

Abstract
Although human influenza B virus (IBV) is a significant human pathogen, its great genetic diversity has limited our ability to universally amplify the entire genome for subsequent sequencing or vaccine production. The generation of sequence data via next-generation approaches and the rapid cloning of viral genes are critical for basic research, diagnostics, antiviral drugs, and vaccines to combat IBV. To overcome the difficulty of amplifying the diverse and ever-changing IBV genome, we developed and optimized techniques that amplify the complete segmented negative-sense RNA genome from any IBV strain in a single tube/well (IBV genomic amplification [IBV-GA]). Amplicons for >1,000 diverse IBV genomes from different sample types (e.g., clinical specimens) were generated and sequenced using this robust technology. These approaches are sensitive, robust, and sequence independent (i.e., universally amplify past, present, and future IBVs), which facilitates next-generation sequencing and advanced genomic diagnostics. Importantly, special terminal sequences engineered into the optimized IBV-GA2 products also enable ligation-free cloning to rapidly generate reverse-genetics plasmids, which can be used for the rescue of recombinant viruses and/or the creation of vaccine seed stock.
INTRODUCTION
Influenza viruses belong to the Orthomyxoviridae family and contain a negative-sense, single-stranded, and segmented RNA genome. They are divided into three types (influenza A, B, and C viruses) originally based on the antigenic specificity of the nucleoprotein (NP) and matrix protein (M) (1, 2). The nature of their segmented genome provides the basis for frequent genetic reassortment between different viruses of the same type, which plays an important role in the evolution of new strains.
Influenza B virus (IBV) is a significant human pathogen associated with excess hospitalizations and mortality (3). IBV and influenza A virus are responsible for epidemics that annually infect 250 to 500 million people, which results in 250,000 to 500,000 deaths and significant socioeconomic losses (4–7). On average, IBVs account for >20% of annual influenza infections, and this has risen to ∼60% in some seasons (8). During the 2012-2013 influenza season in the United States, IBVs caused 60% of influenza-associated pediatric deaths, despite accounting for only 29% of laboratory-confirmed infections (9).
Two antigenically and genetically distinct lineages of IBVs (B/Victoria/2/87-like and B/Yamagata/16/88-like) cocirculated since the 1980s (10, 11), and their genomes are continually changing to evade the human immune response (a phenomenon known as antigenic drift). The current seasonal influenza virus vaccine components typically contain the hemagglutinin (HA) and neuraminidase (NA) antigens of an H1N1 virus, an H3N2 virus, and a B/Victoria or B/Yamagata lineage virus. A key factor that contributes to IBV disease severity in developed countries is the emergence of new IBV strains/lineages, which leads to a substantial mismatch between the emergent strain(s) and the strain chosen as a vaccine seed stock (e.g., mismatched in five out of 10 flu seasons from 2001 to 2011), thus reducing vaccine effectiveness (8). This predicament has led to the recent development of quadrivalent vaccines by some manufacturers in which two IBV strains representing both Yamagata and Victoria lineages are included. However, even within a particular lineage, antigenic drift of the HA/NA genes continues to generate novel variants that escape neutralization by the human immune response, and it is difficult to select/predict the best strain(s) to include in the influenza virus vaccine(s) in order to provide antigenic cross-reactivity against all concurrently circulating Yamagata and/or Victoria strains.
It is critical to conduct genomic studies to improve vaccines, diagnostics, and therapeutics, and to better understand the replication and pathogenesis of IBVs. Complete genome sequencing and analyses of thousands of IBVs are needed to understand the evolution of IBVs and to better predict the lineage and representative strains of IBVs that will predominate in the upcoming influenza seasons. This information is used by various computational approaches that aid in vaccine selection. For example, genomic sequence analysis identifies sequence changes responsible for immune escape by antigenic drift variants, which show increased fitness in the human population and determine the geospatial/temporal gene flow of evolutionarily successful gene segments. Rapid sequencing of a large number of circulating viruses before the biannual WHO Consultation on the Composition of Influenza Virus Vaccines meetings will provide public health officials and scientists with better data sets needed to make the best strain selections for biannual vaccines. Large-scale genomic sequencing of IBVs is central to the development of future genomic diagnostics that simultaneously identify genotypic markers of antigenic escape, antiviral resistance, and/or virulence, and it aids basic research in many critical areas under study. Unfortunately, compared to the >10,000 complete genomes (complete coding regions for all eight segments) of the influenza A viruses (IAVs) available in GenBank, only a few hundred IBV genomes had been deposited as of February 2013. The scarcity of IBV genome sequences has hampered research and novel vaccine development for IBVs, and it prompted us to establish a large-scale project to sequence IBVs as part of the NIH/National Institute of Allergy and Infectious Diseases (NIAID)-sponsored influenza virus genome sequencing project (http://gsc.jcvi.org/projects/msc/influenza/). The major difficulty in high-throughput sequencing for genetically diverse IBVs that continually evolve (i.e., for which oligonucleotide primers need to be continually modified) is the absence of a universal, efficient, robust, and economical method to convert the eight genomic RNA segments to double-stranded DNA (dsDNA) in a single reaction vessel. To address this problem, we developed new IBV genomic amplification (IBV-GA) strategies (IBV-GA/GA1/GA2), which enable sequencing via all next-generation sequencing (NGS) platforms. The IBV-GA approaches developed are also ideally suited for advanced genome-based diagnostics (e.g., pyrosequencing to identify Tamiflu resistance) that are being developed for use in personalized medicine and for the nimble generation of recombinant viruses for vaccines or research.
This study demonstrates universal approaches for amplifying complete segmented genomes of past, present, and future IBVs in a single reaction vessel, irrespective of the virus strain. The optimized technique (IBV-GA2) is robust enough for use on clinical swab material, and it is scalable for use in high-throughput processing. Finally, the IBV-GA/GA2 primers were also designed to facilitate cloning of the IBV genomic segments into our customized reverse-genetics plasmids for virus rescue, which will accelerate vaccine seed stock creation and basic research.
MATERIALS AND METHODS
Cells.
Human embryonic kidney 293T (HEK-293T) cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Madin-Darby canine kidney (MDCK) cells were maintained in minimum essential medium (MEM) supplemented with 5% FBS.
