotinylated PCR product was captured by using streptavidin-coated Sepharose beads (Amersham Biosciences, Little Chalfont, United Kingdom), and the unlabeled strand was denatured and removed by using the vacuum prep tool according to the manufacturer's instructions. After washing, the resulting single-stranded DNA was transferred into a 96-well microtiter plate and used as a template for pyrosequencing with 4 pmol each of sequencing primers per reaction. Nucleotides were added based on the dispensation order generated from the sequence being analyzed from the assay design software.
Nucleotide sequence accession numbers.
The sequences reported in this study have been submitted to GenBank with the following accession numbers: KF448540 to KF448637 for acrR, KF448638 to KF448735 for caiC, KF448736 to KF448833 for citT, KF448834 to KF448931 for corA, KF448932 to KF449029 for cpsG, KF449030 to KF449127 for the IGS between csgB and csgD, KF449128 to KF449225 for cyaA, KF449226 to KF449323 for iagB, KF449324 to KF449421 for marA, KF449422 to KF449519 for marC, KF449520 to KF449617 for narP, KF449618 to KF449715 for the intergenic sequence (IGS) between SEN0084 and SEN0085, KF449716 to KF449813 for SEN0629, KF449814 to KF449911 for SEN0999-SEN1000, KF449912 to KF450009 for SEN1164, KF450010 to KF450107 for SEN2510, KF450108 to KF450205 for the IGS between SEN3612 and ivbL, KF450206 to KF450303 for ssaJ, KF450304 to KF450401 for SEN4255-SEN4256, KF450402 to KF450499 for trxA, KF450500 to KF450597 for wza, KF450598 to KF450695 for yafE, and KF450696 to KF450793 for yddG.
RESULTS
Polymorphism patterns in the S. Enteritidis population.
A total of 160 loci of both chromosomal and plasmid origins were screened among 98 S. Enteritidis isolates with 14 different PTs, including PTs 1, 2, 4, 7, 8, 13 13a, 21, 23, 24, 28, 34, 35, and 4a. Twenty-three loci were found to contain a DNA polymorphism(s), including 19 protein-coding regions and 4 intergenic regions (Table 1). Of the 16,897 sites (excluding sites with gaps) analyzed, 55 were polymorphic, which comprised 17 synonymous and 38 nonsynonymous changes. Most loci (13/23; 56.5%) retained only one nucleotide substitution. Average numbers of nucleotide differences (κ) within each locus ranged from 0.0204 (marA) to 2.0002 (marC). The observed average Tajima D value across loci was 0.5063, indicating that the majority of loci observed here were consistent with neutrality. Ten thousand simulated samples of 23 loci unanimously suggested no significant departure from the neutral equilibrium model. In addition, 43 haplotypes (Hap), with a nucleotide diversity per site (π) of 0.00073, were identified to have a much higher nucleotide diversity than that reported for housekeeping genes (i.e., no nucleotide diversity was noted for housekeeping genes) (24), which explained, in part, the utility of these SNP data for differentiating closely related S. Enteritidis strains. Slight differences between the numbers of polymorphic (or segregating) sites and parsimony-informative (PI) sites (i.e., present in at least two strains) were detected in each individual locus. The locus yielding the most PI sites was narP (7). Conversely, marA retained the fewest PI sites (zero), pointing to a substantial level of convergence within this gene.
TABLE 1.
Polymorphism/indel characteristics and genetic diversity among intergenic and coding regions included in the Salmonella enterica serovar Enteritidis phylogenetic analysis
| Region | No. of sites analyzed | Hap | No. of polymorphic sites | No. of parsimony-informative sites | Avg no. of nucleotide differences (κ) | Nucleotide diversity (π)a | G+C content | Tajima Db | SigDd | K (JC)c |
|---|---|---|---|---|---|---|---|---|---|---|
| Total | 16,897 | 43 | 55 | 41 | 12.3503 | 0.00073 | 0.495 | 0.5063 | NS | 0.005 |
| acrR | 743 | 2 | 1 | 1 | 0.1515 | 0.00020 | 0.456 | −0.2519 | NS | 0.000 |
| caiC | 810 | 5 | 6 | 5 | 1.1037 | 0.00136 | 0.536 | −0.1212 | NS | 0.012 |
| citT | 714 | 2 | 1 | 1 | 0.5043 | 0.00071 | 0.547 | 1.8429 | —e | 0.001 |
| corA | 903 | 2 | 1 | 1 | 0.1515 | 0.00017 | 0.537 | −0.2519 | NS | 0.000 |
| cpsG | 726 | 3 | 2 | 1 | 0.5247 | 0.00072 | 0.630 | 0.54481 | NS | 0.001 |
| csgD-csgB | 570 | 6 | 4 | 3 | 0.9024 | 0.00158 | 0.316 | 0.3252 | NS | 0.005 |
| cyaA | 1,241 | 5 | 3 | 3 | 0.7669 | 0.00062 | 0.537 | 0.5732 | NS | 0.007 |
| iagB | 600 | 2 | 1 | 1 | 0.5043 | 0.00084 | 0.401 | 1.8429 | —e | 0.001 |
| marA | 465 | 2 | 1 | 0 | 0.0204 | 0.00004 | 0.477 | −1.0301 | NS | 0.004 |
| marC | 662 | 5 | 7 | 5 | 2.0002 | 0.00302 | 0.492 | 1.1137 | NS | 0.006 |
| narP | 613 | 4 | 9 | 7 | 1.0856 | 0.0018 | 0.624 | −0.9460 | NS | 0.018 |
| SEN0084-SEN0085 | 652 | 4 | 3 | 1 | 0.3438 | 0.00053 | 0.336 | −0.7366 | NS | 0.033 |
| SEN0629 | 769 | 3 | 2 | 1 | 0.5247 | 0.00068 | 0.560 | 0.5448 | NS | 0.002 |
| SEN0999 | 859 | 2 | 1 | 1 | 0.3030 | 0.00035 | 0.332 | 0.6475 | NS | 0 |
| SEN1164 | 729 | 2 | 1 | 1 | 0.3030 | 0.00042 | 0.513 | 0.6475 | NS | 0.007 |
| SEN2510 | 730 | 2 | 1 | 1 | 0.3030 | 0.00042 | 0.537 | 0.6475 | NS | 0 |
| SEN3612-ivbL | 733 | 2 | 1 | 1 | 0.3030 | 0.00041 | 0.478 | 0.6475 | NS | 0.007 |
| SEN4255-SEN4256 | 708 | 2 | 1 | 1 | 0.3030 | 0.00043 | 0.498 | 0.6475 | NS | 0.003 |
| ssaJ | 625 | 2 | 1 | 1 | 0.3030 | 0.00048 | 0.424 | 0.6475 | NS | 0 |
| trxA | 659 | 2 | 1 | 1 | 0.3030 | 0.00046 | 0.466 | 0.6475 | NS | 0.003 |
| wza | 1,029 | 2 | 1 | 1 | 0.5050 | 0.00049 | 0.551 | 1.8467 | —e | 0.001 |
| yafE | 618 | 3 | 3 | 1 | 0.5047 | 0.00082 | 0.533 | −0.2384 | NS | 0.001 |
| yddG | 741 | 5 | 3 | 2 | 0.6358 | 0.00086 | 0.489 | 0.1674 | NS | 0.003 |
π, average number of nucleotide differences per site between two sequences (equation 10.5 or 10.6 in reference 67) and its sampling variance (equation 10.7 in reference 67).
