

Abstract
A real-time TaqMan PCR assay based on the gene encoding the protein p37 was developed to detect Mycoplasma hyorhinis. Its specificity was validated with 29 epidemiologically unrelated M. hyorhinis strains (28 field strains and one reference strain) and other mycoplasma species or with other microorganisms commonly found in pigs. The estimated detection limit of this qPCR assay was 125 microorganism equivalents/μl. The same 29 epidemiologically unrelated M. hyorhinis strains and four previously fully sequenced strains were typed by two portable typing methods, the sequencing of the p37 gene and a multilocus sequence typing (MLST) scheme. The first method revealed 18 distinct nucleotide sequences and insufficient discriminatory power (0.934). The MLST scheme was developed with the sequenced genomes of the M. hyorhinis strains HUB-1, GDL-1, MCLD, and SK76 and based on the genes dnaA, rpoB, gyrB, gltX, adk, and gmk. In total, 2,304 bp of sequence was analyzed for each strain. MLST was capable of subdividing the 33 strains into 29 distinct sequence types. The discriminatory power of the method was >0.95, which is the threshold value for interpreting typing results with confidence (D = 0.989). Population analysis showed that recombination in M. hyorhinis occurs and that strains are diverse but with a certain clonality (one unique clonal complex was identified). The new qPCR assay and the robust MLST scheme are available for the acquisition of new knowledge on M. hyorhinis epidemiology. A web-accessible database has been set up for the M. hyorhinis MLST scheme at http://pubmlst.org/mhyorhinis/.
INTRODUCTION
Mycoplasmas are commonly described as the simplest and smallest self-replicating bacteria because of their total lack of a cell wall, the paucity of their metabolic pathways, and the small size of their genome (1). While mycoplasmas are considered the simplest free-living organisms, several species are successful pathogens of humans and animals. Mycoplasma hyorhinis, a major contaminant of mammalian tissue cultures in laboratories worldwide, usually infects pigs, leading to respiratory tract disease and inflammation of the chest and joints (2). Polyserositis, arthritis, pneumonia, otitis, and conjunctivitis are clinical disorders associated with M. hyorhinis infection (2). In the porcine respiratory disease complex, M. hyorhinis appears to be frequently associated with Mycoplasma hyopneumoniae, the primary agent of enzootic pneumonia (3). Respiratory diseases remain the most challenging health problems in pig production worldwide (4). For example, in France, a prevalence study showed that 72.4% of slaughter pigs suffered from pneumonia and that 14.4% had pleuritis (5). These lung diseases result in financial losses due to poor growth performance, reduced feeding efficiency, and higher medication costs and have an adverse effect on pig welfare (4). Moreover, the administration of antimicrobials may have a potential negative effect on human health when associated with food-borne contamination by resistant pathogens (even if M. hyorhinis is generally susceptible to the antibiotics used against M. hyopneumoniae) or resistant commensal bacteria (6).
M. hyorhinis is also an infectious agent potentially implicated in human cancers. Accumulating evidence suggests that M. hyorhinis infection in humans does result in clinical outcomes (7–9). Working independently, several groups have detected the M. hyorhinis p37 protein in cancer patients (10–13). p37 is a peptide of 403 amino acids with a molecular mass of 43.5 kDa. An analysis of the protein sequence revealed that p37 has about a 34% to 42% similarity to a periplasmic binding protein-dependent transport system found in Gram-negative bacteria and several species of mycoplasma. Thus, p37 is thought to be part of a high-affinity transport system in M. hyorhinis (10).
M. hyorhinis can be isolated from live pigs (nasal, oropharyngeal, and tracheal swabs or oral fluids) and from dead pigs (tissue or swab samples taken from the synovial fluid of arthritis cases or pleural, pericardial, and peritoneal lesions) (3, 14). Isolation of M. hyorhinis is relatively tedious and time-consuming. Colonies can be observed on solid media around 4 to 15 days after inoculation (14). The difficulty in culturing M. hyorhinis has led to the development of other diagnostic assays. Several specific endpoint PCR tests were developed to detect M. hyorhinis in pure cultures or in cell cultures (15–17). The detection thresholds of these gel-based PCR tests varied from 1,000 to 10,000 microorganisms per reaction. In general, and compared to endpoint PCR, real-time or quantitative PCR (qPCR) tests are usually more sensitive, allowing the detection of as few as 5 copies of a target sequence (18). Moreover, qPCR does not require postamplification manipulations and is extremely fast.
Two old molecular typing schemes were used to determine the relationships between M. hyorhinis strains: restriction endonuclease analysis (REA) (19) and pulsed-field gel electrophoresis (PFGE) (20). No multilocus sequence typing (MLST) scheme has been developed to date for typing M. hyorhinis. MLST is a highly discriminatory and unambiguous method of characterizing bacterial isolates that has now been successfully employed in the characterization of several bacterial species, including Mycoplasma hyopneumoniae, Mycoplasma agalactiae, and Mycoplasma bovis (21–23). MLST is currently regarded as a gold standard for typing and can even replace PFGE (24). It is based on the nucleotide sequences of internal fragments of housekeeping genes, in which mutations are assumed to be largely neutral (25). For each gene fragment, the different nucleotide sequences are assigned allele numbers, and the sequence type (ST) of each isolate is defined by the alleles present at each distinct locus. Isolates that share the same ST are assumed to be members of the same clone; that is, they have a recent common ancestor. An important advantage of MLST is that sequence data are portable and can be readily compared among laboratories. In addition, the data obtained can be used to address questions about the evolutionary and population biology of bacterial species (26).
In the present study, a qPCR assay and two portable typing methods for M. hyorhinis, the sequencing of the p37 gene and an MLST scheme based on the nucleotide sequences of six housekeeping gene fragments, were developed.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Twenty-eight epidemiologically unrelated strains of M. hyorhinis, isolated from 1978 to 2009 in France, from different farms and with different origins were analyzed (Tables 1 and 2). Reference strain ATCC 25021 (isolated in 1955 from a case of pneumonia) was also included in the study. To confirm the identity of the isolates as M. hyorhinis, a conventional PCR (16) and a new qPCR assay, based on the p37 gene, were performed. The specificity of this last assay was tested with a collection of 34 strains of bacterial species other than M. hyorhinis (Table 1). M. hyorhinis, M. hyosynoviae, M. hyopharyngis, M. flocculare, and M. hyopneumoniae were cultivated as previously described (27, 28). Streptococcus sp. and Actinobacillus lignieresii were cultivated on Columbia agar base supplemented with 5% sheep blood (AES Laboratories, Combourg, France). Escherichia coli and Staphylococcus aureus were cultivated as previously described (29). The other bacterial species were cultivated on pleuropneumonialike organism (PPLO) agar (Difco, Cergy Pontoise, France) supplemented with 10 μg/ml nicotinamide dinucleotide (β-NAD), 1 mg/ml glucose, and 5% decomplemented horse serum. All strains were incubated at 37°C in 5% CO2.
TABLE 1.