Viruses and clinical specimens.
IBV samples were received from different laboratories in the United States and other countries for sequencing at the J. Craig Venter Institute (JCVI) as part of the NIH/NIAID-sponsored Influenza Genome Sequencing Project (http://www.niaid.nih.gov/labsandresources/resources/dmid/gsc/influenza/). The samples were confirmed for the presence of IBVs with real-time reverse transcription-PCR (RT-PCR) or other methods in collaborating labs before being submitted to the JCVI. The samples were received in various formats, including (i) nasopharyngeal or oropharyngeal swab specimens collected from patients, (ii) viruses propagated in MDCK or other cells, (iii) viruses propagated in embryonated chicken eggs, and (iv) RNA isolated from virus-containing samples.
Analysis of IBV termini and primer design.
All of the IBV sequences available in the major influenza virus databases (Influenza Research Database [12], Influenza Virus Resource [13], and the EpiFlu database [14]) were downloaded and aligned using the MUSCLE program (15). Eight sequence alignments were generated for each of the genomic RNA segments. Sequence logos based on the nucleotide frequencies at each position were graphed for the 20 nucleotide positions at the 5′ terminus and the 20 nucleotide positions at the 3′ terminus of each alignment using the WebLogo application (Fig. 1) (16).
FIG 1.
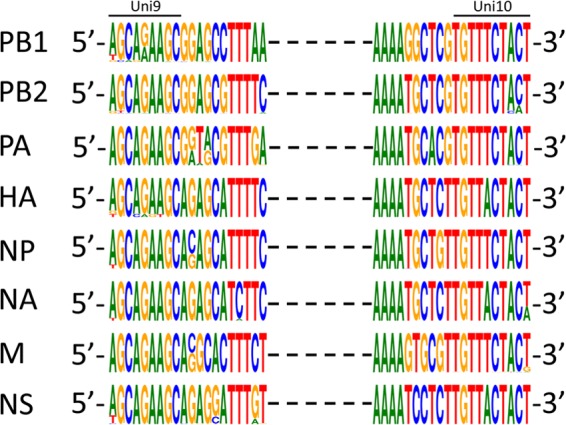
Conserved terminal sequences in the genome of IBVs. All the publicly available sequences (in cDNA sense) of IBVs were aligned for each gene segment. Nucleotides at the 20 terminal positions at each end of each segment were illustrated based on their relative frequencies at a specific position using the WebLogo application (16).
Primers were designed based on the consensus sequences of termini, with consideration of natural variations. Many different versions of primers were tested, including various 5′ tails, various lengths of complementarity, and various ratios of each primer pair. The current optimized versions of the IBV-GA primers and their concentrations are shown in Table 1.
TABLE 1.
Universal IBV-GA2 primer cocktail
| Primer | Sequencesa | Amt (μl) from each 10μM oligo |
|---|---|---|
| B-PBs-UniF | 5′-GGGGGGAGCAGAAGCGGAGC-3′ | 100 |
| B-PBs-UniR | 5′-CCGGGTTATTAGTAGAAACACGAGC-3′ | 100 |
| B-PA-UniF | 5′-GGGGGGAGCAGAAGCGGTGC-3′ | 50 |
| B-PA-UniR | 5′-CCGGGTTATTAGTAGAAACACGTGC-3′ | 50 |
| B-HANA-UniF | 5′-GGGGGGAGCAGAAGCAGAGC-3′ | 100 |
| B-HANA-UniR | 5′-CCGGGTTATTAGTAGTAACAAGAGC-3′ | 100 |
| B-NP-UniF | 5′-GGGGGGAGCAGAAGCACAGC-3′ | 60 |
| B-NP-UniR | 5′-CCGGGTTATTAGTAGAAACAACAGC-3′ | 60 |
| B-M-Uni3F | 5′-GGGGGGAGCAGAAGCACGCACTT-3′ | 30 |
| B-Mg-Uni3F | 5′-GGGGGGAGCAGAAGCAGGCACTT-3′ | 30 |
| B-M-Uni3R | 5′-CCGGGTTATTAGTAGAAACAACGCACTT-3′ | 60 |
| B-NS-Uni3F | 5′-GGGGGGAGCAGAAGCAGAGGATT-3′ | 50 |
| B-NS-Uni3R | 5′-CCGGGTTATTAGTAGTAACAAGAGGATT-3′ | 50 |
| FluB universal primer cocktail | Working stock (mix the primers above)b | 840 |
Underlined nucleotides correspond to IBV genome.
Repeated freezing and thawing of the working stock decreases RT-PCR efficiency.
IBV genomic amplification and optimization.
The majority of the samples required RNA extraction at JCVI. RNA was isolated from 100 to 200 μl of culture supernatant, allantoic fluid, or clinical swab specimens using the RNeasy minikit (Qiagen, Valencia, CA) or ZR-96 viral RNA kit (Zymo Research, Irvine, CA). The RNA was eluted in 30 μl of RNase-free water and served as a template for a one-step RT-PCR amplification of the entire IBV genome in a single reaction.
In the optimization process, multiple primer sets were tested for their ability to amplify the entire genome based on the conserved termini of IBV viral RNA (vRNA), which form the promoter for the viral RNA polymerase. The initial IBV-GA primer set comprised three tailed primers that were complementary or identical to the universally conserved 9 and 10 nucleotides (Uni9 and Uni10) at the termini of all IBV genomic RNA segments. The primers (Uni9/FluB1 [5′-TTGGGGGGGAGCAGAAGC-3′], Uni10/FluB1T [5′-CGCCGGGTTATTAGTAGTAACA-3′], and Uni10/FluB1A [5′-CGCCGGGTTATTAGTAGAAACA-3′]) were mixed at a molar ratio of 2:1:1. The modified IBV-GA1 primer set utilizes four tailed primers (Uni9/FluB1G [5′-GTCAGACTCAGTCAGCAGAAGCG-3′], Uni9/FluB1A [5′-GTCAGACTCAGTCAGCAGAAGCA-3′], Uni10/FluB1T, and Uni10/FluB1A) that were mixed at a molar ratio of 4:6:5:5. The 5′ tails were all designed to increase the annealing temperature in the PCR, and the 5′ tails used for the IBV-GA and IBV-GA2 (Table 1) reactions are identical to the RNA polymerase I promoter or terminator sequences in the reverse-genetics plasmid to enable recombination-based cloning.
