

Abstract
Previous methods of herpes simplex virus 1 (HSV-1) genotype analysis have lacked sufficient discriminatory power for strain analysis within genotypes. The hypervariable reiterative repeat regions in the US1 and US12 introns, known as ReIV, were targeted for strain comparison. PCR methods for these extremely GC-rich target regions were optimized to give reproducible amplicons that were visualized by capillary electrophoresis relative to size standards. Analysis of the size, shape, and pattern of the resulting signatures enabled strain discrimination. Primary clinical specimens were used to develop the assay and the analysis algorithm. A blinded clinical study of 147 in-state and 51 out-of-state samples, including matched specimen-isolate pairs, was then performed. All primary clinical samples had been collected between 2004 and 2011 for viral diagnosis and previously found to be positive for HSV-1 by real-time PCR. The combined database contained patterns from 264 samples collected from 199 patients with a total of 176 unique signatures, none of which were dominant in the population. Matches between the signatures of the more than 50 specimen-isolate pairs were always seen. Signatures also matched across multiple samples collected from individual patients (six such cases), as well as some additional signature matches where epidemiological links were likely. Results were reproducible on repeat testing of individual specimens, even after months in frozen storage. The protocol has multiple potential clinical and public health uses.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) is a significant and prevalent human pathogen capable of causing a range of disease manifestations, most commonly mucocutaneous ulcers, keratitis, and encephalitis (1). After initial infection, the virus establishes latency in the trigeminal ganglia, allowing for symptomatic and asymptomatic recurrences of the disease in the host patient. Viral shedding occurs during reactivation caused by sun exposure, respiratory illness, eczema, emotional and physical stress, gastric upset, fatigue, or injury.
HSV-1 has a linear double-stranded DNA genome of approximately 152,000 bp composed of unique long (UL) and unique short (US) regions bracketed by matching inverted repeat elements. Throughout the HSV-1 genome, microsatellite repeat regions and long variable number tandem repeat regions (VNTRs) <100 and <600 bp in length, respectively, are found. The size of the VNTRs varies between strains of the virus.
HSV-1 strain analysis and genotyping were historically performed by restriction fragment length polymorphism (RFLP) analysis using several different restriction endonucleases (2). RFPL assays were refined by using BamHI, KpnI, and SalI and were used to detect distinct genotypes from Japan, South Korea, China, Sweden, the United States, and Kenya. A distinct geographic distribution of patterns was seen in the different countries (3, 4). In order to differentiate strains with the same RFLP patterns, VNTRs in reiterative repeat region I (ReI), ReIII, ReIV, and ReVII were targeted for variation analysis by Southern hybridization (5, 6).
Hypervariable regions have been investigated as stand-alone targets for the differentiation of clinical strains. The stability of ReI, ReIII, ReIV, and ReVII was investigated by subcloning (7). ReIV and ReVII were found to be stable when subcloned and were suitable for strain comparison of clinical isolates, whereas ReI and ReIII were considered too unstable, exhibiting variable numbers of repeats between the different subclones. ReIV was found to have more discriminating ability than compared ReVII samples from unrelated individuals: ReIV produced a greater variety of results than ReVII while still remaining stable between sequential samples from the same individuals (7). A version of the molecular analysis methods described here, involving amplification of ReIV and accurate capillary electrophoretic sizing of products with specific software, was previously described for the analysis of sequential HSV-1 isolates in cases of recurrent herpetic keratitis (8).
ReIV is located in the introns of HSV-1 genes US1 and US12 (7). These targets are guanine (G) and cytosine (C) rich (65 to 86%) and in some strains contain homopolymeric runs of cytosine and guanine. This base composition is found in sequence repeats throughout the HSV-1 genome (9). These characteristics make the sequencing of ReIV by conventional Sanger dideoxynucleotide sequencing methods extremely challenging and technically impractical. Illumina deep sequencing has been used to sequence the HSV-1 genome, and the difficulty of sequencing through the reiterations is seen in the very low sequence read coverage at those sites (10). Another sequence-based method targeting microsatellite polymorphisms has been used to investigate different HSV-1 strains in viral isolates. While this technique has been used in other organisms, information on the relative stability and suitability of the microsatellite polymorphisms for strain tracking in HSV-1 is not established (11).
In this study, the molecular method was optimized for use on both primary specimens and cultured isolates. In contrast to earlier publications on repeat region analysis, both sample types were extracted with automated instruments and PCR amplification was performed with a new commercial enzyme kit. An algorithm was devised for detailed analysis of the products. Further, a large database containing more than 175 unique signatures was constructed for the comparison of HSV-1 samples with unknown or suspected epidemiological links. Additional potential applications of this assay include distinguishing new infections from reactivations by comparing the strains detected (8, 12, 13) and investigating variable compartmentalized treatment responses, a demonstrated clinical factor in antiviral therapy for herpesviruses (14, 15).
(Some of the data in this report were presented at the Clinical Virology Symposium, Daytona Beach, FL, 8 to 11 May 2011 [abstract T10].)
MATERIALS AND METHODS
Clinical samples.
Initially, a total of 93 primary clinical specimens (lesion, skin, oral, or genital swabs) in M4 universal transport medium (Remel, Lenexa, KS) that were collected between 2006 and 2010 were tested for the development of the molecular assay and the analysis algorithm. Subsequently, a blinded clinical study for verification of the assay and analysis algorithm included the testing of residual portions of 147 samples (lesion, skin, oral, or genital swabs and cultured isolates) collected and received for viral diagnosis between 2004 and 2011. All samples were previously found to be positive for HSV-1 by real-time PCR (16) or conventional culture with confirmation and typing by immunofluorescent staining (MicroTrak HSV1/HSV2 Culture Identification/Typing Test; Trinity Biotech, Wicklow, Ireland). All samples were stored at −70°C after initial testing.