Bacterial strains used to test the specificity of the real-time PCR (qPCR) assay for the detection of M. hyorhinis
| Organism | Strain(s)a | No. tested (n = 63) | qPCR result |
|---|---|---|---|
| Mycoplasma hyorhinis | RS ATCC 25021 and 28 field strains | 29 | + |
| Mycoplasma hyopneumoniae | RS ATCC 25934 and 3 field strains | 4 | − |
| Mycoplasma hyosynoviae | RS ATCC 25591 and 4 field strains | 5 | − |
| Mycoplasma flocculare | RS ATCC 27399 and 2 field strains | 3 | − |
| Mycoplasma hyopharyngis | RS ATCC 51909 | 1 | − |
| Mycoplasma gallisepticum | Field strain | 1 | − |
| Mycoplasma synoviae | Field strain | 1 | − |
| Mycoplasma orale | Field strain | 1 | − |
| Mycoplasma glycophilum | Field strain | 1 | − |
| Mycoplasma genitalium | Field strain | 1 | − |
| Mycoplasma bovis | Field strain | 1 | − |
| Pasteurella multocida subsp. multocida | Field strains | 2 | − |
| Haemophilus parasuis | Field strains | 4 | − |
| Actinobacillus pleuropneumoniae s9 and 11 | RS CVJ 13261, RS 56153 | 2 | − |
| Actinobacillus rossii | RS CCUG 44312 | 1 | − |
| Actinobacillus lignieresii | RS ATCC 49236 | 1 | − |
| Bordetella bronchiseptica | Field strain | 1 | − |
| Escherichia coli | RS ATCC 25922 | 1 | − |
| Staphylococcus aureus | Field strain | 1 | − |
| Streptococcus porcinus | RS ATCC 43138 | 1 | − |
| Streptococcus suis | RS ATCC 43765 | 1 | − |
RS, reference strain; ATCC, American Type Culture Collection, Rockville, USA; CCUG, Culture Collection, University of Göteborg, Sweden.
TABLE 2.
Characteristics of 33 strains belonging to the 18 nucleotide sequences of the p37 gene and to the 29 STs identified in this study
| Strain | Yra | Originb | Source | p37 gene sequence (GenBank accession no.) | p37 group | ST profilec | ST | ST group |
|---|---|---|---|---|---|---|---|---|
| GDL1 | NK | FS | Tissue culture | Seq1 (NC_016829.1) | A2 | 1,1,1,1,1,1 | 1 | a2 |
| MCLD | 2004 | FS | Cell lined | Seq3 (NC_017519.1) | A2 | 1,1,1,1,1,1 | 1 | a2 |
| SK76 | 1973 | FS | Polyserositis | Seq4 (NC_019552.1) | A2 | 2,3,3,1,1,1 | 14 | a3 |
| HUB-1 | NK | FS | Respiratory tract | Seq5 (NC_014448.1) | A1 | 6,5,6,4,2,5 | 27 | b4 |
| ATCC 25021 | 1955 | RSc | Pneumonia | Seq2 (KF806445) | A2 | 1,1,1,1,1,1 | 1 | a2 |
| Mhr39 | 2007 | FS | Pneumonia | Seq16 (KF806452) | B5 | 1,1,6,1,1,1 | 2 | a1 |
| Mhr43 | 2007 | FS | Pneumonia | Seq6 (KF806453) | B1 | 3,4,6,4,1,3 | 10 | b1 |
| Mhr47 | 2007 | FS | Pneumonia | Seq11 (KF806458) | B4 | 1,7,4,3,1,3 | 26 | b4 |
| Mhr55 | 2004 | FS | Pneumonia | Seq10 (KF806457) | B4 | 1,7,2,2,3,1 | 3 | a3 |
| Mhr60 | 2004 | FS | Pneumonia | Seq15 (KF806451) | B5 | 1,1,4,1,1,1 | 17 | b2 |
| Mhr66 | 2004 | FS | Pneumonia | Seq8 (KF806455) | B2 | 10,1,4,1,3,3 | 22 | b3 |
| Mhr120 | 2007 | FS | Pneumonia | Seq7 (KF806454) | B1 | 10,4,2,1,1,3 | 4 | a3 |
| Mhr168 | 2006 | FS | Pneumonia | Seq8 (KF806455) | B2 | 3,1,4,1,1,3 | 12 | b1 |
| Mhr180 | 2007 | FS | Pneumonia | Seq16 (KF806452) | B5 | 1,4,2,1,1,1 | 7 | a3 |
| Mhr30 | 2006 | FS | Trachea | Seq8 (KF806455) | B2 | 1,7,6,4,1,4 | 28 | b4 |
| Mhr48 | 2007 | FS | Trachea | Seq8 (KF806455) | B2 | 3,4,6,1,1,3 | 11 | b1 |
| Mhr97 | 2006 | FS | Trachea | Seq9 (KF806456) | B3 | 1,7,4,4,2,2 | 29 | b4 |
| Mhr100 | 2006 | FS | Trachea | Seq17 (KF806446) | A4 | 1,4,2,7,1,3 | 5 | a3 |
| Mhr104 | 2006 | FS | Trachea | Seq8 (KF806455) | B2 | 9,7,4,6,1,3 | 18 | b3 |
| Mhr113 | 2007 | FS | Trachea | Seq11 (KF806458) | B4 | 1,7,4,1,1,1 | 24 | b4 |
| Mhr123 | 2007 | FS | Trachea | Seq18 (KF806447) | A3 | 1,2,2,1,1,3 | 8 | a3 |
| Mhr160 | 2007 | FS | Trachea | Seq17 (KF806446) | A4 | 4,4,2,1,1,3 | 6 | a3 |
| Mhr164 | 2006 | FS | Trachea | Seq18 (KF806447) | A3 | 5,7,4,1,1,3 | 25 | b4 |
| Mhr52 | 2004 | FS | Nasal cavity | Seq8 (KF806455) | B2 | 10,1,4,5,3,3 | 21 | b3 |
| Mhr204 | 1978 | FS | Nasal cavity | Seq8 (KF806455) | B2 | 8,7,6,1,3,3 | 20 | b3 |
| Mhr211 | 1981 | FS | Nasal cavity | Seq17 (KF806446) | A4 | 1,3,1,1,1,3 | 16 | a3 |
| Mhr9 | 2009 | FS | Polyserositis | Seq14 (KF806450) | B5 | 3,1,6,1,4,3 | 9 | a3 |
| Mhr17 | 2009 | FS | Polyserositis | Seq12 (KF806448) | B5 | 7,1,4,1,3,1 | 23 | b3 |
| Mhr21 | 2009 | FS | Polyserositis | Seq13 (KF806449) | B5 | 8,6,5,1,1,3 | 19 | b3 |
| Mhr141 | 1998 | FS | Polyserositis | Seq18 (KF806447) | A3 | 1,3,1,1,2,3 | 15 | a3 |
| Mhr142 | 1999 | FS | Polyserositis | Seq17 (KF806446) | A4 | 1,3,1,1,1,3 | 16 | a3 |
| Mhr143 | 1999 | FS | Polyserositis | Seq6 (KF806453) | B1 | 1,3,1,1,1,3 | 16 | a3 |
| Mhr208 | 1998 | FS | Polyserositis | Seq16 (KF806452) | B5 | 1,1,1,1,1,3 | 13 | a3 |
NK, not known.