The optimized final IBV-GA2 primer set and protocol were as follows: 3 μl of RNA and 2 μl of the IBV-GA2 universal primer cocktail (Table 1) were used in a 25-μl reaction volume of the SuperScript III one-step RT-PCR system with Platinum Taq high fidelity DNA polymerase (Life Technologies, Grand Island, NY) (Table 2). The temperature cycle parameters were 45°C for 60 min, 55°C for 30 min, 94°C for 2 min, and then 5 cycles of 94°C for 20 s, 40°C for 30 s, and 68°C for 3 min 30 s, followed by 40 cycles of 94°C for 20 s, 58°C for 30 s, and 68°C for 3 min 30 s, with a final extension at 68°C for 10 min.
TABLE 2.
Universal IBV-GA2 RT-PCR setup
| Reagentsa | Amt (μl) |
|
|---|---|---|
| Per reaction | Per 100 reactions | |
| DEPC-treated ddH2O | 7.0 | 700 |
| 2× RT-PCR buffer | 12.5 | 1,250 |
| IBV-GA primer cocktail | 2.0 | 200 |
| RT/HiFi enzyme mix | 0.5 | 50 |
| Viral RNA | 3.0 | Add individually |
DEPC, diethyl pyrocarbonate; ddH2O, double-distilled water. Each aliquot contained the four listed reagents at a total of 22 μl per reaction.
Reverse-genetics plasmids for recombination-based cloning.
The pDZ bidirectional reverse-genetics plasmid (17) was linearized by the endonuclease SapI, and oligonucleotides pDZ-A6-Adapt-F (5′-GGGAGCAGAAGCTGCAGTTTCTACT-3′) and pDZ-A6-Adapt-R (5′-ATTAGTAGAAACTGCAGCTTCTGCT-3′) were annealed and ligated into the linearized pDZ plasmid to generate the plasmid pBZ66A12, resulting in a 22-bp stuffer between the RNA polymerase I promoter and terminator (see Fig. 4A). To accommodate the A/U sequence variation at the sixth position in the 5′ terminus of the vRNAs, the oligonucleotides pDZ-T6-Adapt-F (5′-GGGAGCAGAAGCTGCAGTTACTACT-3′) and pDZ-T6-Adapt-R (5′-ATTAGTAGTAACTGCAGCTTCTGCT-3′) were annealed and ligated into the pDZ to generate pBZ66A13. These modifications create a cloning strategy that can be accomplished using commercially available enzymes (e.g., In-Fusion) and short regions (15 to 19 nucleotides) of identity between the new reverse-genetics plasmids and IBV genomic amplicons (see Fig. 4A).
FIG 4.

Recombination-based cloning of IBV genomic amplicons into reverse-genetics plasmids and the plaque phenotypes of rescued viruses. (A) Schematic diagram of the procedure used to clone genomic amplicons into the IBV recombination-based reverse-genetics plasmid pBZ66A12. To generate the reverse-genetics plasmid, a 22-bp fragment containing 9 bp that correspond to the 5′-termini of all vRNAs, followed by 4 bp (shown in italics) to create a PstI site, followed by 9 bp that correspond to the 3′-termini of all IBV vRNAs, was inserted between an RNA polymerase I promoter (pPolI) and terminator (Pol I-T). The genomic amplicons (1 segment shown) contain dsDNA copies of vRNA segments flanked by 10 (5′) or six (3′) bp that are derived from the 5′ tails incorporated into the primers (see examples in Table 1). The conserved Uni10 and Uni9 regions are shown in bold type. The genomic amplicons and PstI-linearized plasmid are mixed and treated with In-Fusion enzyme(s), and exonuclease activity exposes the complementary nucleotides at the termini leading to annealing and recombination (underlined nucleotides are part of the primer sequences used in the genomic amplification). (B) Plaque phenotypes of wtB/Russia/69 (left) and rB/Russia/69 (right). (C) Plaque phenotypes of wtB/Brisbane/60-10/2010 (left) and rB/Brisbane/60-10/2010 (right).
Nucleotide sequencing, assembly, and deposition.
The products of the various IBV-GA amplicons were successfully sequenced using the Ion Torrent PGM (Life technologies), HiSeq 2000 and MiSeq (Illumina), and 454 (Roche) platforms. The majority of the genomes were sequenced using the Ion Torrent PGM and/or MiSeq platforms. For the Ion Torrent PGM, IBV-GA amplicons were sheared for 15 min, and Ion Torrent-compatible bar-coded adapters were ligated to the sheared DNA using the Ion Xpress Plus fragment library kit (Life Technologies). The bar-coded libraries were pooled and purified with AMPure XP reagent (Agencourt). Quantitative PCR was performed on the pooled bar-coded libraries to assess the quality and to determine the template dilution factor for emulsion PCR. The pool was diluted appropriately and amplified on Ion Sphere particles (ISPs) using the Ion OneTouch instrument (Life Technologies). The pool of viral libraries was enriched for template-positive ISPs on the Ion OneTouch ES instrument (Life Technologies). Sequencing was performed on the Ion Torrent PGM using Ion 316/318 chips.
For MiSeq, the IBV-GA products served as a template for Nextera (Illumina) library construction. Briefly, 25 ng of DNA amplicons was tagmented at 55°C for 5 min. The tagmented DNA was cleaned with the ZR-96 DNA Clean & Concentrator kit (Zymo Research) and eluted in 25 μl resuspension buffer. Illumina sequencing adapters and barcodes were added to the tagmented DNA via PCR amplification. Two-and-a-half microliters of each index primer from the Nextera Index kit (Illumina) was used in each PCR (total volume, 35 μl). Thermal cycling was performed per the Nextera protocol to create a dual indexed library for each sample. After PCR amplification, 10 μl of each library was pooled into a 1.5-ml tube, and the pool was cleaned two times with AMPure XP reagent (Agencourt), 1.2 times the final volume, to remove all leftover primers and small DNA fragments. The purified pool was sequenced on the Illumina MiSeq version 2 instrument, with 250-bp paired-end reads.