Prior to testing, all of the specimens in the clinical study were coded without identifiers by a staff member not otherwise involved in this study. The 147 samples included 51 pairs of primary swab specimens, their corresponding cultured HSV-1 isolates, three pairs of first- and second-passage isolates, two sets of samples each containing two primary swabs and three isolates from a single patient, two HSV-1-negative specimens, and seven additional HSV-1-positive specimens from patients with no known link to any other case in the data bank. Neither the 93 development sample series nor the 147 clinical samples included a temporal series for any patient. Fifty-one coded, HSV-1-positive samples from other states were also assessed to detect geographical differences. These included 20, 7, and 24 aliquots of residual specimen material from Alabama, California, and Wisconsin, respectively. All of the samples, including specimens and isolates, had been stored at −70°C after initial testing.
The anatomical collection sites of the 53 specimens from Alabama, California, and Wisconsin are unknown. However, 71% of the New York samples were genital specimens or cultured viral isolates of specimens collected in the genital region; 19% were skin, vesicle, or lesion swabs or cultured viral isolates from these swabs; and 7% were oral or respiratory swab samples. The remaining 3% of the New York State specimens did not have a specified anatomical collection site.
Demographic information, including age, sex, onset date, submitter, county, and specimen type, was kept in a database separate from the ReIV results. These data were subsequently analyzed to help assess the relevance of matching electrophoretic signal patterns and validate the assay as a molecular epidemiological tool. Other information was deleted to protect patient privacy.
Human subject protection.
This project was approved by the New York State Department of Health Institutional Review Board under study no. 02-054.
Extraction.
Primary swab specimens in M5 medium were extracted with the NucliSENS total nucleic acid kit on the easyMAG instrument (bioMérieux, Durham, NC). Virus cultures were extracted with the QIAamp Viral RNA Minikit on a Qiagen QIAcube (Qiagen, Valencia, CA).
PCR.
PCRs used primers targeting the GC-rich iterative repeat regions in the introns of the US1 and US12 genes of HSV-1 (7, 8). Reaction mixtures included internal and external primer sets for both targets.
Assay optimization included evaluation of new polymerases, independent investigation of PCR additives (17), and optimization of cycling conditions in order to maximize the yield of full-length products while minimizing high-molecular-weight smears and low-molecular-weight bands from either truncated or nonspecific products. Phusion Hot Start II High-Fidelity DNA polymerase was selected over other polymerases (data not shown) with its optimal performance at a 1.5× concentration. Dimethyl sulfoxide (DMSO) was optimized at 3% for US1 and 5% for US12 targets. Various ratios of dGTP to 7-deaza-dGTP were assessed to maintain polymerase reactivity and greatly enhanced product formation. This was achieved by using 0.3 times as much 7-deaza-dGTP as deoxynucleoside triphosphates (dNTPs). Denaturation time, annealing times and temperatures, and extension times were reevaluated and optimized with the different changes to the master mixture.
Amplification with only internal PCR primers generated the cleanest products and produced amplicons suitable for analysis with defined peaks that could be selected with the Peak Scanner software on 83% of the samples. The internal primers for the US1 target were 5′-6-carboxyfluorescein (FAM)–TCCGACGACAGAAACCCACC-3′ and 5′-GTCCCGGAGGACCACAGTGG-3′, and the internal primers for the US12 target were 5′-FAM-TGGTGTCCAGGAAGGTGTCC-3′ and 5′-TTTTTGCACGGGTAAGCAC-3′ (7, 8). The 25-μl PCR mixture included 1× Phusion GC buffer, 200 μM dNTP mixture, 60 μM 7-deaza-dGTP (Roche Diagnostics Corporation, Indianapolis, IN), DMSO (3 or 5% for the US1 or US12 target, respectively), 0.5 μM FAM-tagged forward primer, 0.5 μM reverse primer, 1.5× Phusion Hot Start II High-Fidelity DNA polymerase (Thermo Fisher Scientific Dharmacon Products, Lafayette, CO), and 5 μl of template nucleic acid. The cycling conditions for the US1 PCR were 99°C for 1 min; 40 cycles of 99°C for 30 s, 65°C for 15 s, and 72°C for 25 s; and then 72°C for 7 min. The cycling conditions for the US12 PCR were 99°C for 1 min; 40 cycles of 99°C for 30 s, 60°C for 15 s, and 72°C for 20 s; and then 72°C for 7 min. The relative quality and quantity of the PCR products were assessed on a 2.2% FlashGel DNA Cassette (Lonza, Allendale, NJ) in 1× Tris-acetate-EDTA and SYBR green or SYBR-Safe (Invitrogen, Grand Island, NY). Differences in product size could also be visualized as shown in Fig. 1.
FIG 1.