FS, field strain; RS, reference strain.
Allele numbers for each gene, presented in the following order: dnaA, rpoB, gyrB, gltX, adk, gmk. The sequences are reported in the web-accessible database http://pubmlst.org/mhyorhinis/.
The strain MCLD was isolated from a primary human melanoma cell line.
DNA preparation.
DNA suspensions were prepared as described by Kellog and Kwok (30). Briefly, 1 ml of each mycoplasmal culture was centrifuged (12,000 × g, 4°C, 20 min) and the pellets were resuspended in 800 μl of lysis solution. For the other bacterial species, approximately 10 colonies were placed in 800 μl of lysis solution. Lysates were incubated for 1 h at 60°C and for 10 min at 95°C and stored at −20°C. M. hyorhinis DNA were identified by conventional PCR (16) and qPCR assay. The detection level of the qPCR assay was evaluated using 10-fold dilutions (10.7 ng/μl to 1.07 fg/μl) of chromosomal DNA from M. hyorhinis strain ATCC 25021 (10 runs with one replicate).
qPCR conditions.
The qPCR assay developed in this study permitted detection of M. hyorhinis species. The primers and TaqMan probe were designed using Beacon Designer 6.00 software and guidelines from Premier Biosoft International. The 89-bp PCR product, specific to M. hyorhinis, was obtained with the forward primer Mhr-P37-RT-F and the reverse primer Mhr-P37-RT-R (Table 3) defined on the p37 gene (GenBank accession no. NC_014448.1) (31). The specificity of the primer and probe sequences was tested by homology searches in the nucleotide database (GenBank BLASTN 2.2.14) and in the insilico.ehu.es website (32). The TaqMan probe was labeled at the 5′ end with the reporter dye 6-carboxyfluorescein (FAM) and at the 3′ end with Black Hole Quencher (Sigma-Aldrich, Saint-Quentin Fallavier, France).
TABLE 3.
Primers used for the M. hyorhinis real-time PCR assay, for the sequencing of the p37 gene, and for the MLST scheme
| Gene | Primer | Sequence (5′→3′)a | Start position (in HUB-1 genome) | Amplicon size (bp) | Seq. sizeb (bp) |
|---|---|---|---|---|---|
| p37 gene | Mhr-p37-RT-F | TATCTCATTGACCTTGACTAAC | 768070 | 89 | |
| Mhr-p37-RT-R | ATTTTCGCCAATAGCATTTG | ||||
| Mhr-p37-Probe | FAMa-CATCCTCTTGCTTGACTACTCCTG-BHQ1b | ||||
| p37-A1F | TTTTCCTTGGCCTGATGAAC | 766864 | 836 | 1,278 | |
| p37-A1R | TTCAGATCCACTTGTAAAAATTGC | ||||
| p37-A2F | AGCTTGAACAGTTGGATCGT | 764 | |||
| p37-A2R | GGGTGCTGTAAAAGGCTGAG | ||||
| dnaA | dnaA_F | CCAGAAGTCTTAGGTGGTTTTGA | 612 | 495 | 435 |
| dnaA_R | ATGATCCTTGCCTCCAAAAA | ||||
| rpoB | rpoB_F | CAACGTCAAGCTGTTCCATT | 71189 | 563 | 496 |
| rpoB_R | CCTGCACTAACTTCTGATCCAA | ||||
| gyrB | gyrB_F | CCGATTCTGATGGTTCACAT | 316687 | 399 | 291 |
| gyrB_R | TTTTTGCATATTTTGCATTTTCTT | ||||
| gltX | gltX_F | GCTGAAAGACTCTCAAAATCACC | 415737 | 493 | 370 |
| gltX_R | CAAGCCTTTTTGAAATTAGTTCTTT | ||||
| adk | adk_F | GCGATGGCATCTAATTCTTTT | 488754 | 484 | 277 |
| adk_R | ACTCAGGCAAAGTTTTTAGAACA | ||||
| gmk | gmk_F | TTGCGCCTGTTTCTGTTAAT | 522045 | 499 | 435 |
| gmk_R | AAGAGACAAAAGACCTAATGAAGTAGA |
FAM, 6-carboxyfluorescein, fluorescence reporter dye; BHQ, Black Hole Quencher.
Size of sequenced fragment.
The qPCR mixture contained iQsupermix (20 mmol/liter Tris-HCl, 50 mmol/liter KCl, 3 mmol/liter MgCl2 [pH 8.4], 800 μmol/liter of each deoxyribonucleoside triphosphate, 0.625 units Taq polymerase, and stabilizers) (Bio-Rad, Marnes-La-Coquette, France), 500 nmol/liter of each primer, 300 nmol/liter of probe, and 5 μl of the DNA template. The DNA template was replaced by double-distilled water for the negative control. Amplification was performed with the Chromo4 real-time PCR detection system (Bio-Rad). The reaction procedure consisted of denaturation at 95°C for 3 min and 35 cycles of denaturation at 95°C for 15 s and annealing/extension at 65°C for 60 s.
Sequencing of the p37 gene.
The p37 gene of 29 strains (28 field strains and the reference strain ATCC 25021) was sequenced using the dideoxynucleoside termination chain method described by Sanger (33). Two pairs of primers were designed to fully sequence the 1,278 bp and were named p37-A1F/p37-A1R and p37-A2F/p37-A2R (Table 3). The specificity of the primers was tested by homology searches in the nucleotide database (GenBank BLASTN 2.2.14) and in the insilico.ehu.es website (32).
The PCR mixture contained PCR buffer [20 mM Tris-HCl (pH 8.8), 10 mM (NH4)2SO4, 10 mM KCl, 0.01% Triton X-100, 0.1 mg/ml bovine serum albumin, and 2 mM MgSO4], a 200 μM concentration of each deoxyribonucleoside triphosphate (Eurobio, Les Ulis, France), a 400 nM concentration of each primer, 1.5 units of Taq DNA polymerase (Fidelis; Eurobio) and 5 μl of the DNA template (in a total volume of 50 μl). The DNA template was replaced by double-distilled water for the negative control of the PCR step. Amplification was performed in a G-Storm system (GRI, Massy, France). The reaction procedure consisted of 40 cycles of amplification at 95°C for 30 s, 58°C for 30 s, and 72°C for 1 min. The PCR products were detected in a 2% agarose gel by UV transillumination with GelRed staining (Interchim, Montluçon, France), purified by using QIAquick MinElute gel extraction (Qiagen, Courtaboeuf, France), quantified spectrophotometrically, and sequenced by using the combination of a BigDye Terminator v3.1 cycle sequencing kit (Applied Biosystems, CA, USA) and the Applied Biosystems 3130 genetic analyzer according to the manufacturer's instructions (Applied Biosystems, Courtaboeuf, France). Sequences were checked for accuracy by sequencing twice with the forward and the reverse primers. Sequences were visualized with Chromas software and aligned with Multalin DNA software (http://npsa-pbil.ibcp.fr). For each distinct sequence, an arbitrary number was assigned. A phylogenetic tree was drawn on Phylogeny.fr using the MUSCLE, PhyML, and TreeDyn bioinformatics programs (http://www.phylogeny.fr/version2_cgi/simple_phylogeny.cgi) (34). The “one-click” mode was used.