Tajima's D neutrality test.
K (JC), average number of nucleotide substitutions per site between species in data set 1 and the first sequence in data set 2, with Jukes-Cantor correction.
SigD, significance of Tajima D; NS, not significant (P > 0.10).
—, 0.05 < P < 0.10 (considered not significant).
Incongruence among 23 SNP-containing loci in S. Enteritidis.
Phylogenetic concordance between loci was examined to evaluate the goodness of fit of phylogenetic signals between two independent data matrices. ILD testing of all 23 loci partitioned separately led to a conclusion of incongruence (P = 0.001). In order to isolate the locus responsible for incongruence, each of the 23 loci was partitioned against a combined matrix consisting of the 22 loci (Table 2). All of the loci were congruent with the combined remaining matrix except for three loci, each highly discordant in their signals, including caiC, marC, and narP (P < 0.05). When pairwise ILD comparisons were performed among the three discordant loci, incongruence was observed in the caiC-marC, and caiC-narP comparisons. Although marC failed to yield significant incongruence with narP, the ILD score was between 0.10 and 0.05 (P = 0.092), considered borderline incongruence. These data further supported a “prior-agreement” approach (52), which was subsequently adopted to resolve incongruence and establish a phylogeny representing the backbone loci of S. Enteritidis. As expected, the removal of caiC, marC, and narP from the matrix significantly improved the ILD score (P = 0.239). Thus, congruence was established for the remaining 20 loci of S. Enteritidis.
TABLE 2.
ILD values among 23 loci in S. Enteritidis
| Locus |
P value (1,000 partitions)b |
||||
|---|---|---|---|---|---|
| Remaining matrixa | caiC | marC | narP | Prior agreement | |
| caiC | 0.044 | ||||
| marc | 0.001 | 0.003 | |||
| narP | 0.001 | 0.002 | 0.092 | ||
| acrR | 0.695 | ||||
| citT | 1.000 | ||||
| corA | 0.752 | ||||
| cpsG | 1.000 | ||||
| csgD-csgB | 0.072 | ||||
| cyaA | 0.162 | ||||
| iagB | 0.605 | ||||
| marA | 1.000 | ||||
| SEN0084-SEN0085 | 1.000 | ||||
| SEN0629 | 1.000 | ||||
| SEN0999 | 0.561 | ||||
| SEN1164 | 1.000 | ||||
| SEN2510 | 1.000 | ||||
| SEN3614-ivbL | 1.000 | ||||
| SEN4255-SEN4256 | 1.000 | ||||
| ssaJ | 1.000 | ||||
| trxA | 1.000 | ||||
| wza | 0.875 | ||||
| yafE | 0.442 | ||||
| yddG | 0.107 | ||||
| Prior agreementc | 0.239 | ||||
ILD test conducted between the locus indicated and the remaining 22 loci.
Values in boldface type denote an ILD score of ≤0.05 (incongruence).
Prior-agreement matrix based on concordant ILD scores among all the loci except caiC, marC, and narP.
Phylogenetic analysis of S. Enteritidis isolates in the ID set.
All loci except for caiC, marC, and narP were combined into a single prior-agreement matrix, yielding a total of 14,814 nucleotides for phylogenetic analysis. The GARLI tree with the best log-likelihood score for the prior-agreement matrix is presented in Fig. 2. This tree partitioned most of the 98 S. Enteritidis strains into two distinct clades (clades I and II) and four subclades (Fig. 2). Low bootstrap values (bootstrap values of <80%) for most of the clades/subclades were observed and likely resulted from the limited number of highly conserved nucleotide substitutions that partitioned major S. Enteritidis lineages in the tree. Clade I comprised mostly PT4-like S. Enteritidis strains (40/45; 88.9%), including strains from PT1, PT4, PT7, PT21, and PT35, with the exception of strains 17919_PT23, 22621_PT13a, 23711_PT24, and 18569_PT14b. Clade II was composed mainly of PT8-like S. Enteritidis strains (47/51; 92.2%), including strains from PT2, PT8, PT13, PT13a, PT23, PT24, and PT28, with the exception of strains 30661_PT4a, SE9_PT14b, SE12_PT14b, and SE23_PT14b. All PT13 isolates fell into group IIC, and most PT8 isolates diverged into four subclades (subclades IIA to IID). Surprisingly, three PT8 isolates fell outside these four subclades, indicating the presence of additional lineages of PT8 S. Enteritidis strains. Subclade IID appeared to evolve into a well-supported monophyletic group (bootstrap value, 99%). Perhaps most importantly, the emergence of individual clones of S. Enteritidis (defined by PTs) appeared to be evolutionarily disjunct when viewed in the context of this phylogeny.
FIG 2.