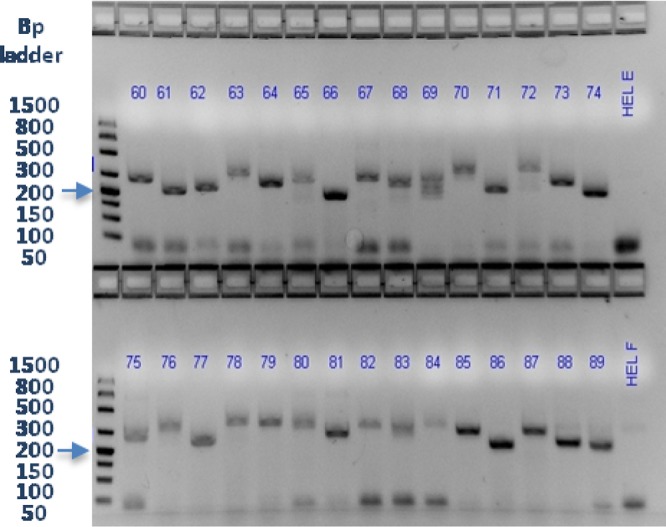
US1 gene target amplicons for 30 specimens from the blinded study and two human HEL controls in a 2.2% agarose FlashGel with SYBR green staining and the image color inverted. Lane numbers refer to samples 60 to 89.
If no product was obtained with the internal primer set for the US1 or US12 assay, then samples were retested in the nested format for that target. The reaction chemistry (other than primers) was the same as that described above. The external primers for the US1 target were 5′-CCACGAAACACAGGGGACGC-3′ and 5′-GGATTCGACCTCAGACTCCA-3′, and the external primers for the US12 target were 5′-ACGCCCCCTTTTATTGATCT-3′ and 5′-CCACGAAACACAGGGGACGC-3′ (8). The cycling conditions for the external US1 PCR were 99°C for 1 min; 40 cycles of 99°C for 30 s, 65°C for 15 s, and 72°C for 30 s; and then 72°C for 7 min. The cycling conditions for the external US12 PCR were 99°C for 1 min; 40 cycles of 99°C for 30 s, 59°C for 15 s, and 72°C for 30 s; and then 72°C for 7 min. A nested PCR was performed with 1 μl of the first-round product as the sample. If the nested PCR also failed to produce an amplicon, the presence of HSV-1 was checked by real-time PCR for HSV-1 (16). This occurred with five specimens in the study, and they were shown to be negative or to contain very low concentrations of HSV-1 DNA (data not shown).
Detection.
The labeled PCR products were separated by high-resolution capillary electrophoresis with the ABI Prism 3130XL Genetic Analyzer (Applied Biosystems, Carlsbad, CA) in the presence of GeneScan-600 LIZ size standards (Applied Biosystems, Carlsbad, CA), which cover 100 to 600 bp. Product peaks were visualized with Peak Scanner software v.1 (Applied Biosystems, Carlsbad, CA), and signal patterns were analyzed with their amplicon size assigned on the basis of the relative mobility of the peaks compared to those of the size standards. The full-length amplicon sizes for each target were documented with single-base-pair resolution. Since the base composition of the amplicons affects their mobility through the capillary electrophoresis tubes, the GC-rich targets generated for ReIV of HSV-1 will migrate differently than the size standards, which have an equal base composition across the four nucleotides. Amplicon sizes are reported on the basis of mobility relative to that of the standards and not on the basis of the absolute number of base pairs; therefore, sizes with a fractional number of base pairs are observed in the ReIV data. The results are rounded to the nearest whole number of base pairs for recording in the database. Any peaks in the electropherograms that were smaller than 100 bp were excluded from the analysis since they are below the expected amplicon size range for ReIV and generally contain unincorporated primers and dye artifacts that interfere with accurate size assignments (18). Visualized by Peak Scanner and assessed while still blind coded, electropherograms were considered to match between samples if the amplicon sizes, shapes, and patterns in both the US1 and US12 products matched. Assays were also performed with a background control sample that was a human origin continuous cell line suspension (HEL). This served as a negative control on the agarose gel and was also carried through capillary electrophoresis to facilitate the exclusion of human background peaks.
RESULTS
Analysis of electrophoretic signal patterns.
The electrophoretic signal patterns generated by the ReIV assay and displayed by the Peak Scanner software for each target (US1 or US12) were visually assessed in a series of analytical steps described here to identify the ReIV signatures of individual samples. The first step in the analysis was to examine the pattern of the full-length amplicon peak and document its length and shape. This full-length peak may have various shapes, including a simple singlet, a doublet, or other various multiplets with increasing numbers of peaks all different in length by 1 bp (Fig. 2).
FIG 2.
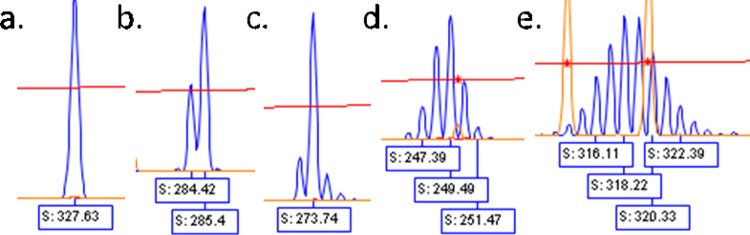
Examples of peak shapes. a, singlet; b, doublet; c to e, multiplets.