MLST.
Six housekeeping genes were chosen for MLST based on the sequenced genomes of M. hyorhinis strains HUB-1, GDL-1, MCLD, and SK76 (31, 35–37). Internal fragments of the chromosomal replication initiation protein (dnaA), RNA polymerase β subunit (rpoB), DNA gyrase subunit B (gyrB), glutamyl-tRNA synthetase (gltX), adenylate kinase (adk), and guanylate kinase (gmk) genes were selected. The primer sequences are listed in Table 3. The specificity of the primers was tested by homology searches in the nucleotide database (GenBank BLASTN 2.2.14) and in the insilico.ehu.es website (32). PCR conditions were the same as for the amplification of the p37 gene. Amplification was performed in a Chromo4 system (Bio-Rad). The reaction procedure consisted of an initial denaturation step at 95°C for 3 min, followed by 35 cycles of amplification at 95°C for 10 s, at 52°C (for the rpoB locus), 55°C (for the gyrB, gltX, adk, and gmk loci), or 58°C (for the dnaA locus) for 20 s, and at 72°C for 30 s. The PCR products were sequenced as previously described for the sequencing of the p37 gene. For each locus, distinct allele sequences were assigned arbitrary allele numbers with no weighting given to the degree of sequence divergence among alleles. For each strain (including the 29 strains of this study and the four sequenced strains HUB-1, GDL-1, MCLD, and SK76), the alleles at each of the six loci (e.g., 1,3,1,1,2,3) defined the allelic profile, or ST (e.g., ST15). The STs were assigned arbitrary numbers in order of description. For diversity analysis, the sequences concatenated head to tail (2,304 bp) of each ST were aligned, and a phylogenetic tree was drawn on Phylogeny.fr with the one-click mode (34). The number of nucleotide polymorphic sites was determined by using specially designed software, BIGSdb, written by Keith Jolley (http://pubmlst.org/software/database/bigsdb/) (38). BURST analysis was carried out to reveal the relationship of MLST sequence types and to analyze clonal complexes (http://pubmlst.org/analysis/). The degree of clonality within the data set was estimated by calculating the index of association (IA). The standardized IA was used to test the null hypothesis of linkage equilibrium for multilocus data and thus determine the relative contribution of mutation and recombination to the diversity seen by MLST. IA is 0 for linkage equilibrium, and a deviation from this indicates a degree of linkage disequilibrium. Analysis was performed with the LIAN program (http://guanine.evolbio.mpg.de/cgi-bin/lian/lian.cgi.pl) by using 10,000 resamplings of the data. The calculation of H (representing genetic diversity) was also performed with LIAN. START2 software was used to calculate the ratio of nonsynonymous to synonymous substitutions (dN/dS) (http://pubmlst.org/software/analysis/start2/).
Statistical analysis and discrimination index.
The relationships between patterns of strains isolated from pathological cases or from clinically healthy pigs were analyzed by using the Fisher exact test (n ≤ 5) or the chi-square test (n > 5) on independence in 2-by-2 tables. Differences were estimated as significant when probabilities (P) were <0.05. The numerical index of discrimination (D) described by Hunter and Gaston (39) was used to evaluate the two typing methods: the sequencing of the p37 gene and the MLST scheme.
RESULTS
Specificity and sensitivity of the qPCR assay.
The specificity of the qPCR assay was assessed by using the different microorganisms listed in Table 1 as DNA templates. The fluorescence emission of the fluorochrome FAM was measured from all M. hyorhinis strains, including reference strains as well as field strains. None of the other bacterial species described in Table 1 showed an amplification product. Similar results were obtained by conventional PCR (16). The threshold line was set at a fluorescence value of 0.065. Under the described trial conditions (5 μl of DNA extract per assay), the detection limit of the PCR test was 537 fg per assay or 107 fg per μl (mean cycle threshold [CT] = 32.8 ± 0.6). Assuming the genome size of M. hyorhinis to be approximately 840 kb (31, 37) and 1 fg DNA to be equivalent to 978 kb (40), 0.86 fg would correspond to approximately one microorganism. Therefore, the estimated detection threshold of our qPCR was 624 microorganism equivalents per assay, or 125 microorganism equivalents per microliter. The slope derived from all positive replicates per log concentration indicates a mean amplification efficiency of 92% (±3% [standard deviation]). The mean correlation coefficient (R2) value was 0.995 (±0.003 [standard deviation]).
Relatedness of M. hyorhinis strains revealed by sequencing of the p37 gene.
The GenBank accession numbers of the locus sequences obtained in this study are listed in Table 2. Eighteen sequences of the p37 gene were identified among the 33 M. hyorhinis strains (field and reference strains), each being present in 1 to 7 strains (Table 2). The index of discrimination (D) for the total population was 0.934. The genetic relationships between the 18 sequences are shown in the dendrogram in Fig. 1. Two groups, A and B, were identified. Group A was divided into four subgroups (A1 to A4); group B was divided into five subgroups (B1 to B5).
FIG 1.
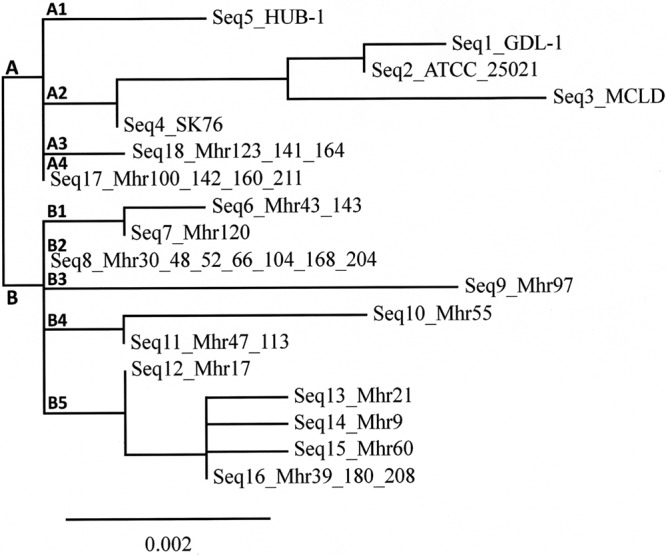
Genetic relationships between 33 M. hyorhinis strains, as estimated by clustering analysis of 18 sequences (Seq) of the p37 gene. The phylogram was constructed using tools at www.phylogeny.fr and the one-click mode.
Relatedness of M. hyorhinis strains revealed by the MLST scheme.
Genetic variation was observed in the six genes analyzed (dnaA, rpoB, gyrB, gltX, adk, and gmk) (Table 4). An online database for the M. hyorhinis MLST scheme, comprising DNA sequences of each allele, was developed by Keith Jolley and sited at the University of Oxford (38) and is available at http://pubmlst.org/mhyorhinis/. The discriminatory ability of the different loci, measured as number of alleles, varied from 4 (adk) to 10 (dnaA) (Table 4). The genetic diversity (H) obtained from the six loci varied from 0.5000 (adk) to 0.7980 (rpoB). Most polymorphisms resulted in synonymous substitutions, with the ratios of nonsynonymous to synonymous substitutions (dN/dS) varying from 0.0614 (for gyrB) to 0.8308 (for gmk) (Table 4). These low ratios indicate a lack or a very limited contribution of environmental selection to the sequence variation in the six housekeeping genes analyzed, which are thus assumed to be suitable for a population genetics study.