The sequence reads from the NGS platforms were sorted by barcode, trimmed, and de novo assembled using the CLC bio clc_novo_assemble program, and the resulting contigs were searched against custom full-length IBV nucleotide databases to find the closest reference sequence for each segment. All sequence reads were then mapped to the selected reference segments using the CLC bio clc_ref_assemble_long program. At loci where both Ion Torrent and Illumina sequence data agreed on a variation (compared against the reference sequence), the reference sequence was updated to reflect the difference. A final mapping of all NGS reads to the updated reference sequences was performed with the CLC bio clc_ref_assemble_long program.
The genomic sequences of >1,000 IBVs have been deposited in the GenBank database (see Table S1 in the supplemental material), following the NIAID/Division of Microbiology and Infectious Diseases (DMID) Data Sharing and Release Guidelines (http://www.niaid.nih.gov/LabsAndResources/resources/dmid/gsc/pages/data.aspx). Phylogenetic analysis is being conducted to gain insight into the evolution of IBV (D. Vijaykrishna, E. Holmes, U. Joseph, M. Fourment, Y. Su, R. Halpin, R. Lee, Y. Deng, V. Gunalan, X. Lin, T. Stockwell, N. Fedorova, B. Zhou, N. Spirason, D. Kuhnert, V. Boskova, T. Stadler, A. Costa, E. Dwyer, Q. Huang, L. Jennings, W. Rawlinson, S. Sullivan, A. Hurt, S. Maurer-Stroh, D. Wentworth, G. Smith, and I. Barr, unpublished data).
Cloning of genomic amplicons into reverse-genetics plasmids.
The recombination-based In-Fusion cloning strategy (18, 19) was used for rapid cloning of the IBV-GA products. The IBV-GA2 amplicons were purified with QIAquick PCR purification kit (Qiagen) and analyzed by agarose gel electrophoresis. Purified amplicons (200 to 400 ng) were mixed with 100 ng of PstI-linearized pBZ66A12 or pBZ66A13 plasmids and incubated in 10 μl In-Fusion mix (In-Fusion HD cloning system; Clontech, Mountain View, CA) at 50°C for 15 min. The mixtures were incubated on ice for 5 min, and 3 μl was used to transform Stellar competent cells (Clontech). The colonies were screened by PCR, and clones representing each vRNA segment were purified and sequenced.
Rescue of IBVs.
Recombinant IBVs were generated by cotransfection of eight reverse-genetics plasmids carrying the cDNA of each gene segment into a 293T/MDCK coculture monolayer, as previously described (20–22). Briefly, 0.6 μg of each plasmid was mixed and incubated with 15 μl of TransIT-LT1 transfection reagent (Mirus Bio, Madison, WI) at 20°C for 20 min and then added to 80% confluent 293T/MDCK cell cocultures in 6-well plates. The transfection plates were incubated at 33°C with 5% CO2 for 12 to ∼24 h, at which time the culture supernatant was replaced with 3 ml of Opti-MEM I medium (Life Technologies) supplemented with 0.3% bovine serum albumin (BSA) fraction V (Life Technologies), 3 μg/ml N-tosyl-l-phenylalanine chloromethyl ketone (TPCK)-trypsin (Worthington, Lakewood, NJ), and 1% antibiotic-antimycotic (Life Technologies). Three days posttransfection, the supernatant was collected and virus was propagated in MDCK cells at 33°C.
RESULTS
Terminal sequences of IBVs are conserved for each segment.
The 5′ and 3′ termini of IBV RNA segments are short conserved complementary regions that form the promoter for the IBV RNA-dependent RNA polymerase. The sequences for each segment were aligned separately using MUSCLE, and conservation of the terminal nucleotides was visualized by creating sequence logos based on the relative frequency of each nucleotide at a specific position (15, 16) (Fig. 1). All the sequences are shown as cDNA (database default format), which is complementary to the negative-sense vRNA. The 9 nucleotides at the 5′ end (5′-AGCAGAAGC-3′) and the 10 nucleotides at the 3′ end (5′-TGTTTCTACT-3′ or 5′-TGTTACTACT-3′) of the cDNA sense genome are conserved, with the exception of position 6 (A/T) from the 3′ terminus. Additional noncoding regions that juxtapose the universal termini are conserved in a segment-specific pattern (Fig. 1). There are some variations in the conserved termini of some segments in the database (Fig. 1). However, many of these appear to be sequencing artifacts, including the primers used for the RT-PCR steps prior to sequencing in the data deposited in the sequence databases.
Universal primer sets amplify IBV genome with high flexibility (IBV-GA/GA1).
We designed tailed primers that were complementary or identical to the universally conserved 9 and 10 nucleotides (Uni9 and Uni10) at the termini of all IBV genomic RNA segments. Primers with various tails at the 5′ ends (to increase the primer annealing temperature in PCR) were tested, and the set with the best sensitivity and specificity was the IBV-GA set comprising the three primers Uni9/FluB1, Uni10/FluB1T, and Uni10/FluB1A. The IBV-GA primer set amplified the full genome of various IBVs, albeit with moderate sensitivity (limit of detection, 103 to 104 50% tissue culture infective dose [TCID50]/RT-PCR) (Fig. 2A). Thus, although this primer set can efficiently amplify the genomes of isolates from cell cultures or embryonated chicken eggs, it was not robust enough to routinely amplify the viral genomes from clinical swab specimens that often contain a lower virus load and many nonviral nucleic acids (data not shown).
FIG 2.