Maximum likelihood tree of S. Enteritidis strains from concatenated molecular phylogenetic analysis of DNA sequences from 20 loci. Branch lengths are estimates of substitutions relative to the tree and are shown as the expected number of substitutions per site. Branch lengths not significantly different from zero are collapsed into polytomies. Bootstrap percentages (out of 1,000 iterations) of >40% are reported above each node. S. Berta strain 20034 is used as an outgroup to root the phylogeny. The brackets to the right of the tree denote the 2 distinct clades and 4 subclades in clade II. All of the human S. Enteritidis isolates in the tree are highlighted in gray. The presence (red cells) or absence (open cells) of phages RE-2010 and ST64B and the 14-kb virulence-related plasmid element are denoted to the right of the tree. The number of clusters (k values ranging from 1 to 5) is inferred from the multilocus data of 98 S. Enteritidis strains by using the model-based clustering method implemented in STRUCTURE. The membership coefficients (Q matrices) for 98 S. Enteritidis strains were visualized by using DISTRUCT (66). Each color represents a different cluster.
Genetic distances observed between the two S. Enteritidis clades and four subclades further supported the phylogenetic partitions revealed in the tree (Fig. 2) and are listed in Table 3. The distance between clades I and II was 13.74 substitutions, while the mean intraclade divergence was 4.42 nucleotide differences. Among the four subclades in clade II, the mean distance was 9.99 differences, while the mean intrasubclade divergence was 0.96 differences. Maximum diversity was observed between clades I and IID (19.34 nucleotide differences), while subclade D was the most distant subclade, showing the greatest divergence from the remaining S. Enteritidis strains residing in clade II (13.97, 16.14, and 9.86 nucleotide differences from subclades A, B, and C, respectively) (Table 3).
TABLE 3.
Sequence divergence within and between S. Enteritidis clades and subclades
| Clade or subclade (no. of strains) | Mean no. of base differences ± SEa |
|||||
|---|---|---|---|---|---|---|
| I | II | IIA | IIB | IIC | IID | |
| I (47) | 0.99 ± 0.41b | |||||
| II (51) | 13.74 ± 4.25 | 7.85 ± 1.79b | ||||
| IIA (4) | 8.61 ± 3.40 | 1.00 ± 0.68b | ||||
| IIB (8) | 10.23 ± 3.64 | 5.06 ± 1.86 | 1.07 ± 0.71b | |||
| IIC (17) | 12.20 ± 4.53 | 6.09 ± 2.24 | 8.82 ± 3.11 | 0.46 ± 0.25b | ||
| IID (18) | 19.34 ± 5.60 | 13.97 ± 3.61 | 16.14 ± 3.89 | 9.86 ± 2.71 | 1.29 ± 0.46b | |
Model-based clustering memberships, recovered when individuals were assigned to one of two clusters (i.e., k = 2) (Fig. 2), matched the relationships presented in the phylogenetic analyses. However, a number of individuals, particularly within PT8, were prone to genomic admixture, where portions of their multilocus genotype resembled those of isolates found in groups I, IIC, and IID (Fig. 2). The unique composition of these individuals may explain, in part, the low bootstrap support as well as the inability to resolve their relationships (Fig. 2). As the value of k increased, group IID became a separate cluster at k values of 3 and 5. Additionally, it was also noted that group I was relatively stable, where all individuals were found within the same cluster, except at a k value of 4, where some individuals were placed in a separate cluster.
Phage and plasmid profiling of S. Enteritidis strains.
The presence or absence of several phages and plasmids, including phage RE-2010, phage ST64B, and a 14-kb plasmid element associated with virulence, was observed among S. Enteritidis strains with different phage types (Fig. 2). Overall, phage RE-2010 was associated with PT8, PT13, PT13a, PT23, PT28, and PT14b, with the exception of one PT14b strain and one PT13a strain (clade II), while phage ST64B was present in PT4, PT1, PT35, and PT21 S. Enteritidis strains (clade I). Notably, both phages were absent from PT4a strains but present in PT34 strains. In the case of the 14-kb plasmid element, 83.7% (82/98) of S. Enteritidis strains retained the virulence-associated plasmid element irrespective of phage type. However, it was also noted that all of the PT21 and PT13 isolates (2 and 10, respectively) lost this 14-kb plasmid element during evolution.
Identification of several “signature” nucleotide substitutions capable of distinguishing S. Enteritidis clades/subclades.
Analysis of the concatenated 20-locus nucleotide sequence alignment by using Winclada (51) revealed the existence of several primary synapomorphic nucleotides capable of differentiating specific clades. The signature synapomorphic substitutions, their positions in the Clustal X alignment, and the S. Enteritidis strain groupings that they distinguish are provided in Table 4. Using S. Berta as the outgroup, all S. Enteritidis strains were defined uniquely by one SNP within the SEN0629 gene at position 248. Clade I and clade II both retained definitive substitutions that were diagnostic for all of the strains residing in each of these particular clades. Specifically, clade I was denoted by two SNPs in the cpsG and SEN0629 genes at positions 155 and 245, respectively. Two SNPs, one within the citT gene at position 393 and one in the wza gene at position 318, uniquely defined clade II, which comprised mostly PT8-like S. Enteritidis strains. Among the four subclades in clade II, subclades C and D were distinguished from subclades A and B by three single nucleotide changes in the cyaA, yafE, and yddG genes. As in clades I and II, clades IIA, IIB, and IID also retained primary signature SNPs uniquely capable of defining these groups. For example, clade IIA was denoted by one single change in the IGS between the csgB and csgD genes, while clade IIB was defined by changes in three separate genes, acrR, corA, and cyaA. Clade IID was the most distant clade from the other subclades, identified by eight additional signature SNPs.
TABLE 4.
Signature nucleotide substitutions unique to individual clades of S. Enteritidis strains
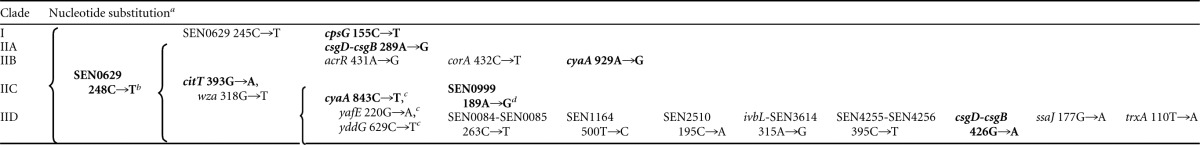
Individual alignment position numbers are given following the individual locus. Except for substitutions linked to footnotes c and d, all other substitutions listed are primary signature substitutions, meaning that the presence of an individual particular sequence change in a strain defines it as being part of this clade. Nucleotide substitutions in boldface type are the selected minimal set of SNPs to form a SNP-based diagnostic scheme for strain evaluation. Each brace applies only to the column in which it appears.