TABLE 4.
Characteristics of the six loci used in the M. hyorhinis typing scheme, including genetic variation
| Locus | Putative gene product | Size of sequenced fragment (bp) | No. of alleles identified | No. of polymorphic nucleotide sites (%) | Genetic diversity | dN/dSa |
|---|---|---|---|---|---|---|
| dnaA | Chromosomal replication initiation protein | 435 | 10 | 9 (2.1) | 0.7512 | 0.0844 |
| rpoB | RNA polymerase β-subunit | 496 | 7 | 6 (1.2) | 0.7980 | 0.1422 |
| gyrB | DNA gyrase subunit B | 291 | 6 | 4 (1.4) | 0.7857 | 0.0614 |
| gltX | Glutamyl-tRNA synthetase | 370 | 7 | 7 (1.9) | 0.5172 | 0.2352 |
| adk | Adenylate kinase | 277 | 4 | 3 (1.1) | 0.5000 | 0.1309 |
| gmk | Guanylate kinase | 435 | 5 | 4 (0.9) | 0.5542 | 0.8308 |
Ratio of nonsynonymous to synonymous mutations.
Twenty-nine sequence types were identified among the 33 M. hyorhinis strains (field and reference strains) (Table 2) each consisting of one to three strains (only for ST1 and ST16). The index of discrimination (D) for the total population was 0.989. The genetic relationships between the 29 STs are shown in the dendrogram in Fig. 2. Two groups, named a and b, were identified. Group a was divided into three subgroups (a1 to a3); group b was divided into four subgroups (b1 to b4).
FIG 2.
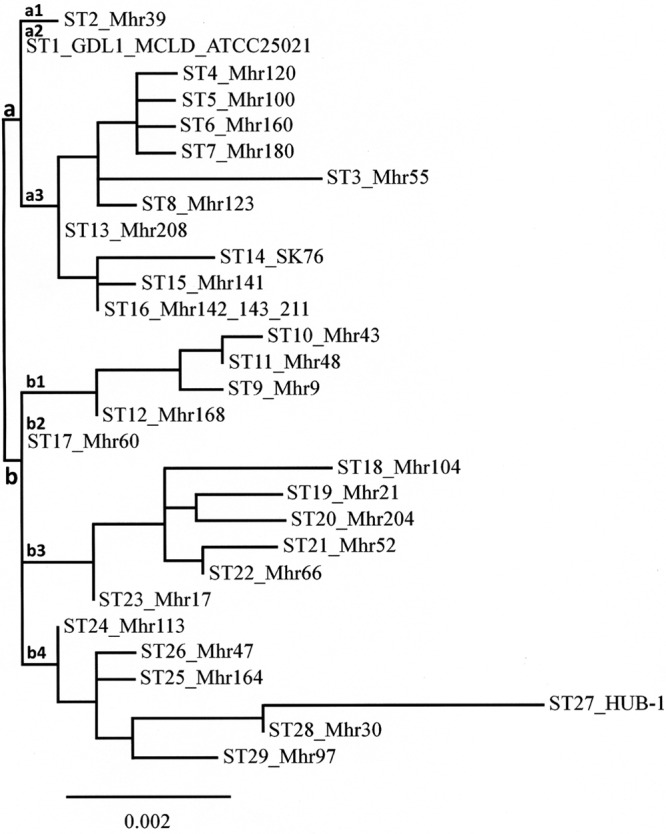
Genetic relationships between 33 M. hyorhinis strains, as estimated by clustering analysis of 29 sequence types (ST) revealed by MLST. The phylogram was constructed using tools at www.phylogeny.fr and the one-click mode.
In order to estimate the relative contributions of recombination and mutation to the genomic evolution of M. hyorhinis, the standardized index of association (IA) was calculated at 0.0191 (at the ST level: one isolate from each ST to avoid bias due to a possible epidemic population structure). This value shows that the genetic variation is roughly in linkage equilibrium and seems due to recombination and not to mutation (41).
The BURST analysis found one clonal complex (Fig. 3). The ancestral ST is the genotype defining the highest number of single-locus variants (SLVs). The ancestral group comprised ST1, which contained three strains (ATCC 25021 [type strain isolated in 1955 from a pig with pneumonia], MCLD [isolated from a primary human melanoma cell line], and GDL-1 [isolated from tissue culture]). SLVs of the ancestral group were ST2 from pneumonia in 2007, ST13 from polyserositis in 1998, and ST17 from pneumonia in 2004. Double-locus variants (DLVs) of the ancestral group were ST7 from pneumonia in 2007, ST16 from polyserositis in 1999 or nasal cavities in 1981, and ST24 from tracheas in 2007. ST3, ST14, ST19, ST20, ST27, ST28, and ST29 were not linked to the clonal complex and were singletons.
FIG 3.
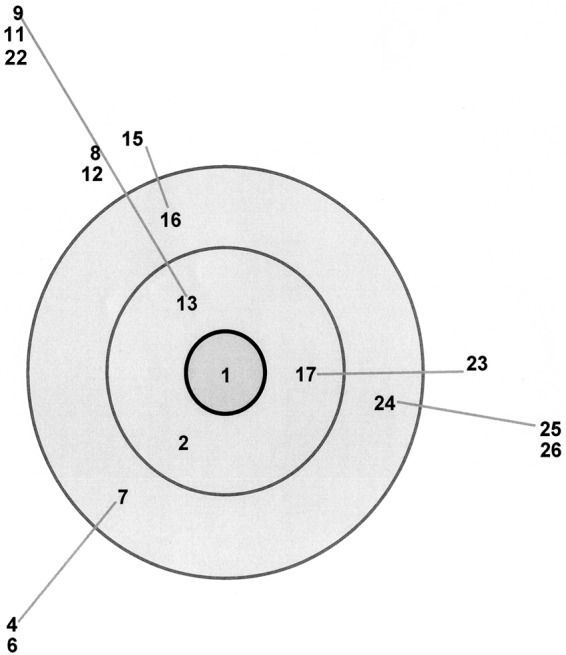
BURST analysis of M. hyorhinis STs. Clonal complex 1 includes ST1 (ancestral genotype), ST2, ST13, and ST17 (descended from single-locus variant genotypes) and ST7, ST24, and ST16 (descended from double-locus variant genotypes). The assigned ancestral genotype is within the central ring (ST1). ST11 and ST22 overlap.
Relationships among p37 gene sequences or STs and the isolation site of M. hyorhinis.
The eight strains obtained from pigs with polyserositis showed eight different p37 gene sequences and seven different STs (Table 2). Most of them were in p37 group B5 (4 out of 8: M. hyorhinis strains 9, 17, 21, and 208) and in ST group a3 (6 out of 8: M. hyorhinis strains 9, 141, 142, 143, 208, and SK76). Relationships between the polyserositis origin and group B5 or a3 were significant (P = 0.042 and 0.035, respectively).