Efficiency of different primer sets in the amplification of IBV genome. The results of the three most efficient primer sets representing the three main stages of the development of the IBV genomic amplification technology are shown after agarose gel electrophoresis. RNA was extracted from 100 μl of 108 TCID50/ml of the B/Brisbane/60-10/2010 virus, eluted in 30 μl of nuclease-free water, and diluted in 1:10 series; 3 μl was used as a template for each RT-PCR. The equivalent amount of virus (TCID50) used in each RT-PCR is shown at the top of each lane. L, 1-kb Plus ladder (Life Technologies); N, negative control (no template). (A) IBV-GA. Three primers (Uni9/FluB1, Uni10/FluB1T, and Uni10/FluB1A) amplified the genome in the range of 106 to 101 TCID50/reaction. (B) IBV-GA2. Four primers (Uni9/FluB1G, Uni9/FluB1A, Uni10/FluB1T, and Uni10/FluB1A) amplified the genome in the range of 106 to 100 TCID50/reaction. (C) Segment-specific. Selected primer sets amplified different groups of genomic segments efficiently using 10 TCID50/reaction. Lanes, left to right: PB1/2-, PA-, HA/NA-, NP-, M-, and NS-specific primers (Table 1) were used. (D) IBV-GA2. Universal primer cocktail comprising the segment-specific primers used to amplify the genome in the range of 106 to 100 TCID50/reaction.
The IBV-GA1 strategy was developed to increase the sensitivity and specificity, and to provide superior control of the ratio of each segment in the multisegment RT-PCR. For this system, an extra nucleotide (G or A) was extended from the 3′ end of the Uni9/FluB1 primer, resulting in two primers (Uni9/FluB1G and Uni9/FluB1A) complementary to the 10 nucleotides at the vRNA 3′ terminus (5′ of cDNA). Based on the sequence alignment (Fig. 1), the Uni9/FluB1G primer is complementary to the polymerase vRNAs (polymerase basic protein 1 [PB1], polymerase basic protein 2 [PB2], and polymerase acidic protein [PA]), and the Uni9/FluB1A primer is complementary to the other five vRNAs (hemagglutinin [HA], nucleoprotein [NP], neuraminidase [NA], matrix protein [M], and nonstructural protein [NS]). By adjusting the ratios of the two Uni9/FluB1G and Uni9/FluB1A primers and the two Uni10/FluB1T and Uni10/FluB1A primers, the relative abundances of the various segments can be controlled. Moreover, the sensitivity for the genomic amplification increased by 10-fold for most of the viruses tested (limit of detection, 102 to 103 TCID50/RT-PCR) (Fig. 2B and data not shown). Although the IBV-GA1 set reduced the amplification of NS and M, the amplicons showed a better representation of the larger gene segments. This IBV-GA1 technology functioned to an acceptable level for many samples; however, it had high failure rate for samples containing <103 infectious units (data not shown).
Optimized universal IBV genomic amplification (IBV-GA2) technology efficiently amplifies genomes of any strain of IBVs from various sources.
To further increase the sensitivity and maximize the flexibility of the genomic amplification technique, the universal primer set was redesigned to add conserved segment-specific nucleotides to their 3′ ends. This generated a primer cocktail (Table 1), which is a combination of 6 segment-specific primer pairs that amplify 8 IBV gene segments in a single reaction, referred to as IBV-GA2. The 6 primer pairs were designed to amplify (i) PB1 or PB2, (ii) PA, (iii) HA or NA, (iv) NP, (v) M, and (vi) NS (Table 1). Using the universal optimized RT-PCR cycling parameters, each segment-specific primer pair robustly amplified the corresponding segment(s) with very high sensitivity (10 TCID50/RT-PCR) (Fig. 2C). The IBV-GA2 technique amplified the complete genome of IBVs in a wide range of virus concentrations (10 to 106 TCID50/RT-PCR) (Fig. 2D). To test the universality of the IBV-GA2 strategy, we applied it to many historic strains and contemporary field isolates and demonstrated that the technology was robust enough to amplify the genome of extremely diverse IBVs (Fig. 3A). The strains analyzed a span of >70 years from the first isolate in 1940 to a recent isolate in 2011, and they constituted various lineages and clades from various continents that were propagated using different substrates (cells or eggs) (Fig. 3A).
FIG 3.
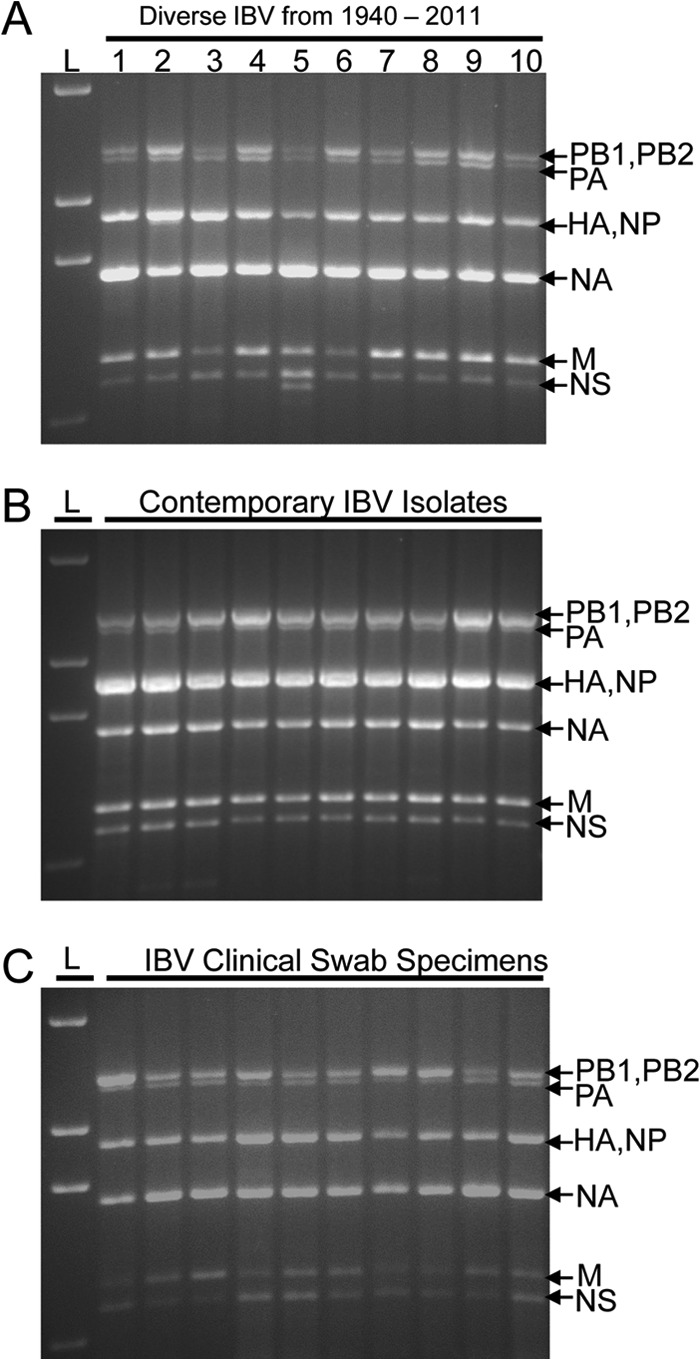
Universal IBV genomic amplification technology (IBV-GA2) amplifies genome of any IBV strain. L, 1-kb Plus ladder (Life Technologies). (A) Illustrates IBV genomes amplified from viruses isolated on different continents from 1940 to 2011. Lane 1, B/Lee/1940; lane 2, B/Russia/1969; lane 3, B/Hong Kong/5/1972; lane 4, B/Panama/45/1990; lane 5, B/Malaysia/11185/1996; lane 6, B/Victoria/143/2002; lane 7, B/England/145/2008; lane 8, B/Wisconsin/01/2010; lane 9, B/Hong Kong/259/2010; lane 10, B/Victoria/804/2011. IBVs in lanes 4 to 8 belong to the Yamagata lineage and viruses in lanes 9 and 10 belong to the Victoria lineage. (B) Representative example of the amplification of genomes from contemporary viruses isolated/cultured in Australia in 2011. (C) Representative example of the IBV-GA2 amplification of viral genomes directly from human swab specimens collected in the United States during the years of 2001 to 2006.