Signature substitution shared by all S. Enteritidis strains compared to the outgroup S. Berta strain.
Shared signature substitution of clades IIC and IID, shared only between clades IIC and IID.
Secondary signature substitution, depending on the presence of the signature substitution for clade II.
Clade IIC did not yield any clade-defining primary substitution(s), although in combination with the presence of any of the signature SNPs defining clade II, clade IIC was capable of being identified by a secondary SNP in the SEN0999 gene at position 189.
Evaluation of a SNP-based diagnostic scheme for S. Enteritidis strains.
A minimal set of SNPs (Table 4) was selected to form a SNP-based diagnostic scheme (Fig. 3) for every clade/subclade described in the phylogenetic analysis reported in Fig. 2. Thirty-one S. Enteritidis strains were chosen to allow evaluation of the scheme in blind strains from nonpoultry sources. SNP calling was made based on the program generated from PyroMark ID software, and all of the pyrosequencing results are summarized in Table 5. The two SNPs at cpsG position 155 and citT position 393 were able to partition these 31 S. Enteritidis strains into two groups. Within group II, 23 S. Enteritidis strains (23/28; 82.1%) were assigned to a specific subgroup based on their SNP profiles. Interestingly, one S. Enteritidis strain that was “atypical” by phage typing was able to be assigned to clade IIC based on the SNP calling decision tree assembled here, suggesting that, like molecular serotyping assays developed previously (53, 54), molecular phage typing will provide rapid and highly accurate calls for this important phenotypic attribute of S. Enteritidis.
FIG 3.

SNP-based diagnostic diagram. A block diagram was made with the minimal set of SNPs (shown in boldface type in Table 4) to delineate each clade/subclade described from the phylogenetic analysis reported in Fig. 2.
TABLE 5.
SNP profiles of 31 non-poultry-related S. Enteritidis strains
| Strain | SNP |
Cluster | PT | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| SEN0629 position 248 | cpsG position 155 | citT position 393 | csgD-csgB position 289 | cyaA position 929 | cyaA position 843 | csgD-csgB position 426 | SEN0999 position 189 | |||
| Reference base | C | C | G | A | A | C | G | A | ||
| 58-6482 | T | C | G | A | A | C | G | A | I | 35 |
| 54-4531 | T | C | A | A | A | C | G | A | II | 8 |
| 50-5646 | T | C | A | A | A | T | A | A | IID | 8 |
| 50-3079 | T | C | A | A | A | T | G | G | IIC | 8 |
| 75-150 | T | C | A | A | A | T | A | A | IID | 8 |
| 75-199 | T | C | A | A | A | T | A | A | IID | 8 |
| 62-1976 | T | C | A | A | G | C | G | A | IIB | 13a |
| 60-2506 | T | C | A | A | A | T | A | A | IID | 8 |
| 50-5306A | T | C | G | A | A | C | G | A | I | 35 |
| 53-407 | T | C | A | A | A | T | G | A | IIC/D | 8 |
| 54-2953 | T | C | A | A | A | T | G | A | IIC/D | 8 |
| 75-970 | T | C | A | A | A | T | G | G | IIC | 2 |
| 76-1594 | T | C | A | A | A | C | G | A | II | 8 |
| 76-2938 | T | G | G | A | A | C | G | A | I | 4 |
| 76-2969 | T | C | A | A | G | C | G | A | IIB | 13a |
| 77-1427 | T | C | A | A | A | T | A | A | IID | 8 |
| 77-3493 | T | C | A | A | A | C | G | A | II | 10 |
| 78-1757 | T | C | A | A | A | T | A | A | IID | 8 |
| 81-2625 | T | C | A | A | A | T | A | A | IID | 8 |
| 22568B | T | C | A | A | A | T | G | G | IIC | Atypical |
| 19755 | T | C | A | A | A | T | A | A | IID | 8 |
| 36388 | T | C | A | A | A | T | G | G | IIC | 13 |
| 36952 | T | C | A | A | A | T | G | G | IIC | 8 |
| 17912 | T | C | A | A | A | T | A | A | IID | 8 |
| 77-2659 | T | C | A | A | A | T | A | A | IID | 8 |
| 76-2651 | T | C | A | A | A | T | A | A | IID | 13a |
| 74-991 | T | C | A | A | A | T | A | A | IID | 2 |
| 75-2325 | T | C | A | A | G | C | G | A | IIB | 8 |
| 36951 | T | C | A | A | A | T | G | G | IIC | 51 |
| 77-0424 | T | C | A | A | A | T | A | A | IID | 8 |
| 76-574 | T | C | A | A | A | T | A | A | IID | 8 |
DISCUSSION
S. Enteritidis strains belonging to multiple PTs, such as PTs 1, 2, 4, 8, 13, 13a, 14b, and 21, have been associated with poultry and poultry-related products for some time (55–57). The evolutionary relationships between multiple PTs of S. Enteritidis strains infecting chicken reproductive tissues remain unclear, since these PTs emerged largely simultaneously in geographically disparate countries (58–62). In line with IS200-defined clonal lineages (3), our multilocus sequence-based phylogeny suggested two major clonal lineages among the 14 PTs studied in our poultry-related S. Enteritidis strain population here. PTs that dominated in western Europe and Asia, including PT1, PT4, and PT21, occurred in clonal lineage I, while PTs that were most common in North America (i.e., PT8, PT13a, and PT13) composed the majority of clonal lineage II (see Table S1 in the supplemental material). Interestingly, clades I and II retained a separate unique SNP profile while diverging from each other (Table 4), suggesting the simultaneous emergence of these two major lineages. Furthermore, the appearance of newly emerged PT14b (10) in both lineages suggested multiple origins for this PT. It was also noteworthy that clade I is more homogeneous in its genetic composition of strains than clade II, as evidenced by model-based clustering analysis as well (Fig. 2), suggesting a more clonal evolutionary relationship among these PTs and a potentially more rapid geographic diversification of this lineage of S. Enteritidis strains. Clade II can be further subdivided into four subgroups, with all the PT13 isolates in clade IIC. A set of S. Enteritidis strains shared the same ancestor and had amply evolved (i.e., clade IID) to form a well-supported monophyletic group away from the rest of clade II, regardless of their PTs. The fact that this subclade contained five major PTs (i.e., PT2, PT8, PT13a, PT28, and PT34) in lineage II, the only exception being PT13, clearly underscores the weak relationship between phage type and phylogenetic signal.