The next step was to assess for any truncated product peaks of >100 bp. These were invariably no greater than 600 bp and could be broadly categorized into the following categories: no pattern, repeating pattern, and complex pattern. No pattern, defined as the presence of only the full-length amplicon, was observed in 53.6% of the samples. In three of the four examples of this shown in Fig. 3a, the second peak in the electropherograms is a commonly observed human background peak (described below). Repeating patterns, observed in 38.1% of the specimens, contained additional peaks that decreased in base pair length by a consistent interval. This interval, or repeat pattern, was commonly observed as 22 to 25 bp in size. Examples of some of these repeating patterns are shown in Fig. 3b and d. These evenly spaced peaks are the same shape as the full-length amplicon peak but with intensities (indicated by peak height) that tend to increase as their length decreases. Other specimens exhibited patterns that are complex, with either no consistent pattern or overlapping patterns, examples of which are shown in Fig. 3c.
FIG 3.

Analysis of electropherogram patterns. (a) No pattern: electropherograms showing only the full-length amplicon signal (100 to 360 bp) without other peaks. The peak (*) at 308.8 bp is a human background signal. (b) Repeating pattern: electropherograms showing different repeat patterns. i, singlet peak at 264 bp and 22-bp repeat pattern; ii, doublet peak at 319 bp and 22-bp repeat pattern with an unlabeled low-intensity doublet at approximately 297 bp; iii, multiplet peak centered around 295 bp and 25-bp repeat pattern with an unlabeled low-intensity multiplet at approximately 270 bp. The human background peak (*) in US12 at 261 bp is also visible. (c) Complex pattern: electropherogram examples. i, varying multiplets; ii, combination of multiplet and singlet peaks; iii, overlapping multiplet patterns. The human background peak at 308.8 bp is also visible in graphs i and ii. (d) Repeating 22-bp pattern in both the US1 and US12 targets of a specimen. Gray lines indicate the alignment.
The human cell control was also evaluated, and correlating cross-reacting peaks (Table 1) were ignored when assigning the categories to the HSV-1 patterns. The presence and intensity of human background peaks vary between samples. The strongest of these background peaks, located at approximately 308.7 bp in the US1 reaction products (Fig. 3a, *), was isolated on an agarose gel from positive specimens and human control cells (Fig. 4) and characterized by Sanger dideoxynucleotide sequencing. The bidirectional product was confirmed as amplified human nucleic acid with a BLASTN (http://blast.ncbi.nlm.nih.gov/) search of the GenBank database, which identified the amplicon sequence as a 98% match to part of human chromosome 4.
TABLE 1.
Relative sizes and frequencies of human background amplicon peaks
| Prominencea | Size(s) (bp) of peak(s) in: |
|
|---|---|---|
| US1 | US12 | |
| High | 308.7 | 260.6 |
| Medium | 287.2 | 465.8 |
| Low | 144.8, 133.4, 161.2, 332.5, 408.4 | 168.4, 198.3, 241, 242.2, 259.5, 286.8, 291.3, 307.4, 349, 391.0 |
Prominence refers to a combination of the frequency and height of a peak in human samples in the database.
FIG 4.
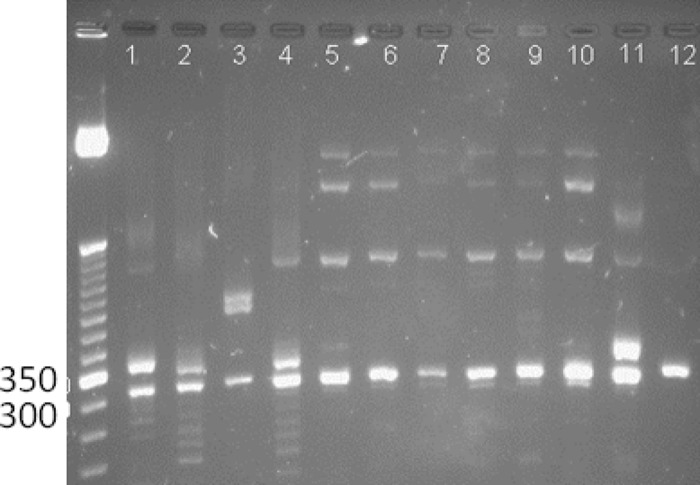
Amplicon products for the US1 target run on a 2% agarose gel with 0.5× Tris-borate-EDTA buffer for isolation of the 308.7-bp peak for dideoxy DNA sequencing. The products in lanes 1 to 6, 10, and 11 are from HSV-positive specimens tested during assay development, and those in lanes 7 to 9 and 12 are from human background controls. The gel was stained with SYBR-Safe. The values on the left are sizes in base pairs.
The full-length amplicon peak size and shape and all of the truncated peaks for both the US1 and US12 targets collectively constitute the ReIV signature for an individual sample. The next step in the analysis of each ReIV signature was to evaluate it against all of the other signatures in the database for potential matches with other specimens. This was done by viewing electropherograms with similar full-length amplicon sizes next to each other in Peak Scanner to allow a visual assessment of each specimen's entire ReIV signature, including pattern and peak shape, in order to assess potential matches. Matching signatures are documented in the database.
Assessment of samples.
All primary specimens were found to have ReIV signatures matching the corresponding cultured isolates. An example is shown in Fig. 5. Additionally, when frozen stored specimens were tested over time, ReIV signatures on the same specimens did not change when repeatedly tested over a 3-month period (data not shown).
FIG 5.

Electropherograms of US1 and US12 targets from two samples with matching signatures and containing 22-bp repeat patterns in both intron targets. When decoded, the samples were identified as a primary specimen (HSV16) and its cultured isolate (HSV94). Human background peaks at 308.7 bp in US1 and ∼260 bp in US12 are visible in the primary specimen (HSV16).
Database.