In collaboration with multiple laboratories throughout the world, the IBV-GA2 technology was applied to amplify the genomes of >1,000 contemporary IBVs. Those samples are different not only in their lineages and originating locations but also in the methods used for specimen collection and virus isolation. Consequently, successful genomic amplification of those samples indicates that this strategy is robust enough to amplify any contemporary IBV. The IBV-GA1/GA2 products were successfully sequenced with various sequencing technologies, including Sanger sequencing and NGS, such as Illumina HiSeq 2000 and MiSeq platforms, Life Technologies Ion Torrent PGM, and Roche 454 pyrosequencing. Furthermore, the termini of the primers need only slight modifications to support direct sequencing via the Pacific Biosciences SMRT sequencing system. The universal and robust nature of the IBV-GA2 technique was also demonstrated by our recent deposition of >1,000 IBV genomes in GenBank (see Table S1 in the supplemental material).
Generation of recombinant IBVs from genomic amplicons using recombination-based cloning strategy.
The generation of recombinant IBVs is critical for basic research and is increasingly important in vaccine seed stock generation. Reverse-genetics systems for IBVs were developed by several groups (23–27) a few years after the development of reverse-genetics systems for IAVs. However, all of these systems rely on type IIS endonuclease (e.g., BsmBI and SapI) digestion of the reverse-genetics plasmid and the generation of specific RT-PCR amplicons containing corresponding restriction sites, which are ligated to the linearized plasmid after restriction digestion. This is a cumbersome approach for generating a genomic set of 8 reverse-genetic clones. Additionally, difficulties arise when the viral genes contain restriction sites (e.g., BsmBI) that the reverse-genetics plasmid employs. In contrast, the DNA generated by the IBV-GA strategies described can be efficiently cloned into our new recombination-based IBV reverse-genetics plasmids (pBZ66A12, pBZ66A13), regardless of the sequence of the virus. To create these plasmids, a bidirectional reverse-genetics plasmid, pDZ (17), was modified by inserting a 22-bp linker to introduce an In-Fusion cloning site between the existing RNA polymerase I promoter and terminator, creating pBZ66A12 (Fig. 4A). To account for the A/U sequence variation at the sixth position of the vRNA 5′ terminal promoter element (T/A sixth from 3′ terminus of the cDNA) of the HA, NA, and NS segments (Fig. 1), pBZ66A13 was also generated.
Amplicons of each of the eight vRNA segments are flanked by the tails in IBV-GA/IBV-GA2 primers (see Materials and Methods; Table 1) that are identical to the RNA polymerase I promoter or terminator used in the reverse-genetics plasmid; thus, they can be cloned separately or simultaneously into our PstI-linearized plasmids (e.g., pBZ66A12) in a single reaction using In-Fusion cloning and other ligase-independent methods. In addition, the segment-specific primers used in the IBV-GA2 universal primer cocktail (Table 1) allow specific genes (such as HA and NA) to be amplified (Fig. 2C) and cloned separately, eliminating the need to screen against all eight segments when only HA and NA are required (e.g., vaccine reassortants).
The genomic amplicons of a lab-adapted B/Russia/69-like virus and the recent isolate B/Brisbane/60-10/2010 virus were cloned into our recombination-based plasmids using the In-Fusion HD cloning system. Clones containing PB1, PB2, PA, NP, and M segments were selected from the pBZ66A12 plasmid, and clones containing HA, NA, and NS segments were selected from the pBZ66A13 plasmid. Sequence-verified plasmids were cotransfected into cocultured 293T/MDCK cells, and the IBVs were rescued successfully. The rescued recombinant viruses (rB/Russia/69 and rB/Brisbane/60-10/2010) were compared to the wild-type isolates (wtB/Russia/69 and wtB/Brisbane/60-10/2010). For both strains, the recombinant viruses replicated to similar titers (data not shown) and formed similar sized plaques as those of their wild-type counterparts (Fig. 4B and C).