It is worth noting that several human S. Enteritidis isolates clustered closely with multiple S. Enteritidis clones isolated from commercial and farm poultry sources (Fig. 2). This correlation also revealed distinct geographic features. For example, human S. Enteritidis strains isolated from Mexico resided in clade I with all the poultry-related S. Enteritidis isolates derived from Mexico, Scotland, and China. Nine human isolates from Iowa and two ground turkey isolates (15845_PT4 and 30663_PT21) from Iowa and Maryland, respectively, were the only exceptions in this “cosmopolitan” cluster of strains (Fig. 2; see also Table S1 in the supplemental material). PT4 and PT4-like phage types are relatively rare in the United States and Canada. Thus, the nine human isolates from Iowa may be best explained as a result of foreign travel with return trips back to the United States (63). In clade II, all of the domestic S. Enteritidis isolates derived from humans clustered with their closely related poultry clones into two predominant groups (clade IIC and clade IID). Two human isolates (22599_PT8 and 17923_PT8) resided outside the four subclades in lineage II, suggesting the presence of other food or environmental sources for these particular strains.
Compared to many other food-borne pathogens, nucleotide sequences in S. Enteritidis were highly conserved, as shown in this study and in other studies as well (24, 29, 64). Among the 160 loci investigated, only 23 loci contained informative SNPs, mostly from genes in COG category F (nucleotide transport and metabolism), K (transcription), Q (secondary metabolites, biosynthesis, transport, and catabolism), or S (unknown function). Among these 23 loci, 9 loci, including 6 coding regions (cpsG, iagB, marA, marC, narP, and wza) and three intergenic regions (SEN0084-SEN0085, SEN0999-SEN1000, and SEN3612-ivbL), were newly discovered in this study to contain polymorphic sites, while the other 14 were also found to contain variable sites that were able to differentiate two S. Enteritidis PT13a strains in a recent study (28). Examination of phage and plasmid profiles revealed several interesting findings. First, the presence or absence of both phages RE-2010 and ST64B may be an important genetic marker to predict PT4a and PT34. Also, the absence of the 14-kb virulence element on the plasmid in certain phage types (i.e., PT21 and PT13) may help to differentiate these phage types from additional PTs that reside within this same clade on the tree.
Several phage types were known to be interconvertible by the gain or loss of a single plasmid (42), which may explain some of the alternative placements of S. Enteritidis isolates in our multilocus phylogenetic tree. For example, the observations that, by acquisition of a plasmid, PT7 can be converted to PT23 (14) and that PT4 can be converted to PT24 (17) would explain why one PT23 isolate and one PT24 isolate studied here were alternatively placed in clade I adjacent to PT7 and PT4 isolates.
Phage typing has been used as an epidemiological tool to track the spread of S. Enteritidis across different geographical regions. However, its suitability for grouping strains in a phylogenetically accurate way is debatable in light of data reported here and elsewhere (30). For example, strains of PT8 were polyphyletic and phylogenetically distributed across all subclades in lineage II, and the multiple origins of PT14b in our multilocus sequence tree suggested their distinct genetic composition despite the same phenotypic feature shared among these S. Enteritidis isolates. Therefore, a SNP typing scheme that can be applied on a global scale to firmly identify all S. Enteritidis clusters is needed. Next-generation genome sequencing is now allowing a rapid expansion of SNPs for S. Enteritidis typing. In this study, a SNP-based diagnostic scheme successfully assigned most non-poultry-related S. Enteritidis isolates into specific clades/subclades in a phylogenetic context, providing core markers for future development of a SNP typing scheme.
One limitation to the WGS-based SNP approach used here was that the emphasis of comparative genomic analysis was only given in nucleotide differences between phage types. As a result, most strain-specific polymorphisms that were found to differentiate two PT13a S. Enteritidis strains in a recent study (28), as well as the variable loci discovered in a recent S. Enteritidis shell egg outbreak (30), were not included here. In addition, SNP markers were obtained originally by comparing only two sequenced S. Enteritidis genomes for each phage type (i.e., PT1, PT21, PT4, PT8, PT13, and PT13a) with reference-based assemblies (the finished S. Enteritidis PT4 strain [strain 125109] from GenBank was used as a reference). Therefore, many informative SNPs may have been overlooked, the highly homogeneous nature of the S. Enteritidis genome notwithstanding (27, 30, 65). For example, no SNP was found between two PT1 and two PT21 S. Enteritidis strains, and this may due to the limited number of genomes compared here or the use of a particular reference method for genome assembly. Thus, future genomic sequencing studies will be required to identify additional SNP markers to expand collapsed branches in the S. Enteritidis strain phylogeny reported here. Despite these caveats, these data provide a genetic solution for the rapid and accurate mediation of phage types among a set of highly clonal S. Enteritidis strains and further underscore the importance of comparative genomics in the diagnostic separation of highly homogeneous Salmonella serovars of public health significance.
Supplementary Material
Footnotes
Published ahead of print 26 February 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JCM.00051-14.