Among the 10 strains isolated from cases of pneumonia, 8 different p37 gene sequences and 10 different STs were identified. In the two dendrograms (Fig. 1 and 2), these strains were not clustered preferentially into a group (P > 0.05). Similar results were obtained for the strains isolated from pig nasal cavities or tracheas (Table 2).
DISCUSSION
M. hyorhinis can be isolated from macroscopic lesions (arthritis, pneumonia, etc.) or from samples collected from live animals (nasal cavities, tonsils, tracheal and bronchiolar mucus, oral fluids), but isolation and identification are tedious and time-consuming. The difficulty in culturing M. hyorhinis has led to the development of other diagnostic assays: three PCR tests were developed to detect this Mycoplasma species in pure cultures (15–17). In our study, a new PCR test—a real-time TaqMan PCR (qPCR)—was developed to rapidly detect small quantities of M. hyorhinis. This test was able to detect all 29 of the M. hyorhinis strains analyzed and did not cross-react with other mycoplasma species or with other microorganisms commonly found in pigs. The estimated detection limits of the qPCR assay were 537 fg per assay (or 107 fg/μl), or 624 microorganism equivalents per assay (or 125 microorganism equivalents/μl). This qPCR assay is more sensitive than the previously developed gel-based PCR tests or qPCR assay (detection thresholds of 1,000 to 10,000 microorganisms per reaction) (15–17, 42). In the future, our qPCR could be developed to quantify M. hyorhinis directly in the specimens.
Two molecular typing methods, PFGE and REA, had already been developed to study intraspecific variations in M. hyorhinis (19, 20). However, they were not applied to many strains for epidemiological studies. Moreover, observed differences in PFGE or REA patterns may be difficult to interpret. In our study, an unambiguous method was performed to characterize M. hyorhinis isolates: the sequencing of the p37 gene, encoding the M. hyorhinis p37 protein, which has been detected in cancer patients by several groups working independently (9, 11). A great degree of variability was observed in M. hyorhinis, because among the 33 epidemiologically unrelated strains analyzed, 18 distinct nucleotide sequences were revealed (55%). However, the discriminatory power was relatively poor (D = 0.934, where D is the probability that the typing system will assign a different type to two unrelated strains randomly sampled in the microbial population of a given taxon). A typing system should achieve a D value of >0.95 for reliable assessment of the clonal relatedness of isolates and for epidemiological investigations (43). Furthermore, single-gene phylogenies may be biased, particularly in populations which exhibit reduced variability or which are not strictly clonal. Therefore, including several phylogenetic markers may provide a closer estimation of species phylogeny. This is why multilocus sequencing could be the new standard in microbial molecular systematics (24). In the present study, a new MLST scheme was developed for the species M. hyorhinis. This MLST scheme of six loci (2,304 nt) offers a highly discriminatory typing method (D = 0.989) and was capable of subdividing 33 strains into 29 distinct sequence types, confirming the great intraspecies variability of M. hyorhinis. Moreover, the MLST scheme is robust, since several strains isolated within 49- and 18-year intervals harbored the same genotype (for ST1, strain ATCC 25021, isolated in 1955, and strain MCLD, isolated in 2004; for ST16, M. hyorhinis strain 211, isolated in 1981, and M. hyorhinis strains 142 and 143, isolated in 1999). The MLST primers were designed to amplify only M. hyorhinis sequences, and their specificity was verified on the insilico.ehu.es website. Therefore, the MLST method could be used for typing M. hyorhinis directly in polycontaminated biological samples or mixed mycoplasma cultures, provided that they contain only one M. hyorhinis strain. The suppression of the isolation stage, considered fastidious due to the long incubation period needed for culturing Mycoplasma (27), would reduce the time required for the typing. Before this possibility can be considered, specificity should be tested on a collection of bacterial strains collected in the same ecological niche as M. hyorhinis and on a collection of bacterial strains phylogenetically close to M. hyorhinis.
MLST has the added advantages of providing meaningful information about population structure. The MLST work here demonstrates that sequence variation also occurs within housekeeping genes, indicating than the core genome is also variable. However, the large majority of variation was synonymous, meaning that there was a smaller amount of variation in the expressed proteins. The large number of synonymous substitutions detected suggested that most nonsilent mutations are eliminated through purifying selection. Analysis was also carried out to determine what the relative contributions of mutation and homologous recombination were in the genetic diversity seen among M. hyorhinis strains. It was found that diversity was probably due to recombination, with mutation playing a much smaller role. Homologous recombination was also found to be high for bacteria with walls, such as Neisseria meningitidis, and for wall-less bacteria, such as Mycoplasma hyopneumoniae and M. hominis (21, 26, 44). Due to the high frequency of recombination observed in M. hyorhinis, it was surprising to observe identical or nearly identical MLST genotypes (which differ by only one allele). The clonality of our M. hyorhinis population was tested by BURST analysis (45). A unique clonal complex was identified (26 strains corresponding to the ancestral group comprising ST1, which contained ATCC 25021, MCLD, and GDL-1), and only seven STs were not linked to the clonal complex and were singletons (ST3, ST14, ST19, ST20, ST27, ST28, and ST29). Clonal groupings were also identified in Helicobacter pylori and Staphylococcus aureus, despite a rich history of interstrain recombination (46, 47). Søgaard et al. (44) showed that the frequency of recombination in M. hominis is not correlated with the level of variability and that recombination does not induce more variation in the genes but rather shuffles the existing mutations, thereby creating new alleles. Recombination in M. hyorhinis is particularly interesting, since no phages or plasmids have been detected in this mycoplasma, and no mechanisms that mediate DNA uptake or recombination have been described. However, Schubert et al. (48) showed that homologous DNA recombination plays a major role in horizontal transfer of genes within the species E. coli, and Sirand-Pugnet et al. (49) demonstrated that a significant number of genes underwent horizontal transfer among different mycoplasma species that share the same ruminant hosts.
In the present work, the strains isolated from cases of pneumonia and from pig nasal cavities and tracheas were not clustered preferentially into phylogenetic groups (P > 0.05). However, significant associations were demonstrated between strains isolated from polyserositis and p37 group B5 and MLST group a3, suggesting low variability between the selected strains and a certain genetic homogeneity. This suggests that an isolate clustering in these groups might pose a high risk of infection for pigs. A complementary study should be performed with a large number of strains isolated from polyserositis to verify this hypothesis. The impact of the genomic particularities revealed by MLST on virulence of strains is worthy of future study.
Because groups associated with polyserositis were observed with both typing methods, similar results were obtained, indicating the robustness of both approaches. This fact is surprising because the genes studied are different: the housekeeping genes are supposed to be under high and constant selective pressure, with mutations accumulating at a defined rate over time, whereas the p37 gene encodes part of a homologous, high-affinity transport system (10) that may be exposed to low and variable selective pressure over time. The p37 gene-based typing method is certainly faster, since only a single locus is sequenced. Data generated by these two typing methods are directly comparable between laboratories and easily shared over the Internet (pubmlst.org, GenBank). In comparison, the MLST scheme has a slightly higher resolution, making it possible to conduct epidemiological investigations and acquire new knowledge with regard to M. hyorhinis infections.