DISCUSSION
Using IBV-GA1/GA2, we successfully amplified and sequenced the genomes of >1,000 IBVs collected over 70 years and from many geographic locations. Amplifying/enriching the viral genome(s) present in samples is needed in order to sequence hundreds of viral genomes simultaneously and efficiently using high-throughput NGS. Prior to the IBV-GA approaches described here, IBV samples were subjected to many RT-PCRs with sequence-specific primers to convert the IBV genomic RNA into overlapping DNA amplicons, and some of the reactions failed due to mismatches between the predesigned primers and the highly variable viral genome. Combining universal IBV-GA2 with NGS creates a sequencing pipeline that is truly IBV sequence independent (i.e., no genomic information is needed a priori). This system has allowed us to double the number of genomes available in GenBank in a short period of time (Fig. 5). We previously developed a similar system for influenza A virus, which we called multisegment RT-PCR (M-RTPCR) (19), and this dramatically reduced costs and increased our throughput (Fig. 5). The IBV-GA2 primer cocktail (composed of multiple segment-specific primers) has very high sensitivity (1 to 10 TCID50/reaction) and efficiently amplifies IBV from clinical swab specimens without the need for virus isolation or culture. This is important because influenza virus RNA polymerase is error prone, and the viruses in an individual exist as a population of genomes (i.e., quasispecies). Laboratory culture conditions used to isolate a virus from a swab specimen often rapidly select for mutants, which are not representative of the original swab material. Therefore, amplification directly from the swab material is critical for an analysis of single nucleotide polymorphisms via deep NGS, accurate antigenic prediction, and the determination of antiviral resistance.
FIG 5.

Complete influenza A and B virus genomes deposited in GenBank through the Influenza Genome Sequencing Project. Graph illustrates the number of full-length influenza A virus (IAV) (blue line) or influenza B virus (IBV) (red line) deposited in GenBank by the NIH/NIAID-sponsored Influenza Genome Sequencing Project since its initiation in 2005. Arrows indicate the initiation of universal genomic amplification procedures for influenza A virus (M-RTPCR) (19) or the influenza B virus (IBV-GA, IBV-GA2).
The primer pairs in the IBV-GA2 cocktail (Table 1) are flexible and can also be used separately to amplify genes of specific interest, such as the HA and NA segments (Fig. 2C). This type of approach facilitates NGS that focuses on antigenic variation and/or drug resistance. For example, the density of viruses analyzed per NGS run might be increased considerably (e.g., 1 lane of MiSeq is needed to sequence ∼93 IBV genomes or ∼420 HA/NA pairs with an average of 200× sequencing coverage). Furthermore, the IBV-GA2 primer pairs (Table 1) in various combinations are ideal for advanced sequence-based diagnostics that are moving into the clinical diagnostic setting. For example, IBV genomic amplicons or specific segments can be rapidly interrogated to simultaneously identify lineage, strain, specific antigenic epitopes, antiviral susceptibility/resistance signatures, and/or pathogenic determinants.
The IBV-GA and IBV-GA1 strategies, which can be used to amplify the IBV genomes from samples with higher titers, facilitate the sequencing of additional nucleotides in the untranslated regions (UTRs). The advantage of IBV-GA/GA1 strategies is that they generate sequence information corresponding to the nearly complete vRNA segment (i.e., very few nucleotides [9 or 10] are incorporated by the primers in a highly conserved region), which is important because the UTRs of influenza virus vRNAs play critical roles in mRNA transcription, vRNA replication, and virion morphogenesis. Thus, it is desirable to determine nonconserved UTR sequences without the need to perform additional techniques, such as rapid amplification of cDNA ends or RNA ligation RT-PCR (28, 29).
The amplicons generated from the IVB-GA reactions are efficiently cloned into our modified reverse-genetics plasmids using recombination-based strategies, without the use of restriction enzymes and ligases that are employed by current strategies. By combining the IBV-GA technique and recombination-based cloning technique, any IBVs can be cloned into reverse-genetics plasmids and rapidly rescued. We previously developed a similar strategy to rescue recombinant IAVs directly from a swab in 10 to 12 days (19), and it dramatically improved our ability to rescue recombinant viruses and engineer vaccines (19–22).
In conclusion, we developed and employed single-reaction universal IBV genomic amplification techniques for NGS and recombinant virus production. The strategy reduces costs, saves time, and conserves original samples while retaining high sensitivity and versatility. Seamless integration of this technology with NGS and recombination-based reverse-genetics plasmids proved its wide application and potential to have a significant impact on IBV research and vaccine production.
Supplementary Material
ACKNOWLEDGMENTS
We thank P. Blair, D. Bucher, and D. Dwyer for providing some of the virus samples used in this study. We also thank Peter Palese and Adolfo García-Sastre for providing the original pDZ reverse-genetics plasmid that we modified to generate our IBV reverse-genetics plasmids.
This study was supported in part with federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under contract HHSN272200900007C. The Melbourne WHO Collaborating Centre for Reference and Research on Influenza is supported by the Australian Government Department of Health.
B.Z., D.E.W., and X.L. have potential conflict of interests, as they have applied for a U.S. patent on some of the techniques described herein.
Footnotes
Published ahead of print 5 February 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JCM.03265-13.