The purposes of the initial study were to develop the assay and test its performance with a large number of primary specimens for the consistent generation of signatures. In reviewing the results, a few matching signatures were observed. There were three instances where two specimens had matching signatures and one instance where three specimens had indistinguishable signatures. The blinded clinical study was designed to assess the utility of the assay for identifying related HSV-1 samples against a background of unrelated samples via the assessment of their ReIV signatures. The samples in this study included pairs of primary clinical specimens and their correlating cultured isolates and some instances of multiple samples from individual patients (Table 2). The signature results for all of the specimens are included in the database.
TABLE 2.
Specimens and isolates from patients with multiple genotyped samples
| Patient set | Sample |
Amplicon size |
Repeat size (bp) |
Peak pattern |
||||
|---|---|---|---|---|---|---|---|---|
| Specimen | Isolate | US1 | US12 | US1 | US12 | US1 | US12 | |
| A | Rectal swab | Yes | 342 | 329 | 24 | 24 | Singlet | Singlet |
| A | Genital swab | Yes | 342 | 329 | 24 | 24 | Singlet | Singlet |
| A | No | From lesion swab | 342 | 329 | 24 | 24 | Singlet | Singlet |
| B | Unidentified swab | Yes | 289 | 282 | 22 | 22 | Singlet | Singlet |
| B | Lesion swab | Yes | 289 | 282 | 22 | 22 | Singlet | Singlet |
| B | Unidentified swab | No | 289 | 282 | 22 | 22 | Singlet | Singlet |
| C | Tracheal swab | No | 360 | 316 | 22 | 22 | Doublet | Complex |
| C | Lymph node biopsy | No | 360 | 316 | 22 | 22 | Doublet | Complex |
The combined database contains signatures from a total of 264 specimens, including 88 from samples in the initial assay development phase, as well as 145 from New York specimens and 51 from non-New York specimens included in the clinical study. The database includes signatures from both primary specimens and cultured viral isolates. It contains 115 different maximum amplicon sizes on the US1 target, ranging from 186 to 582 bp, and 103 different maximum amplicon sizes on the US12 target, ranging from 176 to 517 bp. As shown in Fig. 6a and b, maximum amplicon sizes for both targets, measured in relative numbers of base pairs, were most frequently unique to a single patient. The same amplicon size occurred in two different patients 31 and 22 times for US1 and US12, respectively, and the same size occurred in three or more patients only a few times at each target. Signatures from the samples of approximately 100 patients did not show any repeat patterns in either target (Fig. 6c). Among those that did, the most common repeat size was 22 bp, followed by 24, 23, and 25 bp in both the US1 and US12 introns. Repeats of 20, 27, and 35 bp were seen in one patient each and only in US1 (Fig. 6c). The most commonly observed peak shape was a singlet, which was seen in US1 and US12 of samples from 111 and 135 patients, respectively (Fig. 6d). Doublets were noted in 51 and 31 patients, respectively, while triplets, multiplets, and complex peak patterns were observed less often.
FIG 6.

Frequency of occurrence of characteristics of ReIV signal patterns among patient samples in the database. a and b, US1 and US12 amplicon sizes; c, repeat sizes; d, peak shapes.
A summary of the results for the entire database is shown in Table 3. It is organized by initially grouping the specimens into sets. Within each set, the samples have matching ReIV signatures (amplicon size and signal pattern in both US1 and US12). The sets are further organized into categories defined by a combination of the set and the number of samples within it. For example, one signature pattern with only one sample with that pattern is one category; a set containing two samples with matching signatures is another category. As shown in the top data line of Table 3, for example, there were 114 instances of a single sample within a set (no matching signature). Also shown in Table 3 are the numbers of samples and patients within each set size and category. When the signature analysis was applied to the 264 specimens from 199 patients, 176 unique signature patterns were observed. A total of 159 sets that contained specimens (one, two, or five) from only a single patient had 159 unique signatures in 207 specimens. There were 12 sets, each containing specimens from two patients. These sets contained two, three, four, or six specimens. Another four sets each contained specimens from three patients (total, 14 patients and 17 specimens). Lastly, one set of five specimens from four patients had matching signatures. Results for samples in each category are described below.
TABLE 3.
HSV-1 strain analysis database: summary of ReIV signatures for 264 specimens from 199 patients from combined studiesa
| No. of patients with same Re-IV signature within a setb (A) | No. of specimens with same signature pattern within a setb (B) | No. of times categoryc found in database (C) | Total no. of specimens/categoryc (D) | Total no. of patients/categoryc (E) |
|---|---|---|---|---|
| 1 | 1 | 114 | 114 | 114 |
| 1 | 2 | 44 | 88 | 44 |
| 1 | 5 | 1 | 5 | 1 |
| 1d | 159e | 207f | 159g | |
| 2 | 2 | 6 | 14 | 14 |
| 2 | 3 | 3 | 9 | 6 |
| 2 | 4 | 2 | 8 | 4 |
| 2 | 6 | 1 | 6 | 2 |
| 2d | 14e | 35f | 24g | |
| 3 | 3 | 1 | 3 | 3 |
| 3 | 4 | 2 | 8 | 6 |
| 3 | 6 | 1 | 6 | 3 |
| 3d | 4e | 17f | 14g | |
| 4 | 5 | 1 | 5 | 4 |
| 4d | 1e | 5f | 4g |
In each line, A patients match the signatures in B samples. This category occurs C times in the database for a total of D specimens and E patients. The database contains a total of 176 unique signatures, a total of 264 specimens, and a total of 199 patients.