ACKNOWLEDGMENTS
We thank Pierre-Baptiste David, Claire de Boisseson and Aurélie Leroux for their technical assistance.
The development of the M. hyorhinis MLST website was funded by the Wellcome Trust.
Footnotes
Published ahead of print 12 March 2014
REFERENCES
- 1.Razin S, Yogev D, Naot Y. 1998. Molecular biology and pathogenicity of mycoplasmas. Microbiol. Mol. Biol. Rev. 62:1094–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kobisch M, Friis N. 1996. Swine mycoplasmoses. Rev. Sci. Tech. 15:1569–1605 [DOI] [PubMed] [Google Scholar]
- 3.Thacker EL. 2006. Mycoplasmal diseases, p 701–717 In Straw BE, Zimmerman J, D'Allaire S, Taylor DJ. (ed), Diseases of swine, 9th ed. Iowa State University Press, Ames, IA [Google Scholar]
- 4.Sorensen V, Jorsal SE, Mousing J. 2006. Diseases of the respiratory system, p 149–177 In Straw BE, Zimmerman J, D'Allaire S, Taylor DJ. (ed), Diseases of swine, 9th ed. Iowa State University Press, Ames, IA [Google Scholar]
- 5.Leneveu P, Robert N, Keïta A, Pagot E, Pommier P, Tessier P. 2005. Lung lesions in pigs at slaughter: a 2-year epidemiological study in France. Int. J. Appl. Res. Vet. Med. 3:259–265 http://www.jarvm.com/articles/Vol3Iss3/ISPAIA.pdf [Google Scholar]
- 6.Kobayashi H, Morozumi T, Miyamoto C, Shimizu M, Yamada S, Ohashi S, Kubo M, Kimura K, Mitani K, Ito N, Yamamoto K. 1996. In vitro susceptibility of Mycoplasma hyorhinis and variable mutation of domain II and V in 23S ribosomal RNA. J. Vet. Med. Sci. 58:1107–1111. 10.1292/jvms.58.11_1107 [DOI] [PubMed] [Google Scholar]
- 7.Huang J, Mackillop WJ. 2001. Increased risk of soft tissue sarcoma after radiotherapy in women with breast carcinoma. Cancer 92:172–180. [DOI] [PubMed] [Google Scholar]
- 8.Yang H, Qu L, Ma H, Chen L, Liu W, Liu C, Meng L, Wu J, Shou C. 2010. Mycoplasma hyorhinis infection in gastric carcinoma and its effects on the malignant phenotypes of gastric cancer cells. BMC Gastroenterol. 10:132. 10.1186/1471-230X-10-132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Urbanek C, Goodison S, Chang M, Porvasnik S, Sakamoto N, Li CZ, Boehlein SK, Rosser CJ. 2011. Detection of antibodies directed at M. hyorhinis p37 in the serum of men with newly diagnosed prostate cancer. BMC Cancer 11:233. 10.1186/1471-2407-11-233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dudler R, Schmidhauser C, Parish RW, Wettenhall RE, Schmidt T. 1988. A mycoplasma high-affinity transport system and the in vitro invasiveness of mouse sarcoma cells. EMBO J. 7:3963–3970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ilantzis C, Thomson DM, Michaelidou A, Benchimol S, Stanners CP. 1993. Identification of a human cancer related organ-specific neoantigen. Microbiol. Immunol. 37:119–128. 10.1111/j.1348-0421.1993.tb03188.x [DOI] [PubMed] [Google Scholar]
- 12.Ketcham CM, Anai S, Reutzel R, Sheng S, Schuster SM, Brenes RB, Agbandje-McKenna M, McKenna R, Rosser CJ, Boehlein SK. 2005. p37 induces tumor invasiveness. Mol. Cancer Ther. 4:1031–1038. 10.1158/1535-7163.MCT-05-0040 [DOI] [PubMed] [Google Scholar]
- 13.Goodison S, Nakamura K, Iczkowski KA, Anai S, Boehlein SK, Rosser CJ. 2007. Exogenous mycoplasmal p37 protein alters gene expression, growth and morphology of prostate cancer cells. Cytogenet. Genome Res. 118:204–213. 10.1159/000108302 [DOI] [PubMed] [Google Scholar]
- 14.Clavijo MJ, Rovira A. 20 June 2013. Mycoplasma hyorhinis—not just a commensal. http://www.pig333.com/what_the_experts_say/mycoplasma-hyorhinis-not-just-a-commensal_7288/
- 15.Stemke GW, Phan R, Young TF, Ross RF. 1994. Differentiation of Mycoplasma hyopneumoniae, M. flocculare, and M. hyorhinis on the basis of amplification of a 16S rRNA gene sequence. Am. J. Vet. Res. 55:81–84 [PubMed] [Google Scholar]
- 16.Caron J, Ouardani M, Dea S. 2000. Diagnosis and differentiation of Mycoplasma hyopneumoniae and Mycoplasma hyorhinis infections in pigs by PCR amplification of the p36 and p46 genes. J. Clin. Microbiol. 38:1390–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stakenborg T, Vicca J, Butaye P, Imberechts H, Peeters J, Kruif A, Haesebrouck F, Maes D. 2006. A multiplex PCR to identify porcine mycoplasmas present in broth cultures. Vet. Res. Commun. 30:239–247. 10.1007/s11259-006-3226-3 [DOI] [PubMed] [Google Scholar]
- 18.Klein D. 2002. Quantification using real-time PCR technology: applications and limitations. Trends Mol. Med. 8:257–260. 10.1016/S1471-4914(02)02355-9 [DOI] [PubMed] [Google Scholar]
- 19.Darai G, Zöller L, Matz B, Delius H, Speck PT, Flügel RM. 1982. Analysis of Mycoplasma hyorhinis genome by use of restriction endonucleases and by electron microscopy. J. Bacteriol. 150:788–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barlev NA, Borchsenius SN. 1991. Continuous distribution of Mycoplasma genome sizes. Biomed. Sci. 2:641–645 [PubMed] [Google Scholar]
- 21.Mayor D, Jores J, Korczak BM, Kuhnert P. 2008. Multilocus sequence typing (MLST) of Mycoplasma hyopneumoniae: a diverse pathogen with limited clonality. Vet. Microbiol. 127:63–72. 10.1016/j.vetmic.2007.08.010 [DOI] [PubMed] [Google Scholar]
- 22.McAuliffe L, Gosney F, Hlusek M, de Garnica ML, Spergser J, Kargl M, Rosengarten R, Ayling RD, Nicholas RA, Ellis RJ. 2011. Multilocus sequence typing of Mycoplasma agalactiae. J. Med. Microbiol. 60:803–811. 10.1099/jmm.0.028159-0 [DOI] [PubMed] [Google Scholar]
- 23.Manso-Silván L, Dupuy V, Lysnyansky I, Ozdemir U, Thiaucourt F. 2012. Phylogeny and molecular typing of Mycoplasma agalactiae and Mycoplasma bovis by multilocus sequencing. Vet. Microbiol. 161:104–112. 10.1016/j.vetmic.2012.07.015 [DOI] [PubMed] [Google Scholar]
- 24.Gevers D, Cohan FM, Lawrence JG, Spratt BG, Coenye T, Feil EJ, Stackebrandt E, Van de Peer Y, Vandamme P, Thompson FL, Swings J. 2005. Re-evaluating prokaryotic species. Nat. Rev. Microbiol. 3:733–739. 10.1038/nrmicro1236 [DOI] [PubMed] [Google Scholar]