REFERENCES
- 1.WHO. 1980. A revision of the system of nomenclature for influenza viruses: a WHO memorandum. Bull. World Health Organ. 58:585–591 [PMC free article] [PubMed] [Google Scholar]
- 2.Dowdle WR, Galphin JC, Coleman MT, Schild GC. 1974. A simple double immunodiffusion test for typing influenza viruses. Bull. World Health Organ. 51:213–215 [PMC free article] [PubMed] [Google Scholar]
- 3.Feng L, Shay DK, Jiang Y, Zhou H, Chen X, Zheng Y, Jiang L, Zhang Q, Lin H, Wang S, Ying Y, Xu Y, Wang N, Feng Z, Viboud C, Yang W, Yu H. 2012. Influenza-associated mortality in temperate and subtropical Chinese cities, 2003–2008. Bull. World Health Organ. 90:279–288B. 10.2471/BLT.11.096958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jackson D, Elderfield RA, Barclay WS. 2011. Molecular studies of influenza B virus in the reverse genetics era. J. Gen. Virol. 92:1–17. 10.1099/vir.0.026187-0 [DOI] [PubMed] [Google Scholar]
- 5.WHO. 2009. Influenza (seasonal) fact sheet no. 211. World Health Organization, Geneva, Switzerland: http://www.who.int/mediacentre/factsheets/fs211/en/ [Google Scholar]
- 6.Thompson WW, Shay DK, Weintraub E, Brammer L, Cox N, Anderson LJ, Fukuda K. 2003. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 289:179–186. 10.1001/jama.289.2.179 [DOI] [PubMed] [Google Scholar]
- 7.Molinari NA, Ortega-Sanchez IR, Messonnier ML, Thompson WW, Wortley PM, Weintraub E, Bridges CB. 2007. The annual impact of seasonal influenza in the US: measuring disease burden and costs. Vaccine 25:5086–5096. 10.1016/j.vaccine.2007.03.046 [DOI] [PubMed] [Google Scholar]
- 8.Ambrose CS, Levin MJ. 2012. The rationale for quadrivalent influenza vaccines. Hum. Vaccin. Immunother. 8:81–88. 10.4161/hv.8.1.17623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.CDC. 2013. Influenza activity–United States, 2012–13 season and composition of the 2013–14 influenza vaccine. MMWR Morb. Mortal. Wkly. Rep. 62:473–479 [PMC free article] [PubMed] [Google Scholar]
- 10.Rota PA, Wallis TR, Harmon MW, Rota JS, Kendal AP, Nerome K. 1990. Cocirculation of two distinct evolutionary lineages of influenza type B virus since 1983. Virology 175:59–68. 10.1016/0042-6822(90)90186-U [DOI] [PubMed] [Google Scholar]
- 11.McCullers JA, Saito T, Iverson AR. 2004. Multiple genotypes of influenza B virus circulated between 1979 and 2003. J. Virol. 78:12817–12828. 10.1128/JVI.78.23.12817-12828.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Squires RB, Noronha J, Hunt V, García-Sastre A, Macken C, Baumgarth N, Suarez D, Pickett BE, Zhang Y, Larsen CN, Ramsey A, Zhou L, Zaremba S, Kumar S, Deitrich J, Klem E, Scheuermann RH. 2012. Influenza research database: an integrated bioinformatics resource for influenza research and surveillance. Influenza Other Respir. Viruses 6:404–416. 10.1111/j.1750-2659.2011.00331.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bao YM, Bolotov P, Dernovoy D, Kiryutin B, Zaslavsky L, Tatusova T, Ostell J, Lipman D. 2008. The influenza virus resource at the National Center for Biotechnology Information. J. Virol. 82:596–601. 10.1128/JVI.02005-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bogner P, Capua I, Lipman DJ, Cox NJ, et al. 2006. A global initiative on sharing avian flu data. Nature 442:981. 10.1038/442981a [DOI] [Google Scholar]
- 15.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797. 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crooks GE, Hon G, Chandonia JM, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res. 14:1188–1190. 10.1101/gr.849004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quinlivan M, Zamarin D, García-Sastre A, Cullinane A, Chambers T, Palese P. 2005. Attenuation of equine influenza viruses through truncations of the NS1 protein. J. Virol. 79:8431–8439. 10.1128/JVI.79.13.8431-8439.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sleight SC, Bartley BA, Lieviant JA, Sauro HM. 2010. In-Fusion BioBrick assembly and re-engineering. Nucleic Acids Res. 38:2624–2636. 10.1093/nar/gkq179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou B, Donnelly ME, Scholes DT, St. George K, Hatta M, Kawaoka Y, Wentworth DE. 2009. Single-reaction genomic amplification accelerates sequencing and vaccine production for classical and Swine origin human influenza A viruses. J. Virol. 83:10309–10313. 10.1128/JVI.01109-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou B, Li Y, Halpin R, Hine E, Spiro DJ, Wentworth DE. 2011. PB2 residue 158 is a pathogenic determinant of pandemic H1N1 and H5 influenza a viruses in mice. J. Virol. 85:357–365. 10.1128/JVI.01694-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou B, Li Y, Speer SD, Subba A, Lin X, Wentworth DE. 2012. Engineering temperature sensitive live attenuated influenza vaccines from emerging viruses. Vaccine 30:3691–3702. 10.1016/j.vaccine.2012.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou B, Pearce MB, Li Y, Wang J, Mason RJ, Tumpey TM, Wentworth DE. 2013. Asparagine substitution at PB2 residue 701 enhances the replication, pathogenicity, and transmission of the 2009 pandemic H1N1 influenza A virus. PLoS One 8:e67616. 10.1371/journal.pone.0067616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoffmann E, Mahmood K, Yang CF, Webster RG, Greenberg HB, Kemble G. 2002. Rescue of influenza B virus from eight plasmids. Proc. Natl. Acad. Sci. U. S. A. 99:11411–11416. 10.1073/pnas.172393399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jackson D, Cadman A, Zurcher T, Barclay WS. 2002. A reverse genetics approach for recovery of recombinant influenza B viruses entirely from cDNA. J. Virol. 76:11744–11747. 10.1128/JVI.76.22.11744-11747.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dauber B, Heins G, Wolff T. 2004. The influenza B virus nonstructural NS1 protein is essential for efficient viral growth and antagonizes beta interferon induction. J. Virol. 78:1865–1872. 10.1128/JVI.78.4.1865-1872.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hatta M, Kawaoka Y. 2003. The NB protein of influenza B virus is not necessary for virus replication in vitro. J. Virol. 77:6050–6054. 10.1128/JVI.77.10.6050-6054.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Imai M, Watanabe S, Ninomiya A, Obuchi M, Odagiri T. 2004. Influenza B virus BM2 protein is a crucial component for incorporation of viral ribonucleoprotein complex into virions during virus assembly. J. Virol. 78:11007–11015. 10.1128/JVI.78.20.11007-11015.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szymkowiak C, Kwan WS, Su Q, Toner TJ, Shaw AR, Youil R. 2003. Rapid method for the characterization of 3′ and 5′ UTRs of influenza viruses. J. Virol. Methods 107:15–20. 10.1016/S0166-0934(02)00184-2 [DOI] [PubMed] [Google Scholar]
- 29.de Wit E, Bestebroer TM, Spronken MI, Rimmelzwaan GF, Osterhaus AD, Fouchier RA. 2007. Rapid sequencing of the non-coding regions of influenza A virus. J. Virol. Methods 139:85–89. 10.1016/j.jviromet.2006.09.015 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.