REFERENCES
- 1.CDC. 2013. Incidence and trends of infection with pathogens transmitted commonly through food—Foodborne Diseases Active Surveillance Network, 10 U.S. sites, 1996-2012. MMWR Morb. Mortal. Wkly. Rep. 62:283–287 http://www.cdc.gov/mmwr/preview/mmwrhtml/mm6215a2.htm [PMC free article] [PubMed] [Google Scholar]
- 2.CDC. 2013. Surveillance for foodborne disease outbreaks—United States, 2009-2010. MMWR Morb. Mortal. Wkly. Rep. 62:41–47 http://www.cdc.gov/mmwr/preview/mmwrhtml/mm6203a1.htm [PMC free article] [PubMed] [Google Scholar]
- 3.Stanley J, Baquar N. 1994. Phylogenetics of Salmonella enteritidis. Int. J. Food Microbiol. 21:79–87. 10.1016/0168-1605(94)90202-X [DOI] [PubMed] [Google Scholar]
- 4.Wattiau P, Boland C, Bertrand S. 2011. Methodologies for Salmonella enterica subsp. enterica subtyping: gold standards and alternatives. Appl. Environ. Microbiol. 77:7877–7885. 10.1128/AEM.05527-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hickman-Brenner FW, Stubbs AD, Farmer JJ., III 1991. Phage typing of Salmonella enteritidis in the United States. J. Clin. Microbiol. 29:2817–2823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tighe MK, Savage R, Vrbova L, Toolan M, Whitfield Y, Varga C, Lee B, Allen V, Maki A, Walton R, Johnson C, Dhar B, Ahmed R, Crowcroft NS, Middleton D. 2012. The epidemiology of travel-related Salmonella Enteritidis in Ontario, Canada, 2010-2011. BMC Public Health 12:310. 10.1186/1471-2458-12-310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Threlfall EJ, Hall ML, Rowe B. 1992. Salmonella bacteraemia in England and Wales, 1981-1990. J. Clin. Pathol. 45:34–36. 10.1136/jcp.45.1.34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schroeter A, Ward LR, Rowe B, Protz D, Hartung M, Helmuth R. 1994. Salmonella enteritidis phage types in Germany. Eur. J. Epidemiol. 10:645–648. 10.1007/BF01719587 [DOI] [PubMed] [Google Scholar]
- 9.Brown DJ, Baggesen DL, Hansen HB, Hansen HC, Bisgaard M. 1994. The characterization of Danish isolates of Salmonella enterica serovar Enteritidis by phage typing and plasmid profiling: 1980-1990. APMIS 102:208–214. 10.1111/j.1699-0463.1994.tb04866.x [DOI] [PubMed] [Google Scholar]
- 10.Nygard K, de Jong B, Guerin PJ, Andersson Y, Olsson A, Giesecke J. 2004. Emergence of new Salmonella Enteritidis phage types in Europe? Surveillance of infections in returning travellers. BMC Med. 2:32. 10.1186/1741-7015-2-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson LR, Gould LH, Dunn JR, Berkelman R, Mahon BE. 2011. Salmonella infections associated with international travel: a Foodborne Diseases Active Surveillance Network (FoodNet) study. Foodborne Pathog. Dis. 8:1031–1037. 10.1089/fpd.2011.0854 [DOI] [PubMed] [Google Scholar]
- 12.Molbak K, Neimann J. 2002. Risk factors for sporadic infection with Salmonella enteritidis, Denmark, 1997-1999. Am. J. Epidemiol. 156:654–661. 10.1093/aje/kwf096 [DOI] [PubMed] [Google Scholar]
- 13.Ahmed R, Soule G, Demczuk WH, Clark C, Khakhria R, Ratnam S, Marshall S, Ng LK, Woodward DL, Johnson WM, Rodgers FG. 2000. Epidemiologic typing of Salmonella enterica serotype Enteritidis in a Canada-wide outbreak of gastroenteritis due to contaminated cheese. J. Clin. Microbiol. 38:2403–2406 http://jcm.asm.org/content/38/6/2403.long [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Threlfall EJ, Chart H, Ward LR, de Sa JD, Rowe B. 1993. Interrelationships between strains of Salmonella enteritidis belonging to phage types 4, 7, 7a, 8, 13, 13a, 23, 24 and 30. J. Appl. Bacteriol. 75:43–48. 10.1111/j.1365-2672.1993.tb03405.x [DOI] [PubMed] [Google Scholar]
- 15.Fadl AA, Khan MI. 1997. Genotypic evaluation of Salmonella enteritidis isolates of known phage types by arbitrarily primed polymerase chain reaction. Avian Dis. 41:732–737. 10.2307/1592168 [DOI] [PubMed] [Google Scholar]
- 16.Chart H, Row B, Threlfall EJ, Ward LR. 1989. Conversion of Salmonella enteritidis phage type 4 to phage type 7 involves loss of lipopolysaccharide with concomitant loss of virulence. FEMS Microbiol. Lett. 51:37–40 [DOI] [PubMed] [Google Scholar]
- 17.Frost JA, Ward LR, Rowe B. 1989. Acquisition of a drug resistance plasmid converts Salmonella enteritidis phage type 4 to phage type 24. Epidemiol. Infect. 103:243–248. 10.1017/S0950268800030594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tankouo-Sandjong B, Kinde H, Wallace I. 2012. Development of a sequence typing scheme for differentiation of Salmonella Enteritidis strains. FEMS Microbiol. Lett. 331:165–175. 10.1111/j.1574-6968.2012.02568.x [DOI] [PubMed] [Google Scholar]
- 19.Pang S, Octavia S, Reeves PR, Wang Q, Gilbert GL, Sintchenko V, Lan R. 2012. Genetic relationships of phage types and single nucleotide polymorphism typing of Salmonella enterica serovar Typhimurium. J. Clin. Microbiol. 50:727–734. 10.1128/JCM.01284-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Porwollik S, Santiviago CA, Cheng P, Florea L, Jackson S, McClelland M. 2005. Differences in gene content between Salmonella enterica serovar Enteritidis isolates and comparison to closely related serovars Gallinarum and Dublin. J. Bacteriol. 187:6545–6555. 10.1128/JB.187.18.6545-6555.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stanley J, Jones CS, Threlfall EJ. 1991. Evolutionary lines among Salmonella enteritidis phage types are identified by insertion sequence IS200 distribution. FEMS Microbiol. Lett. 66:83–89 [DOI] [PubMed] [Google Scholar]
- 22.Olsen JE, Skov MN, Threlfall EJ, Brown DJ. 1994. Clonal lines of Salmonella enterica serotype Enteritidis documented by Is200-, ribo-, pulsed-field gel-electrophoresis and RFLP typing. J. Med. Microbiol. 40:15–22. 10.1099/00222615-40-1-15 [DOI] [PubMed] [Google Scholar]