A set is a group of specimens with matching ReIV signatures (amplicon sizes and signal patterns in both the US1 and US12 assays).
A category is the specific combination of the number of patients and the number of specimens within a set.
Subtotal of number of patients per set.
Subtotal of number of different sets.
Subtotal of number of specimens.
Subtotal of number of patients.
Categories with six specimens.
Matching signatures included those from three pairs of primary specimens and the corresponding cultured isolates that had been collected from 18-, 21-, and 23-year-old patients in the same geographic area. The other set of six matching signatures included those from a combination of five primary specimens and cultured isolates from a single patient, together with a primary specimen from another individual with no identifiable epidemiological link.
Categories with five specimens.
Unique ReIV signatures were seen in two sets of five specimens each. A mixture of primary specimens and cultured isolates from a single patient made up the first of these, while the other included a primary-isolate pair from New York and three specimens from Alabama.
Categories with four specimens.
Four sets of four specimens had matching signatures within each set. One set included two primary-isolate pairs from pediatric siblings with the same symptom onset date. Signatures from genital swabs collected from two 17-year-old patients with the same onset date at the same clinic matched each other, as well as the signature from a vesicle swab-isolate pair from a 25-year-old patient from a different area of the state. Another set of four included a genital swab-isolate pair from a 27-year-old patient, a genital swab from a 29-year-old individual from a different region, and a sample from Wisconsin. The fourth set of four matching signatures was obtained from a skin swab-isolate pair and a rectal swab-isolate pair with no apparent epidemiological relatedness.
Categories with three specimens.
Four sets of three specimens had unique signatures. One set contained genital and vesicle swabs from three 20- to 21-year-old students at the same college, and another included a genital swab-isolate pair from a 19-year-old patient and a genital swab from a 21-year-old patient from the adjacent town. The remaining two sets both included a primary-isolate pair and another specimen from cases that were geographically and temporally separated.
Categories with two specimens.
A total of 100 specimens had a unique signature pattern in common with one other specimen in the database. The majority of these comprised 82 primary-specimen and corresponding cultured viral isolate pairs. A pair of primary specimens from different anatomical sites within the same patient also had matching signatures. Two specimens tested during the initial study were included in the blinded clinical study under a different identifier; when unblinded, they were found to have matching signatures. Specimens collected at clinics from 24- and 26-year-old patients weeks apart in adjacent towns also generated matching signatures. The signatures of two Alabama samples matched each other, while that of a third Alabama sample matched the signature of a Wisconsin sample. Three unique signature pairs were also matched between New York samples and Wisconsin specimens.
DISCUSSION
Molecular strain analysis of HSV-1 for the purpose of determining epidemiological relatedness has been a challenging issue for many years. Previous methods such as RFLP analysis have been shown to lack discriminating vigor for the differentiation of HSV-1 strains of a predominant genotype (6). Laboratory methodology for RFLP analysis is time-consuming and labor-intensive; it requires viable virus and the culture of large quantities of virus, followed by many lengthy and subjective procedures. Repeat analysis has been shown to be more discriminatory, allowing the differentiation of strains within single genotypes. The use of automated instrumentation and specialized software allows for single-base-pair resolution and more accurate, objective, reproducible analysis (8). The present study reports an assay and analysis protocol for ReIV signatures in HSV-1 that was developed and validated for use on low-volume positive primary specimens, as well as cultured isolates.
Several components of the assay were investigated and optimized in this study. HotStarTaq DNA polymerase (Qiagen, Valencia, CA) was evaluated with and without a number of commercially available reaction-optimizing additives, but full-length amplicons were difficult to generate because of the GC-rich target sequences. Additionally, terminal adenosines added to amplicons by the polymerase complicated the analysis. Phusion Hot Start II High-Fidelity DNA polymerase, a Pyrococcus-like proofreading polymerase fused to a larger, nonspecific, double-stranded-DNA-binding domain that increases specificity and processivity, has been reported to produce highly accurate results with VNTRs and homopolymeric regions (19). Phire Hot Start DNA polymerase (Thermo Fisher Scientific Dharmacon Products, Lafayette, CO) and Phusion Hot Start II High-Fidelity DNA polymerase were both assessed because of the reported high fidelity facilitated by their DNA-binding domains. Improved full-length amplicon yields were obtained, but better results were obtained with the Phusion polymerase.
HSV-1 genomes are 68 to 86% GC rich, and the GC content of VNTR regions can approach 100% (9). A number of chemicals have been used to facilitate PCR assays of GC-rich regions. Some of these were investigated, and DMSO and 7-deaza-dGTP were included in the final reaction mixture for HSV ReIV. These assays produced distinct electropherograms for the US1 and US12 targets that jointly contain the ReIV signatures and provided more highly detailed information for strain comparison than a single gene or amplicon size alone. The signatures are robust, with reproducible results on repeat testing of stored frozen specimens. ReIV signatures can vary from simple signature patterns showing sharp single peaks on the electropherogram to complex signature patterns with clusters of multiple peaks. Among complex peaks, uniform clusters contain peaks that are 1 bp different in size because of stuttering of the polymerase in homopolymeric regions (20). Another key feature of many of the signatures is evenly spaced repetitive peaks, as shown in Fig. 3b and d. This is proposed to result from skipping of the polymerase on the template because of the structure and composition of the reiterative repeat regions in the introns.