- 25.Selander RK, Caugant DA, Ochman H, Musser JM, Gilmour MN, Whittam TS. 1986. Methods of multilocus enzyme electrophoresis for bacterial population genetics and systematics. Appl. Environ. Microbiol. 51:873–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feil EJ, Maiden MC, Achtman M, Spratt BG. 1999. The relative contributions of recombination and mutation to the divergence of clones of Neisseria meningitidis. Mol. Biol. Evol. 16:1496–1502. 10.1093/oxfordjournals.molbev.a026061 [DOI] [PubMed] [Google Scholar]
- 27.Friis NF. 1975. Some recommendations concerning primary isolation of Mycoplasma suipneumoniae and Mycoplasma flocculare. A survey. Nord. Vet. Med. 27:337–339 [PubMed] [Google Scholar]
- 28.Friis NF, Ahrens P, Hagedorn-Olsen T, Nielsen EO, Kokotovic B. 2003. Mycoplasma hyopharyngis isolation from swine. Acta Vet. Scand. 44:103–104. 10.1186/1751-0147-44-103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Le Minor L, Véron M. 1990. Bactériologie médicale. Flammarion, Paris, France [Google Scholar]
- 30.Kellog DE, Kwok S. 1990. Detection of human immunodeficiency virus, p 339–343 In Innis MA, Gelfand DH, Sninsky JJ, White TJ. (ed), PCR protocols: a guide to methods and applications. Academic Press, San Diego, CA [Google Scholar]
- 31.Liu W, Fang L, Li S, Li Q, Zhou Z, Feng Z, Luo R, Shao G, Wang L, Chen H, Xiao S. 2010. Complete genome sequence of Mycoplasma hyorhinis strain HUB-1. J. Bacteriol. 192:5844–5845. 10.1128/JB.00946-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bikandi J, San Millán R, Rementeria A, Garaizar J. 2004. In silico analysis of complete bacterial genomes: PCR, AFLP-PCR, and endonuclease restriction. Bioinformatics 20:798–799. 10.1093/bioinformatics/btg491 [DOI] [PubMed] [Google Scholar]
- 33.Sanger F. 1981. Determination of nucleotide sequences in DNA. Science 214:1205–1210. 10.1126/science.7302589 [DOI] [PubMed] [Google Scholar]
- 34.Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard J-F, Guindon S, Lefort V, Lescot M, Claverie J-M, Gascuel O. 2008. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 36:W465–W469. 10.1093/nar/gkn180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kornspan JD, Lysnyansky I, Kahan T, Herrmann R, Rottem S, Nir-Paz R. 2011. Genome analysis of a Mycoplasma hyorhinis strain derived from a primary human melanoma cell line. J. Bacteriol. 193:4543–4544. 10.1128/JB.05505-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Calcutt MJ, Foecking MF, Rosales RS, Ellis RJ, Nicholas RA. 2012. Genome sequence of Mycoplasma hyorhinis strain GDL-1. J. Bacteriol. 194:1848. 10.1128/JB.00033-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goodison S, Urquidi V, Kumar D, Reyes L, Rosser CJ. 2013. Complete genome sequence of Mycoplasma hyorhinis strain SK76. Genome Announc. 1:e00101–12. 10.1128/genomeA.00101-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jolley KA, Maiden MC. 2010. BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11:595. 10.1186/1471-2105-11-595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hunter PR, Gaston MA. 1988. Numerical index of the discriminatory ability of typing systems: an application of Simpson's index of diversity. J. Clin. Microbiol. 26:2465–2466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dolezel J, Bartos J, Voglmayr H, Greilhuber J. 2003. Nuclear DNA content and genome size of trout and human. Cytometry A 51:127–128. 10.1002/cyto.a.10013 [DOI] [PubMed] [Google Scholar]
- 41.Haubold B, Travisano M, Rainey PB, Hudson RR. 1998. Detecting linkage disequilibrium in bacterial populations. Genetics 150:1341–1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Clavijo MJ, Oliveira S, Rovira A. 2012. Development of a quantitative PCR assay for the detection of Mycoplasma hyorhinis, poster BP-360, p 717 Proc. 22nd Int. Pig Vet. Society Congr. [Google Scholar]
- 43.Struelens MJ, the Members of the European Study Group on Epidemiological Markers (ESGEM), of the European Society for Clinical Microbiology and Infectious Diseases (ESCMID) 1996. Consensus guidelines for appropriate use and evaluation of microbial epidemiologic typing systems. Clin. Microbiol. Infect. 2:2–11. 10.1111/j.1469-0691.1996.tb00193.x [DOI] [PubMed] [Google Scholar]
- 44.Søgaard IZ, Boesen T, Mygind T, Melkova R, Birkelund S, Christiansen G, Schierup MH. 2002. Recombination in Mycoplasma hominis. Infect. Genet. Evol. 1:277–285. 10.1016/S1567-1348(02)00036-9 [DOI] [PubMed] [Google Scholar]
- 45.Feil EJ, Spratt BG. 2001. Recombination and the population structures of bacterial pathogens. Annu. Rev. Microbiol. 55:561–590. 10.1146/annurev.micro.55.1.561 [DOI] [PubMed] [Google Scholar]
- 46.Achtman M, Azuma T, Berg DE, Ito Y, Morelli G, Pan ZJ, Suerbaum S, Thompson SA, van der Ende A, van Doorn LJ. 1999. Recombination and clonal groupings within Helicobacter pylori from different geographical regions. Mol. Microbiol. 32:459–470. 10.1046/j.1365-2958.1999.01382.x [DOI] [PubMed] [Google Scholar]
- 47.Day NP, Moore CE, Enright MC, Berendt AR, Smith JM, Murphy MF, Peacock SJ, Spratt BG, Feil EJ. 2001. A link between virulence and ecological abundance in natural populations of Staphylococcus aureus. Science 292:114–116. 10.1126/science.1056495 [DOI] [PubMed] [Google Scholar]
- 48.Schubert S, Darlu P, Clermont O, Wieser A, Magistro G, Hoffmann C, Weinert K, Tenaillon O, Matic I, Denamur E. 2009. Role of intraspecies recombination in the spread of pathogenicity islands within the Escherichia coli species. PLoS Pathog. 5:e1000257. 10.1371/journal.ppat.1000257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sirand-Pugnet P, Lartigue C, Marenda M, Jacob D, Barré A, Barbe V, Schenowitz C, Mangenot S, Couloux A, Segurens B, de Daruvar A, Blanchard A, Citti C. 2007. Being pathogenic, plastic, and sexual while living with a nearly minimal bacterial genome. PLoS Genet. 3:e75. 10.1371/journal.pgen.0030075 [DOI] [PMC free article] [PubMed] [Google Scholar]