- 23.Urwin R, Maiden MC. 2003. Multi-locus sequence typing: a tool for global epidemiology. Trends Microbiol. 11:479–487. 10.1016/j.tim.2003.08.006 [DOI] [PubMed] [Google Scholar]
- 24.Noda T, Murakami K, Asai T, Etoh Y, Ishihara T, Kuroki T, Horikawa K, Fujimoto S. 2011. Multi-locus sequence typing of Salmonella enterica subsp. enterica serovar Enteritidis strains in Japan between 1973 and 2004. Acta Vet. Scand. 53:38. 10.1186/1751-0147-53-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Achtman M. 2008. Evolution, population structure, and phylogeography of genetically monomorphic bacterial pathogens. Annu. Rev. Microbiol. 62:53–70. 10.1146/annurev.micro.62.081307.162832 [DOI] [PubMed] [Google Scholar]
- 26.Filliol I, Motiwala AS, Cavatore M, Qi W, Hazbon MH, Bobadilla del Valle M, Fyfe J, Garcia-Garcia L, Rastogi N, Sola C, Zozio T, Guerrero MI, Leon CI, Crabtree J, Angiuoli S, Eisenach KD, Durmaz R, Joloba ML, Rendon A, Sifuentes-Osornio J, Ponce de Leon A, Cave MD, Fleischmann R, Whittam TS, Alland D. 2006. Global phylogeny of Mycobacterium tuberculosis based on single nucleotide polymorphism (SNP) analysis: insights into tuberculosis evolution, phylogenetic accuracy of other DNA fingerprinting systems, and recommendations for a minimal standard SNP set. J. Bacteriol. 188:759–772. 10.1128/JB.188.2.759-772.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olson AB, Andrysiak AK, Tracz DM, Guard-Bouldin J, Demczuk W, Ng LK, Maki A, Jamieson F, Gilmour MW. 2007. Limited genetic diversity in Salmonella enterica serovar Enteritidis PT13. BMC Microbiol. 7:87. 10.1186/1471-2180-7-87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guard J, Morales CA, Fedorka-Cray P, Gast RK. 2011. Single nucleotide polymorphisms that differentiate two subpopulations of Salmonella Enteritidis within phage type. BMC Res. Notes 4:369. 10.1186/1756-0500-4-369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Octavia S, Lan R. 2007. Single-nucleotide-polymorphism typing and genetic relationships of Salmonella enterica serovar Typhi isolates. J. Clin. Microbiol. 45:3795–3801. 10.1128/JCM.00720-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Allard MW, Luo Y, Strain E, Pettengill J, Timme R, Wang C, Li C, Keys CE, Zheng J, Stones R, Wilson MR, Musser SM, Brown EW. 2013. On the evolutionary history, population genetics and diversity among isolates of Salmonella Enteritidis PFGE pattern JEGX01.0004. PLoS One 8:e55254. 10.1371/journal.pone.0055254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Timme RE, Allard MW, Luo Y, Strain E, Pettengill J, Wang C, Li C, Keys CE, Zheng J, Stones R, Wilson MR, Musser SM, Brown EW. 2012. Draft genome sequences of 21 Salmonella enterica serovar Enteritidis strains. J. Bacteriol. 194:5994–5995. 10.1128/JB.01289-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ward LR, de Sa JD, Rowe B. 1987. A phage-typing scheme for Salmonella enteritidis. Epidemiol. Infect. 99:291–294. 10.1017/S0950268800067765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Darling AC, Mau B, Blattner FR, Perna NT. 2004. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14:1394–1403. 10.1101/gr.2289704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739. 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vaidya G, Lohman DJ, Meier R. 2011. SequenceMatrix: concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 27:171–180. 10.1111/j.1096-0031.2010.00329.x [DOI] [PubMed] [Google Scholar]
- 36.Librado P, Rozas J. 2009. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452. 10.1093/bioinformatics/btp187 [DOI] [PubMed] [Google Scholar]
- 37.Tajima F. 1983. Evolutionary relationship of DNA sequences in finite populations. Genetics 105:437–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pritchard JK, Stephens M, Donnelly P. 2000. Inference of population structure using multilocus genotype data. Genetics 155:945–959 http://www.genetics.org/content/155/2/945.long [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hubisz MJ, Falush D, Stephens M, Pritchard JK. 2009. Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Resour. 9:1322–1332. 10.1111/j.1755-0998.2009.02591.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Swofford DL. 2002. PAUP*. Phylogenetic analysis using parsimony, version 4. Sinauer Associates, Sunderland, MA [Google Scholar]
- 41.Frati F, Simon C, Sullivan J, Swofford DL. 1997. Evolution of the mitochondrial cytochrome oxidase II gene in Collembola. J. Mol. Evol. 44:145–158. 10.1007/PL00006131 [DOI] [PubMed] [Google Scholar]
- 42.Sullivan J, Markert JA, Kilpatrick CW. 1997. Phylogeography and molecular systematics of the Peromyscus aztecus species group (Rodentia: Muridae) inferred using parsimony and likelihood. Syst. Biol. 46:426–440. 10.1093/sysbio/46.3.426 [DOI] [PubMed] [Google Scholar]
- 43.Swofford DL, Olsen GJ, Waddell PJ, Hillis DM. 1996. Phylogenetic inference, p 407–514 In Hillis DM, Moritz C. (ed), Molecular systematics. Sinauer Associates, Sunderland, MA [Google Scholar]
- 44.Yang Z, Goldman N, Friday A. 1995. Maximum-likelihood trees from DNA-sequences—a peculiar statistical estimation problem. Syst. Biol. 44:384–399. 10.1093/sysbio/44.3.384 [DOI] [Google Scholar]
- 45.Zwickl DJ. 2006. Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion. Ph.D. thesis The University of Texas, Austin, TX [Google Scholar]
- 46.Felsenstein J. 1985. Confidence-limits on phylogenies—an approach using the bootstrap. Evolution 39:783–791. 10.2307/2408678 [DOI] [PubMed] [Google Scholar]