When multiple peaks are present but do not display a discernible pattern, it may be indicative of the presence of more than one strain, a high background of human products, or potentially other nonspecific PCR products. In practice, provided the HSV titer in the sample is not low, the interference of nonspecific products in this assay has been found to be minimal. While mixed infections have been reported in immunocompetent individuals, they are more commonly encountered in immunosuppressed patients; therefore, the degree to which the analysis of mixed infections is problematic will depend on the patient population under investigation. Future experiments will attempt to confirm the ability of this assay to identify mixed infections by separating the strains by plaque purification and reevaluating the resulting electropherograms. Ratios of experimentally mixed strains will also be tested.
Analysis of the test results was performed by a scientist who, throughout the study, remained blinded to the minimal demographic information that had been recorded about the specimens. The matching of each signature to others in the database was assessed while all of the samples were still coded. When the available demographic information was correlated with the assay results, signatures from all primary specimens matched those from their cultured isolates, as did the signatures from retesting of thawed stored samples. No culture-induced differences in signatures were observed anywhere in the database, with the exception of human background peaks, readily identified in primary specimens and not seen in cultured isolates. However, these peaks are not considered part of the true HSV ReIV signature and therefore do not represent real differences between specimen and culture signatures.
Matching ReIV signatures were seen in specimens collected from pediatric siblings with the same disease onset date, in specimens from two 17-year-old patients attending the same clinic with the same disease onset date, in three student patients 20 to 21 years old attending the same college, in three 18- to 23-year-old patients in the same geographic area, and in a 19-year-old patient and a 21-year-old patient in adjacent townships. In each of these five situations, the signatures did not match those from any other samples in the database, with the exception of the 17-year-old patients, where the patterns were matched by those of samples collected from a 25-year-old patient elsewhere in New York. Minimal demographic information was retained in the database, so epidemiological links cannot be definitively confirmed. In the absence of epidemiological data that could potentially link specimens, additional confirmation of matching strains may be required. Additional confirmation of molecular relatedness in such cases by ReVII analysis (7), thymidine kinase sequencing (8), or other methods may be required.
Samples received from Alabama contained one group of three and one group of two specimens with matching ReIV signatures, although the epidemiological relatedness between any of the samples is unknown. Five of the signatures observed in New York samples were also seen in samples from Wisconsin, and one signature in a sample from Wisconsin was seen in a sample from Alabama. Interestingly, studies that used RFLP analysis have noted geographic differences between HSV-1 isolates from different countries or regions (4, 21). Overall, however, sample numbers from outside New York would need to be larger to allow further assessment of this aspect of the analysis.
With 115 different amplicon sizes for the US1 target, 103 different amplicon sizes for the US12 target, and numerous signal peak patterns, producing a combined signature database of 176 unique signatures from 264 samples, this study provides one of the largest reference sources of its type ever reported for this kind of analysis of clinical specimens. No dominant or commonly occurring signature was observed at any time during the analysis, supporting its potential use for various clinical and epidemiological purposes. There are multiple potential applications of the technique. Different strains from distinct anatomical sites within a patient may be identified; this is particularly useful if the strains are responding variably to treatment. The method could also be used to assist with the investigation of outbreaks or infection sources and for cases of clinical or public health interest. Since the signatures are reproducible and for any sample they are stable over time in frozen storage, the database provides a long-term reference source for epidemiological studies. Additional signatures will be added as work in this area proceeds.
ACKNOWLEDGMENTS
We thank the Wadsworth Center Virology Laboratory, specifically, Meghan Fuschino, Jennifer Laplante, and Michael Popowich, for providing coded archived samples and correlating historical test results; Daryl Lamson for sequence analysis of the human background peak; and the Wadsworth Center Applied Genomic Technologies Core for running the ABI Prism 3130XL Genetic Analyzer and performing Sanger dideoxynucleotide sequencing. We thank Morris S. Jones, California Department of Public Health; Fred Lakeman, University of Alabama; and Eric Reisdorf and Audrey Prieve, Wisconsin State Laboratory of Hygiene, for sharing samples and Andrea Habura and Kimberly Musser for their assistance with reagent samples during development. We are very grateful to Georges M. G. M. Verjans, April Burch, and Andrea Habura for helpful scientific discussions and suggestions during this study.
Footnotes
Published ahead of print 5 March 2014
REFERENCES
- 1.Kolb AW, Adams M, Cabot EL, Craven M, Brandt CR. 2011. Multiplex sequencing of seven ocular herpes simplex virus type-1 genomes: phylogeny, sequence variability, and SNP distribution. Invest. Ophthalmol. Vis. Sci. 52:9061–9073. 10.1167/iovs.11-7812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Norberg P. 2010. Divergence and genotyping of human alpha-herpesviruses: an overview. Infect. Genet. Evol. 10:14–25. 10.1016/j.meegid.2009.09.004 [DOI] [PubMed] [Google Scholar]
- 3.Sakaoka H, Saito H, Sekine K, Aomori T, Grillner L, Wadell G, Fujinaga K. 1987. Genomic comparison of herpes simplex virus type 1 isolates from Japan, Sweden and Kenya. J. Gen. Virol. 68:749–764. 10.1099/0022-1317-68-3-749 [DOI] [PubMed] [Google Scholar]
- 4.Sakaoka H, Kurita K, Iida Y, Takada S, Umene K, Kim Y, Ren C, Nahmias A. 1994. Quantitative analysis of genomic polymorphism of herpes simplex virus type 1 strains from six countries: studies of molecular evolution and molecular epidemiology of the virus. J. Gen. Virol. 75:513–527. 10.1099/0022-1317-75-3-513 [DOI] [PubMed] [Google Scholar]
- 5.Umene K, Yoshida M. 1989. Reiterated sequences of herpes simplex virus type 1 (HSV-1) genome can serve as physical markers for the differentiation of HSV-1 strains. Arch. Virol. 106:218–299 [DOI] [PubMed] [Google Scholar]
- 6.Umene K, Sakaoka H. 1991. Homogeneity and diversity of genome polymorphism in a set of herpes simplex virus type 1 strains classified as the same genotypic group. Arch. Virol. 119:53–65. 10.1007/BF01314323 [DOI] [PubMed] [Google Scholar]
- 7.Maertzdorf J, Remeijer L, van der Lelij A, Buitenwerf J, Niesters HGM, Osterhaus ADME, Verjans GMGM. 1999. Amplification of reiterated sequences of herpes simplex virus type 1 (HSV-1) genome to discriminate between clinical HSV-1 isolates. J. Clin. Microbiol. 37:3518–3523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duan R, de Vries D, van Dun JM, van Loenen FB, Osterhaus ADME, Remeijer L, Verjans GMGM. 2009. Acyclovir susceptibility and genetic characteristics of sequential herpes simplex virus type 1 corneal isolates from patients with recurrent herpetic keratitis. J. Infect. Dis. 200:1402–1414. 10.1086/606028 [DOI] [PubMed] [Google Scholar]
- 9.Ouyang Q, Zhao X, Feng H, Tian Y, Li D, Li M, Tan Z. 2012. High GC content of simple sequence repeats in herpes simplex virus type 1 genome. Gene 499:37–40. 10.1016/j.gene.2012.02.049 [DOI] [PubMed] [Google Scholar]
- 10.Szpara M, Parsons L, Enquist L. 2010. Sequence variability in clinical and laboratory isolates of herpes simplex virus 1 reveals new mutations. J. Virol. 84:5303–5313. 10.1128/JVI.00312-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deback C, Boutolleau D, Depienne C, Luyt C, Bonnafous P, Gautheret-Dejean A, Garrigue I, Agut H. 2009. Utilization of microsatellite polymorphism for differentiating herpes simplex virus type 1 strains. J. Clin. Microbiol. 47:533–540. 10.1128/JCM.01565-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Umene K, Yamanaka F, Oohashi S, Koga C, Kameyama T. 2007. Detection of differences in genomic profiles between herpes simplex virus type 1 isolates sequentially separated from the saliva of the same individual. J. Clin. Virol. 39:266–270. 10.1016/j.jcv.2007.05.012 [DOI] [PubMed] [Google Scholar]
- 13.Roest RW, Carman WF, Maertzdorf J, Scoular A, Harvey J, Kant M, van der Meijden WI, Verjans GMGM, Osterhaus ADME. 2004. Genotypic analysis of sequential genital herpes simplex virus type 1 (HSV-1) isolates of patients with recurrent HSV-1 associated genital herpes. J. Med. Virol. 73:601–604. 10.1002/jmv.20132 [DOI] [PubMed] [Google Scholar]
- 14.Brink AATP, van Gelder M, Wolffs PF, Bruggeman CA, van Loo IHM. 2011. Compartmentalization of acyclovir-resistant varicella zoster virus: implications for sampling in molecular diagnostics. Clin. Infect. Dis. 52:982–987. 10.1093/cid/cir079 [DOI] [PubMed] [Google Scholar]
- 15.Hamprecht K, Eckle T, Prix L, Faul C, Einsele H, Jahn G. 2003. Ganciclovir-resistant cytomegalovirus disease after allogeneic stem cell transplantation: pitfalls of phenotypic diagnosis by in vitro selection of an UL97 mutant strain. J. Infect. Dis. 187:139–143. 10.1086/346240 [DOI] [PubMed] [Google Scholar]
- 16.Dupuis M, Hull R, Wang H, Nattanmai S, Glasheen B, Fusco H, Dzigua L, Markey K, Tavakoli N. 2011. Molecular detection of viral causes of encephalitis and meningitis in New York State. J. Med. Virol. 83:2172–2181. 10.1002/jmv.22169 [DOI] [PubMed] [Google Scholar]
- 17.Kaiser R, Tremblay P-B, Roots I, Brockmöller J. 2002. Validity of PCR with emphasis on variable number of tandem repeats analysis. Clin. Biochem. 35:49–56. 10.1016/S0009-9120(02)00273-4 [DOI] [PubMed] [Google Scholar]
- 18.Applied Biosystems. 2014. DNA fragment analysis by capillary electrophoresis user manual (PN 4474504 rev. A). Life Technologies, Carlsbad, CA [Google Scholar]
- 19.Fazekas AJ, Steeves R, Newmaster SG. 2010. Improving sequencing quality from PCR products containing long mononucleotide repeats. Biotechniques 48:277–285. 10.2144/000113369 [DOI] [PubMed] [Google Scholar]
- 20.Applied Biosystems. 2004. Evaluating genetic analysis systems: microsatellite analysis. Applied Biosystems, Foster City, CA [Google Scholar]
- 21.Eda H, Ozawa S, Yoshino K, Yanagi K. 2007. Contrasting geographic distribution profiles of the herpes simplex virus type 1 BgOL and BgKL variants in Japan suggest dispersion and replacement. J. Clin. Microbiol. 45:771–782. 10.1128/JCM.01236-06 [DOI] [PMC free article] [PubMed] [Google Scholar]