- 47.Myers DS, Bazinet AL, Cummings MP. 2008. Expanding the reach of grid computing: combining Globus- and BOINC-based systems, p 71–85 In Talbi EG, Zomaya A. (ed), Grids for bioinformatics and computational biology. Wiley book series on parallel and distributed computing. John Wiley & Sons, New York, NY [Google Scholar]
- 48.Myers DS, Cummings MP. 2003. Necessity is the mother of invention: a simple grid computing system using commodity tools. J. Parallel Distrib. Comput. 63:578–589. 10.1016/S0743-7315(03)00004-2 [DOI] [Google Scholar]
- 49.Cummings MP, Handley SA, Myers DS, Reed DL, Rokas A, Winka K. 2003. Comparing bootstrap and posterior probability values in the four-taxon case. Syst. Biol. 52:477–487. 10.1080/10635150390218213 [DOI] [PubMed] [Google Scholar]
- 50.Farris JS, Kallersjo M, Kluge AG, Bult C. 1994. Testing significance of incongruence. Cladistics 10:315–319. 10.1111/j.1096-0031.1994.tb00181.x [DOI] [Google Scholar]
- 51.Nixon KC. 1999. Winclada (beta), version 0.9.9. K. C. Nixon, Ithaca, NY [Google Scholar]
- 52.Brown EW, Kotewicz ML, Cebula TA. 2002. Detection of recombination among Salmonella enterica strains using the incongruence length difference test. Mol. Phylogenet. Evol. 24:102–120. 10.1016/S1055-7903(02)00222-1 [DOI] [PubMed] [Google Scholar]
- 53.Fitzgerald C, Collins M, van Duyne S, Mikoleit M, Brown T, Fields P. 2007. Multiplex, bead-based suspension array for molecular determination of common Salmonella serogroups. J. Clin. Microbiol. 45:3323–3334. 10.1128/JCM.00025-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McQuiston JR, Waters RJ, Dinsmore BA, Mikoleit ML, Fields PI. 2011. Molecular determination of H antigens of Salmonella by use of a microsphere-based liquid array. J. Clin. Microbiol. 49:565–573. 10.1128/JCM.01323-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lukinmaa S, Schildt R, Rinttila T, Siitonen A. 1999. Salmonella enteritidis phage types 1 and 4: pheno- and genotypic epidemiology of recent outbreaks in Finland. J. Clin. Microbiol. 37:2176–2182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Altekruse SF, Bauer N, Chanlongbutra A, DeSagun R, Naugle A, Schlosser W, Umholtz R, White P. 2006. Salmonella Enteritidis in broiler chickens, United States, 2000-2005. Emerg. Infect. Dis. 12:1848–1852. 10.3201/eid1212.060653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kang ZW, Jung JH, Kim SH, Lee BK, Lee DY, Kim YJ, Lee JY, Won HK, Kim EH, Hahn TW. 2009. Genotypic and phenotypic diversity of Salmonella Enteritidis isolated from chickens and humans in Korea. J. Vet. Med. Sci. 71:1433–1438. 10.1292/jvms.001433 [DOI] [PubMed] [Google Scholar]
- 58.Wall PG, Ward LR. 1999. Epidemiology of Salmonella enterica serovar Enteritidis phage type 4 in England and Wales, p 19–25 In Saeed AM, Gaste RE, Potter ME, Wall PG. (ed), Salmonella enterica serovar Enteritidis in humans and animals: epidemiology, pathogenesis, and control. Iowa State University Press, Ames, IA [Google Scholar]
- 59.Grimont PAD, Bouvet P, Grimont F, Desenclos J-C. 1999. Salmonella enterica serovar Enteritidis in France, p 43–49 In Saeed AM, Gaste RE, Potter ME, Wall PG. (ed), Salmonella enterica serovar Enteritidis in humans and animals: epidemiology, pathogenesis, and control. Iowa State University Press, Ames, IA [Google Scholar]
- 60.Tschape H, Liesegang A, Gericke B, Prager R, Rabsch W, Helmuth R. 1999. Ups and downs of Salmonella enterica serovar Enteritidis in Germany, p 51–61 In Saeed AM, Gaste RE, Potter ME, Wall PG. (ed), Salmonella enterica serovar Enteritidis in humans and animals: epidemiology, pathogenesis, and control. Iowa State University Press, Ames, IA [Google Scholar]
- 61.Gerner-Smidt P, Wegener HC. 1999. Salmonella enterica serovar Enteritidis in Denmark, p 63–69 In Saeed AM, Gaste RE, Potter ME, Wall PG. (ed), Salmonella enterica serovar Enteritidis in humans and animals: epidemiology, pathogenesis, and control. Iowa State University Press, Ames, IA [Google Scholar]
- 62.Angulo FJ, Swerdlow DL. 1999. Epidemiology of human Salmonella enterica serovar Enteritidis infections in the United States, p 33–41 In Saeed AM, Gaste RE, Potter ME, Wall PG. (ed), Salmonella enterica serovar Enteritidis in humans and animals: epidemiology, pathogenesis, and control. Iowa State University Press, Ames, IA [Google Scholar]
- 63.Kimura AC, Reddy V, Marcus R, Cieslak PR, Mohle-Boetani JC, Kassenborg HD, Segler SD, Hardnett FP, Barrett T, Swerdlow DL, Emerging Infections Program FoodNet Working Group 2004. Chicken consumption is a newly identified risk factor for sporadic Salmonella enterica serotype Enteritidis infections in the United States: a case-control study in FoodNet sites. Clin. Infect. Dis. 38:S244–S252. 10.1086/381576 [DOI] [PubMed] [Google Scholar]
- 64.Usera MA, Popovic T, Bopp CA, Strockbine NA. 1994. Molecular subtyping of Salmonella enteritidis phage type 8 strains from the United States. J. Clin. Microbiol. 32:194–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Botteldoorn N, Van Coillie E, Goris J, Werbrouck H, Piessens V, Godard C, Scheldeman P, Herman L, Heyndrickx M. 2010. Limited genetic diversity and gene expression differences between egg- and non-egg-related Salmonella Enteritidis strains. Zoonoses Public Health 57:345–357. 10.1111/j.1863-2378.2008.01216.x [DOI] [PubMed] [Google Scholar]
- 66.Rosenberg NA. 2004. DISTRUCT: a program for the graphical display of population structure. Mol. Ecol. Notes 4:137–138. 10.1046/j.1471-8286.2003.00566.x [DOI] [Google Scholar]
- 67.Nei M. 1987. Molecular evolutionary genetics. Columbia University Press, New York, NY [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.